

Abstract
Allosteric coupling between protein domains is fundamental to many cellular processes. For example, Hsp70 molecular chaperones use ATP binding by their actin-like N-terminal ATPase domain to control substrate interactions in their C-terminal substrate-binding domain, a reaction that is critical for protein folding in cells. Here, we generalize the statistical coupling analysis to simultaneously evaluate co-evolution between protein residues and functional divergence between sequences in protein sub-families. Applying this method in the Hsp70/110 protein family, we identify a sparse but structurally contiguous group of co-evolving residues called a ‘sector’, which is an attribute of the allosteric Hsp70 sub-family that links the functional sites of the two domains across a specific interdomain interface. Mutagenesis of Escherichia coli DnaK supports the conclusion that this interdomain sector underlies the allosteric coupling in this protein family. The identification of the Hsp70 sector provides a basis for further experiments to understand the mechanism of allostery and introduces the idea that cooperativity between interacting proteins or protein domains can be mediated by shared sectors.
Keywords: allostery, chaperone, co-evolution, SCA, sector
Introduction
Allosteric coupling, the process by which spatially distant sites on proteins functionally interact, is a defining biological property of many proteins, but the underlying structural basis remains difficult to understand (Smock and Gierasch, 2009). The central problem is the difficulty of detecting the pattern of cooperative functional interactions between amino-acid residues in protein structures. One approach to this problem is to analyze the correlated evolution of amino acids in a protein family—the expected statistical signature of conserved cooperative actions of amino acids (e.g. Lockless and Ranganathan, 1999; Kass and Horovitz, 2002; Liu et al, 2008). Recently, an approach for the global analysis of correlated evolution in protein families has been introduced (Estabrook et al, 2005; Russ et al, 2005; Socolich et al, 2005; Lee et al, 2008), and the results imply a new and potentially general architecture of amino-acid interactions within protein domains. The basic finding is that most residues evolve nearly independently, whereas a small fraction of residues is collectively coupled to form functional units called sectors (Halabi et al, 2009). A characteristic of sectors is structural connectivity; a contiguous system of sector residues within the protein core often connects distant surfaces in the three-dimensional structure. Thus, at least within single protein domains, sectors provide a structural basis for explaining functional properties of proteins such as allosteric coupling.
However, the principle of co-evolution of protein residues that underlies the sectors is not limited to the coupling of amino acids within a single domain. Indeed, allosteric coupling (or signal transmission) between two or more protein domains is a common finding in studies of cellular function. This suggests the existence of sectors—units of evolutionary selection—that are shared between different non-homologous protein domains. For example, a sector spanning two domains could couple a functional site on one protein domain to a functional site on a second protein domain. Such sectors could explain conserved aspects of allosteric coupling.
The Hsp70 molecular chaperones—a large and diverse family of allosteric two-domain proteins—present an excellent case study to test this concept. Hsp70 proteins interact with substrate proteins at a C-terminal substrate-binding domain, but both the affinity and kinetics of substrate binding are controlled by the activity of an N-terminal nucleotide-binding domain (Figure 1A). Specifically, exchange of ADP for ATP in the N-terminal domain reduces the binding affinity for substrates at the C-terminal domain and is accompanied by significant conformational change and interdomain docking (Mayer and Bukau, 2005; Rist et al, 2006; Swain et al, 2007; Bertelsen et al, 2009). The structure of the ATP-bound state of the Escherichia coli Hsp70, DnaK, is yet unsolved, but we made a model by homology-based methods and simulated-annealing molecular dynamics using the crystal structure of ATP-bound Hsp110 from yeast (Liu and Hendrickson, 2007) (Figure 1B; Supplementary Figure 8). This model illustrates the large conformational change in the substrate-binding domain associated with ATP binding in the nucleotide-binding domain, and indicates the expected interaction surface between the two Hsp70 domains. The allosteric cycle is completed when the intrinsic ATPase activity of the nucleotide-binding domain reverses the conformational rearrangement, returning the Hsp70 to an ADP-bound configuration suitable for another round of substrate binding and release.
Figure 1.

A model for Hsp70 interdomain allostery. (A) In the ADP-bound state, nucleotide-binding and substrate-binding domains tumble independently of one another, the hydrophobic interdomain linker is relatively exposed, the β-sandwich sub-domain is relatively ordered, the lid sub-domain is closed, and substrate binds with high affinity (PDB codes 1DKG and 1DKZ). (B) ATP binding is accompanied by conformational changes within the nucleotide-binding domain, domain docking with participation of the interdomain linker, opening of the lid sub-domain, reduction in order at the substrate-binding site within the β-sandwich, and loss of substrate-binding affinity.
The overall family of Hsp70-like proteins that comprises the Hsp70s and the Hsp110s, homologs that contain both domains and as indicated above, are regarded as structural models for Hsp70s (Easton et al, 2000). However, despite their sequence similarity, the Hsp110 proteins have evolved to be non-allosteric, such that the nucleotide-binding domain remains stably bound to ATP and does not appear to regulate the substrate-binding domain. Consistent with these findings, Hsp110s are incapable of folding substrate proteins on their own through cycles of nucleotide exchange and hydrolysis (Shaner and Morano, 2007).
Here, we present a new application of the statistical coupling analysis (SCA) to sequences of the Hsp70-like family in which we take advantage of the functional divergence of Hsp70s and Hsp110s to reveal patterns of co-evolution that can be associated with interdomain allostery (see Box 1). The identification of a group of co-evolving residues that show structural contiguity between the two Hsp70 domains provides testable hypotheses about allosteric function in these molecular chaperones and introduces methods that may be applicable for more generally characterizing co-evolution within and between protein domains.
SCA overview.
