

Abstract
Synthetic lethal interaction between oncogenic Ha-ras and loss of PKC has been demonstrated. Recently, the authors reported that the concurrent knockdown of PKC α and β, via upregulating PKC δ, sensitizes cells with aberrant Ras signaling to apoptosis. As a continuation of the study, using shRNA, the authors demonstrate that loss of PKC δ causes a lethal reaction in NIH3T3/Hras or prostate cancer DU145 cells that overexpress JNK. In this apoptotic process, PKC α and β are upregulated and then associated with RACK1 (an adaptor for activated PKC) and JNK. Immunoblotting analysis shows that JNK is phosphorylated, accompanied with caspase 8 cleavage. The inhibition of JNK abrogates this apoptotic process triggered by PKC δ knockdown. Interestingly, without blocking PKC δ, the concurrent overexpression of wt- or CAT-PKC α and β is insufficient to induce apoptosis in the cells. Together with the authors’ previous findings, the data suggest that PKC α/β and δ function oppositely to maintain a balance that supports cells expressing v-ras to survive and prevents them from being eliminated through oncogenic stress-induced apoptosis.
Keywords: PKC, JNK, RACK1, Ras, synthetic lethality
Introduction
PKC consists of more than 10 isoforms that differ in their structures, cellular functions, and tissue distributions.1-4 PKC α, βI, βII, and γ belong to the conventional PKC isoforms, the activities of which depend on calcium and diacylglycerol (DAG). The unconventional PKC subgroup (PKC δ, ε, η, and θ) does not rely on calcium for the activation. The atypical group of PKC isozymes (PKC ζ and λ/υ) requires neither DAG nor calcium for the function. The role of PKC in the regulation of apoptosis remains unclear and controversial. PKC α and β are known for promoting cell proliferation and survival.5-7 In human tonsil epithelial cells, PKC α and β were upregulated during the apoptotic process.8 PKC α was demonstrated to translocate to the mitochondria and induce cell death in human prostate cancer cells.9 PKC δ often participates in the regulation of apoptosis in various types of cells and animal models.3,10-13 We recently demonstrated that the concurrent inhibition of PKC α and β, via upregulating PKC δ, rendered cells susceptible to apoptosis.14 However, the suppression of PKC δ by rottlerin was also shown to sensitize various cells expressing a mutated ras to apoptosis.15 Here, using the shRNAs, we thoroughly investigated the role of PKC δ in this synthetic lethality.
The structures of PKC isoforms are very similar and consist of 4 conserved domains (C1-C4) and 5 variable sites (V1-V5).1,7 The C2 domain exists in classic PKC isoforms only. Such a regulatory domain is responsible for their binding to receptors for activated C kinases (RACKs), which define the localization of PKCs upon activation.16,17 In response to mitogenic stimulation, studies showed that PKC binds to RACK1 that, as an adaptor, further recruits and activates JNK. RACK1 is a highly conserved protein and belongs to a family of proteins containing different numbers of structural Trp-Asp repeats. RACK1 was shown to serve both as an anchor protein for PKCs and as a scaffold protein recruiting PKCs and other proteins into a signaling complex.16
JNK is a stress-related kinase and activated by various extracellular stimuli, such as growth factors, ultraviolet (UV) radiation, oncogenic or genotoxic stress, and cytokines.18-20 Activation of JNK results in the phosphorylation of c-Jun at serines 63 and 73.21,22 Using genetic alternative mouse models, it was demonstrated that JNK mutant mice exhibit decreased activation-induced T-cell death.23 The requirement of JNK for the induction of apoptosis was exhibited.20,24 Ras was reported to take part in some cellular stress responses, resulting in the activation of JNK (e.g., the UV response).25 In apoptosis elicited by PKC suppression in cells expressing oncogenic Ha-ras, JNK was a key player.26,27 Although JNK and c-Jun often play important roles in the regulation of cell proliferation,18,28 their participation in apoptosis depends on cell types and contexts imposed on cells.
In the tumorigenic process, cancer cells often require secondary dependencies on other growth-related factors. Alterations of these factors in tumor cells may cause an oncogene-mediated lethal reaction.29-31 Recently, using large-scale siRNA screens, it was identified that K-ras, together with alternations of kinases that are within the same pathway, parallel with, or distal to the Ras signaling pathway but functionally connected, is synthetically lethal.32,33 Using RNAi screening, CARD11 (a NFκB regulator) was identified as being critical for the subset of diffuse large B lymphoma.34 Indeed, cancer cells become heavily dependent on basic cellular activities, such as those regulated by PKC, to avoid oncogenic stress-induced apoptosis.14,32,33 The mechanisms of such lethal interaction remain largely unknown.
In this study, we demonstrated that genetic knockdown of PKC δ in cells expressing v-ras or overexpressing JNK caused the upregulation of PKC α and β. The PKCs subsequently bound to RACK1, which further recruited JNK to the complex, which initiated an apoptotic process. In conjunction with our previous findings of the induction of apoptosis by concurrent knockdown of PKC α and β, the data suggest the existence of a balance oppositely controlled by PKC α/β and δ to facilitate cancer cells to maintain homeostasis for survival, and perturbation of the balance triggers an apoptotic crisis.
