

Abstract
Myeloma is a malignant proliferation of monoclonal plasma cells. Although morphologically similar, several subtypes of the disease have been identified at the genetic and molecular level. These genetic subtypes are associated with unique clinico-pathological features and dissimilar outcome. At the top hierarchical level, myeloma can be divided into hyperdiploid and non-hyperdiploid subtypes. The latter is mainly composed of cases harboring IgH translocations, generally associated with more aggressive clinical features and shorter survival. The three main IgH translocations in myeloma are the t(11;14)(q13;q32), t(4;14)(p16;q32) and t(14;16)(q32;q23). Trisomies and a more indolent form of the disease characterize hyperdiploid myeloma. A number of genetic progression factors have been identified including deletions of chromosomes 13 and 17 and abnormalities of chromosome 1 (1p deletion and 1q amplification). Other key drivers of cell survival and proliferation have also been identified such as nuclear factor- B-activating mutations and other deregulation factors for the cyclin-dependent pathways regulators. Further understanding of the biological subtypes of the disease has come from the application of novel techniques such as gene expression profiling and array-based comparative genomic hybridization. The combination of data arising from these studies and that previously elucidated through other mechanisms allows for most myeloma cases to be classified under one of several genetic subtypes. This paper proposes a framework for the classification of myeloma subtypes and provides recommendations for genetic testing. This group proposes that genetic testing needs to be incorporated into daily clinical practice and also as an essential component of all ongoing and future clinical trials.
Keywords: multiple myeloma, genetics, cytogenetics, molecular, prognosis, gene expression profiling
Introduction
This paper is a first attempt at creating an international consensus classification of multiple myeloma (MM). Undoubtedly this paper will present a working classification for MM, and will be updated as additional information becomes available regarding the underlying biological and genetic composition of the disease. The goals of this paper are: (i) to provide a biological classification of MM based on known genetic subtypes with associated clinicopathological associations; (ii) to establish the prognostic value of known and new genetic factors for MM outcome, including the information generated through genomic tools; (iii) and lastly, to provide a framework for evaluation of new markers capable of serving as predictive for efficacy of novel therapeutics.
Multiple myeloma is a clonal late B-cell disorder in which malignant plasma cells (PCs) expand and accumulate in the bone marrow, leading to cytopenias, bone resorption and the production (in most cases) of the characteristic monoclonal protein.1 MM is a heterogeneous disease with some patients dying within a few weeks of diagnosis, whereas others live for longer than 10 years. The reason for this heterogeneity is compound and involves interaction between host factors and features intrinsic to disease biology. It is increasingly evident that the underlying genetic features of the tumor cells largely dictate the clinical heterogeneity of MM. The advent of interphase molecular cytogenetics and genomics has unraveled a complexity hereto underappreciated for MM oncogenomics.
Throughout this paper we will review current knowledge regarding the effect those factors have in determining the likelihood of a better or worse outcome for patients with new diagnosis MM (prognosis). However, the validity of most prognostic factors has been tested predominantly in the new diagnosis setting, and little validation exists for the same factors in the case of relapsed and refractory disease. Moreover, the value of different prognostic factors possibly changes with advancing stages of the disease (that is, first relapse versus second and subsequent relapses). Biological factors that can predict outcome at diagnosis possibly will have much lessened effect when tested in patients receiving third-line chemotherapy. For instance, although it is generally accepted that patients entered into clinical trials for relapsed/refractory disease carry the worst outcome, this is generally not true if one estimates survival since the time of diagnosis. Patients with the most dire host factors, or the most aggressive biological variants of MM, will not live for long enough to be enrolled in these types of clinical trials. Lastly, it is important to stress that different stages of clonal evolution can be categorized as all being ‘new diagnosis MM.’ Some patients with new diagnosis MM may be a slow progressive evolution from monoclonal gammopathy of undetermined significance (MGUS) (for example, evolving anemia over several months), whereas others may be associated with features of high clonal aggressiveness (for example, PC leukemia or extramedullary plasmacytomas) (Figure 1). Although both can be clinically categorized as ‘new diagnosis MM’ they clearly present two different biological states of evolution of the monoclonal PCs.