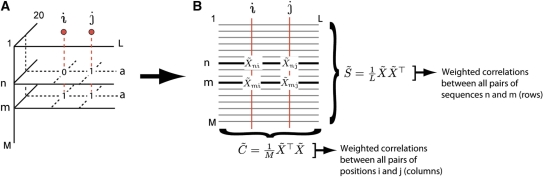
The aim of SCA is to examine the joint conservation of all pairs of amino-acid positions in a protein family to identify sectors—groups of sequence positions that mutually co-evolve in a protein family. As previously described (Halabi et al, 2009), the basic process is to start with a large and diverse multiple sequence alignment (MSA) of a protein family comprising M sequences by L positions, and to compute an L × L-weighted correlation matrix (C˜, the SCA matrix) that describes the co-evolution of all pairs of sequence positions. When sequences are diverged to such an equal extent (i.e. homogeneously) that distinct sub-families are not clearly evident, sector identification amounts to identifying positions that group in the top eigenmodes of the SCA correlation matrix. Here, we extend this approach to the case of ‘inhomogeneous’ sequence alignments in which functionally distinct sub-families of sequences can be identified in the MSA. Such functional structure in alignments can facilitate sector identification because of mathematical methods that provide a direct mapping between patterns of sequence divergence and patterns of positional covariation.
Step 1: Definition of alignment and correlation matrices
In general, an MSA can be described as a three-dimensional binary tensor Xsia (M × L × 20) whose elements are 1 if sequence s contains amino-acid a at position i and 0 if not (A). To use the mathematical methods below, we reduce the MSA to an M × L two-dimensional binary matrix Xsi by only including the terms in Xsia representing the most prevalent amino-acid at each position, a process we term the ‘binary approximation’ of the MSA. We next compute a weighted, normalized alignment
where ϕi is related to the conservation of position i in the MSA and is the weighting function used in the current implementation of SCA (B, and see SOM and Materials and methods), and 〈Xsi〉s represents the average value of Xsi over all sequences. In effect, weighting by ϕi provides a measure of the significance of amino-acid occurrences and correlation in the MSA. From the X˜ matrix representation of the MSA, two correlation matrices can then be computed: 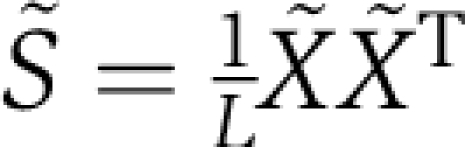 , which is a ϕi-weighted version of a sequence correlation matrix, and
, which is a ϕi-weighted version of a sequence correlation matrix, and 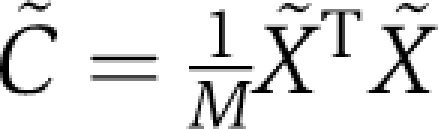 , which is a ϕi-weighted version of a positional correlation matrix (i.e. the SCA matrix).
, which is a ϕi-weighted version of a positional correlation matrix (i.e. the SCA matrix).
The use of the binary approximation of the MSA is a necessary simplification for usage of the specific mathematical methods for sector analysis in this work (described below). Generalization of the methods to consider the full alignment will be a subject of future work. However, we note that for instances such as the Hsp70/110 family in which the function of interest (e.g. allostery) is a property of a major sub-family, sector identification is robust to the binary approximation (Halabi et al, 2009).
Step 2: Mapping modes of sequence covariation and positional covariation
To relate the divergence of sub-families of sequences to the correlated evolution of groups of positions, we use the method of singular value decomposition. In this method, the M × L binary matrix X˜ can be written as a product of three matrices: X˜=UΣVT, where U is an M × M matrix whose columns contain the eigenvectors of S˜, the sequence correlation matrix, and V is an L × L matrix whose columns contain the eigenvectors of C˜, the SCA positional correlation matrix. Σ is a diagonal M × L matrix of so-called ‘singular values’ that are related to the eigenvalues of the S˜ and C˜ matrices.

The important concept is that if an eigenmode of the sequence correlation matrix (a column of U, ∣Un〉) reveals a separation of two classes of sequences, then the corresponding eigenmode of the positional correlation matrix (a column of V, ∣Vn〉) will reveal the positions that primarily contribute to this sequence divergence. However, in general, the eigenvectors of the S˜ or C˜ matrix need not represent statistically independent modes of sequence correlation or of positional correlation. For example, examination of the top three modes of the U matrix for the Hsp70/110 family (∣U1…3〉, C) reveals the existence of distinct sub-families of sequences (in different colors), but fails to clearly separate these sub-families along the orthogonal eigenvectors. To better represent the divergence of the sub-families, we used a simple implementation of independent component analysis (ICA), a method specifically designed to transform the k top eigenmodes of a correlation matrix into k maximally independent components. Application of ICA to the significant top eigenmodes of the Hsp70/110 family (see SOM and Materials and methods) indeed shows the separation of the sequences in the MSA into a few major sub-families that now largely separate along orthogonal independent components (∣U1…3S〉), D).
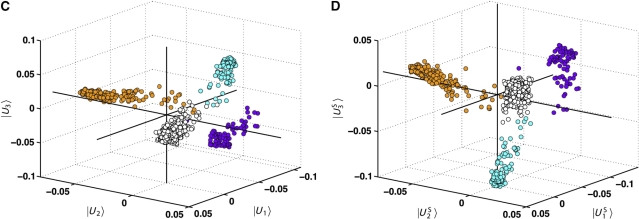
Application of the same ICA-transformation computed for the U matrix to the k top eigenmodes of the C˜ matrix (∣V1…k〉) then provides a corresponding transformation to define the positions responsible for the directions of sequence variation observed in panel D. Thus, we can identify correlated groups of sequence positions that are responsible for the divergence of groups of sequences.
In the main paper, Figure 2A shows the first independent component of the S˜ matrix for the family of Hsp70/110 proteins (∣U1S〉), which reveals a clear separation of the family into two groups—one that includes the allosteric Hsp70-like proteins (white, orange, and cyan in panels C and D, and black in Figure 2A) and one that includes the non-allosteric Hsp110-like proteins (purple, panels C and D, and gray in Figure 2A). The corresponding first independent component of the positional correlation matrix C˜ (∣V1S〉) reveals the positions most responsible for this sequence divergence (Figure 2B), and identifies the allosteric sector.