Results
Suppression of PKC δ induces apoptosis in NIH3T3/Hras cells
Hyperactive Ras can initiate apoptosis under certain circumstances.25,32,33 Using shRNAs, we recently demonstrated that the concurrent knockdown of PKC α and β caused apoptosis in cells with aberrant Ras signaling.14 To further investigate the mechanisms of this apoptotic process, the shRNAs against different PKC isoforms were used to identify their effects on the induction of apoptosis in NIH3T3/Hras cells. The shRNAs targeting PKC α, β, δ, ε, η, or θ and control scrambled (sc) shRNAs were transiently infected into NIH3T3 cells. Subsequently, the expressions of these isoforms were examined by immunoblotting (Fig. 1A). The shRNAs, but not the sc-shRNAs, blocked the expressions of targeted PKCs. The amount of PKC isoforms in the cells after being infected with the shRNAs was quantified. The expression of PKCs in the ras transfectants after the infection of the shRNAs was also examined, and a similar result was obtained (data not shown). Real-time PCR was also performed to test the effect of the shRNAs on the gene expression of PKCs (Fig. 1B). The results were consistent, in which the shRNAs effectively knocked down PKCs. Each shRNA did not interfere with the expression of other PKC isoform genes.
Figure 1.
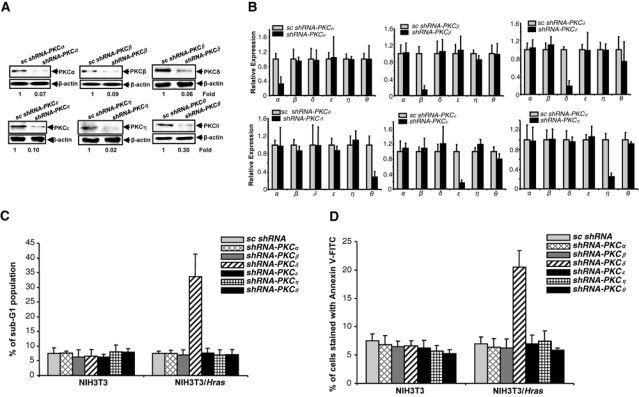
Knockdown of PKC δ induces apoptosis in NIH3T3/Hras cells. (A) After the infection of the shRNAs, the expression of PKC isoforms in NIH3T3 cells was tested by immunoblotting. The equal loading of the samples was normalized by β-actin. The relative expression level of PKC isoforms was analyzed by Image J software. (B) mRNAs were extracted from the cells infected with shRNAs, and real-time PCR was performed to analyze each PKC isoform expression. The error bars represent the standard deviation (SD) from 3 independent experiments (P values < 0.05 were significant). (C) After being infected with the shRNAs, the cells were subjected to DNA fragmentation analysis. The error bars represent the SD from 5 independent experiments (P values < 0.05 were significant). (D) Following treatments, Annexin V-FITC analysis was performed. The error bars represent the SD from 5 independent experiments (P values < 0.05 were significant).
To test which PKC isoforms, after being knocked down, were able to elicit apoptosis, we performed a DNA fragmentation assay (Fig. 1C). Knockdown of PKC α, β, ε, η, or θ alone by the shRNAs had no apoptotic effect on either NIH3T3 or NIH3T3/Hras cells. However, the infection with shRNA-PKC δ induced more than 25% of NIH3T3/Hras cells to undergo apoptosis. A similar result was obtained from the Annexin V-FITC apoptotic assay (Fig. 1D). Thus, it appears that the knockdown of PKC δ sensitizes NIH3T3/Hras cells to apoptosis.
Upregulation of PKC α and β in NIH3T3/Hras cells after knocking down PKC δ
PKC δ has been suggested to be a tumor suppressor via regulating proapoptotic or growth restriction activity in many types of cells.3,4 It was reported that the concurrent knockdown of PKC α and β by the shRNAs upregulated PKC δ in cells with aberrant Ras signaling to induce apoptosis.14 However, it was also reported that the blockade of PKC δ by rottlerin rendered various cells harboring v-ras to cell death.15 The data suggest the existence of the cooperation among PKC α/β and δ. To test this, we first examined whether the expression of the conventional PKC isoforms in NIH3T3 or NIH3T3/Hras cells was affected by knocking down PKC δ. Immunoblotting was conducted in the cells after being infected with shRNA-PKC δ (Fig. 2A). The level of PKC α or β was increased in NIH3T3/Hras cells (about 2- to 3-fold) following knockdown of PKC δ, which was not detected in the parental cells or affected by the introduction of the shRNAs targeting other PKC isoforms. The relative expression of each PKC isoform after PKC δ knockdown in the cells was quantified and indicated. To confirm the results obtained from immunoblotting, we performed semi-quantitative PCR (Fig. 2B) and real-time PCR (Fig. 2C) analyses. In concordance with our immunoblotting results, the knockdown of PKC δ elevated the expressions of PKC α and β but not other PKC isoform genes. The upregulation of PKC α and β occurred only in NIH3T3/Hras cells but not in parental cells.
Figure 2.

Concurrent upregulation of PKC α and β in NIH3T3/Hras cells following knockdown of PKC δ. (A) After the infection with shRNA-PKCδ or sc-shRNA, the expression of PKC isoforms in the cells was analyzed by immunoblotting. The equal loadings of total proteins were normalized by β-actin. The relative expression level of PKC isoforms was compared with Image J software. (B, C) After infection with the shRNAs, mRNAs were subjected to (B) semi-quantitative PCR or (C) real-time PCR. The error bars represent the standard deviation (SD) from 3 independent experiments (P values < 0.05 were significant). (D) Lysates were subjected to immunoprecipitation with either anti-PKC α or β antibody. The immunoprecipitates were then analyzed with a PKC enzymatic kit. The error bars represent SD from 3 independent experiments (P values <0.05 were significant).
