

Abstract
Analysis of parallel genotyping and expression profiling data has shown that mRNA expression levels are highly heritable. Currently, only a tiny fraction of this genetic variance can be mechanistically accounted for. The influence of trans-acting polymorphisms on gene expression traits is often mediated by transcription factors (TFs). We present a method that exploits prior knowledge about the in vitro DNA-binding specificity of a TF in order to map the loci (‘aQTLs’) whose inheritance modulates its protein-level regulatory activity. Genome-wide regression of differential mRNA expression on predicted promoter affinity is used to estimate segregant-specific TF activity, which is subsequently mapped as a quantitative phenotype. In budding yeast, our method identifies six times as many locus-TF associations and more than twice as many trans-acting loci as all existing methods combined. Application to mouse data from an F2 intercross identified an aQTL on chromosome VII modulating the activity of Zscan4 in liver cells. Our method has greatly improved statistical power over existing methods, is mechanism based, strictly causal, computationally efficient, and generally applicable.
Keywords: gene expression, gene regulatory networks, genetic variation, quantitative trait loci, transcription factors
Introduction
Understanding how phenotype relates to genotype, in terms of the myriad molecular processes that govern the behavior of cells and organisms, is one of the central goals of biology. Genome-wide messenger RNA expression levels constitute an intermediate molecular phenotype of great utility. They can be readily measured using modern genomics technologies, and provide high-dimensional information about the cellular state. In recent years, the use of parallel genotyping and expression profiling on segregating populations has enabled researchers to ask quantitative questions regarding the genetics of genome-wide expression in a variety of organisms (Jansen and Nap, 2001; Brem et al, 2002; Cheung et al, 2003; Schadt et al, 2003). These studies have revealed that steady-state mRNA abundance for individual genes is highly heritable, and can be treated as a quantitative genetic trait. Expression quantitative trait loci (eQTLs), whose allelic variation influences the expression level of individual genes, have successfully been mapped in a number of model organisms, from yeast to mouse (Brem et al, 2002). Local eQTL linkages to polymorphisms in cis-regulatory regions frequently occur (Ronald et al, 2005). However, trans-acting polymorphisms at distal loci can influence the expression of large numbers of genes in countless ways by changing the state and/or connectivity of the gene regulatory network of the cell (Yvert et al, 2003). It is therefore expected that such polymorphisms account for much of the genetic variance of gene expression.
Perhaps the simplest method for mapping trans-acting loci is to identify eQTL ‘hotspots’ that influence the expression of a disproportionate number of genes (Brem et al, 2002). A number of such hotspots have been identified in yeast and other organisms (Rockman and Kruglyak, 2006). The genes that link to a particular hotspot are often enriched for specific biological functions, and tend to be controlled through the same regulatory subnetwork (Brem et al, 2002; Zhu et al, 2008). A different approach has been to map trans-acting loci for sets of coexpressed genes identified using hierarchical clustering (Yvert et al, 2003) or more sophisticated module inference algorithms (Lee et al, 2006). However, methods based on coexpression are most useful when a relatively small number of cell state parameters are perturbed and the expression of large subsets of genes changes in a coherent way. One expects them to be less naturally suitable for analyzing natural gene expression variation, where the segregation of alleles in a genetic cross causes a very large number of cell state parameters to be independently perturbed. Indeed, with some exceptions, the number of genes in genetic coexpression modules is very small (Yvert et al, 2003; Lee et al, 2006). Principal component analysis (PCA) (Biswas et al, 2008) of the matrix of genes by segregants, and extensions of PCA that incorporate qualitative information about regulatory network topology (Kliebenstein et al, 2006; Sun et al, 2007; Ye et al, 2009), have also been applied to map trans-acting loci. Although these methods all improve upon single-gene based approaches, the lion's share of the heritable variation in gene expression remains to be accounted for.
We here present a transcription-factor-centric and sequence-based method for the dissection of genetic expression variation. A key feature of our approach is the use of quantitative prior information about the DNA-binding specificity of transcription factors (TFs) in the form of position-specific affinity matrices (Bussemaker et al, 2007). These matrices are used to predict the affinity with which each TF binds to the promoter region of each gene. We use a linear regression model motivated by a biophysical description of gene expression regulation (Bussemaker et al, 2001, 2007) to explain the genome-wide transcriptional response to the genetic perturbations in each segregant in terms of changes in ‘hidden’ TF activity. Treating the latter as a quantitative trait allows us to map the activity quantitative trait loci (‘aQTLs’) whose allelic status modulates the regulatory activity of specific TFs.
As we will demonstrate below, our method has a greatly improved statistical power to detect regulatory mechanisms underlying the heritability of genome-wide mRNA expression. Specifically, it identified six times as many locus-TF associations from a genetic cross between two haploid yeast strains as all existing methods combined. This includes novel trans-acting polymorphisms in the TF-encoding gene STB5, RFX1, and HAP4. We also identified 20 previously unknown trans-acting loci. Furthermore, for many of the 13 known eQTL hotspots in yeast, our method implicated several TFs that were not previously known to mediate the effect of inheritance of these loci on gene expression levels. We validated our ability to predict locus-TF associations in yeast using gene expression profiles for allele replacement strains. Finally, application to mouse data identified an aQTL modulating the activity of a specific TF in liver cells, demonstrating that our method also works in higher eukaryotes.
Results
We applied our method in two different organisms: budding yeast and mouse. For yeast, the data set we used (Smith and Kruglyak, 2008) covers 108 haploid segregants from a cross between two haploid strains of Saccharomyces cerevisiae—a lab strain (BY) and a wild isolate from a vineyard (RM). It includes two-color DNA microarray measurements for each gene of the mRNA abundance in each individual segregant relative to a pooled reference consisting of equals amounts of mRNA from both parental strains, and genotyping information at 2957 genomic marker locations. The mouse data set consisted of gene expression levels in the liver cell lines of an F2 intercross population between C57BL/6J and DBA/2J (BXD) consisting of 111 animals (Schadt et al, 2003), and the genotyping at 139 microsatellite markers uniformly distributed over the mouse genome (Drake et al, 2001).