Results
A sector associated with Hsp70 allostery
To identify sectors in Hsp70, we used the SCA to compute a weighted correlation matrix, C˜, that describes the co-evolution of every pair of amino-acid positions in the Hsp70/110 family. The essence of sector identification is to analyze the non-random correlations in the C˜ matrix to find collectively evolving groups of residues. One approach to do this is spectral decomposition, in which sectors are defined by the pattern of residue contribution to the top few eigenmodes of the C˜ matrix (Halabi et al, 2009). Importantly, sector identification by this method proceeds without presupposing the function of sectors; such properties are then assigned through experimental study.
Analysis of the Hsp70/110 protein family suggests a more targeted strategy for analysis of the C˜ matrix in which we take advantage of the functional divergence of allosteric mechanism between Hsp70 and Hsp110 proteins to guide sector identification. The basic idea is to simultaneously evaluate the pattern of divergence between sequences in a protein family and the pattern of co-evolution between amino-acid positions (Casari et al, 1995; Lichtarge et al, 1996). This can be performed in the framework of SCA using a mathematical method known as singular value decomposition (see Box 1). If the pattern of sequence divergence classifies members of a protein family according to distinctions in a functional mechanism (e.g. allostery), then we can identify the group of co-evolving residues that correspond to this mechanism. We describe this approach here in context of the Hsp70/110 family (see Box 1, Materials and methods, and SOM for additional details). Given a weighted binarized sequence alignment (X˜) comprised of M sequences (rows) and L positions (columns), we can compute the following two correlation matrices:
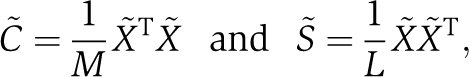 |
where C˜ is the SCA correlation matrix between positions and S˜ is a correlation matrix between sequences. The singular value decomposition of X˜ is:
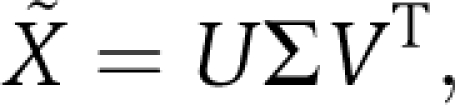 |
in which columns of U are eigenvectors of S˜, columns of V are eigenvectors of C˜, and Σ is related to the eigenvalues of these matrices. Importantly, this decomposition allows a direct mapping between each principal axis of sequence variation (a column in U) and the corresponding principal axis of positional co-evolution (the same column in V). If functionally distinct sequences segregate along an axis of sequence variation, then the positions that underlie this divergence are defined in the corresponding axis of positional co-evolution.
Examination of the top principal axes of sequence variation for the Hsp70/110 family (see Materials and methods) shows in fact a clear separation of the allosteric (Hsp70) and non-allosteric (Hsp110) members into two distinct clusters (Figure 2A). This axis of separation between the two sub-families is identified in an unbiased manner using an algorithm for independent component analysis (see Materials and methods; Supplementary information). The corresponding axis of the C˜ matrix of positional correlations reveals a protein sector comprising a small fraction of Hsp70/110 positions (∼20%, 115 sector out of 605 total residues) that underlie the separation of Hsp70 and Hsp110 family members (Figure 2B). The sector positions derive roughly equally from the nucleotide-binding domain (in blue, 56 positions; Figure 2B) and the substrate-binding domain (in green, 59 positions; Figure 2B) showing co-evolution of residues in both domains to form a single unit of evolutionary selection. Consistent with the finding that allosteric coupling is a property of the Hsp70 sub-family, positions comprising this sector are more conserved within the Hsp70 sub-family than in the Hsp110 sub-family (Supplementary Figure 5). Taken together, these results define an interdomain sector in the Hsp70 sub-family that is associated with the allosteric mechanism.
Figure 2.
Identification of an allosteric sector in Hsp70 proteins. (A) A histogram of the Hsp70/110 sequences projected on a top axis of sequence variation derived from singular value decomposition and independent component analysis (see Box 1 Materials and methods). This axis separates family into two sub-families that correspond to the allosteric Hsp70s and the non-allosteric Hsp110s. (B) A histogram of Hsp70 residues projected on the corresponding axis of positional co-evolution, showing that a small fraction of residues is largely responsible for the sequence divergence shown in panel A (115 sector out of 605 total residues or ∼20%). This defines the Hsp70 sector. The sector nearly equally comprises residues from the nucleotide-binding domain (blue, 56 residues) and the substrate-binding domain (green, 59 residues). (C) The Hsp70 sector mapped onto a model of the ATP-bound state of E. coli DnaK. The sector forms a physically contiguous group of residues that connect the ATP-binding site in the nucleotide-binding domain (pale blue) to the substrate-binding pocket of the substrate-binding domain (yellow) through the binding interface between the two domains. Sector positions are represented as spheres and colored as in panel B. Source data is available for this figure at www.nature.com/msb.

Structural interpretation of the Hsp70 sector
What is the structural interpretation of this Hsp70 sector? NMR (Swain et al, 2007; Bertelsen et al, 2009) and tryptophan fluorescence (Moro et al, 2003) data in DnaK, the E. coli Hsp70, show that in the ADP-bound state, the nucleotide-binding and substrate-binding domains are dissociated and largely independent. In contrast, upon ATP binding, the nucleotide-binding domain undergoes conformational rearrangement, participates in the interdomain interface, and promotes substrate release from the substrate-binding domain (Wilbanks et al, 1995; Moro et al, 2003; Mayer and Bukau, 2005; Swain et al, 2007).
