

Abstract
Genome-scale metabolic reconstructions can serve as important tools for hypothesis generation and high-throughput data integration. Here, we present a metabolic network reconstruction and flux-balance analysis (FBA) of Plasmodium falciparum, the primary agent of malaria. The compartmentalized metabolic network accounts for 1001 reactions and 616 metabolites. Enzyme–gene associations were established for 366 genes and 75% of all enzymatic reactions. Compared with other microbes, the P. falciparum metabolic network contains a relatively high number of essential genes, suggesting little redundancy of the parasite metabolism. The model was able to reproduce phenotypes of experimental gene knockout and drug inhibition assays with up to 90% accuracy. Moreover, using constraints based on gene-expression data, the model was able to predict the direction of concentration changes for external metabolites with 70% accuracy. Using FBA of the reconstructed network, we identified 40 enzymatic drug targets (i.e. in silico essential genes), with no or very low sequence identity to human proteins. To demonstrate that the model can be used to make clinically relevant predictions, we experimentally tested one of the identified drug targets, nicotinate mononucleotide adenylyltransferase, using a recently discovered small-molecule inhibitor.
Keywords: flux-balance analysis, Plasmodium falciparum metabolism, systems biology
Introduction
Malaria is an ancient disease, which can be dated back to 2800 BC (Nerlich et al, 2008), and remains one of the most severe public health challenges worldwide. Currently, about half of the Earth's population is at risk from this infectious disease according to the World Health Organization (WHO, 2008). Malaria inflicts acute illness on hundreds of millions of people worldwide and leads to at least one million deaths annually (Baird, 2005; WHO, 2008). It ranks as a leading cause of death and disease in many developing countries, where the most affected groups are young children and pregnant women (WHO, 2008). The disease is transmitted to humans by the female Anopheles mosquito and is caused by at least five species of Plasmodium parasites. The life cycle of the parasite is highly complex and includes various hosts and tissue types. During a blood meal, sporozoites are transmitted from the mosquito to humans and initiate infection in the liver where they reproduce prolifically but are asymptomatic. In the next stage of infection, the parasites are released from the liver cyst into the bloodstream in the form of merozoites, where they invade red blood cells (RBCs) and reproduce asexually (Aly et al, 2009). The destruction of RBCs coupled with the significant load imposed on the host metabolism is ultimately responsible for the major clinical symptoms of malaria, which are often fatal (Haldar and Mohandas, 2009).
Although several anti-malarial drugs are currently available, most of them are losing efficacy due to acquired drug resistance in the most lethal causative agent, Plasmodium falciparum (Wongsrichanalai et al, 2002; Mackinnon and Marsh, 2010). The loss of drug efficiency in resistant strains poses a great threat to malaria control and has been linked to increases in worldwide malaria mortality (Hyde, 2007). There is an urgent need for new anti-malarial drugs coupled with better administration strategies. Understanding the molecular mechanisms and interactions of the parasite's cellular components is essential for identification of new drug targets, especially given the difficulties associated with in vivo drug testing (Liu et al, 2008).
Various systems biology approaches have been applied to improve our understanding of P. falciparum physiology and to facilitate drug development (Dharia et al, 2010). The sequencing of the P. falciparum genome has provided researchers with a complete collection of parasite proteins and likely regulatory interactions (Gardner et al, 2002; Carvalho and Menard, 2005). Several large-scale transcriptome (Bozdech et al, 2003; Le Roch et al, 2003; Llinas et al, 2006; Hu et al, 2010), proteome (Florens et al, 2002; Lasonder et al, 2002, 2008) and metabolome (Olszewski et al, 2009) analyses have been conducted in order to dissect functional interactions and define essential biological pathways. In addition to experimental studies, several databases have been developed to integrate functional knowledge of the parasite and its metabolism. For example, PlasmoCyc is an integrated database that links genomic data, protein annotation, enzymatic reactions, and pathway information (Yeh et al, 2004); the Malaria Parasite Metabolic Pathway Database (MPMP), on the other hand, is a manually curated resource that assembles annotated enzymes into likely metabolic pathways (Ginsburg, 2010).
A stoichiometric representation of metabolism can be effectively used to study functional properties of biochemical networks using a growing number of computational methods (Price et al, 2004). For example, flux-balance analysis (FBA) considers steady-state distributions of metabolic fluxes satisfying a set of biophysical constrains, such as bounds and mass balance of fluxes (Orth et al, 2010). Given the applied constraints, a likely distribution of fluxes in the network can be obtained by maximizing an appropriate objective function (e.g. biomass production) (Varma and Palsson, 1994) or applying minimal perturbation principles (Segre et al, 2002; Shlomi et al, 2005). The analysis of flux-balanced genome-scale metabolic networks is useful not only for the discovery of essential genes and potential drug targets, but also as a tool to better understand species-specific biology (Breitling et al, 2008; Oberhardt et al, 2009). For example, among other applications, these models have been used to identify minimal media requirements for growth (Chavali et al, 2008), explore metabolic weaknesses in bacterial pathogens (Navid and Almaas, 2009), integrate gene expression and other types of data (Colijn et al, 2009), and investigate objective functions important under different growth conditions (Schuetz et al, 2007). Given the complex life cycle of Plasmodium, a flux-balanced model of this organism is of direct relevance to the ongoing search to identify new therapeutic drug targets.
In this study, we reconstructed the genome-scale flux-balanced metabolic network of P. falciparum and used it to perform a systems-level analysis of the parasite's metabolism. On the basis of in silico gene deletions, we identified potential new anti-malarial drug targets with low sequence identity to human proteins. One of these targets, nicotinate mononucleotide adenylyltransferase (NMNAT), was experimentally tested in a growth inhibition assay using a recently discovered small-molecule inhibitor. We also illustrate, using a previously published methodology, how the reconstructed metabolic model can be used to integrate flux analysis with expression data to more accurately simulate the physiology of this complex eukaryotic pathogen.
Results
Scale of the reconstructed flux-balanced metabolic network
The reconstructed flux-balanced model is based on gene-reaction associations reported in public domain databases as well as on a careful literature analysis. We used well-curated microbial metabolic models and enzyme databases to determine the stoichiometry of most reactions. To produce a functional reconstruction, we also searched the literature for missing steps necessary for the model to produce a set of required biomass components (see Materials and methods). The model accounts for 366 genes, corresponding to 7% of all genes identified in P. falciparum. Compared with 61 metabolic models of various organisms compiled by Feist et al (2009), our model ranks 10th in terms of the smallest number of genes. Not surprisingly, the other metabolic models with small gene numbers include many parasitic/symbiotic species, such as Mycoplasma genitalium, Buchnera aphidicola, Haemophilus influenzae, and Helicobacter pylori. The P. falciparum network also includes 616 metabolites and 1001 reactions, 657 of which are metabolic transformations (Table I). In addition, there are 233 reactions corresponding to transport between different cellular compartments and 111 input–output exchange reactions that allow extracellular metabolites to enter and end products to be excreted from the network.
Table 1. Characteristics of the reconstructed metabolic network of P. falciparum.
| Reactions | 1001 | Metabolites | 616 |
| Cytosolic reactions | 503 | Cytosolic metabolites | 537 |
| Mitochondrial reactions | 49 | Mitochondrial metabolites | 83 |
| Apicoplast reactions | 105 | Apicoplast metabolites | 135 |
| Transport reactions | 233 | Extracellular metabolites | 159 |
| Cytosolic transport reactions | 132 | Genes | 366 |
| Mitochondrial transport reactions | 51 | ||
| Apicoplast transport reactions | 50 | ||
| Exchange reactions | 111 |
The metabolic reconstruction includes four distinct compartments: parasite cytosol, mitochondria, apicoplast (a non-photosynthetic plastid), and the extracellular space (representing the host cell cytosol and host serum). The majority of all reactions (50%) occur in the cytosol. The apicoplast accounts for 10% of all reactions, such as the synthesis of isopentenyl diphosphate, fatty acids, and heme (Ralph et al, 2004). A special reaction is included in the model to account for the biomass components and essential metabolites needed for growth (Supplementary Table S1). Supplementary information provides a complete list of all network reactions and metabolite abbreviations.
Excluding metabolite-exchange reactions, 74% of the reactions in the model are directly associated with P. falciparum genes, which compares well to other models of eukaryotes such as the iND750 yeast model (70%) (Duarte et al, 2004) and the iAC560 model for Leishmania major (63%) (Chavali et al, 2008). The remaining reactions include spontaneous transformations that can proceed without enzymatic catalysis and reactions required for the proper functioning of the metabolic model. Intracellular and inter-compartmental transport reactions, most of which are not currently associated with any gene, account for about 6% and 15% of all reactions in the model, respectively (Figure 1A). Most of the transporter proteins in Plasmodium spp. are currently uncharacterized. However, it is well established that the parasite significantly modifies the permeability of the host cell membrane (Kirk et al, 1999; Martin et al, 2005) and several metabolic processes occur across different organelles. For instance, such metabolic pathways as heme biosynthesis and antioxidant defense have been shown to involve both host and parasite enzymes localized to multiple intracellular compartments (Bonday et al, 1997; Koncarevic et al, 2009). Given the importance of metabolite exchange, many transport reactions were included in the model, although the identities of the corresponding genes remain unknown (Figure 1A).
Figure 1.

Annotation of reactions in the genome-scale metabolic model of P. falciparum. (A) Number of orphan (non-gene associated) reactions in P. falciparum grouped by metabolic processes. (B) Reactions grouped by Enzyme Commission (EC) classifications. (C) Reactions grouped by metabolic processes in P. falciparum and S. cerevisiae (Duarte et al, 2004).