Figure 1.
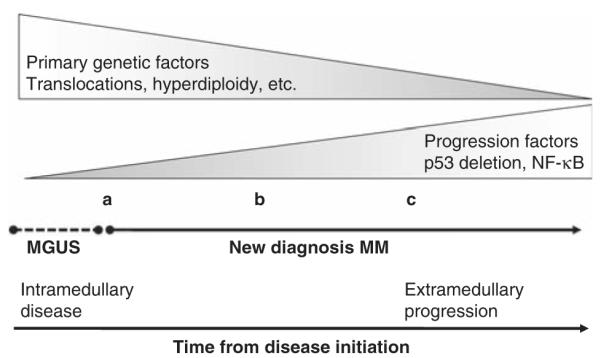
Relationship between clonal evolution of plasma cells and time of diagnosis. The picture depicts the biology and genetic heterogeneity of patients with a clinical diagnosis of ‘new diagnosis myeloma.’ In some cases (a) the situation involves a slow progression from MGUS with gradual development of mild anemia, incipient evidence of bone disease and slowly emerging need for treatment. In some other individuals (b) myeloma presents with frank clinical features of aggressive disease (for example, bone lesions, anemia and other). Furthermore, in some individuals (c) the disease presents with very aggressive features, including extramedullary disease, multiple plasmacytomas and other complicating features. In these three scenarios the clinical diagnosis is of ‘new diagnosis myeloma’, yet the biological and genetic features are quiet different.
The purpose of this proposed classification is not merely to provide prognostic estimates; the ability to accurately prognosticate is one of the several features to be discussed here. There may be subgroups of the disease that have no known or demonstrable prognostic associations, but are perceived at the biological level to be unique. These associations, though supporting unique biology, should not be considered prerequisite for creation of a biological subtype of the disease. Conversely, there may be biological factors or gene expression signatures capable of discerning prognosis, which may not yet be explained by unified biological concepts, but have the ability to discern patients with clearly dissimilar outcomes. In this paper, we will not attempt to discuss all available prognostic models for MM, but rather focus on the prognostic implications of the genetic derangements of MM and discuss the power of genomics to unravel the prognostic subcategories of the disease.
There are many reasons why an accurate prognostic determination is paramount for clinical practice and research (Table 1). It allows the physician to engage in a more direct discussion with the patient regarding disease threat and likelihood of survival. This risk stratification also allows for a more rational selection and sequencing of therapy approaches. This prognostic classification is essential for better understanding the composition of patients entered into clinical trials, and also allows, albeit with the usual statistical limitations, cross comparison of different clinical trial populations. Indirectly, this prognostic classification can also provide relevance to new biological factors proposed as significant in disease pathogenesis (but, as will be shown below, biological factors that are considered crucial in the pathogenesis do not need to necessarily have prognostic associations).
Table 1.
FISH markers and association with outcome for patients with MM
| Level | FISH tests | Testing frequency |
Validation |
|---|---|---|---|
| Minimal proposed testing (essential testing) | |||
| Established markers | t(4;14)(p16;q32) | Once | Validated by several studies |
| t(14;16)(q32;q23) | Once | ||
| 17p13 | May be repeated | ||
| Expanded panel | |||
| Markers with modest effects |
Hyperdiploidy | Once | Weak effects when used alone. The first two may portend a more favorable outcome |
| t(11;14)(q13;q32) | Once | ||
| Chromosome 13 | May be repeated | ||
| Other | Other translocations | Once | Rare events and not routinely tested |
| Chromosome 1 | 1q amplification | May be repeated | Although conflicting studies seem to predict outcome |
| 1p deletion | |||
| aCGH derived markers |
12p deletion | ||
| 5q amplification | May be repeated | Data not validated yet | |
Abbreviations: FISH, fluorescent in situ hybridization; MM, multiple myeloma.