To examine the spatial arrangement of the Hsp70 sector in the ATP-bound state, we represented sector residues on the Sse1-derived model for the DnaK Hsp70 (Supplementary Figure 7). Consistent with a function in allosteric coupling, residues comprising the Hsp70 sector form a physically contiguous network of atoms linking the ATP-binding site to the substrate-binding site, passing through the interdomain interface (Figure 2C). The physical connectivity is remarkable given that only a small fraction of overall Hsp70 residues is involved (Figure 2B). Prior work showed that sparse but connected clusters of amino acids forming sectors link distantly positioned functional sites within individual protein domains (Lockless and Ranganathan, 1999; Socolich et al, 2005; Halabi et al, 2009). This work extends this result to show that functionally coupled but non-homologous protein domains can share a single sector that connects their respective functional sites through a protein–protein interface.
Functional studies of Hsp70 allostery
Does the Hsp70 sector represent the mechanism of allosteric coupling between the nucleotide-binding domain and substrate-binding domain? A number of biochemical and genetic studies on a variety of Hsp70s provide a basis for this assessment. Within the nucleotide-binding domain, sector positions include catalytic residues E171 and D201, the mutation of which impairs ATP-induced conformational change (Johnson and McKay, 1999), and T199, the mutation of which stabilizes ATP-induced conformational change (Buchberger et al, 1995), among other sites, making direct contact with bound nucleotide (Figure 3A). Studies of isolated bacterial Hsp70 nucleotide-binding domains have shown ATP-dependent reorientation of all four sub-domains (Zhang and Zuiderweg, 2004; Bhattacharya et al, 2009), and the sector spans all of the sub-domain interfaces. In particular, actin and Hsp70 retain sequence conservation at nucleotide-binding loops and adjacent crossing helices that form an interface between sub-domains 1A and 2A (Figure 1A) (Bork et al, 1992). Actin responds to bound nucleotide through an ATP-dependent shearing motion between sub-domains 1A and 2A (Schuler, 2001), and this structural region is a focal point of Hsp70 sector mapping. These findings are consistent with the view that at least in part, the co-evolution of sector positions may be related to the anisotropic physical coupling of amino acids within the protein structure. A similar empirical relationship has been noted between distributed physical interactions and the sector-mediating specificity in the S1A serine proteases (Halabi et al, 2009).
Figure 3.

Evidence for a function of the Hsp70 sector in allosteric coupling. Examination of sector positions in the nucleotide-binding domain (blue) and in the substrate-binding domain (green) reveals many experimentally characterized sites implicated in allostery (colored in red; see text for details) (A) In the ATP-binding site, sector residues include determinants of nucleotide hydrolysis and nucleotide-mediated conformational change. (B) At nucleotide-binding domain surface contacting the substrate-binding domain, a patch of sector residues includes many known to perturb allostery upon mutation. (C) In the substrate-binding domains, the sector contains residues both proximal and distal from the substrate-binding site, and includes the functionally critical interdomain linker (390–392). (D) At the substrate-binding domain surface that comprises the interface with the nucleotide-binding domain, sector residues include many that display defects in allostery upon mutation. (E) Across the interdomain interface, mutation at residues Q152 and F216 (not shown) partially suppress the loss-of-function mutations at position 462 on the substrate-binding domain. Also shown are positions 415 and 326, sector residues that contact across the interdomain interface and are previously untested with regard to allosteric function.
Moreover, the crossing helices form a solvent-accessible cleft between sub-domains 1A and 2A in actin-like nucleotide-binding domains. In actin, the cleft mediates interaction with allosteric effector proteins (Dominguez, 2004), whereas in Hsp70/110, the cleft is proposed to act as an intramolecular-binding surface for the interdomain linker (Jiang et al, 2007; Liu and Hendrickson, 2007; Swain et al, 2007). In the allosteric Hsp70 sector that our analysis describes, the cleft surface is lined with sector residues (e.g. Y145, D148, K155, E217 and V218; see Figure 3B), and mutation at these sites is reported to perturb Hsp70 allostery (Gassler et al, 1998; Vogel et al, 2006). The conserved interdomain linker sequence motif 389VLLL392 in the sector stimulates ATPase activity when present on truncated nucleotide-binding domain constructs (Swain et al, 2007) and its mutation impairs interdomain allostery in full-length Hsp70 (Figure 3C and D) (Laufen et al, 1999; Vogel et al, 2006). Binding of the linker to the cleft below the crossing helices is postulated to be important to the formation of the domain-docked state, bringing the substrate-binding domain into proximity to the nucleotide-binding domain (Swain et al, 2007).
Sector positions within the substrate-binding domain comprise a structurally contiguous set of atoms that extends from the substrate-binding site through the protein core to a solvent-exposed region that includes the interdomain linker (Figure 3C and D). The functional significance of the sector is supported by several previous observations. Sector residue K414 is centered in the domain surface patch, making multiple interdomain contacts in the docked state. Previous work showed that this residue has a critical function in allosteric signal transmission as mutation at this site blocked interdomain docking and allostery (Montgomery et al, 1999). A substrate-binding domain sector position (I462) has been shown to have an epistatic relationship with sector positions in the nucleotide-binding domain (Q152 and F216): mutation of I462 to Asn is lethal in the yeast Hsp70 Ssc1, but is partially suppressed by nucleotide-binding domain mutations Q152L or F216L (Figure 3E) (Davis et al, 1999). There is also evidence that structural regions not essential for allosteric coupling are not involved in the interdomain sector; the substrate-binding domain lid is nearly absent in sector residues, and a DnaK variant lacking the lid retains core allosteric function (Swain et al, 2006). Interestingly, several sector positions within the substrate-binding domain experience large NMR chemical shift changes upon binding of a peptide substrate (S398, T403, G405, T428, D431, I438, F457, L459, G468) (Swain et al, 2006). In addition, mutation of sector residues far from the substrate-binding site (S398, G400, G443, E444, L459) reduces substrate-binding affinity (Figure 3C and D) (Burkholder et al, 1996).
The physical and functional connectivity of a single co-evolutionary sector across domains provides strong support for the proposal that the sector mediates the allosteric coupling central to the basic biological activity of Hsp70.