Transferases and hydrolases comprise the largest fraction of enzymatic reactions in the network (Figure 1B). In terms of specific metabolic processes (Figure 1C), most reactions in the network are related to lipid metabolism, followed by transport and exchange reactions. In comparison with Saccharomyces cerevisiae, a free-living eukaryote of similar genome size, the most significant metabolic difference is the fraction of reactions involved in amino-acid metabolism (Figure 1C). About 20% of the reactions in the iND750 yeast metabolic network (Duarte et al, 2004) are responsible for amino-acid pathways; in contrast, this fraction is only 7% in P. falciparum. Amino-acid biosynthesis pathways are absent in P. falciparum metabolism because of the unique ability of the parasite to catabolize the erythrocyte hemoglobin (Francis et al, 1997) and to scavenge free amino acids from the host serum (human stages) or hemolymph (mosquito stages).
Analysis of in silico single and double gene deletions
We simulated gene deletions using FBA of the reconstructed metabolic network. Even though sugars other than glucose do not support P. falciparum growth in culture (Saito et al, 2002), in performing the in silico deletions we initially allowed all exchange reactions to carry non-zero metabolic fluxes, thereby permitting the potential import and utilization of other hexoses. Purines, such as inosine and adenosine, which are not normally included for in vitro culture but can be imported in vivo (LeRoux et al, 2009), were also made available to the network. We note that genes identified as essential when all exchange reactions are allowed will also be essential under more specific (constrained) conditions. The phenotypic effects of in silico deletions were classified into four groups: lethal, growth reducing (growth between 0 and 95% of the wild-type network), slight growth reducing (growth between 95 and 100%), and with no effect. About 15% of all single gene deletions (Table II) were lethal, ∼1% were growth reducing, and 3.5% were slightly growth reducing.
Table 2. Total number of single and non-trivial double deletion phenotypes.
| Predicted phenotypea | Single deletion (# genes) | Single deletion (%) | Double deletion (# non-trivial pairs) | Double deletions (%) |
|---|---|---|---|---|
| aSingle and double gene deletion predictions were classified into lethal, growth reducing (growth between 0 and 95% of wild type), slight growth reducing (growth between 95 and 100%), and no effect. | ||||
| No effect (NE) | 295 | 80.60 | 43 160+4974 (trivial GR, SGR) | 99.85 |
| Lethal (L) | 55 | 15.02 | 16 | 0.03 |
| Growth reducing (GR) | 3 | 0.82 | 48 | 0.10 |
| Slight growth reducing (SGR) | 13 | 3.55 | 7 | 0.01 |
| Total | 366 | 100.0 | 48 205 | 100.0 |
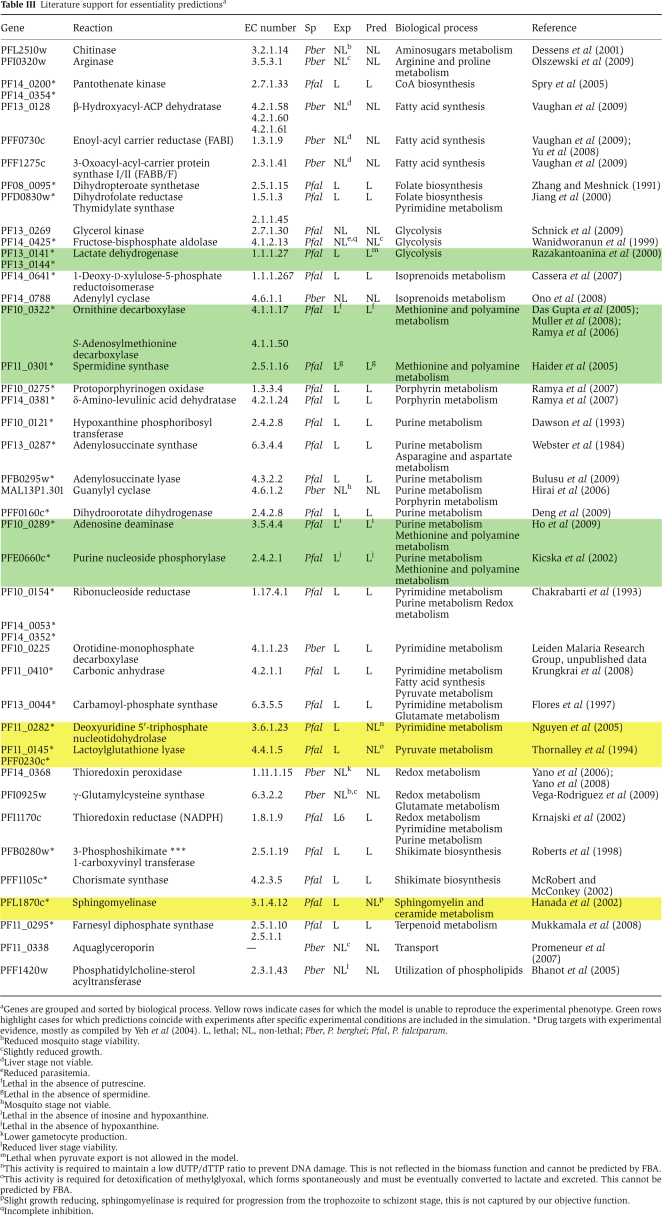
Out of 366 genes in the P. falciparum metabolic network, 55 genes were predicted to be essential for growth (Supplementary Table S2). To assess the accuracy of these predictions, we compiled a list of experimentally validated gene knockouts and phenotypes resulting from targeted inhibitions of enzymatic activities with drugs (Table III). In the computational analysis, we assumed that the drug treatments resulted in a complete inhibition of targeted enzymatic activities. In this way, the available drug phenotypes were simulated with computational deletions of the corresponding genes. In total, 14 metabolic gene knockouts and 25 drug inhibition phenotypes for genes were retrieved from the literature for P. falciparum and Plasmodium berghei, a murine malaria parasite commonly used in experimental studies (Janse and Waters, 1995; Janse et al, 2006).
Table 3. Literature support for essentiality predictionsa.
|
The FBA analysis was able to achieve 100% accuracy for predictions of both essential and non-essential gene knockouts (14 cases in total). In contrast, about 70% accuracy was achieved for phenotypes resulting from drug inhibitions of metabolic enzymes. Interestingly, all mispredicted drug phenotypes (eight cases) involved genes for which the computational analysis predicted a non-zero growth phenotype, whereas corresponding drugs were lethal to the parasite in experimental studies. In three cases, inconsistencies between the FBA predictions and experimental drug inhibitions can be explained by considering functions that are not explicitly represented in our model. These included the degradation of spontaneously forming toxic metabolites (e.g. methylglyoxal), and the synthesis of metabolites that are involved in the progression between the intraerythrocytic stages (e.g. sphingolipid, ceramide) (Hanada et al, 2002). The remaining discrepancies (five cases) can be resolved by taking into account specific literature-based evidence (Table III, green rows), that is, by considering nutrient availabilities and directionality of exchange reactions. Interestingly, in one case, the source of discrepancy between the model and experiments was clearly related to off-target drug effects. Specifically, the inhibitor of enoyl-acyl carrier reductase (FabI), triclosan, has been shown to kill P. falciparum in vitro and in vivo (Surolia and Surolia, 2001) despite the fact that its presumed target, PfFabI, can be deleted with no apparent blood-stage phenotype (Surolia and Surolia, 2001; Yu et al, 2008; Vaughan et al, 2009); this deletion phenotype is correctly predicted by our model.
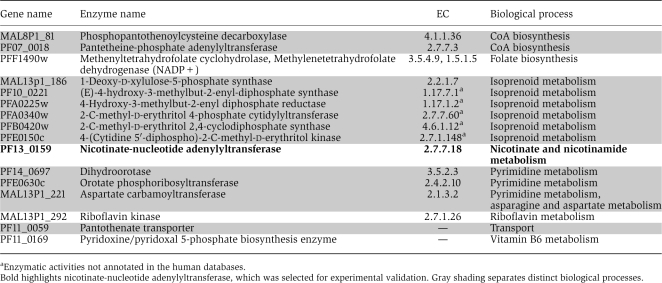
For 15 metabolic genes identified in our analysis as essential, knockout experiments or drug inhibition assays in P. falciparum/P. berghei are already available in the literature (see Table III; Supplementary Figure S2). The remaining predictions include 24 genes coding for proteins with relatively low sequence identity (20–40%) to human transcripts (Supplementary Figure S1; see Materials and methods), and 16 genes with no significant sequence identity to any human protein (BLAST E-value>10−2). This last group comprises six genes associated with isoprenoid metabolism, three genes involved in nucleotide metabolism, and genes related to CoA, shikimate, and folate biosynthesis (Table IV). Nine of the genes with no homology to human proteins are homologous to plant proteins; this is consistent with the essential functions of apicoplast-associated genes in the Apicomplexa (Lim and McFadden, 2010). These 40 enzymes are of immediate interest as potential drug targets, as low homology to human proteins suggests that side effects for drugs targeting these enzymes may be minimized or avoided. Interestingly, 5 of the 16 enzymes with no detectable homology correspond to enzymatic activities (Enzyme Commission (EC) numbers) that are unlikely to be present in human metabolism according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al, 2008), HumanCyc (Romero et al, 2005), and UniProt (Uniprot.Consortium, 2010) databases. Among the predicted essential genes with low but significant sequence identity to human transcripts, only aminodeoxychorismate lyase/synthetase (2.6.1.85, 4.1.3.38, PFI1100w) is associated with EC numbers not reported in human metabolism. In addition to genes predicted as essential, nine internal metabolic reactions with no associated network genes (orphan reactions) were also predicted to be essential for growth. Four of these reactions are associated with the shikimate biosynthetic pathway; three with ubiquinone metabolism, one with nicotinamide, and one with porphyrin metabolism (Supplementary Table S3).