Although undoubtedly a large fraction of disease heterogeneity can be determined by the genetic subtypes of MM,2,3 an important component in determining outcome is related to host features. The contribution of these factors will not be discussed here further, but some general considerations apply. For instance, genetics alone cannot fully explain outcome heterogeneity and it is likely that host factors, such as performance status, comorbidities (for example, renal function) and age, have a predominant role in the prognosis determination in the immediate period after diagnosis.2,3 It has been recently shown that, despite being enriched for higher-risk genetic subtypes, younger patients live longer, presumptively as a consequence of their ability to better tolerate treatment.4 With passage of time it is also likely that progression (also called secondary events) have important roles in determining the fate of patients.
Classification of MM
Multiple myeloma is not one disease but rather many, with each one of the subtypes largely defined by the specific genetic and cytogenetic aberrations.2,3,5–7 These groups can now be considered unique entities and unlikely to lose their classification status over time. Linked to these groups are others that may depict only a small fraction of cases, and for whom there is no known prognostic implications. These subgroups will be shown in association with the larger ones, but highlighted as still under study and subject to further classification. As we have previously proposed, classifications can be proposed at three levels.
Biological genetic classification
This classification scheme is mostly driven by biology-based considerations. Usually, the groups created will have unique clinical and prognostic implications, but this is not a prerequisite. Classic examples of this include the hyperdiploid versus non-hyperdiploid classification, specific chromosome translocations, and so on.8–10 Assuming that no changes in the etiology of the disease occur over time, similar proportion of patients with the different subtypes will be diagnosed in future years. Ethnicity or age may result in different prevalence of these primary genetic subtypes, but for the most part the distributions within one of those groups should remain similar. This classification can be considered as enduring, as the biological basis for its classification will be more stable and less likely to evolve. The subgroups will be defined primordially by a primary genetic abnormality that will be associated with a constellation of other genetic changes associated with clone progression and evolution. The ultimate application of a predictive classification would be on the basis of the development of targeted therapies against these primary genetic changes, such as is possible for bcr-abl inhibition of chronic myeloid leukemia cells with imatinib. Such therapies do not yet exist for MM.
Prognostic classification
A prognostics classification incorporates classifiers capable of discriminating outcome of patients, and usually, albeit with dissimilar penetrance, in groups of patients treated with multiple treatment modalities3,6,7 (Table 2). In addition, factors capable of serving as prognostic determinants will usually be associated with baseline clinicopathological features of disease aggressiveness (such as high β2-microglobulin, high proliferation rates, extramedullary disease, hypercalcemia, elevated lactate dehydrogenase and so forth). As an example, the t(4;14)(p16;q32) has always been associated with more aggressive disease at baseline, and in the series of patients treated with conventional or high-dose chemotherapy, it discerns patients with shortened survival.3,6,7,11,12 Recently it has been shown that the prognostic significance of t(4;14)(p16;q32) may be ameliorated or eliminated in patients treated with bortezomib-based combinations.13,14 It is also postulated that refinements of classifications beyond those carried out merely by t(4;14) (p16;q32) using other genomic tools gene expression profiling (GEP) (Tables 3 and 4) or clinical parameters (for example, β2-microglobulin) may modulate the prognostic significance of the abnormality.6,15 These considerations will be discussed in greater detail under the t(4;14)(p16;q32) heading.
Table 2.
Emerging genetic tests: clinical and research recommendations
| Level | FISH tests | Testing frequency |
|---|---|---|
| Established markers |
Minimal FISH panel (clinic) | Once (baseline) |
| GEP | May be repeated | |
| Suggested tests | aCGH/SNP | Once |
| Predictive markers | Once | |
| Serial GEP | Repeated |
Abbreviations: FISH, fluorescent in situ hybridization; SNP, single nucleotide polymorphism.
Table 3.