Direct experimental analysis of the interdomain sector
Knowledge of the interdomain sector provides new hypotheses for further experimental testing. For example, residues D326 and N415 are sector positions that display interdomain contact, but no previous experiment has tested their involvement in interdomain allostery (Figure 3; Supplementary Figure 7). Therefore, we made conservative mutations based on amino-acid frequencies at these positions in Hsp70 sequences and measured the effect on interdomain allostery both in vivo and in vitro. A direct test for the influence of sector mutants on organism fitness is provided by the ability of DnaK to promote E. coli growth at elevated temperature (Bukau and Walker, 1990). For example, strains of E. coli in which the chromosomal copy of DnaK is deleted grow very weakly after heat shock, but are rescued by expression of wild-type DnaK from a plasmid (Figure 4A). In contrast, the D326V or N415G DnaK variants fail to complement the DnaK knockout strain upon heat shock, showing that these positions are critical for Hsp70 activity.
Figure 4.

Experimental test of sector-based predictions of allosteric mechanism. (A) A stress-response assay, showing that though wild-type DnaK efficiently rescues growth in an E. coli dnaK knockout strain at heat-shock conditions (43°C), DnaK variants D326V and N415G do not. (B) Fluorescence of the sole tryptophan residue in DnaK is diagnostic for ATP-dependent interdomain docking. DnaK D326V and N415G display normal tryptophan fluorescence in the absence of ATP, but only a partial conversion of the ensemble to the ATP state based on blue shift and intensity (wild type, black; D326V, blue; N415G, red). (C) The sector mutations distal from the ATP-binding site cause elevated basal ATPase activities (gray) and reduced stimulation by the substrate peptide p5 (p5-stimulated rates in white; fold stimulation below plot). (D) These data can be explained by a two-state model in which sector mutants are partially defective in the formation of the docked, ATP-bound Hsp70 conformation.
The origin of these cellular defects was investigated by purifying the mutant DnaK proteins and using biochemical tests of allosteric function in vitro. DnaK D326V and N415G are soluble, natively folded and thermally stable in the absence of ATP and substrate (Supplementary Figure 9). The fluorescence of the sole intrinsic tryptophan residue in DnaK is diagnostic for ATP-dependent interdomain docking because it displays a characteristic blue shift and intensity quench upon interdomain interaction (Moro et al, 2003). DnaK D326V and N415G show the same tryptophan fluorescence spectrum as wild-type DnaK in the absence of nucleotide, indicating that W102 is in its normal chemical environment in the undocked state. However, the extents of W102 fluorescence blue shifting and intensity quenching upon addition of ATP are reduced relative to wild type (Figure 4B). The same trends are observed for wild type and mutants when W102 accessibility is assessed by acrylamide quenching (Supplementary Figure 10). These findings are characteristic of a specific defect in ATP-induced conformational change and domain docking in the point mutants. In addition, functional Hsp70 allostery entails an approximately seven-fold stimulation of ATPase activity upon binding of peptide to the substrate-binding domain. Relative to wild-type DnaK, D326V and N415G show significantly elevated basal ATPase rates and only approximately three-fold stimulation by peptide (Figure 4C). Given these data and the knowledge that ATP hydrolysis is the rate-limiting step of the reaction cycle (McCarty et al, 1995), the likely interpretation is that the sector mutants shift the normal DnaK conformational equilibrium from the ATP-induced, domain-docked state to the more independent domain arrangement characteristic of the ADP state (Figure 4D).
These findings are consistent with the hypothesis that these two sector positions are important for stabilizing the interdomain interface and mediating allosteric communication between domains. More generally, these data provide further evidence that Hsp70 sector analysis has predictive value in describing an interdomain allosteric network.
Discussion
In summary, we show that sequence analysis alone of the Hsp70/110 molecular chaperone family identifies a group of co-evolving residues, a sector, that is responsible for the core function of the Hsp70 proteins—allosteric coupling between distantly positioned functional sites on two distinct protein domains. As per previous reports (Lockless and Ranganathan, 1999; Socolich et al, 2005; Halabi et al, 2009), the sector is sparse, such that only a small fraction of total amino acids in the protein are involved, and physically contiguous, so that the ATP-binding site on the nucleotide-binding domain is connected to the substrate-binding site on the substrate-binding domain through a continuous network of interacting amino acids. The identification of the sector provides a clear basis for directing new experiments toward a more complete understanding of the mechanism and evolutionary divergence of allostery in these proteins. For example, Hsp70s use diverse co-chaperones in team-assisted functions and many sector positions emerging as an allosteric surface for interdomain allostery in Hsp70s also have a function in J-domain binding and J-mediated ATPase stimulation (Gassler et al, 1998; Suh et al, 1998; Vogel et al, 2006; Jiang et al, 2007).
This work adds an important new finding with regard to the concept of protein sectors. Previous work showed that multiple quasi-independent sectors are possible within a single protein domain, each of which contributes to a different aspect of function (Halabi et al, 2009). Here, we show that a single sector can also exist to functionally couple two different, non-homologous protein domains. This result emphasizes the point that sectors are simply defined as units of selection, without regard to hierarchies of structural organization. An interesting possibility that follows is that sectors could physically join and co-evolve across protein–protein interfaces in order to mediate the coupling of activities between proteins—the essence of signal transmission and allosteric regulation. Indeed, this idea has been recently used to design a synthetic two-domain allosteric protein (Lee et al, 2008), and a similar concept was used to map regions involved in controlling the specificity of bacterial two-component signaling systems (Skerker et al, 2008). It will be interesting to further test the notion that interaction between protein sectors is a process through which allostery between proteins might evolve.