Table 4. Predicted essential genes with no homologs in the human genome.
|
One metabolic pathway of significant interest in the parasite is the mitochondrial tricarboxylic acid (TCA) cycle. In most free-living microbes, this pathway fully oxidizes available carbon sources to carbon dioxide, in the process generating high-energy phosphate bonds (ATP/GTP). Within the Plasmodium species, however, the nature and function of the TCA cycle remains unclear (van Dooren et al, 2006). In the malaria parasites, the sole pyruvate dehydrogenase complex, which normally provides the key link between glycolysis and TCA metabolism, localizes not to the mitochondrion but to the apicoplast. In that compartment it is likely to be used primarily to generate acetyl-CoA for lipogenesis (Foth et al, 2005). Incorporating this fact into our model, we find that the only TCA cycle enzyme predicted to be essential is the 2-oxoglutarate dehydrogenase complex. This enzyme converts 2-oxoglutarate into succinyl-CoA, which is required for heme biosynthesis. Intriguingly, metabolic-labeling experiments indicate that the TCA cycle of the parasite also reduces 2-oxoglutarate to malate, generating acetyl-CoA from citrate cleavage (Olszewski et al, 2010). A non-zero flux through this reaction is observed in our model when additional constraints are applied to mitochondrial transport reactions.
We also extended the computational analysis of essential Plasmodium genes to pairs of genes that are not essential on their own, but are lethal if deleted simultaneously, that is synthetic lethal enzyme pairs with non-trivial genetic interactions (Dixon et al, 2009) (see Table II). In total, deletion of 16 gene pairs gave rise to such synthetic lethality in the unconstrained model. The enzymes that were essential upon double deletions participate in glycolysis, metabolism of nucleotides, lipids, porphyrin, the pentose phosphate cycle, and transport of NO2 and phosphate (Supplementary Table S4).
The analysis of essential genes in the S. cerevisiae metabolic network iND750 (Duarte et al, 2004) can be used to put the results of the P. falciparum in silico deletions into an appropriate context. To make the proper comparison, we only considered deletions of genes carrying non-zero metabolic fluxes in wild-type models of both networks; the focus on non-zero fluxes is necessary to prevent the difference in network sizes (S. cerevisiae 750 genes/1266 reactions, P. falciparum 366 genes/1001 reactions) from biasing the results. When both networks were allowed to simultaneously use all carbon sources, the fraction of essential genes associated with non-zero fluxes in P. falciparum was 37%, whereas in S. cerevisiae it was 5% (Fisher's exact test, P-value<10−10). The fraction of essential genes was also significantly higher in the parasite when single carbon sources were used in both models. For example, when glucose was used as the sole source of carbon, 50% of genes were essential in the parasite versus 26% in yeast (P-value=10−6). To understand whether the significantly higher fraction of essential genes in P. falciparum arises exclusively from a smaller number of isoenzymes in that network (111 in P. falciparum versus 276 in S. cerevisiae) or if it is also related to the inherent differences in the networks’ architectures, we performed deletions of all isoenzymes associated with metabolic reactions, instead of individual gene deletions. As a result, when networks used all carbon sources, 39% of the reactions with non-zero fluxes were essential in P. falciparum, compared with only 6% in S. cerevisiae (P-value<10−10). In a glucose minimal media, 62% of the reactions are essential in the parasite and 47% in yeast (P-value<10−10). These results demonstrate that a significantly smaller genetic robustness of the parasite's network arises, at least in part, because of a paucity of alternative metabolic pathways (Wagner, 2005). The lower redundancy of the P. falciparum network is likely to be a consequence of the adaptation to the relatively homeostatic and nutrient-rich environments of the hosts in which it proliferates (Gardner et al, 2002).
Resolution of disagreements between in silico predictions and experimental data
Although the presented metabolic model achieves a high accuracy in predicting phenotypes of the experimental knockouts and drug inhibition assays, it is the disagreement between the model and experiments that often leads to model improvement (Thiele and Palsson, 2009). Thus, it is important to discuss the inconsistencies between modeling and experimental results, which were corrected in the process of model construction. In four cases, the disagreements between the predicted gene essentiality and experimental results reported in the literature were resolved through additional flux constraints. The adjustments included purine nucleoside phosphorylase (PFE0660c), adenosine deaminase (PF10_0289), ornithine decarboxylase (PF10_0322), and lactate dehydrogenase (PF13_0141/PF13_0144). The additional constraints applied to the network were based either on specific experimental conditions or known details of Plasmodium spp. physiology (Table III, green rows). For example, lactate is believed to be the main byproduct of glucose metabolism in P. falciparum (Vaidya and Mather, 2009), and lactate dehydrogenase is an essential enzyme that regenerates nicotinamide adenine dinucleotide (NAD+) from NADH (Berwal et al, 2008). In contrast, in our initial model, pyruvate was exported as the primary glycolysis byproduct and NAD+ was regenerated through the transformation of pyrroline-5-carboxylate to proline (EC: 1.5.1.2). As there is no evidence of extensive pyruvate export in Plasmodium, a constraint was added to the corresponding pyruvate exchange reaction. As a result, we observed reduction of pyruvate to lactate, followed by lactate export. In the adjusted model, lactate dehydrogenase carried a non-zero flux and was correctly predicted to be essential for growth. In the other three cases, constraints on exchange fluxes were added to reproduce the composition of the media used in drug inhibition experiments. Specifically, purine nucleoside phosphorylase has been shown to be essential if hypoxanthine is not available in the environment (Kicska et al, 2002), whereas adenosine deaminase is essential in the absence of both inosine and hypoxanthine (Ho et al, 2009). When the fluxes through the inosine and hypoxantine exchange reactions were set to zero, the experimentally observed knockout phenotypes were reproduced. Similarly, ornithine decarboxylase was correctly predicted to be essential without putrescine in the growth media (Das Gupta et al, 2005).
In two cases, the inability of our initial model to reproduce experimental results was due to reactions involving metabolic dead ends; that is metabolites that are either only produced or only consumed in the network (Reed et al, 2003). In the first case, the model was not able to synthesize spermidine. The synthesis of spermidine from putrescine by spermidine synthase was accompanied by the production of 5-methylthioadenosine (MTA) (Haider et al, 2005), which was a metabolic dead end in the initial model. Consequently, the spermidine synthesis caused MTA to accumulate, violating the steady-state assumption of the constraint-based approach. However, although it is known that in Plasmodium spp. MTA is first converted to 5-methylthioinosine by adenosine deaminase and then recycled into methionine and hypoxanthine (Ting et al, 2005), not all enzymes involved in these reactions have been fully characterized. We addressed this problem by including in the model the PfADA and PfPNP activities, responsible for the hypoxanthine synthesis from MTA (Ting et al, 2005), and an additional hypothetical reaction that converts the resulting by-product, 5-methylthioribose-1-PO4, to methionine in order to represent the methionine salvage pathway. The second case was related to the folate biosynthesis pathway. In this pathway, the reaction catalyzed by 6-pyruvoyltetrahydropterin synthase (4.2.3.12), in addition to the folate biosynthesis intermediate 6-hydroxymethyl-7,8-dihydropterin, is known to produce a small amount of 6-pyruvoyl-5,6,7,8-tetrahydropterin (6PTHP) (Hyde et al, 2008). Initially, both products were included in the same reaction and, because 6PTHP represented another metabolic dead end, the cell was not able to synthesize folate. We resolved this problem by including in the model separate reactions for each of the two alternative products.
The total number of remaining dead-end metabolites in the final model (266) is comparable to that of other recently published genome-scale metabolic networks; for example the iBsu1103 model for Bacillus subtilis (270 dead-end metabolites; Henry et al, 2009), the iAC560 model of L. major (261; Chavali et al, 2008), or the iND750 S. cerevisiae model (194; Duarte et al, 2004). The remaining dead ends in the Plasmodium network include 109 metabolites that are consumed but are not currently produced or imported in the model, 80 metabolites that are produced but not consumed, and 79 metabolites, associated with reversible reactions, that can be either exclusively consumed or produced. As protein synthesis is not explicitly included in the metabolic model, a significant number (42) of the remaining dead-end compounds correspond to tRNAs; this compares to 68 dead-end tRNAs in the iND750 yeast network. Among the other functional categories associated with a large number of the metabolic dead ends are lipid metabolism (45%), transport reactions (15%), the metabolism of carbohydrates (5%), amino acids (8%), and nucleotides (5%) (Supplementary information). More precise measurements of the P. falciparum biomass composition, for example a detailed lipid composition of the parasite membrane (Hsiao et al, 1991; Mi-Ichi et al, 2006), can be used in the future to significantly shrink the pool of the remaining dead-end metabolites.
Validation of the predicted drug target nicotinate nucleotide adenylyltransferase
The most urgent motivation for flux-balance reconstruction of the pathogen metabolism is to facilitate drug development. To illustrate the potential of the model to make clinically relevant predictions, we experimentally tested a predicted target for which candidate drugs have been reported in other microbial species. The ideal drug target will be an essential enzyme with no homolog in the human genome and in a pathway not currently targeted by any pharmaceutical. On the basis of these criteria, we selected for validation NMNAT (PlasmoDB ID PF13_0159, EC 2.7.7.18) (Table IV). This enzyme, a member of the plasmodial NAD synthesis and recycling pathway, catalyzes the conversion of nicotinic acid mononucleotide to nicotinic acid adenine dinucleotide (Figure 2A). NMNAT has recently been the focus of novel anti-microbial agent development because of structural and metabolic differences between the enzyme in microbial and human cells (Magni et al, 2009). As NAD(P) is one of the most promiscuous redox molecules in metabolism, as well as a cofactor for important histone regulatory proteins such as sirtuins (Merrick and Duraisingh, 2007), inhibition of NAD(P) synthesis and recycling should have a profound impact on parasite metabolism. However, to the best of our knowledge, this pathway has not been previously targeted by pharmaceutical interventions in P. falciparum.
Figure 2.