Comparison between TC and UAMS GEP-derived classification (major subtypes)
| Group | TC | UAMS | |
|---|---|---|---|
| Categories associated with a defined genetic lesion | |||
| Cyclin D translocation | 11q13 CCND1 (15%) | CD-1 | |
| 6p21 CCND3 (2%) | CD-2 | ||
| 12p13 CCND2 (<1%) | |||
| MMSET translocation | 4p16 (15%) | MS | Three quarters express FGFR3 |
| MAF translocation | 16q23 C-maf (5%) | MF | Shared gene expression profile with expression of ITGB7 |
| 20q11 MAFB (2%) | |||
| 8q24 MAFA (<1%) | |||
| Hyperdiploid | D1 (30%) | HY | D1+D2 presumed to be progression from D1 |
| D1+D2 (8%) | |||
| RB deletion | None (2%) | ||
Abbreviations: ITGB, integrin beta; RB, retinoblastoma; MAFA and MAFB, masculo aponeurotic fibrosarcoma, UAMS, University of Arkansas for Medical Science.
Adapted from Chng et al.86
Table 4.
Concordance between TC and UAMS GEP-derived classification
| Subtypes | 4p16 | MAF | 6p21 | 11q13 | D1 | D1+D2 | D2 | None | All cases |
|---|---|---|---|---|---|---|---|---|---|
| MS | 68 | 68 | |||||||
| MF | 37 | 37 | |||||||
| CD-1 | 2 | 22 | 2 | 1 | 27 | ||||
| CD-2 | 1 | 3 | 50 | 4 | 1 | 2 | 61 | ||
| HY | 1 | 1 | 106 | 5 | 2 | 1 | 116 | ||
| LB | 1 | 1 | 8 | 8 | 39 | 1 | 58 | ||
| PR | 6 | 2 | 4 | 10 | 9 | 13 | 3 | 47 | |
| All cases | 74 | 40 | 7 | 78 | 130 | 23 | 57 | 5 | 414 |
Abbreviation: UAMS, University of Arkansas for Medical Science.
Adapted from Chng et al.86
Predictive classification
Accurate predictive classifications for MM do not yet exist and will not be discussed in further detail in this paper. Although dissimilar outcomes can be associated with the utilization of some of the aforementioned, a predictive factor should be able to discriminate with great accuracy the clinical benefit of a specific therapy intervention. The value of such factors should be such that it allows selection and elimination of therapies for subgroups of patients. For instance, if we had an effective inhibitor of a cyclin D gene that only worked against a certain subtype of the disease (those expressing this cyclin D gene), the predictive marker would be clearly useful. The treatment would only be given to patients with this abnormality and not to those without it, perhaps even if no other treatments were available. Another situation may arise in which subsets of MM cases (coming from the many genetic subtypes) depend on a specific growth factor (for example, interleukin-6 or hepatocyte growth factor) and for which a specific intervention may be beneficial (but not for cases without these abnormalities). Interestingly, the presence or lack of dependency on these growth factors could confer no specific prognostic associations and not be dictated by the major biological or prognostic classification, and yet be a very useful predictive classifier.
Although most therapeutics available against MM target specific vulnerabilities of the cells, most are not based on the targeting of specific genetic markers of subsets of the disease, or targeting abnormal pathways in subsets of patients. When such therapies become available the emergence of correct prediction will be paramount for proper selection of treatments. A classic example would be the use of endocrine-based therapies for receptor-positive breast cancer or the use of trastuzomab against HER-2-neu breast cancer. Some clinical trials in MM are underway or have been completed using therapies that target specific pathways known to be activated by identifiable genetic derangements. The first example was the phase 1 trial of TKI-258, an fibroblast growth factor 3 (FGFR3) inhibitor.16 If such therapy would become of clinical use, then it would most likely be indicated only for cases of t(4;14)(p16;q32) MM. The parallel development of biomarkers capable of enriching groups of patients likely to have higher clinical benefit would be beneficial.