Because the expression of PKC α and β in NIH3T3/Hras cells was increased after knocking down PKC δ, the activities of these two PKC isoforms were tested using a PKC enzymatic activity kit. Following knockdown of PKC δ, lysates were immunoprecipitated with an anti-PKC α or β (Fig. 2D) antibody and then subjected to the kinase assay. PKC α or β activity was increased only in NIH3T3/Hras cells lacking PKC δ. Overall, the data implicated that the knockdown of PKC δ upregulated not only the expression of PKC α and β but also the activities of these two isozymes in NIH3T3/Hras cells.
JNK forms a complex with PKC α, β and RACK1 after knocking down PKC δ in NIH3T3/Hras cells
JNK is one of the Ras downstream effectors and often participates in stress-related responses.12,18-20,25 This kinase was shown to participate in apoptosis triggered by PKC suppression in cells with aberrant Ras signaling.26,27 To test whether JNK is activated following PKC δ knockdown in NIH3T3/Hras cells, cell lysates were prepared and immunoblotted with the anti-phosphorylated-JNK antibody (Fig. 3A). A baseline phosphorylated JNK1 was revealed by the antibody in parental cells and untreated NIH3T3/Hras cells. The suppression of PKC δ by the shRNA greatly enhanced the amount of phosphorylated JNK1 in NIH3T3/Hras cells. The phosphorylation of c-Jun (a JNK substrate) was also tested. The antibody against Ser63 or Ser73 of c-Jun was employed. Consistently, the increased amounts of phosphorylated c-Jun at Ser63 and Ser73 were recognized by the antibodies only in NIH3T3/Hras cells following PKC δ knockdown but not in parental cells.
Figure 3.
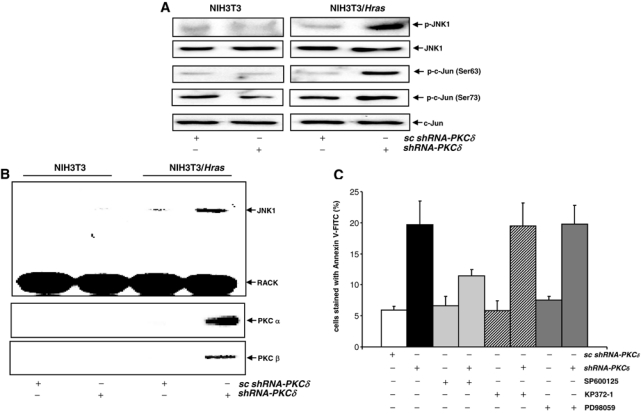
JNK is activated in NIH3T3/Hras cells following PKC δ knockdown. (A) After knocking down PKC δ, cell lysates were immunoblotted with the anti-phosphorylated-JNK antibody or anti-phosphorylated-c-Jun (Ser63 or Ser73) antibodies. Equal loadings of total proteins were normalized by JNK1 or c-Jun. (B) With or without being infected with shRNA-PKC δ or scRNA, cell lysates were subjected to immunoprecipitation with the anti-RACK1 antibody. The precipitates were then immunoblotted with anti-JNK, RACK1, and the PKC α or δ antibody, respectively. (C) Following knockdown of PKC δ, in the presence or absence of SP600125 (a JNK inhibitor, 5 μM), KP372-1 (an Akt inhibitor, 0.1 μM), or PD98059 (a MAPK inhibitor, 5 μM), an Annexin V apoptotic assay was performed. The error bars are the standard deviation (SD) from 5 independent experiments (P values < 0.05 were significant).
RACK1 is an ATF2 binding protein and serves as an anchor protein to recruit activated PKC (especially PKC α or β) and other intracellular signal transducers.16,28,35 Upon mitogenic stimulation, RACK1 was shown to bind to activated PKC α and β and recruit JNK to the complex.28 Thus, we tested whether such a signaling complex might be formed in our experimental setting. Co-immunoprecipitation and immunoblotting were conducted (Fig. 3B). Following the suppression of PKC δ by the shRNA, JNK1 and PKC α and β were pulled down together with RACK1 in NIH3T3/Hras cells but not from parental cells. Furthermore, the addition of JNK inhibitor abrogated the recruitment of JNK to the complex (data not shown).
To determine the role of JNK in this apoptotic process, we infected NIH3T3/Hras cells with shRNA-PKC δ, in the presence of a JNK inhibitor (SP600125), KP372-1 (an Akt inhibitor), or PD98059 (a MAPK inhibitor), and the onset of apoptosis was analyzed by an Annexin V assay (Fig. 3C). PKC δ suppression initiated programmed cell death in the cells, which was significantly blocked by the JNK inhibitor. In comparison, the addition of the inhibitors to block Akt or the MAPK pathway did not affect the magnitude of apoptosis. However, the incomplete suppression of the apoptotic process by the JNK inhibitor indicates the involvement of other apoptotic pathways.