Small-molecule inhibition of the parasite nicotinate mononucleotide adenylyltransferase (NMNAT). (A) Schematic of the P. falciparum NAD(P) synthesis and recycling pathway determined from the genome sequence. Nicotinamide (NM) and nicotinic acid (NA) can be scavenged from the host. Compound 1_03 is an inhibitor targeting NMNAT. (B) Compound 1_03 causes growth arrest of intraerythrocytic P. falciparum. Cultures were resuspended in niacin-free medium containing 0 or 100 μM of compound 1_03 at early ring stage and observed for 66 h (see Materials and methods). Untreated parasites undergo normal development and reinvasion, whereas drug-treated parasites arrest at the trophozoite (‘troph’) stage and do not reinvade. NM, nicotinamide; NA, nicotinic acid; NaMN, nicotinate mononucleotide; NaAD, nicotinate adenine dinucleotide; NAD(P)+, nicotinamide adenine dinucleotide (phosphate), reduced; NMase, nicotinamidase; NPRT, nicotinate phosphoribosyltransferase; NMNAT, nicotinate mononucleotide adenylyltransferase; NADS, NAD synthase; NADK, NAD kinase.
Recently, Sorci et al (2009) used a combination of in silico structure modeling and enzyme inhibition assays to identify several classes of small molecules that inhibit bacterial (Escherichia coli and B. subtilis) but not human NMNAT. Several of these drug candidates were able to inhibit bacterial growth in culture. We tested two of the designed candidate compounds (1_03 and 3_02), representing two different chemotypes, for their ability to inhibit P. falciparum growth using both the SYBR Green I fluorescence assay (Smilkstein et al, 2004) to measure DNA synthesis, and microscopic examination of morphological effects. The two compounds were tested at a range of concentrations for growth inhibitory effects in nicotinamide-free culture medium, so as not to rescue any metabolic blocks induced by the drugs. Nicotinamide removal has been previously shown not to affect normal growth in culture (Divo et al, 1985), which we confirmed before running our growth assay experiments (data not shown). Although the compound 3_02 did not significantly affect parasite's growth at moderate concentrations (MIC50 >100 μM), the compound 1_03 exerted an inhibitory effect in a growth assay (MIC50 = 50 μM; Supplementary Figure S2) comparable to that previously observed for bacteria (MIC50 >80 μM for E. coli, MIC50 =10 μM for B. subtilis). At 100 μM, the compound 1_03 completely blocked host cell escape and reinvasion by arresting parasites in the trophozoite growth stage (Figure 2B). Importantly, the human NMNAT isoforms are insensitive to the compound at least up to the concentrations used in our assay (Sorci et al, 2009). This suggests that the parasite NMNAT enzyme and, more generally, the NAD(P) synthesis pathway are indeed potentially effective and druggable targets. The experimental results also highlight the ability of our model to identify promising candidates for pharmaceutical intervention.
Prediction of metabolite concentration changes based on expression data
Genome-scale metabolic networks can be used not only to predict the effects of gene deletions, but also as a tool for the integration of diverse genomic and physiological data (Breitling et al, 2008; Oberhardt et al, 2009). For example, information on nutrient availability, uptake rates, and maximal rates of internal reactions can be used to further constrain the space of feasible metabolic fluxes. To illustrate the ability of the model to combine genomic data, we investigated whether available gene-expression data sets can be used to predict shifts in concentrations of external metabolites caused by the P. falciparum exchange fluxes at different developmental stages. Investigation of the exchange fluxes is essential for understanding perturbations caused by parasitic infections in the metabolic state of their host tissues, and, consequently, main mechanisms of pathogenesis. As the FBA operates in the space of the fluxes and not in the space of metabolic concentrations, the model cannot be directly used to predict absolute concentration changes. Nevertheless, it is possible to use the model to investigate the direction of concentration changes for external metabolites, following the simple logic that an increase in the uptake rate or decrease in the excretion (output) rate should lead to a drop in the concentration of the corresponding external metabolite; similarly, an uptake rate decrease or excretion rate increase should increase the metabolite concentrations.
Although gene-expression level does not perfectly correlate with the flux through the associated enzyme (Daran-Lapujade et al, 2004; Shlomi et al, 2008), the recent study by Colijn et al (2009) demonstrated that mRNA abundance data, if used as additional constraints on maximal reaction fluxes, can significantly improve stoichiometric model predictions. For instance, if the expression level of a particular enzyme is low, it is unlikely that the enzyme will be used by the metabolic network to carry a large flux. Consequently, it should be possible to use gene-expression data to obtain a more accurate view of the in vivo metabolic state. To test this, we used DNA microarray results collected from synchronized cultures of the P. falciparum 3D7 strain during the RBC phase of the parasite's life cycle (Llinas et al, 2006; Olszewski et al, 2009). The expression data were collected at the ring, trophozoite, and schizont developmental stages (see Materials and methods). Following Colijn et al (2009), the maximum flux allowed through enzymes was constrained proportionally to the relative expression level of the corresponding genes.
We compared the accuracy of our predictions to the experimentally measured metabolic changes in Plasmodium-infected RBCs (Olszewski et al, 2009). In Figure 3, we show the predicted and experimentally measured changes, indicating either an increase or decrease in metabolic concentrations for the transition from the ring to trophozoite and from trophozoite to schizont stages. The predicted shifts in metabolic concentrations agree with the experimental results in 70% of the measurements (binomial, P-value=9 × 10−4). In addition, we found a significant correlation between the magnitudes of the change in metabolite concentrations and the predicted flux values (Pearson's correlation: 0.34, P-value=6 × 10−3, Spearman's correlation: 0.25, P-value=0.04).
Figure 3.

Comparison between the predicted and experimentally measured shifts in metabolite concentrations in infected red blood cells. UP/DOWN indicates direction of experimentally measured changes in metabolic concentrations in infected versus uninfected cells. Blue color indicates agreement between experiment and predictions, whereas yellow indicates disagreement. In most cases (70%, P-value=9 × 10−4), the shifts in metabolic concentrations from one stage to the next can be predicted based on changes in the P. falciparum metabolic exchange fluxes. The in silico predictions of exchange fluxes were made based on the expression-constrained flux-balance analysis (Colijn et al, 2009). Briefly, for genes with available mRNA-expression data, the maximum flux through the associated metabolic reactions was constrained proportionally to their expression level; with the highest expression value corresponding to the maximum allowed flux.
In order to further investigate the statistical significance of the results, we repeated flux predictions after randomly shuffling expression values between P. falciparum genes. In only 2% of these random trials, the accuracy of the predictions made with the shuffled data were higher than those obtained using the original expression values (Supplementary Figure S3). To explore the effects of multiple optimal FBA solutions (Mahadevan and Schilling, 2003) on the prediction accuracy, we used the centering hit-and-run algorithm (Kaufman and Smith, 1998), implemented in the COBRA toolbox (Becker et al, 2007), to randomly sample the solution space associated with the expression constraints. The 70% accuracy value, obtained for a single solution, is close to the mean of solutions sampled from alternative optima (mean 0.69, s.d. 0.05; see Supplementary Figure S3). Moreover, there is a significant difference (Mann–Whitney U, P-value<10−10) between the results for randomized expression values and those based on the multiple alternative optima. These results illustrate the ability of the model, with appropriate constraints, to predict physiological changes unrelated to gene knockouts. It also suggests that expression and metabolomics measurements, which are being rapidly accumulated for various stages of parasite growth (Winzeler, 2008; Kafsack and Llinas, 2010), can be integrated with the model to gain a better understanding of the P. falciparum physiology.
Discussion
The presented flux-balanced model can serve as a valuable tool for quantitative predictions of P. falciparum metabolic states under various growth conditions and perturbations. The results of in silico gene deletions demonstrate that the model achieves high accuracy in reproducing available experimental measurements. In addition, our analysis suggests several dozen essential metabolic targets for therapeutic intervention. Although several studies that assemble and analyze plasmodial metabolic pathways have been performed previously (Yeh et al, 2004; Ginsburg, 2006, 2009, 2010), our contribution is important because the genome-scale model can be used to investigate and predict genetic perturbations from a network-level perspective.
Interestingly, our results suggest a limited degree of robustness in the P. falciparum network, which should lead to a relatively high-success rate for inhibitors of metabolic genes. It is possible that the small robustness of the reconstructed model, for example in comparison with the yeast metabolic network, is due primarily to unannotated P. falciparum genes without significant homology to known enzymes in other organisms. To investigate this possibility further, we used the available collection of single metabolic gene knockouts/inhibitions in P. falciparum or P. berghei (Table III) and all metabolic gene knockouts in S. cerevisiae (Giaever et al, 2002). We calculated the fraction of orthologs for essential metabolic knockouts in the parasite, which are also essential in yeast, and, vice versa, the fraction of orthologs for essential metabolic knockouts in yeast, which are essential in the parasite (see Supplementary Table S5). Interestingly, while only about half of the orthologs for essential metabolic genes in P. falciparum are also essential in S. cerevisiae, all essential metabolic genes we analyzed in yeast are essential in the parasite (Supplementary Table S5; Fisher's exact test, P=0.04). This result independently supports the conclusion of the flux-balance simulations about the relatively small robustness of the P. falciparum network.
We anticipate several immediate extensions of our work. First, the presented network can be used for effective integration of multiple genomic data types. For example, known regulatory interactions can be incorporated into the model (Covert and Palsson, 2002). Accurate measurements of gene expression (Hu et al, 2010), key protein–DNA regulatory interactions (De Silva et al, 2008), and post-translational modifications (Chung et al, 2009) in the parasite will be especially important for modeling the dynamic behavior of the network under varying environmental conditions. Second, it will be important to model exchanges and interactions between the metabolic networks of the parasite and its hosts. The analysis of the combined parasite–host metabolic network should significantly improve understanding of the P. falciparum vulnerabilities. For example, several host cell enzymes are actively used by the parasite during its life cycle (Dhanasekaran et al, 2004; Ting et al, 2005). Although we did not consider these human enzymes in our analysis, they can be easily included in future applications of the model. The available global flux-balanced metabolic human network (Duarte et al, 2007), metabolic network specifically active in the liver (Zhao et al, 2010), and well-curated models of human RBC metabolism (Joshi and Palsson, 1989a, 1989b, 1990a, 1990b; Jamshidi et al, 2001) make such combined analyses possible. Third, it will be interesting to reconstruct stoichiometric metabolic networks for other clinically relevant Plasmodium species (e.g. P. vivax, P. malariae, P. ovale, and P. knowlesi) as well as the important model species P. berghei and Plasmodium yoelii. The comparative analysis of these networks may reveal important physiological and evolutionary differences between Plasmodium spp., and also help in the identification of common metabolic drug targets.