Materials and methods
Multiple sequence alignment
Hsp70/110 sequences were obtained by combining the non-redundant results of PSI-BLAST (Altschul et al, 1997) searches queried with E. coli DnaK, human Hsc70, and yeast Sse1. Sequences were aligned automatically (Thompson et al, 1994) and by manual structure-based methods (Doolittle et al, 1996). Non-Hsp70/110 sequences were removed based on their anomalous length or sequence identity. Any sequence sharing >95% similarity to another sequence was removed to distribute sampling. The final alignment was large (926 sequences) and diverse (unconserved sites approached random amino-acid distributions).
Statistical coupling analysis
As in previous work (Halabi et al, 2009), the alignment is binarized in an M by L matrix X with Xsi=1, if the most frequent amino-acid at position i is present in sequence s, and Xsi=0 otherwise; M is here the number of sequences (rows of X) and L the number of positions (columns of X). The definition of the SCA matrix C˜ involves position-specific weights ϕi that quantify the degree of conservation of each position i: 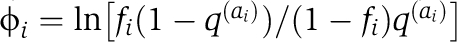 , where fi represents the frequency of the prevalent amino-acid ai at position i, and
, where fi represents the frequency of the prevalent amino-acid ai at position i, and 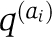 a background frequency for this amino-acid. A weighted alignment X˜ is defined with X˜si=ϕi(X˜si−fi). The SCA matrix C˜ is the L by L matrix of correlations between positions given by C˜=X˜TX˜/M, where X˜T denotes the transpose of X˜. Similarly, S˜=X˜X˜T/L gives an M by M matrix of correlations between sequences. The eigenvectors of C˜ and S˜ form the columns of two orthogonal matrices, V and U, which are related through the singular value decomposition of X˜: X˜=UΣVT, where Σ is a diagonal matrix.
a background frequency for this amino-acid. A weighted alignment X˜ is defined with X˜si=ϕi(X˜si−fi). The SCA matrix C˜ is the L by L matrix of correlations between positions given by C˜=X˜TX˜/M, where X˜T denotes the transpose of X˜. Similarly, S˜=X˜X˜T/L gives an M by M matrix of correlations between sequences. The eigenvectors of C˜ and S˜ form the columns of two orthogonal matrices, V and U, which are related through the singular value decomposition of X˜: X˜=UΣVT, where Σ is a diagonal matrix.
An independent component analysis provides a linear transformation Ws that maps the top three eigenvectors of S˜ into three maximally independent axes of sequence variations (see Supplementary information for algorithmic details). One of these three directions, the M-dimensional vector 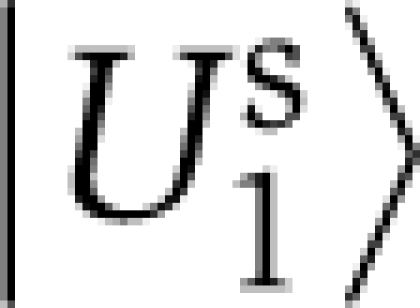 , is found to discriminate the non-allosteric sequences from the rest of the sequences in the alignment (Figure 2A). Applying the same linear transformation Ws to the top three eigenvectors of C˜ defines a direction of positional variations, the L-dimensional vector
, is found to discriminate the non-allosteric sequences from the rest of the sequences in the alignment (Figure 2A). Applying the same linear transformation Ws to the top three eigenvectors of C˜ defines a direction of positional variations, the L-dimensional vector 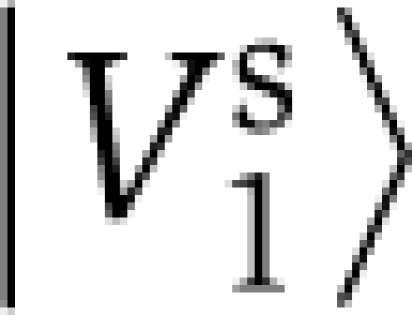 , which indicates the positions underlying the discrimination. The allosteric sector is defined as the positions i making significant contribution to
, which indicates the positions underlying the discrimination. The allosteric sector is defined as the positions i making significant contribution to 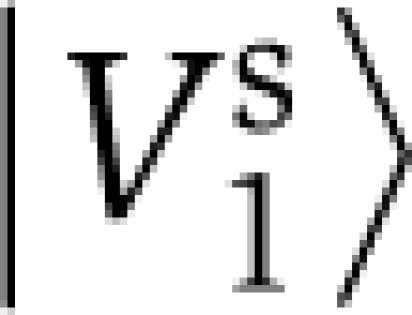 , that is
, that is 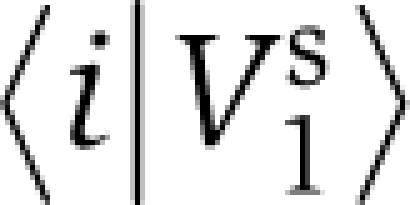 ⩾ε, where ε=0.05 corresponds to a threshold of statistical significance (Figure 2B).
⩾ε, where ε=0.05 corresponds to a threshold of statistical significance (Figure 2B).
Structural modeling
The ATP-bound Saccharomyces cerevisiae Sse1 structure and a sequence alignment between Sse1 (Hsp110) and DnaK (Hsp70) (Liu and Hendrickson, 2007) were used to generate a homology model of DnaK(ATP) using Modeller (Sali and Blundell, 1993). Molecular dynamics simulations were carried out using the Gromacs platform and Gromos96 force field (Hess et al, 2008). The structural model was truncated at residue 531, and ATP, magnesium, and potassium ions coordinated in the active site were included. An ATP topology file provided by an earlier study (Colombo et al, 2008) was used. The system was solvated in a box with at least 12 Å spacing from protein atoms to the edge of the box. Net charge was neutralized with potassium ions and energy was minimized by steepest descents followed by short position-restrained molecular dynamics to equilibrate water molecules at 300 K. In the production of molecular dynamics simulation, a simulated-annealing protocol cycled through temperature gradients based on previous work on a smaller system (Lindorff-Larsen et al, 2005): 300–400 K over 150 ps, 400–350 K over 150 ps, 350–300 K over 500 ps, and 300 K held for 100 ps. Berendsen temperature coupling, Parrinello–Rahman pressure coupling, and a periodic boundary condition were used. All trajectory analysis was performed within the Gromacs package. Atomic RMSD fluctuations were analyzed by principal component analysis, and cosine content of the first component indicated that motions within the RMSD plateau region were dominated by diffusion (Hess, 2002). To avoid over-interpretation, structural clustering was performed on the trajectory such that the entire RMSD plateau region was defined as a single cluster (4.7–53 ns) to determine a median structure, and correlated motions within this region were not investigated further on the basis of cooperativity. PyMol was used for molecular visualization (Delano, 2002).