Induction of apoptosis in prostate cancer cells after PKC δ knockout
To further test the synthetic lethal interaction between loss of PKC δ and v-ras, we used human prostate cancer cells. DU145 cells expressed a higher amount of JNK (3.5-fold) than LNCaP cells (Fig. 4A, left panels). The introduction of shRNA-PKC δ dramatically blocked PKC δ expression in DU145 cells (Fig. 4A, middle panels). v-Ha-ras was also stably introduced into DU145 cells (DU145/Hras), and an elevated expression of Ras was detected in the cells (Fig. 4A, right panels). Next, the expression of PKC α and β after the knockdown of PKC δ was examined in LNCaP and DU145 cells (Fig. 4C, left panels). The expression of these two PKC isoforms was increased upon the introduction of shRNA-PKC δ. The phosphorylation status of JNK in the cells was also tested (Fig. 4B, right panels). With or without PKC δ knockdown, JNK was not activated in LNCaP cells. In comparison, a relatively high baseline of phosphorylated JNK1 existed in untreated DU145 or DU145/Hras cells. After blockade of PKC δ by the shRNA, a significantly increased amount of phosphorylated JNK was detected in both cells. Subsequently, the induction of apoptosis in LNCaP, DU145, or DU145/Hras cells was analyzed by the Annexin V assay (Fig. 4C). About 35% of DU145/Hras underwent apoptosis following PKC δ knockdown. In comparison, less DU145 cells (about 20%) underwent apoptosis. The suppression of PKC δ was not apoptotic to LNCaP cells. The data further suggest that JNK is a major player in this apoptotic process. The data also indicate that oncogenic Ras recruits multiple pathways (besides JNK) to execute a full magnitude of cell death program.
Figure 4.
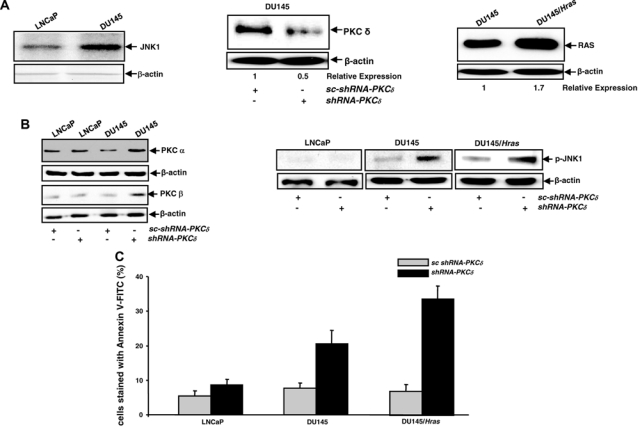
Prostate cancer DU145 or DU145/Hras cells are susceptible to apoptosis following PKC δ knockdown. (A) The expression of JNK in prostate cancer: the cells were analyzed by immunoblotting. Equal loadings of the samples were normalized by β-actin (left panels). After the infection of shRNA-PKC δ, the expression of PKC δ in DU145 cells was analyzed by immunoblotting. The relative expression level of PKC isoforms was analyzed by Image J software (middle panels). After the stable infection of v-Ha-ras, the expression of Ras in DU145 or DU145/Hras cells was examined by immunoblotting (right panels). Equal loadings of total proteins were normalized by β-actin, and the relative expression of Ras was analyzed by Image J. (B) After the knockdown of PKC δ, PKC α or β expression (left panels) and phosphorylated JNK (right panels) were examined by immunoblotting. Equal loading of total proteins was normalized by β-actin. (C) LNCaP, DU145, or DU145/Hras cells, after being infected with the shRNAs, were subjected to Annexin V-FITC analysis. The error bars represent the standard deviation (SD) from 5 independent experiments (P values <0.05 were significant).
Caspase 8 is activated upon the knockdown of PKC δ
Some caspase family members were shown to be activated by JNK in cells expressing oncogenic ras for the induction of apoptosis.27 To determine if caspase 8 was activated in our experimental setting, the cleavage of caspase 8 was tested by immunoblotting. The active, cleaved form of caspase 8 was detected in NIH3T3/Hras and DU145 cells following the suppression of PKC δ, which was absent in parental or untreated cells (Fig. 5A). To further determine whether caspase 8 activity is upregulated in NIH3T3/Hras cells after PKC δ inhibition, we performed a caspase activity assay (Fig. 5B). The activity of this protease was increased after PKC δ suppression. JNK inhibitor blocked shRNA–PKC δ–induced caspase 8 activity. The addition of Akt or the MAPK inhibitor had no role in caspase 8 activity, indicating that JNK functions upstream of caspase 8.
Figure 5.
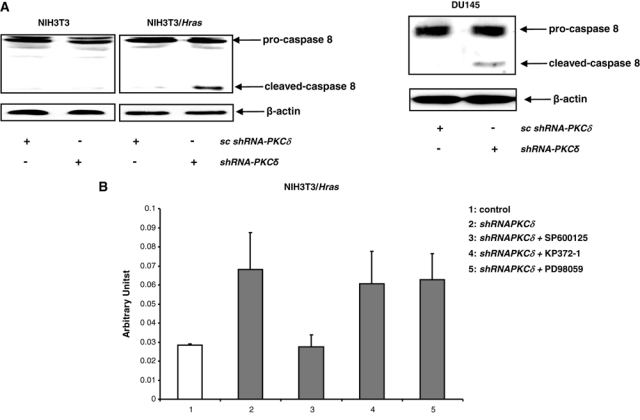
Caspase 8 is activated after the knockdown of PKC δ. (A) After the infection of shRNA-PKC δ, cell lysates were immunoblotted with anti–caspase 8 antibody. Equal loadings of total proteins were normalized by β-actin. (B) In the presence or absence of SP600125, KP372-1, or PD98059, caspase 8 activity was measured following knockdown of PKC δ. The error bars represent the standard deviation (SD) from 3 independent experiments (P values < 0.05 were significant).