Inferring segregant-specific TF activities
Figure 1 provides an overview of our computational procedure. As inputs, it requires: (i) the nucleotide sequence of the cis-regulatory region associated with each gene; (ii) a weight matrix for each TF, used to predict the strength with which the TF binds to each cis-regulatory region; (iii) a matrix containing continuous values, whose rows correspond to genes and whose columns contain the genome-wide mRNA expression profile of a particular segregant; and (iv) a genotype matrix containing binary values, whose rows correspond to genetic markers, and whose columns specify from which parent each marker was inherited in a particular segregant. As cis-regulatory sequence, we used 600 bp upstream of each open reading frame. We previously demonstrated that when the binding specificity of a TF is known, quantitative changes in its regulatory activity can be inferred by performing genome-wide linear regression of differential mRNA expression on the predicted in vitro binding affinity of cis-regulatory regions (Foat et al, 2008). The biophysical foundation that underlies this regression approach requires the binding specificity of each TF to be represented as a position-specific affinity matrix (PSAM) (Foat et al, 2005). We used an existing compendium of position weight matrices (PWMs) for yeast TFs (MacIsaac et al, 2006), converting each PWM to an approximate PSAM by assuming base frequencies to be proportional to relative binding affinities at each position within the binding site (Bussemaker et al, 2007). Each PSAM was then used to estimate the segregant-specific promoter affinity for all genes (Figure 1A). With only a few exceptions, these promoter affinity profiles are not correlated between TFs (Supplementary Figure S1). This allowed us to estimate the segregant-specific regulatory activity of most TFs in an independent manner. For each segregant, genome-wide linear regression of differential mRNA expression on segregant-specific promoter affinity for each TF was performed (Figure 1B). The coefficients from this fit represent protein-level TF activities, which we treat as a quantitative phenotype. Whenever the distribution of TF activity depends on the inheritance at a particular genomic position, this indicates the presence of an aQTL (Figure 1C). Details are provided in the Materials and methods section.
Figure 1.

Overview of our transcription-factor-centric approach to detecting trans-acting sequence variation. (A) We construct a matrix containing the promoter-binding affinity for each combination of the upstream non-coding sequence of a particular gene and the position-specific affinity matrix (PSAM) of a particular transcription factor (TF). (B) The promoter-binding affinity matrix is interpreted as a regulatory connectivity matrix and used to infer a matrix containing the regulatory activity of each TF in each segregant. For each segregant independently, multivariate genome-wide linear regression of segregant-specific differential mRNA expression on the matrix of promoter affinity for all TFs is performed. The coefficients from this linear fit represent (differential) protein-level TF activities. (C) For each TF independently, we treat the inferred activity as a quantitative phenotype and use genetic linkage analysis across all segregants to identify loci that genetically modulate TF activity. Whenever TF activity is statistically associated with genotype at a particular genetic marker, this shows as a high log-odds (LOD) score indicating the presence of a TF activity quantitative trait locus, or ‘aQTL’.
TF activity is a highly heritable quantitative trait
To establish that the TF activities inferred by our regression procedure are meaningful, we calculated their heritability h2 (see Materials and methods). Encouragingly, we found that the activity of 102 of the 123 TFs tested is heritable at a false discovery rate (FDR) of 5% corresponding to h2>80.4%. In general, the heritability of the inferred TF activity is higher than that of the mRNA expression level of the gene encoding the TF (Supplementary Figure S2). Figure 2 shows differences in TF activity between the BY and RM parental strains as estimated by applying the regression procedure of Figure 1 to the average differential mRNA expression profile between BY and RM (Smith and Kruglyak, 2008). Hap1p is the factor whose regulatory activity is the most strongly modulated between the BY and RM strains. Indeed, it is known that a Ty1 insertion in the HAP1 coding region occurs in BY and other derivatives of the lab strain S288C (Gaisne et al, 1999) and that this insertion is absent in RM (Brem et al, 2002). Overall, 46 TFs are more active in RM, whereas 56 are more active in BY, at a 5% FDR. Merely comparing the two parental strains, however, does not reveal which loci are responsible for the differences in TF activity. Only genetic mapping to quantitative trait loci can provide that information.
Figure 2.

Inferred differences in TF activity between the BY and RM parental strains. Shown are the t-values corresponding to the regression coefficients in a multivariate linear model that predicts genome-wide differential mRNA expression from predicted binding affinity of upstream promoter regions.
Identifying aQTLs: genomic loci that modulate TF activity
The regression procedure of Figure 1 takes into account prior information about the connectivity of the transcriptional network of the cell in a way that allows us to directly treat TF activity as a quantitative trait. To identify aQTLs for each TF, we used composite interval mapping (CIM) (Zeng, 1994), which accounts for linkage between neighboring markers and has significantly better spatial resolution than single-marker methods (Supplementary Table S1; Supplementary Figure S3). This is important, as even the aQTL regions detected using CIM typically encompass 20–30 genes, and our goal is to uncover trans-acting causal mutations in individual genes or even nucleotides. Figure 3 provides an overview of the TF-locus associations identified using our method. To control for multiple testing, we use a log-odds (LOD) score threshold (red line in Figure 1C) corresponding to a 5% FDR (see Materials and methods and Supplementary Figure S6). We identified a single aQTL for 55 and multiple aQTLs for 22 of the 123 TFs analyzed. Together, the mapped aQTLs cover several dozen distinct genomic loci (Supplementary Table S2). Note that all aQTLs are by definition trans-acting from the point of view of the mRNA expression level of individual genes, as the trait analyzed is the ‘hidden’ regulatory activity of each TF.
Figure 3.

Overview of the trans-acting genetic modulators of TF activity mapped using our method. All transcription factors that have at least one significant aQTL region at a 5% FDR are shown. Transcription factors are sorted according to the chromosomal position of their maximum LOD score. Putative causal gene assignments are indicated in green (local aQTL: TF encoded by gene in aQTL) or red (protein–protein interaction identified between TF and gene in aQTL).
Validation of aQTL-TF linkages using an IRA2 allele swap
To the extent that aQTLs act independently, the regulatory consequences of allelic variation at a particular locus should be independent of the genetic background in which it occurs. To validate our method, we therefore analyzed gene expression profiles of allele replacement strains from a previous study (Smith and Kruglyak, 2008). According to our analysis, chromosome 15 contains an aQTL that influences the activity of several dozen distinct TFs (Figure 3; Supplementary Table S2). Among the 19 genes in this region is IRA2, which encodes a GTPase-activating protein that negatively regulates Ras proteins and thereby controls intracellular cAMP levels (Tanaka et al, 1990). The coding region of IRA2 is highly polymorphic (Smith and Kruglyak, 2008). We analyzed the gene expression profile of a BY strain carrying the RM allele of the IRA2 coding region, and vice versa, and found that the activity of Adr1p, Cha4p, and Msn4p was significantly affected by the allele replacement (Supplementary Figure S4; P-value 3.3 × 10−16, 1.1 × 10−10, and 1.6 × 10−5, respectively, see Materials and methods). Each of these TFs was indeed predicted by our method to link to the IRA2 locus. Consistently, cAMP-dependent protein kinase is known to influence Adr1p activity (Cherry et al, 1989) and regulate subcellular localization of Msn4p, which influences its activity (Gorner et al, 1998). Altogether, there are 30 TFs with an aQTL region containing the IRA2 gene. They do not all need to be influenced by the polymorphism(s) in its coding region; additional causal polymorphisms in nearby genes, modulating other subsets of the 30 TFs, may well exist. It is therefore not surprising that the activity of only 3 out of 30 TFs was significantly affected by the IRA2 allele replacement. On the other hand, we do not expect any TF whose activity does not link to the IRA2 locus to be affected by the allele replacement. Indeed, as can be seen from Supplementary Figure S4, our method achieved 100% specificity in this regard: none of the 93 TFs whose aQTL(s) do not contain IRA2 showed a change in regulatory activity.