Heat-shock assay
Plasmid pMS119 containing a wild-type E. coli dnaK gene insertion was used as a template for site-directed mutagenesis (Montgomery et al, 1999). Plasmids were transformed into temperature-sensitive E. coli BB1553 cells (ΔdnaK52, sidB1) (Bukau and Walker, 1990). Single colonies were grown overnight in LB in the presence of antibiotics at 30°C, and each growth's optical density at 600 nm was normalized to 0.2 by dilution with LB media. Growths were serially diluted 10-fold in water pre-equilibrated at 43°C, spotted onto growth media plates at 43°C, and placed in an incubator at the same temperature for 15 h. Leaky expression of the pMS119 tac promoter was sufficient to achieve nearly optimal growth rescue by plasmid-encoded DnaK in LB media without IPTG induction.
Purification of proteins and peptides
E. coli DnaK was prepared similarly as previously described (Montgomery et al, 1999), except that E. coli BB1553 cells were used and grown at 30°C. In a modified two-column purification, the first anion exchange column was used with buffers at pH 7.4. In the second column, DnaK was eluted from ATP agarose with 2 mM ADP. KCl replaced NaCl in all purification buffers. Crude p5 peptide (CLLLSAPRR) was purchased from Genscript and purified by HPLC using a diphenyl column with elution at ∼30% acetronitrile and 70% water; mass spectrometry confirmed the identity of the peptide.
ATPase activity
Steady-state ATPase activity was measured in an enzyme-coupled system as previously described (Montgomery et al, 1999). The ATPase activity of 1 μM DnaK plus or minus 100 μM p5 at 30°C was measured on a Biotek Gen5 platereader using Costar 3631 plates. Measurements were taken 3–5 times for each DnaK and auto-hydrolysis sample.
W102 fluorescence
DnaK W102 fluorescence and acrylamide quenching were measured similarly as previously described (Moro et al, 2003). Measurements were taken at room temperature in a Photon Technology International fluorometer at 295 nm excitation wavelength with 4 nm slit widths on both excitation and emission sides. For each sample, spectra were averaged over 10 acquisitions and normalized to an intensity of 1.0 before the addition of ATP. Faster 15 s scans showed the same spectral trends, indicating that ATP hydrolysis was not a complicating factor during the measurement.
Circular dichroism
Measurements were taken as previously described (Montgomery et al, 1999) using a Jasco J-715 spectrophotomer. Wavelength scans were measured at 30°C using 2 μM DnaK in 10 mM potassium phosphate buffer at pH 7.6. Temperature melts were measured at 222 nm using 2 μM DnaK in 10 mM potassium phosphate buffer, 1 mM MgCl2 and 1 mM ADP, pH 7.6.
Supplementary Material
Supplementary Information
Acknowledgments
This study was supported by grants from NIH (LMG), the Robert A Welch foundation (RR), the Green Center for Systems Biology (RR), and a Simons Foundation fellowship from Rockefeller University (OR). Computational resources were funded by NSF and NIH.
Footnotes
The authors declare that they have no conflict of interest.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ERP (2009) Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci USA 106: 8471–8476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Kurochkin AV, Yip GNB, Zhang Y, Bertelsen EB, Zuiderweg ERP (2009) Allostery in Hsp70 chaperones is transduced by subdomain rotations. J Mol Biol 388: 475–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P, Sander C, Valencia A (1992) An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc Natl Acad Sci USA 89: 7290–7294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Theyssen H, Schroder H, McCarty JS, Virgallita G, Milkereit P, Reinstein J, Bukau B (1995) Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J Biol Chem 270: 16903–16910 [DOI] [PubMed] [Google Scholar]
- Bukau B, Walker GC (1990) Mutations altering heat shock specific subunit of RNA polymerase suppress major cellular defects of E. coli mutants lacking the DnaK chaperone. EMBO J 9: 4027–4036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkholder WF, Zhao X, Zhu X, Hendrickson WA, Gragerov A, Gottesman ME (1996) Mutations in the C-terminal fragment of DnaK affecting peptide binding. Proc Natl Acad Sci USA 93: 10632–10637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casari G, Sander C, Valencia A (1995) A method to predict functional residues in proteins. Nat Struct Biol 2: 171–178 [DOI] [PubMed] [Google Scholar]
- Colombo G, Morra G, Meli M, Verkhivker G (2008) Understanding ligand-based modulation of the Hsp90 molecular chaperone dynamics at atomic resolution. Proc Natl Acad Sci USA 105: 7976–7981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JE, Voisine C, Craig EA (1999) Intragenic suppressors of Hsp70 mutants: interplay between the ATPase- and peptide-binding domains. Proc Natl Acad Sci USA 96: 9269–9276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano WL (2002) The PyMol Molecular Graphics System. Palo Alto, CA, USA: Delano Scientific [Google Scholar]
- Dominguez R (2004) Actin-binding proteins—a unifying hypothesis. Trends Biochem Sci 29: 572–578 [DOI] [PubMed] [Google Scholar]
- Doolittle RF, Abelson JN, Simon MI (1996) Computer methods for macromolecular sequence analysis. In Methods Enzymol, Doolittle RF (ed), Vol. 266, pp 497–598. San Diego: Academic Press [Google Scholar]