Requirement of PKC a suppression for the induction of apoptosis in NIH3T3/Hras cells
Because suppression of PKC δ by the shRNA upregulated the expression of PKC α and β, we then tested if overexpression of these two PKC isoforms would render the cells susceptible to apoptosis. WT (wild-type)–PKC α, WT-PKC β, CAT (constitutively active)–PKC α, or CAT-PKC β with HA was transiently introduced into the cells. The overexpression of these exogenous proteins in NIH3T3/Hras cells was analyzed using an anti-HA (hemagglutinin) antibody (Fig. 6A). The expression of the exogenous proteins in the parental cells was also examined, and similar expression patterns were observed (data not shown). Subsequently, an Annexin V assay was performed in the cells with or without knocking down PKC δ (Fig. 6B). Ectopic expression of wild-type or constitutively active PKC α and PKC β, alone or in combination, was not apoptotic to NIH3T3/Hras cells. However, following knockdown of PKC δ, with more than 25% of NIH3T3/Hras cells ectopically expressing WT- or CAT-PKC α, β alone became apoptotic, the magnitude of which was further increased by cotransfection of WT- or CAT-PKC α plus β. The results suggest that PKC δ downregulation perturbs its negative influence on PKC α or β in cells expressing aberrant Ras and initiates an apoptotic crisis.
Figure 6.
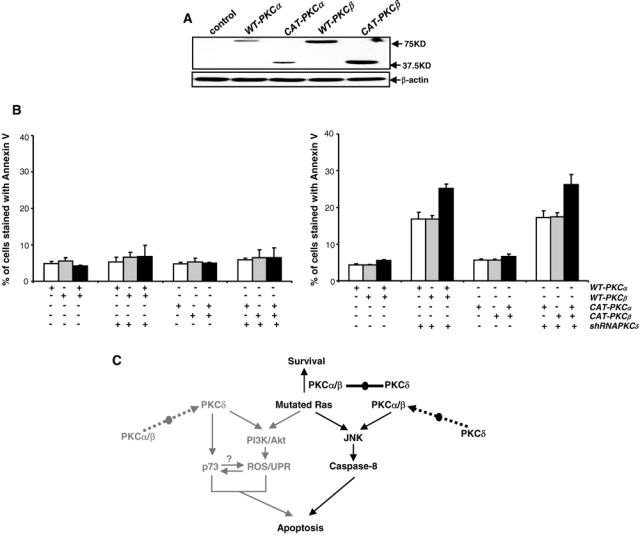
Overexpression of WT- or CAT-PKC α and PKC β is insufficient for the induction of apoptosis without aberrant Ras. (A) NIH3T3/Hras cells were transfected with WT- or CAT-PKC α or β. Subsequently, the expression of exogenous proteins was tested by immunoblotting with an anti-HA antibody. Equal loadings of total proteins were normalized by β-actin. (B) With or without PKC δ knockdown, the cells transiently transfected with WT- or CAT-PKC α, β, alone or in combination, were subjected to an Annexin V-FITC assay. The error bars represent the standard deviation (SD) from 5 independent experiments (P values <0.05 were significant). (C) Hypothetic regulatory network of oncogenic Ras and PKC isoforms (α/β and δ) for the induction of apoptosis. PKC α/β and δ function oppositely to coop with aberrant Ras signaling. Once the balance is perturbed, an apoptotic crisis occurs. Signaling pathways in gray are being published and in dark color are being investigated in the current study.
Discussion
PKC regulates diverse cellular activities, including proliferation, differentiation, and apoptosis.1,7,36 Studies showed that PKC family members are positively or negatively involved in the regulation of apoptosis induced by death receptors, genotoxic agents, anticancer drugs, and cell toxins.4,36 PKC α, β, ε, and ζ are known to be pro-survival in many experimental settings.3,36 The predominant role of PKC δ was found to be tumor suppressive.3,37 However, it was also demonstrated that various types of cells expressing oncogenic Ha- or K-ras became apoptotic after suppression of PKC δ, suggesting that this PKC isozyme is crucial for survival.15 To understand the mechanisms of how loss of PKC δ sensitizes cells with mutated Ras to apoptosis, we used the shRNAs to genetically co–knock down PKC α and β and recently demonstrated that PKC δ was upregulated, resulting in apoptosis.14 In this study, by genetic knockdown of PKC δ, we showed that PKC α and β were upregulated, which formed a complex with RACK1 and JNK. Subsequently, caspase 8 was cleaved, resulting in the induction of apoptosis. The suppression of JNK abrogated this synthetic lethal reaction in the cells expressing v-ras. Interestingly, the concurrent overexpression of WT- or CAT-PKC α plus β in the presence of oncogenic Ras, without knocking down PKC δ, is not lethal to the cells. It is well known that PKC α and β participate in cell growth promotion or tumorigenesis.1,7 However, the ability of these PKC isoforms to induce apoptosis has been reported to be crucial in various types of cells.9,38,39 By genetically intervening into these PKC isoforms, our current findings reveal a possible mechanism balanced by PKC α/β and δ, which facilitates cells to overcome oncogenic stress. Once the balance is perturbed, an apoptotic crisis is initiated.