Novel trans-acting polymorphisms in transcription-factor genes
Of the aQTL linkages we detected, only four—those of Hap1p, Stb5p, Rfx1p, and Hap4p—are local (Figure 3, green boxes). The probability that a locus showing aQTL linkage encompasses the gene encoding the TF itself by chance is typically <1% (it equals the ratio of the number of genes in the aQTL and the total number of genes). Therefore, whenever such local linkage happens, it is highly likely that the causal polymorphism resides in the coding region or regulatory region of the TF gene. The aQTL profile for Hap1p is shown in Figure 1C, and the polymorphism in HAP1 that gives rise to it was already discussed above.
Stb5p is a C2H2 zinc finger protein that serves as an activator of multidrug resistance genes (Kasten and Stillman, 1997). A significant difference in Stb5p activity exists between the BY and RM strains (Figure 4A), and this activity is highly heritable (h2=95%). We detected highly significant local linkage (LOD score=10.84; Q-value=2.69 × 10−8) between Stb5p activity and the allelic status of the STB5 locus (Figure 4B). Alignment of the BY and RM protein sequences for Stb5p revealed five amino-acid mutations (see Supplementary Table S3), all of which occur outside the DNA-binding domain. We found no nucleotide differences in the 5′ and 3′ untranslated regions or <1 kb upstream of the transcription start site of STB5. Consistently, the mRNA expression level of the STB5 gene is not significantly correlated with the activity of Stb5p (r=0.18; P-value>0.05). Furthermore, CIM analysis of the mRNA expression level of the STB5 gene did not reveal any local eQTL linkage (Supplementary Figure S5). The power of our aQTL approach is further underscored by the fact that no eQTL hotspot has been detected at the STB5 locus (Brem et al, 2002). It will be interesting to further dissect the post-translational mechanism(s) by which the sequence differences between the BY and RM alleles of Stb5p cause a difference in its regulatory activity.
Figure 4.

(A) Inferred activity of Stb5p in parental strains and segregants. The first and second columns show the activity of Stb5 in six replicates of a BY-reference comparison and six replicates of a RM-reference comparison. The third and fourth columns show the activity of Stb5p for segregants that inherited the BY and RM allele, respectively, at the STB5 locus. (B) LOD score profile for the activity of Stb5p. An asterisk denotes the STB5 locus.
Rfx1p is a major transcriptional repressor of the DNA damage response. The RM allele of the RFX1 gene contains a premature stop codon. Consistently, genes whose promoter is predicted to be bound by Rfx1p tend to be more highly expressed in the BY strain than in the RM strain (Figure 2).
The last local aQTL we discovered was for Hap4p, a subunit of the heme-activated, glucose-repressed Hap2p/3p/4p/5p CCAAT binding complex. Consistently, the mRNA expression level of the HAP4 gene is highly correlated with the activity of Hap4p (r=0.79).
CDC28 antagonistically modulates Fkh1 and Fkh2
Chromosome II contains an ‘aQTL hotspot’ whose allelic status influences the activity of no fewer than 15 distinct TFs (Figure 3), including Fkh1p and Fkh2p. The locus contains the CDC28 gene, which encodes a cyclin-dependent kinase. Phosphorylation by Cdc28p is known to regulate the activity of Fkh2 by promoting interaction with a coactivator (Pic-Taylor et al, 2004). On the basis of the aQTL mapping to the CDC28 locus in combination with high-throughput evidence of their physical interaction (Ho et al, 2002) with Cdc28p (Supplementary Table S3), we predict that Fkh1p is also post-translationally modulated by Cdc28p. The sign of the aQTL linkage to the CDC28 locus for Fhk2p is the opposite of that for Fkh1p (Figure 5A): whereas the transcriptional targets of Fkp1p are more highly expressed in segregants carrying the BY allele at the CDC28 locus, the opposite is true for the targets of Fkh2p (Figure 5B). The same pattern holds for the inferred difference in TF activity between the two parental strains (Figure 2). The antagonism between Fkh1p and Fkh2p is consistent with previously observed differences in function between the two factors (Hollenhorst et al, 2001; Morillon et al, 2003). These two TFs have similar sequence specificity, and consequently their total promoter affinity profiles are correlated across genes (r=0.72; see also Supplementary Figure S1B). Nevertheless, we were able to detect the opposite influence of the CDC28 polymorphism on their activity because our method uses multivariate regression, which forces TFs with correlated promoter affinity profiles to compete for the same differential mRNA expression signal. When we analyze each TF separately using a univariate model, the CIM regression coefficients for Fkh1p and Fkh2p (incorrectly) have the same sign. This example underscores the importance of our affinity-based quantification of the matrix of regulatory connectivities between TFs and their target genes.
Figure 5.

(A) Activity of Fkh1p and Fkh2p across all segregants. The activity of Fkh1p is negatively correlated with that of Fkh2p. The yellow dots correspond to segregants carrying the BY allele at the CDC28 locus, the green dots to those carrying the RM allele. (B) Schematic diagram illustrating the antagonistic modulation of Fkh1p and Fkh2p by Cdc28p. Although the transcriptional targets of Fkh1p are more highly expressed in segregants carrying the BY allele at the CDC28 locus, the opposite is true for the targets of Fkh1p.