- Easton DP, Kaneko Y, Subjeck JR (2000) The Hsp110 and Grp1 70 stress proteins: newly recognized relatives of the Hsp70s. Cell Stress Chaperones 5: 276–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estabrook RA, Luo J, Purdy MM, Sharma V, Weakliem P, Bruice TC, Reich NO (2005) Statistical coevolution analysis and molecular dynamics: identification of amino acid pairs essential for catalysis. Proc Natl Acad Sci USA 102: 994–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassler CS, Buchberger A, Laufen T, Mayer MP, Schroder H, Valencia A, Bukau B (1998) Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc Natl Acad Sci USA 95: 15229–15234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halabi N, Rivoire O, Leibler S, Ranganathan R (2009) Protein sectors: evolutionary units of three-dimensional structure. Cell 138: 774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B (2002) Convergence of sampling in protein simulations. Phys Rev E Stat Nonlin Soft Matter Phys 65: 031910. [DOI] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4: 435–447 [DOI] [PubMed] [Google Scholar]
- Jiang J, Maes EG, Taylor AB, Wang L, Hinck AP, Lafer EM, Sousa R (2007) Structural basis of J cochaperone binding and regulation of Hsp70. Mol Cell 28: 422–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ER, McKay DB (1999) Mapping the role of active site residues for transducing an ATP-induced conformational change in the bovine 70-kDa heat shock cognate protein. Biochemistry 38: 10823–10830 [DOI] [PubMed] [Google Scholar]
- Kass I, Horovitz A (2002) Mapping pathways of allosteric communication in GroEL by analysis of correlated mutations. Proteins 48: 611–617 [DOI] [PubMed] [Google Scholar]
- Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B (1999) Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci USA 96: 5452–5457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Natarajan M, Nashine VC, Socolich M, Vo T, Russ WP, Benkovic SJ, Ranganathan R (2008) Surface sites for engineering allosteric control in proteins. Science 322: 438–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge O, Bourne HR, Cohen FE (1996) An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol 257: 342–358 [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Best RB, Depristo MA, Dobson CM, Vendruscolo M (2005) Simultaneous determination of protein structure and dynamics. Nature 433: 128–132 [DOI] [PubMed] [Google Scholar]
- Liu Q, Hendrickson WA (2007) Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell 131: 106–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Eyal E, Bahar I (2008) Analysis of correlated mutations in HIV-1 protease using spectral clustering. Bioinformatics 24: 1243–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless SW, Ranganathan R (1999) Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286: 295–299 [DOI] [PubMed] [Google Scholar]
- Mayer M, Bukau B (2005) Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 62: 670–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty JS, Buchberger A, Reinstein J, Bukau B (1995) The role of ATP in the functional cycle of the DnaK chaperone system. J Mol Biol 249: 126–137 [DOI] [PubMed] [Google Scholar]
- Montgomery DL, Morimoto RI, Gierasch LM (1999) Mutations in the substrate binding domain of the Escherichia coli 70 kDa molecular chaperone, DnaK, which alter substrate affinity or interdomain coupling. J Mol Biol 286: 915–932 [DOI] [PubMed] [Google Scholar]
- Moro F, Fernandez V, Muga A (2003) Interdomain interaction through helices A and B of DnaK peptide binding domain. FEBS Lett 533: 119–123 [DOI] [PubMed] [Google Scholar]
- Rist W, Graf C, Bukau B, Mayer MP (2006) Amide hydrogen exchange reveals conformational changes in Hsp70 chaperones important for allosteric regulation. J Biol Chem 281: 16493–16501 [DOI] [PubMed] [Google Scholar]
- Russ WP, Lowery DM, Mishra P, Yaffe MB, Ranganathan R (2005) Natural-like function in artificial WW domains. Nature 437: 579–583 [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815 [DOI] [PubMed] [Google Scholar]
- Schuler H (2001) ATPase activity and conformational changes in the regulation of actin. Biochim Biophys Acta 1549: 137–147 [DOI] [PubMed] [Google Scholar]
- Shaner L, Morano KA (2007) All in the family: atypical Hsp70 chaperones are conserved modulators of Hsp70 activity. Cell Stress Chaperones 12: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerker JM, Perchuk BS, Siryapron A, Lubin EA, Ashenberg O, Gouilian M, Laub MT (2008) Rewiring the specificity of two-component signal transduction systems. Cell 133: 1043–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smock RG, Gierasch LM (2009) Sending signals dynamically. Science 324: 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socolich M, Lockless SW, Russ WP, Lee H, Gardner KH, Ranganathan R (2005) Evolutionary information for specifying a protein fold. Nature 437: 512–518 [DOI] [PubMed] [Google Scholar]
- Suel GM, Lockless SW, Wall MA, Ranganathan R (2003) Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol 10: 59–69 [DOI] [PubMed] [Google Scholar]
- Suh W-C, Burkholder WF, Lu CZ, Zhao X, Gottesman ME, Gross CA (1998) Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc Natl Acad Sci USA 95: 15223–15228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM (2007) Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell 26: 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain JF, Schulz EG, Gierasch LM (2006) Direct comparison of a stable isolated Hsp70 substrate-binding domain in the empty and substrate-bound states. J Biol Chem 281: 1605–1611 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel M, Bukau B, Mayer MP (2006) Allosteric regulation of Hsp70 chaperones by a proline switch. Mol Cell 21: 359–367 [DOI] [PubMed] [Google Scholar]
- Wilbanks SM, Chen L, Tsuruta H, Hodgson KO, McKay DB (1995) Solution small-angle X-ray scattering study of the molecular chaperone Hsc70 and its subfragments. Biochemistry 34: 12095–12106 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zuiderweg ERP (2004) The 70-kDa heat shock protein chaperone nucleotide-binding domain in solution unveiled as a molecular machine that can reorient its functional subdomains. Proc Natl Acad Sci USA 101: 10272–10277 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information