Dual functions of PKC isoforms have been shown in different experimental settings,3,36 suggesting the functional complexity of PKC isozymes. Long-term phorbol ester treatment to downregulate PKC or transient exposure to PKC inhibitors was able to induce cells expressing v-ras to undergo apoptosis.27 The blockade of PKC α sensitized salivary epithelial cells to PKC δ–induced apoptosis, in which JNK was required.40 Recently, we demonstrated that the concurrent suppression of PKC α/β conferred cells expressing oncogenic ras or Akt the susceptibility to apoptosis, via upregulating PKC δ.41 Our current study again demonstrated that in the presence of aberrant Ras signaling, loss of PKC δ causes the increase of PKC α/β expression and activation of JNK, resulting in apoptosis. In doxorubicin-induced cytotoxicity, JNK activated c-Jun/ATF2 to upregulate PKC δ transcripts.42 It is possible that JNK, downstream of Ras, affects the activity of transcription factors and further the expression of PKC α and β.
RACK1 has been implicated in the regulation of conventional PKC isoforms for cell growth or tumorigenesis.16,17 The RACK1-regulated signal transducer module was shown to be involved in JNK activation in melanoma cells.28 JNK often functions downstream of Ras and participates in the regulation of stress-mediated apoptosis in different types of cells.18-20 Depletion of nerve growth factor from PC12 cells could activate JNK, resulting in apoptosis.42 JNK activity has been indicated to be crucial for initiating caspase cascade following the suppression of PKC in cells expressing oncogenic ras.27 The association of PKC α/β, RACK1, and JNK in our current experimental setting provides a plausible explanation for how active PKC α and β use scaffold protein RACK1 to activate JNK and caspase 8 for initiating a cell death program.
The study using rottlerin to suppress PKC δ demonstrated that PI3K (phosphatidylinositol 3-kinase) was necessary for the induction of apoptosis in various types of cells expressing v-ras (including NIH3T3 cells ectopically expressing v-Ha-ras).15 However, our study, employing the shRNA to genetically knock down PKC δ, showed that PKC α and β were upregulated, accompanied with the activation of JNK and caspase 8 in cells expressing oncogenic ras. It is conceivable that chemical inhibitors often are less specific to target a particular kinase, which may contribute to the discrepancies of these two different experimental settings, even when the same cell line is being used.
Ras governs signaling pathways that are crucial regulators in controlling normal cell growth and tumor transformation. In tumorigenesis, it is crucial for cells to adapt to stress rendered by oncogenes or oncogene-mediated signaling. To survive, a proper coordination of Ras with other signaling regulators (such as PKC) becomes crucial and pivotal. Disruption of such cooperation may trigger a clash among these signaling regulators in cells, resulting in an apoptotic crisis. Ras mutations occur in more than 30% of human malignancies. Therefore, targeting oncogenic Ras is a very attractive strategy for the development of new anticancer therapies. Studies have highlighted the respective roles of Ras in the induction of apoptosis.41,43-45 High-throughput RNA interference screens identified various kinases that are essential for tumor cells harboring mutated K-ras to survive.32,33 A synthetic lethal interaction occurs when the expression of these kinases is altered in the presence of K-ras mutations. Our current study, together with previously reported results,14 demonstrates a possible mechanism regulating the synthetic lethal interaction between oncogenic Ras and loss of PKC (Fig. 6C). Once the balance maintained by PKC α/β and δ is perturbed, aberrant Ras becomes apoptotic. Thus, these PKC isoforms can be potential molecular targets for therapeutic intervention into cancers harboring an active ras.
Furthermore, genetic knockdown of PKC δ is able to sensitize prostate cancer DU145 cells that express a high level of JNK to apoptosis. We reported that concurrent inhibition of PKC α and β renders prostate cancer PC3 cells susceptible to apoptosis, via the Akt pathway. It appears that aberrant Ras downstream effectors not only participate in the process of tumorigenesis but also are involved in the regulation of apoptosis. With increasing attention to search the vulnerabilities of cancers harboring Ras or Ras-governed signaling effectors, our study adds new knowledge for targeting different oncogenic molecules to develop new strategies that preferentially activate a suicide program in the tumor cells and keep surrounding normal cells intact.
Materials and Methods
Cells and reagents
Murine fibroblasts NIH3T3, human prostate cancer LNCaP, and DU145 cells were purchased from ATCC (Manassas, VA) and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% serum (Sigma-Aldrich, St. Louis, MO). The cells were stably infected with a retroviral vector inserted with v-Ha-ras and selected in the growth medium containing geneticin. Anti-PKC α, β, η; Ras; phospho-JNK; RACK1; and caspase 8 antibodies were purchased from BD Biosciences (San Jose, CA). Anti-PKC δ, ε, θ; JNK1; and c-Jun antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-phospho-Ser63- and Ser73-c-Jun antibodies were from Cell Signaling Technology (Danvers, MA).
The oligonucleotides targeting PKC isozymes were ligated to lentiviral small-hairpin vector pLL3.7. The shRNA sequences of murine PKC α, β, and δ already have been reported.17 The shRNA sequence for murine PKC ε is 5′-gcaaggaagggattatgaa-3′, PKC η is 5′-gtgaacggacataagttca-3′, and PKC θ is 5′-cgagaaaccatgttccata-3′. The shRNA sequence for human PKC δ is 5′-ggtcctgggcaaaggcagctt-3′. The constructs containing wt-PKC α, β and CAT-PKC α or β were from Dr. Soh (Inha University, South Korea).