An aQTL on chromosome VII controlling Zscan4 activity in mouse liver cells
To determine whether our method could map aQTLs for mammalian TFs, we applied it to parallel genotyping and liver cell expression data for an F2 mouse population (Schadt et al, 2003). Weight matrices derived from protein-binding microarray (PBM) data for 104 mouse TFs were used (Badis et al, 2009). The model we used to analyze the yeast segregants contains ‘cis’ coefficients, which explicitly model changes in expression because of allelic variation in promoter sequence, in addition to the ‘trans’ coefficient that model the changes in TF activity. However, we found that a simpler ‘trans-only’ model performed equally well in terms of mapping aQTLs when applied to the yeast segregant data (Supplementary Figure S7). This gave us confidence to use a ‘trans-only’ model in mouse, where the density of markers is too low to assign gene-specific promoter sequences. We identified an aQTL for Zscan4, a TF containing four zinc finger domains and a SCAN domain, which is also known as the leucine-rich region (Williams et al, 1995) (Figure 6). Using a multivariate linear model to analyze the homozygous C57BL/6J (BB), homozygous DBA/2J (DD), and heterozygous (BD) genotype at the aQTL locus (Figure 6A), we found the behavior of the aQTL to be additive and show no significant dominant effect (see Materials and methods). A highly significant linkage (LOD score=10.8) with Zscan4 activity occurs between 43 and 66 cM on mouse chromosome 7 (Figure 6B). This region contains >500 genes, which makes it difficult to predict the causal polymorphism. Limited information is available about protein–protein interaction (PPI) for mouse, and we could not detect any direct interaction between genes within this region and Zscan4p. However, our result demonstrates that TF activity can also be inferred and mapped in mammalian cells using our method, and provides a starting point for further dissection of trans-acting regulatory variation mediated by Zscan4p.
Figure 6.

(A) Inferred activity of Zscan4p across all F2 mouse population. Each column shows the activity of Zscan4 in homozygous C57BL/6J (BB), heterozygous (BD), and homozygous DBA/2J (DD) mice at aQTL positions, respectively. (B) LOD score profile for Zscan4p.
Discussion
We have presented a transcription-factor-centric method for identifying trans-acting genetic modulators of gene expression using parallel genotyping and mRNA expression phenotyping data. Our approach is based on the idea of treating the genotype-specific regulatory activity of each TF as a quantitative trait. It exploits prior information about the network of interactions between TFs and their target genes to infer genotype-specific TF activities from genome-wide measurements of mRNA expression. Our method has greatly increased statistical power to detect locus-TF associations. It is sensitive even to a relatively subtle influence of genotype-specific TF activity on mRNA expression because it is based on a statistical analysis across both genes and segregants. The fact that TF activity is not a gene-specific phenotype allows us to make the rather crude assumption that the strength of the regulatory connectivity between TF and target gene is proportional to in vitro promoter affinity. In reality, many of the predicted binding sites in promoter regions are not functional, due to complex interactions with nucleosomes and other chromatin-associated factors. It is remarkable that our method works in spite of this complexity.
Application of our aQTL method to a data set for 108 haploid segregants from a cross between two yeast strains (Smith and Kruglyak, 2008) demonstrated a dramatic increase in statistical power to uncover the regulatory mechanisms underlying genetic variation in gene expression levels. The results are summarized in Supplementary Table S2. We identified a total of 103 locus-TF associations, a more than six-fold improvement over the 17 locus-TF associations identified by several existing methods (Brem et al, 2002; Yvert et al, 2003; Lee et al, 2006; Smith and Kruglyak, 2008; Zhu et al, 2008). The total number of distinct genomic loci identified as an aQTL for one or more TFs equals 31, which includes 11 of the 13 previously identified eQTL hotspots (Smith and Kruglyak, 2008). Thus, our method identifies 20 novel trans-acting polymorphisms: almost double the number of known such loci in yeast. For many of the eQTL hotspots, it also implicated several TFs not previously known to mediate the influence of these loci on genome-wide mRNA expression.
Our regression procedure fully accounts for post-translational regulation of TF activity at the protein level, as we do not use the mRNA expression level of either the gene encoding the TF or one its upstream modulators as a surrogate for regulatory activity. Indeed, the correlation between the protein-level regulatory activity of a TF and its expression at the mRNA level across a large number of experimental conditions in yeast was recently found to often be quite poor (Boorsma et al, 2008). The present study confirms this observation: Only one third of TFs analyzed show a significant (<5% FDR) correlation between mRNA expression and activity (Supplementary Figure S6). Moreover, only 12 of the 103 TF-locus associations could be confirmed when mRNA expression level was used as a proxy inferred protein-level TF activity.
We also applied our aQTL method to the earlier yeast segregant data set of (Brem and Kruglyak, 2005). This confirmed the dramatic increase in statistical power afforded by our approach (see Supplementary Table S5). We detected a total of 79 locus-TF associations, which again is a more than six-fold improvement over the 14 locus-TF associations detected from these data by several existing methods (Lee et al, 2006; Sun et al, 2007; Zhu et al, 2008; Ye et al, 2009) combined. Furthermore, 28 of these 79 locus-TF associations were also detected using the data of (Smith and Kruglyak, 2008). This degree of reproducibility strongly validates our method: given that the number of possible such associations equals the number of TFs (123) times the number of markers (∼3000) divided by the average number of genes per locus (∼20), we would expect this overlap to be ∼0.4 by random chance. There is also no reason to expect complete overlap, as the data sets were similar but not identical. Indeed, although 13 eQTL hotspots have been identified in each respective data set, only 8 of these are the same (Smith and Kruglyak, 2008; Zhu et al, 2008).
Our findings are consistent with previous observations (Yvert et al, 2003) that most trans-acting variation in yeast does not map to TF genes, but to upstream modulators of TF activity. Indeed, of the total of 103 TF-locus associations shown in Figure 3 only four are local. We confirmed that HAP1 is directly affected by a sequence polymorphism, and discovered novel trans-acting polymorphisms in the TF-encoding gene STB5, RFX1, and HAP4. Unexpectedly, our analysis revealed loci on chromosomes II and XV that are informative for a large number of TFs (‘aQTL hotspots’). We stress that this cannot be accounted for in terms of correlated profiles of promoter affinity across genes, as we found these to be largely independent between TFs (cf. Supplementary Figure S1A). Rather, this phenomenon seems to point to one-to-many relationships between signal transduction pathways and TFs. For instance, our method predicts that genetic variation at the locus on chromosome II encoding the cyclin-dependent kinase CDC28 changes the activity of multiple cell cycle associated TFs (Ace2p, Fkh1p, Fkh2p, and Swi5p). At the same time, distinct polymorphisms at the same aQTL could be responsible for modulating different subsets of linked TFs. Evidence for this is our observation that allele replacement at the IRA2 locus on chromosome XV only affected a small subset of the TFs whose activity is linked to this aQTL (cf. Supplementary Figure S4).