Taking into account the global health burden of malaria, it is essential to develop and implement new effective pharmaceuticals as quickly as possible. Systems biology approaches can be used to significantly facilitate drug identification and development (Yao and Rzhetsky, 2008; Kuhn et al, 2010). To date, we have only begun to see the application of such integrative methods in the context of malaria research (reviewed in Dharia et al, 2010). We believe that the presented network represents an important step in this direction. The experimental validation of a candidate drug, compound 1_03, targeting the parasite NMNAT illustrates the ability of the model to speed up development of novel anti-malaria targets and pharmaceuticals. Importantly, although the identified compound is available, it has not been previously tested against Plasmodium spp. Even though 1_03 inhibited parasite growth only at relatively high concentrations (MIC50=50 μM), these were comparable to the inhibitory concentrations for the bacteria against which the drug was initially developed (Sorci et al, 2009). The incomplete growth inhibition at lower compound concentrations could be explained by incomplete drug inhibition. Our model predicts linear decrease in the P. falciparum biomass production as the level of NMNAT inhibition increases; for example, 90% inhibition results in 90% growth decrease. As Sorci et al initially screened for compounds that could selectively inhibit pure NMNAT enzyme, these compounds have not been optimized for cellular permeability, accumulation, or other pharmacokinetic parameters, and thus should primarily serve as a structural basis for further malaria drug development.
Future improvements to the reconstructed P. falciparum metabolic network, including adding experimental details for missing activities and precise metabolic measurements necessary to describe the growth-related objective function, will lead to a better understanding of parasite physiology. Ultimately, such models should significantly accelerate the identification of desperately needed new drug leads against this devastating disease.
Materials and methods
Genome-scale metabolic reconstruction
The reconstruction process started with the identification of enzyme-coding genes in the P. falciparum genome. We considered a variety of resources, including PlasmoDB (Aurrecoechea et al, 2009), the MPMP (Ginsburg, 2006), PlasmoCyc (Yeh et al, 2004), and KEGG (Kanehisa et al, 2008). The identified enzymes were mapped to the corresponding metabolic reactions by consulting several well-studied metabolic models, including the iAF1260 model for E. coli (Feist et al, 2007), the iND750 model for S. cerevisiae (Duarte et al, 2004), the genome-scale human metabolic network by Duarte et al (2007), and the KEGG database (Kanehisa et al, 2008). On the basis of this set of enzymatic activities and their stoichiometry, we used FBA to see if the network was able to produce a set of basic biomass components; e.g. amino acids, lipids, nucleotides, and cofactors. For each metabolite that the network was unable to synthesize, we searched the literature for relevant publications concerning pathways and genes associated with the metabolite production or transport (relevant publications are included as notes in Supplementary information). Network enzymes were assigned to different cellular compartments based on experimental evidence, when available, and on computationally predicted localization information (Waller et al, 1998; Ralph et al, 2004; Chan et al, 2006; van Dooren et al, 2006). Transport and exchange reactions reported in the literature or in databases such as PlasmoDB and MPMP were initially included in the model. We added additional transport reactions required for production of the biomass components. All metabolic and transport reactions were used to formulate a stoichiometric flux-balance model (Edwards et al, 1999). The model was improved following an iterative procedure as previously described (Feist et al, 2009; Thiele and Palsson, 2010).
The assembled network was manually inspected and compared with the MPMP (Ginsburg, 2010). Metabolic network gaps (Kharchenko et al, 2006) were identified and included in the assembled network model (Mullin et al, 2006; Quashie et al, 2008). The reactions for which no literature support is available and which are not essential for the biomass production were removed from the network. Additional adjustments related to reaction directionalities and metabolite availabilities were made following the computational analyses described in this work.
As the P. falciparum biomass objective function cannot be completely established based on the available literature, in our calculations we used a modified version of the yeast objective function reported in the iND750 model (Duarte et al, 2004). The objective function modifications included the lipid composition, which was adjusted as reported for Plasmodium (Hsiao et al, 1991), and amino acid and nucleotide compositions adjusted based on the proteome and genome sequences weighted by available expression data (Llinas et al, 2006). In particular, the percent prevalence of each ribonucleotide and amino acid across all open reading frames (ORFs) was calculated as the relative frequency of each monomer; the counts at each ORF were multiplied by the ORF's expression level (when available). The percent prevalence of the dNTPs was derived from the genome A+T content of 80.6%. These percentages were converted to mmol/gDW as described (Chavali et al, 2008). Systems Biology Research Tool (Wright and Wagner, 2008) was used to perform FBA (Edwards et al, 1999) of the network, including single and double in silico deletions of network enzymes. The reconstructed network was able to either synthesize or import all the biomass components presented in Supplementary Table S1. The assembled metabolic model is available as an Excel spreadsheet (Supplementary information) and in the Systems Biology Markup Language (SBML) format (Supplementary information). The SBML model was submitted to the BioModels database (Le Novere et al, 2006) with accession number MODEL1007060000.
Parasite culture, growth, and drug inhibition assays
The cultures of P. falciparum were maintained and synchronized by standard methods (Trager and Jensen, 1976; Lambros and Vanderberg, 1979). Briefly, RBCs, infected by P. falciparum (3D7 strain), were grown in the RPMI 1640 culture medium supplemented with sodium carbonate (2 mg/ml), hypoxanthine (100 μM), Albumax II (0.25%), and gentamycin (50 μg/ml) in a humidified incubator at 5% CO2, 6% O2, and 37°C. The growth synchronizations were carried out by incubating parasite-infected RBCs in phosphate-buffered saline (PBS) containing 5% w/v sorbitol for 5 minutes at room temperature, washing once with sorbitol-free PBS, and resuspending in culture medium.
The compounds 1_03 and 3_02 were acquired from ChemDiv (http://chemdiv.emolecules.com; ChemDiv IDs 8003-6329 and 5350-0029, respectively) and resuspended at 100 mM in DMSO. Growth inhibition studies were carried out using the SYBR Green I fluorescence assay (Smilkstein et al, 2004). Briefly, synchronized parasite cultures (early ring stage, 1% parasitemia, 1% hematocrit, 100 μl total volume) were suspended in nicotinamide-free RPMI 1640 containing 0.1% DMSO and differing concentrations of drug in 96-well plates. After 72 h incubation, the plates were frozen at −80°C overnight, then thawed and mixed with 100 μl lysis buffer (20 mM Tris–HCl, pH 7.5; 5 mM EDTA; 0.008% w/v saponin; 0.08% v/v Triton X-100; 1 × SYBR Green I) per well, incubated 1 h at room temperature and quantified using a BioTek SynergyMX plate reader (excitation 488 nm, emission 522 nm). The concentrations 0, 0.1, 1, 5, 10, 50, 100, and 250 μM were tested in triplicate in two independent growth assays.
Using an expression-constrained network to predict shifts in external metabolite concentrations
In the expression-constrained flux analysis, we used P. falciparum expression data (Olszewski et al, 2009) as described previously in Colijn et al (2009). Specifically, for genes with available mRNA-expression data, the maximum flux through the associated metabolic reactions was constrained proportionally to their expression level; with the highest expression value corresponding to the maximum possible metabolic flux. In order to obtain absolute expression values, rather than the ratios between the microarray intensities at a given time point and those of a pooled sample, we multiplied each ratio with the average sum of the median intensity across the full intraerythrocytic developmental cycle (Llinas et al, 2006).
The ring, throphozoite, and schizont developmental stages in cultures were defined for hours 1–18, 19–30, and 31–48 after synchronization with D-sorbitol, respectively. In the analysis, we used intracellular RBC metabolite concentration data obtained by Olszewski et al (2009). We compared the changes in metabolite abundances between infected and uninfected RBCs, from one development stage to the next. For each metabolite, the predicted concentration changes were considered to agree with experimental data, if the metabolite consumption in the network increased (or the metabolite production decreased) when the experimentally measured metabolite concentration decreased, or alternatively, if consumption of the metabolite decreased (or production increased) when the metabolite concentration increased.
Supplementary Material
Supplementary figures S1–3, Supplementary tables S1–5
Acknowledgments
We thank the members of DV and ML groups for stimulating discussions and comments on the manuscript. Research was funded in part by NIH grant GM079759 and BU GC207272NGC to DV, and a National Centers for Biomedical Computing (MAGNet) grant U54CA121852 to Columbia University. ML is funded in part by the Burroughs Welcome Fund, an NIH Director's New Innovators award (1DP2OD001315-01) and receives support from the Center for Quantitative Biology (P50 GM071508). KO is supported by an NSF Graduate Research Fellowship.
Footnotes
The authors declare that they have no conflict of interest.