DNA fragmentation analysis
After treatments, cells were fixed in 70% ethanol containing RNAase, stained with propidium iodide, and analyzed by a flow cytometer for DNA content assessment.
Annexin V assay
After treatments, cells were prepared using the Annexin V-FITC Apoptosis Kit I (BD Biosciences). Samples were then analyzed by a flow cytometer.
PKC activity analysis
The PKLight HTS Protein Kinase Assay kit (Lonza Rockland, Rockland, ME) was used. Cells lysates were immunoprecipitated with corresponding antibodies. Immunoprecipitates were incubated with PKC substrate peptide and adenosine triphosphate (ATP) in the kinase buffer. The intensity of the luminescence in the samples was measured by a microplate luminometer.
Immunoprecipitation and immunoblot analyses
Following treatments, cells were lysed and subjected to immunoprecipitation. Subsequently, immunoprecipitates were blotted with corresponding antibodies. The blots were analyzed by Image J software for densitometry comparison.
Semi-quantitative RT-PCR and real-time PCR
Total RNAs were isolated and reversely transcribed. cDNAs were subjected to semi-quantitative PCR in an icycler (Bio-Rad, Hercules, CA). The real-time PCR analysis was performed on a 7300 Real-Time PCR system (Applied Biosystems, Foster City, CA). β-actin was used as control. The primers used were as follows: 5′-agaagggcacatcaaaatcg-3′ and 5′-acgcccaccaatctacagac-3′ for PKC α, 5′-ctccattcctgcttccagac-3′ and 5′-aacagaccgatggcaatctc-3′ for PKC β, 5′-cacgagtttatcgccacctt-3′ and 5′-cggccgataatcttgtcaat-3′ for PKC δ, 5′-aggtcaatggccacaagttc-3′ and 5′-agctcatgacatcgcttgtg-3′ for PKC ε, 5′-atattcggtgtcaggcgaac-3′ and 5′-ctttccctgccgtcttagtg-3′ for PKC η, and 5′-tggtggaaaagagggttctg-3′ and 5′-tggcaactttggatgtggta-3′ for PKC θ.
Caspase 8 activity assay
A caspase 8 assay kit (BioVision, Mountain View, CA) was used to measure caspase 8 activity. Briefly, cell lysates were incubated with IEPD-p-nitroanilide as a substrate. The enzyme activity was then determined.
Acknowledgments
We thank Dr. Luo (Boston University School of Medicine, Boston, MA) for providing various reagents. We also thank Dr. Soh (Inha University, South Korea) for providing the PKC constructs.
Footnotes
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This study is supported by the NIH (RO1CA100498) and DOD (W81XWH-04-1-0246).
References
- 1. Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J 1995;9:484-96 [PubMed] [Google Scholar]
- 2. Spitaler M, Cantrell DA. Protein kinase C and beyond. Nat Immunol 2004;5:785-90 [DOI] [PubMed] [Google Scholar]
- 3. Reyland ME. Protein kinase C isoforms: multi-functional regulators of cell life and death. Front Biosci 2009;14:2386-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gutcher I, Webb PR, Anderson NG. The isoform-specific regulation of apoptosis by protein kinase C. Cell Mol Life Sci 2003;60:1061-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol 2000;279:429-38 [DOI] [PubMed] [Google Scholar]
- 6. Meinhardt G, Roth J, Totok G. Protein kinase C activation modulates pro- and anti-apoptotic signaling pathways. Eur J Cell Biol 2000;79:824-33 [DOI] [PubMed] [Google Scholar]
- 7. Newton AC. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev 2001;101:2353-64 [DOI] [PubMed] [Google Scholar]
- 8. Knox KA, Johnson GD, Gordon J. A study of protein kinase C isozyme distribution in relation to Bcl-2 expression during apoptosis of epithelial cells in vivo. Exp Cell Res 1993;207:68-73 [DOI] [PubMed] [Google Scholar]
- 9. Powell CT, Brittis NJ, Stec D, Hug H, Heston WD, Fair WR. Persistent membrane translocation of protein kinase C alpha during 12-0-tetradecanoylphorbol-13-acetate-induced apoptosis of LNCaP human prostate cancer cells. Cell Growth Differ 1996;7:419-28 [PubMed] [Google Scholar]
- 10. Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, et al. Proteolytic activation of protein kinase C delta by an ICE/CED 3-like protease induces characteristics of apoptosis. J Exp Med 1996;184:2399-404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khwaja A, Tatton L. Caspase-mediated proteolysis and activation of protein kinase Cdelta plays a central role in neutrophil apoptosis. Blood 1999;94:291-301 [PubMed] [Google Scholar]
- 12. Matassa AA, Carpenter L, Biden TJ, Humphries MJ, Reyland ME. PKCdelta is required for mitochondrial-dependent apoptosis in salivary epithelial cells. J Biol Chem 2001;276:29719-28 [DOI] [PubMed] [Google Scholar]
- 13. Majumder PK, Mishra NC, Sun X, Bharti A, Kharbanda S, Saxena S, et al. Targeting of protein kinase C delta to mitochondria in the oxidative stress response. Cell Growth Differ 2001;12:465-70 [PubMed] [Google Scholar]