In an effort to uncover further specific molecular mechanisms underlying the aQTL linkages summarized in Figure 3, we supplemented our genetic analysis with knowledge about physical and genetic PPIs; see Materials and methods for details. The information provided by PPI and aQTL is highly complementary. On the one hand, aQTL linkage can only implicate relatively large genomic regions, not individual genes, as genetic modulators of TF activity. On the other hand, although PPI data can connect a TF to a putative modulator of its activity, it would be questionable to conclude that the interaction corresponds to a functional regulatory network connection without the strict causality and directionality associated with aQTL linkage. In all cases, the probability that a gene within the aQTL region encodes one of the direct interactors of the TF by chance is <3% (see Materials and methods and Supplementary Table S4). Therefore, most of these genes (aQTG) are expected to encode direct or indirect modulators of the TF's activity. We were able to implicate a non-coding polymorphism in the CDC28 gene as a plausible genetic factor underlying the major eQTL hotspot on chromosome II (in addition to the experimentally validated trans-acting polymorphism in the AMN1 gene in the same region (Yvert et al, 2003)) and make a strong prediction that the functionally distinct cell cycle regulators Fkh1p and Fkh2p are modulated by the cyclin-dependent kinase Cdc28p in an antagonistic manner.
Extensive transgressive segregation has been previously identified for the expression levels of individual genes (Brem and Kruglyak, 2005). However, when we tested for the same phenomenon at the level of TF activity (see Materials and methods), we were only able to detect transgressive segregation for Ecm22p and Tec1p (Supplementary Figure S8); in both cases, the effects of two aQTLs for same TF cancel each other in both parental strains, and no differential activity between RM and BY could be observed (Figure 2). Presumably, much of the transgressive segregation at the level of individual genes is due to the fact that positive and negative contributions from different TFs can cancel each other. Our multivariate modeling of each individual gene's expression level in terms of the activity of multiple TFs accounts for such compensation explicitly, and hence the transgression is much less prevalent for aQTLs than for eQTLs.
In our approach, ‘phenotype space’ is reduced from that of all genes to that of all TFs. Rather than mapping the measured mRNA expression level of individual genes to eQTLs, we map the inferred activity of each TF to ‘aQTLs.’ This enhances statistical power in two distinct ways. First, it improves the signal-to-noise ratio for the quantitative trait itself, as the activity of each TF is estimated from the mRNA expression levels of its many targets. Second, the severity of the multiple-testing problem associated with QTL mapping because of the large number of marker/trait combinations is greatly reduced. Running in only seconds on a single processor, our algorithm is also computationally efficient.
It is important to emphasize that in our method the molecular identity of a TF is only defined through the PSAM that parameterizes its DNA-binding specificity. The sequence-to-affinity model for each TF needs to be specific enough to allow differentiation from all other TFs. We found that in the case of the budding yeast S. cerevisiae this condition generally holds. Given the rapid pace at which in vitro DNA-binding data is currently being generated for mammalian TFs (Badis et al, 2009), together with the demonstrated ability of regression-based models to infer TF activity in human cells (Das et al, 2006), we expect application of our method also to be feasible in higher eukaryotes.
Taken together, our results underscore the value of explicitly treating TF activity as a quantitative trait from a systems biology perspective as a promising strategy for increasing the statistical power of genome-wide linkage and association studies. More generally, our method is applicable whenever a matrix of connection strengths between regulators and targets, independent of the phenotype matrix, is available as prior information. There are several directions in which this approach can be extended. First, the use of more sophisticated methods for causal gene identification (Sun et al, 2007; Suthram et al, 2008; Lee et al, 2009) is likely to uncover additional molecular mechanisms. It will also be interesting to analyze to what extent the connectivity between the TF and their genetic modulators depends on the nutrient condition in which the yeast cells are grown (Smith and Kruglyak, 2008). Furthermore, aQTLs provide a novel vantage point for analyzing locus–locus interactions. Finally, it should be interesting to analyze to what extent genetic variation in steady-state gene expression levels because of post-transcriptional regulation of mRNA stability (Foat et al, 2005; Lee et al, 2009) is amenable to dissection using the method introduced in this paper.
Materials and methods
Gene expression and genotyping data
We analyzed genome-wide mRNA expression data from a study performed by Smith and Kruglyak (2008), which used two-color cDNA arrays. The data (GEO accession number GSE9376) cover a genetic cross between two haploid yeast strains—a laboratory strain (BY4716) and a natural isolate (RM11-1a). The data set includes six biological replicates of the BY parental strain, six replicates of the RM parental strain, and one replicate for each of 108 haploid segregants grown in two different conditions, with glucose and ethanol as the carbon source, respectively. For the present study, we only used data for the glucose condition. The study used a reference design in which all hybridizations were performed using equal amounts of mRNA from both parents (BY and RM) grown in both conditions as a reference. Log2-ratios, averaged over a dye swap, were used for all further analysis.
For comparison, we also analyzed genome-wide mRNA expression data for yeast segregants from a cross between BY and RM strains (GEO accession number GSE1990) from an earlier study performed by Brem and Kruglyak (2005). Following these authors, we excluded ORFs rejected by Kellis et al (2003). The data set covers 6 biological replicates of the BY parental strain, 12 replicates of the RM parental strain, and 1 replicate for each of 112 haploid segregants. The study used the BY material as a reference. Log2-ratios, averaged over the dye swap, were used for all further analysis. In addition, we averaged log-ratios for 13 ORFs that were spotted twice. Finally, we normalized each array by subtracting the mean log-ratio. For each of the segregants whose expression levels were determined, 2957 markers were genotyped by Brem and Kruglyak (2005), who kindly made this data available to us.
We also analyzed previously published F2 mouse genome-wide expression data (Drake et al, 2001; Schadt et al, 2003) (GEO accession GSE2008). The data set contains genome-wide oligonucleotide microarrays profiled using liver tissue from 111 F2 mice, which were constructed from two standard inbred strains, C57BL/6J and DBA/2J. The F2 mice fed an atherogenic diet for 4 months beginning at 12 months of age. This study used a common pool created from equal portions of RNA from each of the samples as a reference. Following the previous study, expression changes between each sample and a reference were quantified as expression log10-ratios between normalized, background-corrected intensity values for the two channels. The F2 intercross mice were genotyped at 139 microsatellite markers uniformly distributed over the mouse genome.
Genome sequence of BY and RM strains
We obtained RM11-1a sequence data from the Broad Institute (http://www.broad.mit.edu) and BY4716 sequence data from the Saccharomyces Genome Database (SGD; http://www.yeastgenome.org).