References
- Aly AS, Vaughan AM, Kappe SH (2009) Malaria parasite development in the mosquito and infection of the mammalian host. Annu Rev Microbiol 63: 195–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Nayak V, Pennington C, Pinney DF et al. (2009) PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res 37: D539–D543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird JK (2005) Effectiveness of antimalarial drugs. N Engl J Med 352: 1565–1577 [DOI] [PubMed] [Google Scholar]
- Becker SA, Feist AM, Mo ML, Hannum G, Palsson BO, Herrgard MJ (2007) Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat Protoc 2: 727–738 [DOI] [PubMed] [Google Scholar]
- Berwal R, Gopalan N, Chandel K, Prasad GB, Prakash S (2008) Plasmodium falciparum: enhanced soluble expression, purification and biochemical characterization of lactate dehydrogenase. Exp Parasitol 120: 135–141 [DOI] [PubMed] [Google Scholar]
- Bhanot P, Schauer K, Coppens I, Nussenzweig V (2005) A surface phospholipase is involved in the migration of plasmodium sporozoites through cells. J Biol Chem 280: 6752–6760 [DOI] [PubMed] [Google Scholar]
- Bonday ZQ, Taketani S, Gupta PD, Padmanaban G (1997) Heme biosynthesis by the malarial parasite. Import of delta-aminolevulinate dehydrase from the host red cell. J Biol Chem 272: 21839–21846 [DOI] [PubMed] [Google Scholar]
- Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, DeRisi JL (2003) The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol 1: E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling R, Vitkup D, Barrett MP (2008) New surveyor tools for charting microbial metabolic maps. Nat Rev Microbiol 6: 156–161 [DOI] [PubMed] [Google Scholar]
- Bulusu V, Srinivasan B, Bopanna MP, Balaram H (2009) Elucidation of the substrate specificity, kinetic and catalytic mechanism of adenylosuccinate lyase from Plasmodium falciparum. Biochim Biophys Acta 1794: 642–654 [DOI] [PubMed] [Google Scholar]
- Carvalho TG, Menard R (2005) Manipulating the Plasmodium genome. Curr Issues Mol Biol 7: 39–55 [PubMed] [Google Scholar]
- Cassera MB, Merino EF, Peres VJ, Kimura EA, Wunderlich G, Katzin AM (2007) Effect of fosmidomycin on metabolic and transcript profiles of the methylerythritol phosphate pathway in Plasmodium falciparum. Mem Inst Oswaldo Cruz 102: 377–383 [DOI] [PubMed] [Google Scholar]
- Chakrabarti D, Schuster SM, Chakrabarti R (1993) Cloning and characterization of subunit genes of ribonucleotide reductase, a cell-cycle-regulated enzyme, from Plasmodium falciparum. Proc Natl Acad Sci USA 90: 12020–12024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan M, Tan DS, Wong SH, Sim TS (2006) A relevant in vitro eukaryotic live-cell system for the evaluation of plasmodial protein localization. Biochimie 88: 1367–1375 [DOI] [PubMed] [Google Scholar]
- Chavali AK, Whittemore JD, Eddy JA, Williams KT, Papin JA (2008) Systems analysis of metabolism in the pathogenic trypanosomatid Leishmania major. Mol Syst Biol 4: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DW, Ponts N, Cervantes S, Le Roch KG (2009) Post-translational modifications in Plasmodium: more than you think!. Mol Biochem Parasitol 168: 123–134 [DOI] [PubMed] [Google Scholar]
- Colijn C, Brandes A, Zucker J, Lun DS, Weiner B, Farhat MR, Cheng TY, Moody DB, Murray M, Galagan JE (2009) Interpreting expression data with metabolic flux models: predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput Biol 5: e1000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert MW, Palsson BO (2002) Transcriptional regulation in constraints-based metabolic models of Escherichia coli. J Biol Chem 277: 28058–28064 [DOI] [PubMed] [Google Scholar]
- Daran-Lapujade P, Jansen ML, Daran JM, van Gulik W, de Winde JH, Pronk JT (2004) Role of transcriptional regulation in controlling fluxes in central carbon metabolism of Saccharomyces cerevisiae. A chemostat culture study. J Biol Chem 279: 9125–9138 [DOI] [PubMed] [Google Scholar]
- Das Gupta R, Krause-Ihle T, Bergmann B, Muller IB, Khomutov AR, Muller S, Walter RD, Luersen K (2005) 3-Aminooxy-1-aminopropane and derivatives have an antiproliferative effect on cultured Plasmodium falciparum by decreasing intracellular polyamine concentrations. Antimicrob Agents Chemother 49: 2857–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, Cochran DA, Emmerson BT, Gordon RB (1993) Inhibition of Plasmodium falciparum hypoxanthine-guanine phosphoribosyltransferase mRNA by antisense oligodeoxyucleotide sequence. Mol Biochem Parasitol 60: 153–156 [DOI] [PubMed] [Google Scholar]
- Deng X, Gujjar R, El Mazouni F, Kaminsky W, Malmquist NA, Goldsmith EJ, Rathod PK, Phillips MA (2009) Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J Biol Chem 284: 26999–27009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva EK, Gehrke AR, Olszewski K, Leon I, Chahal JS, Bulyk ML, Llinas M (2008) Specific DNA-binding by apicomplexan AP2 transcription factors. Proc Natl Acad Sci USA 105: 8393–8398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessens JT, Mendoza J, Claudianos C, Vinetz JM, Khater E, Hassard S, Ranawaka GR, Sinden RE (2001) Knockout of the rodent malaria parasite chitinase pbCHT1 reduces infectivity to mosquitoes. Infect Immun 69: 4041–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran S, Chandra NR, Chandrasekhar Sagar BK, Rangarajan PN, Padmanaban G (2004) Delta-aminolevulinic acid dehydratase from Plasmodium falciparum: indigenous versus imported. J Biol Chem 279: 6934–6942 [DOI] [PubMed] [Google Scholar]
- Dharia NV, Chatterjee A, Winzeler EA (2010) Genomics and systems biology in malaria drug discovery. Curr Opin Invest Drugs 11: 131–138 [PubMed] [Google Scholar]
- Divo AA, Geary TG, Davis NL, Jensen JB (1985) Nutritional requirements of Plasmodium falciparum in culture. I. Exogenously supplied dialyzable components necessary for continuous growth. J Protozool 32: 59–64 [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, Boone C (2009) Systematic mapping of genetic interaction networks. Annu Rev Genet 43: 601–625 [DOI] [PubMed] [Google Scholar]
- Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson BO (2007) Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci USA 104: 1777–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte NC, Herrgard MJ, Palsson BO (2004) Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Res 14: 1298–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JS, Ramakrishna R, Schilling CH, B.O. P (1999) Metabolic flux balance analysis. In Metabolic Engineering, Lee Sypet (ed), pp 13–57. New York, NY: Marcel Dekker Inc [Google Scholar]
- Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BO (2007) A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol 3: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM, Herrgard MJ, Thiele I, Reed JL, Palsson BO (2009) Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol 7: 129–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, Witney AA, Wolters D, Wu Y, Gardner MJ, Holder AA, Sinden RE, Yates JR, Carucci DJ (2002) A proteomic view of the Plasmodium falciparum life cycle. Nature 419: 520–526 [DOI] [PubMed] [Google Scholar]
- Flores MV, Atkins D, Wade D, O′Sullivan WJ, Stewart TS (1997) Inhibition of Plasmodium falciparum proliferation in vitro by ribozymes. J Biol Chem 272: 16940–16945 [DOI] [PubMed] [Google Scholar]
- Foth BJ, Stimmler LM, Handman E, Crabb BS, Hodder AN, McFadden GI (2005) The malaria parasite Plasmodium falciparum has only one pyruvate dehydrogenase complex, which is located in the apicoplast. Mol Microbiol 55: 39–53 [DOI] [PubMed] [Google Scholar]
- Francis SE, Sullivan DJ Jr, Goldberg DE (1997) Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol 51: 97–123 [DOI] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ et al. (2002) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419: 498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A et al. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391 [DOI] [PubMed] [Google Scholar]
- Ginsburg H (2006) Progress in in silico functional genomics: the malaria Metabolic Pathways database. Trends Parasitol 22: 238–240 [DOI] [PubMed] [Google Scholar]
- Ginsburg H (2009) Caveat emptor: limitations of the automated reconstruction of metabolic pathways in Plasmodium. Trends Parasitol 25: 37–43 [DOI] [PubMed] [Google Scholar]
- Ginsburg H (2010) Malaria Parasite Metabolic Pathways, http://sites.huji.ac.il/malaria/ [DOI] [PubMed] [Google Scholar]
- Haider N, Eschbach ML, Dias Sde S, Gilberger TW, Walter RD, Luersen K (2005) The spermidine synthase of the malaria parasite Plasmodium falciparum: molecular and biochemical characterisation of the polyamine synthesis enzyme. Mol Biochem Parasitol 142: 224–236 [DOI] [PubMed] [Google Scholar]
- Haldar K, Mohandas N (2009) Malaria, erythrocytic infection, and anemia. Hematology Am Soc Hematol Educ Program 2009: 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Palacpac NM, Magistrado PA, Kurokawa K, Rai G, Sakata D, Hara T, Horii T, Nishijima M, Mitamura T (2002) Plasmodium falciparum phospholipase C hydrolyzing sphingomyelin and lysocholinephospholipids is a possible target for malaria chemotherapy. J Exp Med 195: 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CS, Zinner JF, Cohoon MP, Stevens RL (2009) iBsu1103: a new genome-scale metabolic model of Bacillus subtilis based on SEED annotations. Genome Biol 10: R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai M, Arai M, Kawai S, Matsuoka H (2006) PbGCbeta is essential for Plasmodium ookinete motility to invade midgut cell and for successful completion of parasite life cycle in mosquitoes. J Biochem 140: 747–757 [DOI] [PubMed] [Google Scholar]