- 14. Zhu T, Tsuji T, Chen CY. Roles of PKC isoforms in the induction of apoptosis elicited by aberrant Ras. Oncogene 2009. Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xia S, Forman LW, Faller DV. Protein kinase C delta is required for survival of cells expressing activated p21RAS. J Biol Chem 2007;282:13199-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Cloning of an intracellular receptor for protein kinase C: a homolog of the beta subunit of G proteins. Proc Natl Acad Sci USA 1994;91:839-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schechtman D, Mochly-Rosen D. Adaptor proteins in protein kinase C-mediated signal transduction. Oncogene 2001;20:6339-47 [DOI] [PubMed] [Google Scholar]
- 18. Buchner K. The role of protein kinase C in the regulation of cell growth and in signalling to the cell nucleus. J Cancer Res Clin Oncol 2000;126:1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mayer C, Grummt I. Cellular stress and nucleolar function. Cell Cycle 2005;4:1036-8 [DOI] [PubMed] [Google Scholar]
- 20. Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene 2008;27:6245-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Whitmarsh AJ, Davis RJ. Role of mitogen-activated protein kinase kinase 4 in cancer. Oncogene 2007;26:3172-84 [DOI] [PubMed] [Google Scholar]
- 22. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol 2007;19:142-9 [DOI] [PubMed] [Google Scholar]
- 23. Han J, Shui JW, Zhang X, Zheng B, Han S, Tan TH. HIP-55 is important for T-cell proliferation, cytokine production, and immune responses. Mol Cell Biol 2005;25:6869-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sluss HK, Barrett T, Derijard B, Davis RJ. Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol Cell Biol 1994;14:8376-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994;76:1025-37 [DOI] [PubMed] [Google Scholar]
- 26. Chen CY, Liou J, Forman LW, Faller DV. Differential regulation of discrete apoptotic pathways by Ras. J Biol Chem 1998;273:16700-9 [DOI] [PubMed] [Google Scholar]
- 27. Chen CY, Juo P, Liou JS, Li CQ, Yu Q, Blenis J, et al. The recruitment of Fas-associated death domain/caspase-8 in Ras-induced apoptosis. Cell Growth Differ 2001;12:297-306 [PubMed] [Google Scholar]
- 28. Lopez-Bergami P, Habelhah H, Bhoumik A, Zhang W, Wang LH, Ronai Z. RACK1 mediates activation of JNK by protein kinase C [corrected]. Mol Cell 2005;19:309-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997;278:1064-8 [DOI] [PubMed] [Google Scholar]
- 30. Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005;5:689-98 [DOI] [PubMed] [Google Scholar]
- 31. Sawyers CL. Finding and drugging the vulnerabilities of RAS-dependent cancers. Cell 2009;137:796-8 [DOI] [PubMed] [Google Scholar]
- 32. Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009;137:821-34 [DOI] [PubMed] [Google Scholar]
- 33. Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009;137:835-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006;441:106-10 [DOI] [PubMed] [Google Scholar]
- 35. Ron D, Jiang Z, Yao L, Vagts A, Diamond I, Gordon A. Coordinated movement of RACK1 with activated betaIIPKC. J Biol Chem 1999;274:27039-46 [DOI] [PubMed] [Google Scholar]
- 36. Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer 2007;7:281-94 [DOI] [PubMed] [Google Scholar]
- 37. Afrasiabi E, Ahlgren J, Bergelin N, Tornquist K. Phorbol 12-myristate 13-acetate inhibits FRO anaplastic human thyroid cancer cell proliferation by inducing cell cycle arrest in G1/S phase: evidence for an effect mediated by PKCdelta. Mol Cell Endocrinol 2008;292:26-35 [DOI] [PubMed] [Google Scholar]
- 38. Yin L, Bennani-Baiti N, Powell CT. Phorbol ester-induced apoptosis of C4-2 cells requires both a unique and a redundant protein kinase C signaling pathway. J Biol Chem 2005;280:5533-41 [DOI] [PubMed] [Google Scholar]
- 39. Paone A, Starace D, Galli R, Padula F, De CP, Filippini A, et al. Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC-alpha-dependent mechanism. Carcinogenesis 2008;29:1334-42 [DOI] [PubMed] [Google Scholar]
- 40. Matassa AA, Kalkofen RL, Carpenter L, Biden TJ, Reyland ME. Inhibition of PKCalpha induces a PKCdelta-dependent apoptotic program in salivary epithelial cells. Cell Death Differ 2003;10:269-77 [DOI] [PubMed] [Google Scholar]
- 41. Guo J, Zhu T, Luo LY, Huang Y, Sunkavalli RG, Chen CY. PI3K acts in synergy with loss of PKC to elicit apoptosis via the UPR. J Cell Biochem 2009;107:76-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995;270:1326-31 [DOI] [PubMed] [Google Scholar]
- 43. Downward J. Ras signalling and apoptosis. Curr Opin Genet Dev 1998;8:49-54 [DOI] [PubMed] [Google Scholar]
- 44. Ma P, Magut M, Faller DV, Chen CY. The role of Ras in T lymphocyte activation. Cell Signal 2002;14:849-59 [DOI] [PubMed] [Google Scholar]
- 45. Smakman N, van den Wollenberg DJ, Elias SG, Sasazuki T, Shirasawa S, Hoeben RC, et al. KRAS(D13) promotes apoptosis of human colorectal tumor cells by ReovirusT3D and oxaliplatin but not by tumor necrosis factor–related apoptosis-inducing ligand. Cancer Res 2006;66:5403-8 [DOI] [PubMed] [Google Scholar]