Defining genotype-specific promoter sequences
To define genotype-specific promoter sequences, we first identified pairs of genes orthologous between BY and RM. We aligned coding sequences of RM genes to the BY strains using BLAST in Bioperl (Altschul et al, 1997), and chose the best BLAST hits to identify the orthologous genes. Then, we obtained 600 bp upstream sequences of each orthologous pair to define BY and RM-specific promoter sequence. For segregants, we determined whether the promoter sequence of a particular gene was inherited from BY or RM strains. To this end, we first identified all genetic markers located within the 600 bp upstream of each open reading frame. If no genetic marker within 600 bp could be found, we selected the marker closest to the upstream region. The genotype of the selected markers was used to assign either the BY or RM promoter sequence to the gene. If multiple markers with inconsistent genotypes were selected, we discarded the gene.
Inferring segregant-specific TF activities
We downloaded a collection of 124 PWMs from a study by MacIsaac et al (2006) (we excluded Hap3, as it has the exact same PWM as Hap5). Next, we used the convert2psam utility from the REDUCE Suite version 2.0 software package (see http://bussemakerlab.org) to convert each PWM to a PSAM (Foat et al, 2005, 2006; Bussemaker et al, 2007). Pseudo-counts equal to one were added to the PWM at each position, and the resulting base counts were divided by that of the most frequent base at each position to get an estimate for the relative affinity associated with each point mutation away from the optimal-binding sequence (Foat et al, 2008). The resulting PSAM collection was used to infer genotype-specific changes in TF activity.
The occupancy Nϕg of the upstream region Ug of gene g by TF ϕ depends on the nuclear concentration [ϕ] of the TF and on the landscape of binding affinity across Ug. Both these quantities are genotype specific. At non-saturating concentrations of the TF, the occupancy in genotype G can be approximated by the product of concentration and affinity (Foat et al, 2006):
The total promoter affinity Kϕg(G) depends on the segregant-specific upstream sequence Ug(G), and is given by:
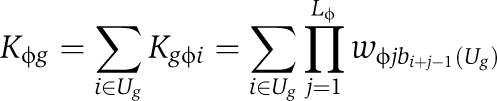 |
Here, Kgϕi represents the binding affinity (relative to the optimal DNA sequence) between TF ϕ and the DNA in a window of length Lϕ starting at position i within Ug. Assuming independence between nucleotide positions, we approximate Kgϕi by a product of position-specific relative affinities wϕjb. Finally, bi(Ug) denotes the base identity at nucleotide position i within Ug.
We assume that when steady-state mRNA abundances are being compared between genotype G and reference genotype Gref, the expression log2-ratio for gene g, to linear approximation, is proportional to the difference in promoter occupancy:
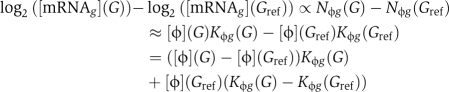 |
All total promoter affinities are known, so we can use the differential mRNA abundances to estimate coefficients βcis≡[ϕ](Gref) and βtrans≡[ϕ](G)−[ϕ](Gref). This motivated us to fit the following multivariate linear model to each segregant:
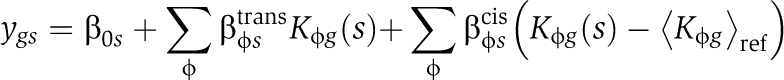 |
Here ygs represents mRNA expression log-ratios for gene g in segregant s. For the segregant data of Smith and Kruglyak (2008), whose used a pool of equals amounts of parental strains as their reference sample, 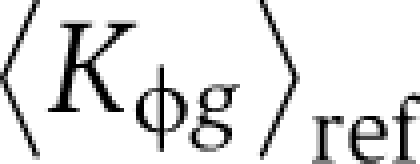 equals the average of BY and RM promoter affinities, whereas for that of Brem and Kruglyak (2005), who used the BY strain as their reference,
equals the average of BY and RM promoter affinities, whereas for that of Brem and Kruglyak (2005), who used the BY strain as their reference, 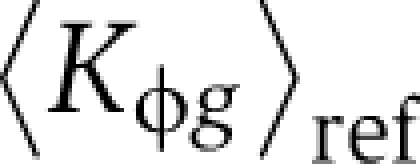 equals the BY promoter affinity. The intercept β0s absorbs any normalization differences that may occur. The genome-wide affinity profiles for several PSAMs are highly correlated (e.g. Msn2 and Msn4, Ino2 and Ino4). To avoid any problems resulting from such multicollinearity, we used ridge regression, which minimizes the residual sum of squares subject to a penalty proportional to the L2-norm of the coefficients, and gives a slightly biased but more precise estimator of coefficients than ordinary least squares (Hoerl and Kennard, 1970). We also fit the above model in ‘trans-only’ mode (βcis≡0).
equals the BY promoter affinity. The intercept β0s absorbs any normalization differences that may occur. The genome-wide affinity profiles for several PSAMs are highly correlated (e.g. Msn2 and Msn4, Ino2 and Ino4). To avoid any problems resulting from such multicollinearity, we used ridge regression, which minimizes the residual sum of squares subject to a penalty proportional to the L2-norm of the coefficients, and gives a slightly biased but more precise estimator of coefficients than ordinary least squares (Hoerl and Kennard, 1970). We also fit the above model in ‘trans-only’ mode (βcis≡0).
To infer segregant-specific TF activities in mouse, we downloaded PWMs defined by Badis et al (2009) who used PBM technology to determine the in vitro DNA-binding specificities of 104 different mouse TFs. We estimated PSAM and total promoter affinity from PWMs using 1000 bp upstream sequence of C57BL/6J strain by the same procedure explained above. We obtained C57BL/6J mouse genome sequence from UCSC Genome Browser (http://genome.ucsc.edu/).
Heritability
We calculated the heritability of the activity of each TF as follows:
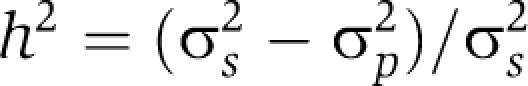 |
Here σs2 and σp2 are the variance of the linear regression coefficient from the ridge regression across the segregants, and the pooled variance of the parental strains, respectively. To determine the statistical significance of the heritability, we performed ridge regression after independent random permutation of expression log-ratios (parents and segregants combined) for each gene (1000 samples) and used the resulting empirical null distribution to compute a FDR.
aQTL mapping in yeast
To detect significant genetic contributions to TF activity by specific loci, we performed a split of the segregants by each specific marker and tested for a difference between the two distributions of ridge regression coefficients using Welch's t-test and the non-parametric Wilcoxon–Mann–Whitney test. We also used CIM, which uses multivariate regression on multiple markers for increased precision of QTL mapping (Zeng, 1994), as implemented in the R/qtl package (Broman et al, 2003). Statistical significance was determined by performing independent random permutation of expression log-ratios (segregants only) for each gene. The FDR corresponding to a given LOD score threshold was computed as the ratio of the number of linkages above threshold averaged over 20 randomized data sets, and the number of transcripts with detected linkage. We also estimated the FDR using the standard Benjamini–Hochberg procedure (Benjamini and Hochberg, 1995). For the CIM method, a 5% FDR based on the empirical permutation test corresponded to a LOD score >4.49 (Supplementary Figure S9).