- Ho MC, Cassera MB, Madrid DC, Ting LM, Tyler PC, Kim K, Almo SC, Schramm VL (2009) Structural and metabolic specificity of methylthiocoformycin for malarial adenosine deaminases. Biochemistry 48: 9618–9626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao LL, Howard RJ, Aikawa M, Taraschi TF (1991) Modification of host cell membrane lipid composition by the intra-erythrocytic human malaria parasite Plasmodium falciparum. Biochem J 274 (Part 1): 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Cabrera A, Kono M, Mok S, Chaal BK, Haase S, Engelberg K, Cheemadan S, Spielmann T, Preiser PR, Gilberger TW, Bozdech Z (2010) Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat Biotechnol 28: 91–98 [DOI] [PubMed] [Google Scholar]
- Hyde JE (2007) Drug-resistant malaria—an insight. FEBS J 274: 4688–4698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JE, Dittrich S, Wang P, Sims PF, de Crecy-Lagard V, Hanson AD (2008) Plasmodium falciparum: a paradigm for alternative folate biosynthesis in diverse microorganisms? Trends Parasitol 24: 502–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamshidi N, Edwards JS, Fahland T, Church GM, Palsson BO (2001) Dynamic simulation of the human red blood cell metabolic network. Bioinformatics 17: 286–287 [DOI] [PubMed] [Google Scholar]
- Janse CJ, Ramesar J, Waters AP (2006) High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc 1: 346–356 [DOI] [PubMed] [Google Scholar]
- Janse CJ, Waters AP (1995) Plasmodium berghei: the application of cultivation and purification techniques to molecular studies of malaria parasites. Parasitol Today 11: 138–143 [DOI] [PubMed] [Google Scholar]
- Jiang L, Lee PC, White J, Rathod PK (2000) Potent and selective activity of a combination of thymidine and 1843U89, a folate-based thymidylate synthase inhibitor, against Plasmodium falciparum. Antimicrob Agents Chemother 44: 1047–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Palsson BO (1989a) Metabolic dynamics in the human red cell. Part I--A comprehensive kinetic model. J Theor Biol 141: 515–528 [DOI] [PubMed] [Google Scholar]
- Joshi A, Palsson BO (1989b) Metabolic dynamics in the human red cell. Part II--Interactions with the environment. J Theor Biol 141: 529–545 [DOI] [PubMed] [Google Scholar]
- Joshi A, Palsson BO (1990a) Metabolic dynamics in the human red cell. Part III--Metabolic reaction rates. J Theor Biol 142: 41–68 [DOI] [PubMed] [Google Scholar]
- Joshi A, Palsson BO (1990b) Metabolic dynamics in the human red cell. Part IV--Data prediction and some model computations. J Theor Biol 142: 69–85 [DOI] [PubMed] [Google Scholar]
- Kafsack BF, Llinas M (2010) Eating at the table of another: metabolomics of host-parasite interactions. Cell Host Microbe 7: 90–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36: D480–D484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman DE, Smith RL (1998) Direction choice for accelerated convergence in hit-and-run sampling. Oper Res 46: 84–95 [Google Scholar]
- Kharchenko P, Chen L, Freund Y, Vitkup D, Church GM (2006) Identifying metabolic enzymes with multiple types of association evidence. BMC Bioinformatics 7: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kicska GA, Tyler PC, Evans GB, Furneaux RH, Schramm VL, Kim K (2002) Purine-less death in Plasmodium falciparum induced by immucillin-H, a transition state analogue of purine nucleoside phosphorylase. J Biol Chem 277: 3226–3231 [DOI] [PubMed] [Google Scholar]
- Kirk K, Staines HM, Martin RE, Saliba KJ (1999) Transport properties of the host cell membrane. Novartis Found Symp 226: 55–66; discussion 66–73 [DOI] [PubMed] [Google Scholar]
- Koncarevic S, Rohrbach P, Deponte M, Krohne G, Prieto JH, Yates J III, Rahlfs S, Becker K (2009) The malarial parasite Plasmodium falciparum imports the human protein peroxiredoxin 2 for peroxide detoxification. Proc Natl Acad Sci USA 106: 13323–13328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krnajski Z, Gilberger TW, Walter RD, Cowman AF, Muller S (2002) Thioredoxin reductase is essential for the survival of Plasmodium falciparum erythrocytic stages. J Biol Chem 277: 25970–25975 [DOI] [PubMed] [Google Scholar]
- Krungkrai J, Krungkrai SR, Supuran CT (2008) Carbonic anhydrase inhibitors: inhibition of Plasmodium falciparum carbonic anhydrase with aromatic/heterocyclic sulfonamides-in vitro and in vivo studies. Bioorg Med Chem Lett 18: 5466–5471 [DOI] [PubMed] [Google Scholar]
- Kuhn M, Campillos M, Letunic I, Jensen LJ, Bork P (2010) A side effect resource to capture phenotypic effects of drugs. Mol Syst Biol 6: 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambros C, Vanderberg JP (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65: 418–420 [PubMed] [Google Scholar]
- Lasonder E, Ishihama Y, Andersen JS, Vermunt AM, Pain A, Sauerwein RW, Eling WM, Hall N, Waters AP, Stunnenberg HG, Mann M (2002) Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature 419: 537–542 [DOI] [PubMed] [Google Scholar]
- Lasonder E, Janse CJ, van Gemert GJ, Mair GR, Vermunt AM, Douradinha BG, van Noort V, Huynen MA, Luty AJ, Kroeze H, Khan SM, Sauerwein RW, Waters AP, Mann M, Stunnenberg HG (2008) Proteomic profiling of Plasmodium sporozoite maturation identifies new proteins essential for parasite development and infectivity. PLoS Pathog 4: e1000195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Novere N, Bornstein B, Broicher A, Courtot M, Donizelli M, Dharuri H, Li L, Sauro H, Schilstra M, Shapiro B, Snoep JL, Hucka M (2006) BioModels Database: a free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res 34: D689–D691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, De La Vega P, Holder AA, Batalov S, Carucci DJ, Winzeler EA (2003) Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301: 1503–1508 [DOI] [PubMed] [Google Scholar]
- LeRoux M, Lakshmanan V, Daily JP (2009) Plasmodium falciparum biology: analysis of in vitro versus in vivo growth conditions. Trends Parasitol 25: 474–481 [DOI] [PubMed] [Google Scholar]
- Lim L, McFadden GI (2010) The evolution, metabolism and functions of the apicoplast. Philos Trans R Soc Lond B Biol Sci 365: 749–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Mu J, Jiang H, Su XZ (2008) Effects of Plasmodium falciparum mixed infections on in vitro antimalarial drug tests and genotyping. Am J Trop Med Hyg 79: 178–184 [PMC free article] [PubMed] [Google Scholar]
- Llinas M, Bozdech Z, Wong ED, Adai AT, DeRisi JL (2006) Comparative whole genome transcriptome analysis of three Plasmodium falciparum strains. Nucleic Acids Res 34: 1166–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon MJ, Marsh K (2010) The selection landscape of malaria parasites. Science 328: 866–871 [DOI] [PubMed] [Google Scholar]
- Magni G, Di Stefano M, Orsomando G, Raffaelli N, Ruggieri S (2009) NAD(P) biosynthesis enzymes as potential targets for selective drug design. Curr Med Chem 16: 1372–1390 [DOI] [PubMed] [Google Scholar]
- Mahadevan R, Schilling CH (2003) The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab Eng 5: 264–276 [DOI] [PubMed] [Google Scholar]
- Martin RE, Henry RI, Abbey JL, Clements JD, Kirk K (2005) The ‘permeome’ of the malaria parasite: an overview of the membrane transport proteins of Plasmodium falciparum. Genome Biol 6: R26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRobert L, McConkey GA (2002) RNA interference (RNAi) inhibits growth of Plasmodium falciparum. Mol Biochem Parasitol 119: 273–278 [DOI] [PubMed] [Google Scholar]
- Merrick CJ, Duraisingh MT (2007) Plasmodium falciparum Sir2: an unusual sirtuin with dual histone deacetylase and ADP-ribosyltransferase activity. Eukaryot Cell 6: 2081–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi-Ichi F, Kita K, Mitamura T (2006) Intraerythrocytic Plasmodium falciparum utilize a broad range of serum-derived fatty acids with limited modification for their growth. Parasitology 133: 399–410 [DOI] [PubMed] [Google Scholar]
- Mukkamala D, No JH, Cass LM, Chang TK, Oldfield E (2008) Bisphosphonate inhibition of a Plasmodium farnesyl diphosphate synthase and a general method for predicting cell-based activity from enzyme data. J Med Chem 51: 7827–7833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller IB, Das Gupta R, Luersen K, Wrenger C, Walter RD (2008) Assessing the polyamine metabolism of Plasmodium falciparum as chemotherapeutic target. Mol Biochem Parasitol 160: 1–7 [DOI] [PubMed] [Google Scholar]
- Mullin KA, Lim L, Ralph SA, Spurck TP, Handman E, McFadden GI (2006) Membrane transporters in the relict plastid of malaria parasites. Proc Natl Acad Sci USA 103: 9572–9577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navid A, Almaas E (2009) Genome-scale reconstruction of the metabolic network in Yersinia pestis, strain 91001. Mol Biosyst 5: 368–375 [DOI] [PubMed] [Google Scholar]
- Nerlich AG, Schraut B, Dittrich S, Jelinek T, Zink AR (2008) Plasmodium falciparum in ancient Egypt. Emerg Infect Dis 14: 1317–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen C, Kasinathan G, Leal-Cortijo I, Musso-Buendia A, Kaiser M, Brun R, Ruiz-Perez LM, Johansson NG, Gonzalez-Pacanowska D, Gilbert IH (2005) Deoxyuridine triphosphate nucleotidohydrolase as a potential antiparasitic drug target. J Med Chem 48: 5942–5954 [DOI] [PubMed] [Google Scholar]
- Oberhardt MA, Palsson BO, Papin JA (2009) Applications of genome-scale metabolic reconstructions. Mol Syst Biol 5: 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski KL, Mather MW, Morrisey JM, Garcia BA, Vaidya AB, Rabinowitz JD, Llinas M (2010) Branched tricarboxylic acid metabolism in Plasmodium falciparum. Nature 466: 774–778 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Olszewski KL, Morrisey JM, Wilinski D, Burns JM, Vaidya AB, Rabinowitz JD, Llinas M (2009) Host-parasite interactions revealed by Plasmodium falciparum metabolomics. Cell Host Microbe 5: 191–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Cabrita-Santos L, Leitao R, Bettiol E, Purcell LA, Diaz-Pulido O, Andrews LB, Tadakuma T, Bhanot P, Mota MM, Rodriguez A (2008) Adenylyl cyclase alpha and cAMP signaling mediate Plasmodium sporozoite apical regulated exocytosis and hepatocyte infection. PLoS Pathog 4: e1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Thiele I, Palsson BO (2010) What is flux balance analysis? Nat Biotechnol 28: 245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price ND, Reed JL, Palsson BO (2004) Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat Rev Microbiol 2: 886–897 [DOI] [PubMed] [Google Scholar]
- Promeneur D, Liu Y, Maciel J, Agre P, King LS, Kumar N (2007) Aquaglyceroporin PbAQP during intraerythrocytic development of the malaria parasite Plasmodium berghei. Proc Natl Acad Sci USA 104: 2211–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quashie NB, Dorin-Semblat D, Bray PG, Biagini GA, Doerig C, Ranford-Cartwright LC, De Koning HP (2008) A comprehensive model of purine uptake by the malaria parasite Plasmodium falciparum: identification of four purine transport activities in intraerythrocytic parasites. Biochem J 411: 287–295 [DOI] [PubMed] [Google Scholar]
- Ralph SA, van Dooren GG, Waller RF, Crawford MJ, Fraunholz MJ, Foth BJ, Tonkin CJ, Roos DS, McFadden GI (2004) Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat Rev Microbiol 2: 203–216 [DOI] [PubMed] [Google Scholar]
- Ramya TN, Mishra S, Karmodiya K, Surolia N, Surolia A (2007) Inhibitors of nonhousekeeping functions of the apicoplast defy delayed death in Plasmodium falciparum. Antimicrob Agents Chemother 51: 307–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramya TN, Surolia N, Surolia A (2006) Polyamine synthesis and salvage pathways in the malaria parasite Plasmodium falciparum. Biochem Biophys Res Commun 348: 579–584 [DOI] [PubMed] [Google Scholar]
- Razakantoanina V, Nguyen Kim PP, Jaureguiberry G (2000) Antimalarial activity of new gossypol derivatives. Parasitol Res 86: 665–668 [DOI] [PubMed] [Google Scholar]
- Reed JL, Vo TD, Schilling CH, Palsson BO (2003) An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol 4: R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts F, Roberts CW, Johnson JJ, Kyle DE, Krell T, Coggins JR, Coombs GH, Milhous WK, Tzipori S, Ferguson DJ, Chakrabarti D, McLeod R (1998) Evidence for the shikimate pathway in apicomplexan parasites. Nature 393: 801–805 [DOI] [PubMed] [Google Scholar]
- Romero P, Wagg J, Green ML, Kaiser D, Krummenacker M, Karp PD (2005) Computational prediction of human metabolic pathways from the complete human genome. Genome Biol 6: R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Maeda T, Nakazawa M, Takeuchi T, Nozaki T, Asai T (2002) Characterisation of hexokinase in Toxoplasma gondii tachyzoites. Int J Parasitol 32: 961–967 [DOI] [PubMed] [Google Scholar]
- Schnick C, Polley SD, Fivelman QL, Ranford-Cartwright LC, Wilkinson SR, Brannigan JA, Wilkinson AJ, Baker DA (2009) Structure and non-essential function of glycerol kinase in Plasmodium falciparum blood stages. Mol Microbiol 71: 533–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz R, Kuepfer L, Sauer U (2007) Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol Syst Biol 3: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segre D, Vitkup D, Church GM (2002) Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci USA 99: 15112–15117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomi T, Berkman O, Ruppin E (2005) Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc Natl Acad Sci USA 102: 7695–7700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomi T, Cabili MN, Herrgard MJ, Palsson BO, Ruppin E (2008) Network-based prediction of human tissue-specific metabolism. Nat Biotechnol 26: 1003–1010 [DOI] [PubMed] [Google Scholar]
- Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M (2004) Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother 48: 1803–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorci L, Pan Y, Eyobo Y, Rodionova I, Huang N, Kurnasov O, Zhong S, MacKerell AD Jr, Zhang H, Osterman AL (2009) Targeting NAD biosynthesis in bacterial pathogens: structure-based development of inhibitors of nicotinate mononucleotide adenylyltransferase NadD. Chem Biol 16: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spry C, Chai CL, Kirk K, Saliba KJ (2005) A class of pantothenic acid analogs inhibits Plasmodium falciparum pantothenate kinase and represses the proliferation of malaria parasites. Antimicrob Agents Chemother 49: 4649–4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surolia N, Surolia A (2001) Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat Med 7: 167–173 [DOI] [PubMed] [Google Scholar]
- Thiele I, Palsson BO (2009) A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc 5: 93–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I, Palsson BO (2010) A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc 5: 93–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley PJ, Strath M, Wilson RJ (1994) Antimalarial activity in vitro of the glyoxalase I inhibitor diester, S-p-bromobenzylglutathione diethyl ester. Biochem Pharmacol 47: 418–420 [DOI] [PubMed] [Google Scholar]
- Ting LM, Shi W, Lewandowicz A, Singh V, Mwakingwe A, Birck MR, Ringia EA, Bench G, Madrid DC, Tyler PC, Evans GB, Furneaux RH, Schramm VL, Kim K (2005) Targeting a novel Plasmodium falciparum purine recycling pathway with specific immucillins. J Biol Chem 280: 9547–9554 [DOI] [PubMed] [Google Scholar]
- Trager W, Jensen JB (1976) Human malaria parasites in continuous culture. Science 193: 673–675 [DOI] [PubMed] [Google Scholar]
- Uniprot.Consortium (2010) The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res 38: D142–D148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya AB, Mather MW (2009) Mitochondrial evolution and functions in malaria parasites. Annu Rev Microbiol 63: 249–267 [DOI] [PubMed] [Google Scholar]
- van Dooren GG, Stimmler LM, McFadden GI (2006) Metabolic maps and functions of the Plasmodium mitochondrion. FEMS Microbiol Rev 30: 596–630 [DOI] [PubMed] [Google Scholar]
- Varma A, Palsson BO (1994) Metabolic flux balancing—basic concepts, scientific and practical use. Bio-Technology 12: 994–998 [Google Scholar]
- Vaughan AM, O′Neill MT, Tarun AS, Camargo N, Phuong TM, Aly AS, Cowman AF, Kappe SH (2009) Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell Microbiol 11: 506–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Rodriguez J, Franke-Fayard B, Dinglasan RR, Janse CJ, Pastrana-Mena R, Waters AP, Coppens I, Rodriguez-Orengo JF, Srinivasan P, Jacobs-Lorena M, Serrano AE (2009) The glutathione biosynthetic pathway of Plasmodium is essential for mosquito transmission. PLoS Pathog 5: e1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2008) World Malaria Report 2008. Geneva: World Health Organization [Google Scholar]
- Wagner A (2005) Robustness and Evolvability in Living Systems. Princeton: Princeton University Press
- Waller RF, Keeling PJ, Donald RG, Striepen B, Handman E, Lang-Unnasch N, Cowman AF, Besra GS, Roos DS, McFadden GI (1998) Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc Natl Acad Sci USA 95: 12352–12357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanidworanun C, Nagel RL, Shear HL (1999) Antisense oligonucleotides targeting malarial aldolase inhibit the asexual erythrocytic stages of Plasmodium falciparum. Mol Biochem Parasitol 102: 91–101 [DOI] [PubMed] [Google Scholar]
- Webster HK, Whaun JM, Walker MD, Bean TL (1984) Synthesis of adenosine nucleotides from hypoxanthine by human malaria parasites (Plasmodium falciparum) in continuous erythrocyte culture: inhibition by hadacidin but not alanosine. Biochem Pharmacol 33: 1555–1557 [DOI] [PubMed] [Google Scholar]
- Winzeler EA (2008) Malaria research in the post-genomic era. Nature 455: 751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongsrichanalai C, Pickard AL, Wernsdorfer WH, Meshnick SR (2002) Epidemiology of drug-resistant malaria. Lancet Infect Dis 2: 209–218 [DOI] [PubMed] [Google Scholar]
- Wright J, Wagner A (2008) The systems biology research tool: evolvable open-source software. BMC Syst Biol 2: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano K, Komaki-Yasuda K, Tsuboi T, Torii M, Kano S, Kawazu S (2006) 2-Cys peroxiredoxin TPx-1 is involved in gametocyte development in Plasmodium berghei. Mol Biochem Parasitol 148: 44–51 [DOI] [PubMed] [Google Scholar]
- Yano K, Otsuki H, Arai M, Komaki-Yasuda K, Tsuboi T, Torii M, Kano S, Kawazu S (2008) Disruption of the Plasmodium berghei 2-Cys peroxiredoxin TPx-1 gene hinders the sporozoite development in the vector mosquito. Mol Biochem Parasitol 159: 142–145 [DOI] [PubMed] [Google Scholar]
- Yao L, Rzhetsky A (2008) Quantitative systems-level determinants of human genes targeted by successful drugs. Genome Res 18: 206–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh I, Hanekamp T, Tsoka S, Karp PD, Altman RB (2004) Computational analysis of Plasmodium falciparum metabolism: organizing genomic information to facilitate drug discovery. Genome Res 14: 917–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Kumar TR, Nkrumah LJ, Coppi A, Retzlaff S, Li CD, Kelly BJ, Moura PA, Lakshmanan V, Freundlich JS, Valderramos JC, Vilcheze C, Siedner M, Tsai JH, Falkard B, Sidhu AB, Purcell LA, Gratraud P, Kremer L, Waters AP et al. (2008) The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell Host Microbe 4: 567–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Meshnick SR (1991) Inhibition of Plasmodium falciparum dihydropteroate synthetase and growth in vitro by sulfa drugs. Antimicrob Agents Chemother 35: 267–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Geng C, Tao L, Zhang D, Jiang Y, Tang K, Zhu R, Yu H, Zhang WD, He F, Li Y, Cao Z (2010) Reconstruction and analysis of human liver-specific metabolic network based on CNHLPP data. J Proteome Res 9: 1648–1658 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures S1–3, Supplementary tables S1–5