aQTL mapping in mouse
In the case of mouse data analysis, aQTL mapping was conducted using a linear model. First, we constructed explanatory variables for the additive and dominance terms for each marker from the estimated genotype probabilities and used them in the regression analysis. Linkages were identified by comparing the likelihood, maximized as a function of the regression coefficients, for the following multivariate linear model
to the likelihood for the null model βϕstrans=β0. Here, the dependent variable βϕstrans represents the TF activity as estimated using the affinity-based model defined above in ‘trans-only’ mode (βcis≡0). The independent variables Xmsadd (taking values 0, 1, and 2, for (diploid) genotypes BB, BD, and DD, respectively) and Xmsdom (taking values 0, 1, and 1, for the same respective genotypes) represent additive and dominant terms for each marker, respectively. The LOD score was defined as the log10 of the likelihood ratio between the two models. The FDR was computed using the same procedure described above; an FDR <5% based on empirical permutation test corresponded to a LOD score >4.21.
Protein–protein interaction data
To identify putative causal genes from the aQTL regions of each specific TF, we used three different types of PPI data: (i) physical and genetic interactions in the BioGRID database (Stark et al, 2006), (ii) interactions between chromatin modifiers and associated TFs (Steinfeld et al, 2007), and (iii) kinase–TF interactions (Ptacek et al, 2005). We computed the expected number of direct interactors among the genes in the aQTL region for a specific TF based on the total number of interactors of the TF genome wide, the number of genes in the aQTL, and the total number of genes. Statistical significance was computed using Fisher's exact test.
Validation of predicted locus-TF associations
We downloaded gene expression profiles obtained by Smith and Kruglyak (2008) for a strain carrying the RM allele of IRA2 in the BY4742 background (RM@IRA2), a strain carrying the BY allele of IRA2 in the RM11-1a background (BY@IRA2), and six replicates each of the BY and RM parental strains (GEO accession number GSE9376). We only used the data for cells grown in glucose as the carbon source. The reference sample used in all cases was pooled parental mRNA (see above). Therefore, to obtain an estimate for the differential expression between RM@IRA2 and BY, we subtracted the mean log-ratio of the BY replicates from the RM@IRA2 log-ratios,
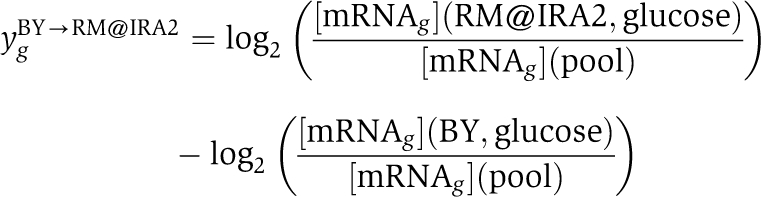 |
and performed multivariate (ridge) regression of these values on the BY promoter affinities for all TFs. We also performed the equivalent analysis where the roles of RM and BY were reversed. Finally, to average over strain background, we took the difference between the two regression coefficients for each TF to be our statistic for differential activity. To determine statistical significance, we performed 1000 random permutations of all genes to determine the standard error of an empirical null distribution, and used it to compute a P-value. A FDR of 5% corresponded to a P-value of 10−4.20.
Supplementary Material
Supplementary tables and figures
Compressed TAR file containing position-specific affinity matrices (PSAMs) for yeast transcription factors (TFs), which we used to infer genotype-specific TF activities
Compressed TAR file containing position-specific affinity matrices (PSAMs) for mouse transcription factors (TFs), which we used to infer genotype-specific TF activities
Matrix containing inferred regulatory TF activities for each yeast segregant
Matrix containing inferred regulatory TF activities for each F2 mouse
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grants R01HG003008 and U54CA121852. We thank the members of the Bussemaker and Pe’er laboratories for discussions, Jonas Maaskola for assistance and discussions during the early stages of this project, and Leonid Kruglyak, Matthew Rockman, and Bas van Steensel for a critical reading of the paper. Author contributions: HJB designed and supervised the study. EL implemented all algorithms and performed all computations. EL and HJB analyzed the results. EL and HJB wrote the paper.
Footnotes
The authors declare that they have no conflict of interest.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, Kuznetsov H, Wang CF, Coburn D, Newburger DE, Morris Q, Hughes TR, Bulyk ML (2009) Diversity and complexity in DNA recognition by transcription factors. Science 324: 1720–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B (Methodol) 57: 289–300 [Google Scholar]
- Biswas S, Storey JD, Akey JM (2008) Mapping gene expression quantitative trait loci by singular value decomposition and independent component analysis. BMC Bioinformatics 9: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorsma A, Lu XJ, Zakrzewska A, Klis FM, Bussemaker HJ (2008) Inferring condition-specific modulation of transcription factor activity in yeast through regulon-based analysis of genomewide expression. PLoS ONE 3: e3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem RB, Kruglyak L (2005) The landscape of genetic complexity across 5700 gene expression traits in yeast. Proc Natl Acad Sci USA 102: 1572–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem RB, Yvert G, Clinton R, Kruglyak L (2002) Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755 [DOI] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890 [DOI] [PubMed] [Google Scholar]
- Bussemaker HJ, Foat BC, Ward LD (2007) Predictive modeling of genome-wide mRNA expression: from modules to molecules. Annu Rev Biophys Biomol Struct 36: 329–347 [DOI] [PubMed] [Google Scholar]
- Bussemaker HJ, Li H, Siggia ED (2001) Regulatory element detection using correlation with expression. Nat Genet 27: 167–171 [DOI] [PubMed] [Google Scholar]
- Cherry JR, Johnson TR, Dollard C, Shuster JR, Denis CL (1989) Cyclic AMP-dependent protein kinase phosphorylates and inactivates the yeast transcriptional activator ADR1. Cell 56: 409–419 [DOI] [PubMed] [Google Scholar]
- Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS (2003) Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet 33: 422–425 [DOI] [PubMed] [Google Scholar]
- Das D, Nahle Z, Zhang MQ (2006) Adaptively inferring human transcriptional subnetworks. Mol Syst Biol 2: 2006.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake TA, Schadt E, Hannani K, Kabo JM, Krass K, Colinayo V, Greaser LE III, Goldin J, Lusis AJ (2001) Genetic loci determining bone density in mice with diet-induced atherosclerosis. Physiol Genomics 5: 205–215 [DOI] [PubMed] [Google Scholar]
- Foat BC, Houshmandi SS, Olivas WM, Bussemaker HJ (2005) Profiling condition-specific, genome-wide regulation of mRNA stability in yeast. Proc Natl Acad Sci USA 102: 17675–17680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foat BC, Morozov AV, Bussemaker HJ (2006) Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics 22: e141–e149 [DOI] [PubMed] [Google Scholar]
- Foat BC, Tepper RG, Bussemaker HJ (2008) TransfactomeDB: a resource for exploring the nucleotide sequence specificity and condition-specific regulatory activity of trans-acting factors. Nucleic Acids Res 36: D125–D131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaisne M, Becam AM, Verdiere J, Herbert CJ (1999) A ‘natural’ mutation in Saccharomyces cerevisiae strains derived from S288c affects the complex regulatory gene HAP1 (CYP1). Curr Genet 36: 195–200 [DOI] [PubMed] [Google Scholar]
- Gorner W, Durchschlag E, Martinez-Pastor MT, Estruch F, Ammerer G, Hamilton B, Ruis H, Schuller C (1998) Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev 12: 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C et al. (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180–183 [DOI] [PubMed] [Google Scholar]
- Hoerl AE, Kennard RW (1970) Ridge regression: biased estimation for nonorthogonal problems. Technometrics 12: 55–67 [Google Scholar]
- Hollenhorst PC, Pietz G, Fox CA (2001) Mechanisms controlling differential promoter-occupancy by the yeast forkhead proteins Fkh1p and Fkh2p: implications for regulating the cell cycle and differentiation. Genes Dev 15: 2445–2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RC, Nap JP (2001) Genetical genomics: the added value from segregation. Trends Genet 17: 388–391 [DOI] [PubMed] [Google Scholar]
- Kasten MM, Stillman DJ (1997) Identification of the Saccharomyces cerevisiae genes STB1-STB5 encoding Sin3p binding proteins. Mol Gen Genet 256: 376–386 [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES (2003) Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423: 241–254 [DOI] [PubMed] [Google Scholar]
- Kliebenstein DJ, West MA, van Leeuwen H, Loudet O, Doerge RW, St Clair DA (2006) Identification of QTLs controlling gene expression networks defined a priori. BMC Bioinformatics 7: 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Dudley AM, Drubin D, Silver PA, Krogan NJ, Pe’er D, Koller D (2009) Learning a prior on regulatory potential from eQTL data. PLoS Genet 5: e1000358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Pe’er D, Dudley AM, Church GM, Koller D (2006) Identifying regulatory mechanisms using individual variation reveals key role for chromatin modification. Proc Natl Acad Sci USA 103: 14062–14067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E (2006) An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics 7: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillon A, O’Sullivan J, Azad A, Proudfoot N, Mellor J (2003) Regulation of elongating RNA polymerase II by forkhead transcription factors in yeast. Science 300: 492–495 [DOI] [PubMed] [Google Scholar]
- Pic-Taylor A, Darieva Z, Morgan BA, Sharrocks AD (2004) Regulation of cell cycle-specific gene expression through cyclin-dependent kinase-mediated phosphorylation of the forkhead transcription factor Fkh2p. Mol Cell Biol 24: 10036–10046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptacek J, Devgan G, Michaud G, Zhu H, Zhu X, Fasolo J, Guo H, Jona G, Breitkreutz A, Sopko R, McCartney RR, Schmidt MC, Rachidi N, Lee SJ, Mah AS, Meng L, Stark MJ, Stern DF, De Virgilio C, Tyers M et al. (2005) Global analysis of protein phosphorylation in yeast. Nature 438: 679–684 [DOI] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L (2006) Genetics of global gene expression. Nat Rev Genet 7: 862–872 [DOI] [PubMed] [Google Scholar]
- Ronald J, Brem RB, Whittle J, Kruglyak L (2005) Local regulatory variation in Saccharomyces cerevisiae. PLoS Genet 1: e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, Ruff TG, Milligan SB, Lamb JR, Cavet G, Linsley PS, Mao M, Stoughton RB, Friend SH (2003) Genetics of gene expression surveyed in maize, mouse and man. Nature 422: 297–302 [DOI] [PubMed] [Google Scholar]
- Smith EN, Kruglyak L (2008) Gene-environment interaction in yeast gene expression. PLoS Biol 6: e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M (2006) BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34: D535–D539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinfeld I, Shamir R, Kupiec M (2007) A genome-wide analysis in Saccharomyces cerevisiae demonstrates the influence of chromatin modifiers on transcription. Nat Genet 39: 303–309 [DOI] [PubMed] [Google Scholar]
- Sun W, Yu T, Li KC (2007) Detection of eQTL modules mediated by activity levels of transcription factors. Bioinformatics 23: 2290–2297 [DOI] [PubMed] [Google Scholar]
- Suthram S, Beyer A, Karp RM, Eldar Y, Ideker T (2008) eQED: an efficient method for interpreting eQTL associations using protein networks. Mol Syst Biol 4: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Nakafuku M, Tamanoi F, Kaziro Y, Matsumoto K, Toh-e A (1990) IRA2, a second gene of Saccharomyces cerevisiae that encodes a protein with a domain homologous to mammalian ras GTPase-activating protein. Mol Cell Biol 10: 4303–4313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AJ, Khachigian LM, Shows T, Collins T (1995) Isolation and characterization of a novel zinc-finger protein with transcription repressor activity. J Biol Chem 270: 22143–22152 [DOI] [PubMed] [Google Scholar]
- Ye C, Galbraith SJ, Liao JC, Eskin E (2009) Using network component analysis to dissect regulatory networks mediated by transcription factors in yeast. PLoS Comput Biol 5: e1000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvert G, Brem RB, Whittle J, Akey JM, Foss E, Smith EN, Mackelprang R, Kruglyak L (2003) Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat Genet 35: 57–64 [DOI] [PubMed] [Google Scholar]
- Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136: 1457–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang B, Smith EN, Drees B, Brem RB, Kruglyak L, Bumgarner RE, Schadt EE (2008) Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat Genet 40: 854–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary tables and figures
Compressed TAR file containing position-specific affinity matrices (PSAMs) for yeast transcription factors (TFs), which we used to infer genotype-specific TF activities
Compressed TAR file containing position-specific affinity matrices (PSAMs) for mouse transcription factors (TFs), which we used to infer genotype-specific TF activities
Matrix containing inferred regulatory TF activities for each yeast segregant
Matrix containing inferred regulatory TF activities for each F2 mouse