Currently available therapies for MM are not thought to be targeted therapies and thus used in all suitable candidates. Yet only a fraction of cases exhibit responses to single agents, suggesting that subgroups of MM may be more or less responsive to specific therapies. The group from the University of Arkansas has been developing assays, using GEP, to predict patients who are likely to derive benefit from bortezomib early in the course of therapy, and are able to identify patterns associated with disease responsiveness (after drug administration).17 In another example, our group has made the first observation that patients with relapsed and regractory (RR) MM with non-canonical nuclear factor kappa B (NF-κB) activation seem to have heightened susceptibility to bortezomib treatment (before drug administration).18 Although this observation is preliminary and needs validation, it also suggests that the identification of patients likely to benefit from bortezomib is possible. A minority of cases exhibits long-lasting disease control and remission with minimal toxicity. The identification of this subset of the disease should be a top priority of ongoing research, given the long duration of clinical benefit for patients who still have many other treatment options available in the future, including HDT and bortezomib.
Lastly, the difficulty in creating clinical applications of predictive factors in MM is likely to be challenging, unless the observations are so absolute that they clearly show no or minimal likelihood of response to a specific treatment. Given that many MM patients will commonly receive three or more lines of treatment, it may be that predictive schemes may help in selecting the sequence of treatments to be administered, more than providing data to make decisions about the use (or not) of a specific therapy. For example, if a marker shows that certain subsets of patients have a likelihood of response to bortezomib of only 15%, although those without the marker have a response rate of 70%, should we withhold therapy for those in the former group if no other options exist? Probably not, and thus one may only want to use either bortezomib as the last option or as part of a combination that can elicit added value to early introduction of bortezomib. Furthermore, the predictive value will be specific to the treatments as used in the clinic, distinguishing the use of single agents versus those same agents in combination. Clinically, it is well known that a patient who has failed an Immunomodulatory drug (IMiD) and who has also failed treatment with bortezomib may respond to both when given in combination.
Biological genetic classification
Primary genetic events
A biological classification of MM is unlikely to change dramatically, given the current knowledge of disease pathogenesis19 (Tables 3 and 4).
Hyperdiploid and nh-MM
Overall MM is broadly divided at the top level into two major categories, hyperdiploid MM (h-MM) (harboring numerous chromosomal trisomies and a low prevalence of IgH translocations) and non-hyperdiploid MM (nh-MM) (encompassing hypodiploid, pseudodiploid and near tetraploid MM, and highly enriched for IgH translocations).8,9,20 The dichotomy has been validated in multiple series and is observed when patients are studied through karyotypes, interphase fluorescent in situ hybridization (FISH) or other genomic tools. The ploidy categories are stable over time such that patients with h-MM will usually remain hyperdiploid over the course of the disease.21 The dichotomy into hyperdiploidy has not been documented by at least two groups at the stage of MGUS, indicating that two fundamentally different pathogenesis pathways exist for MM, h-MM and nh-MM.22,23 It should be noted that although hyperdiploidy is mainly a feature of h-MM, and is predominant in cases with multiple trisomies and no IgH translocations, cases with primary IgH translocations and hyperdiploidy do exist (and are presumed to have an unfavorable outcome). It is notable that the majority of the human MM cell lines (HMCLs) belong to the nh-MM and just data emerging regarding the description of h-MM cell lines.24
The broad classification of the disease does not have major implications at the clinical level. However, patients with h-MM have a tendency towards a more favorable outcome and more commonly are elderly individuals, slightly more common among males, and have a higher incidence of MM bone disease. A recent genomic-based classification derived from array based comparative genomic hybridization (aCGH) identified this dichotomy and further subclassified h-MM and nh-MM as the major branches of the disease.10 In addition, albeit with a small group of patients studied, the authors subdivided h-MM into those with chromosome 13 deletion and chromosome 1 abnormalities and those without them. Patients with h-MM and chromosome 13 deletions had a reported shorter survival.10 In another larger series, the prognostic significance of chromosome 13 among h-MM was not apparent. More recently, the French group IFM did show that most of the prognostic value of h-MM was related to the gain of chromosome 5.25 Within the h-MM, GEP can identify four recurrent groups associated with an NF-κB/anti-apoptosis signature, an interleukin-6/HGF signature, a cancer testis antigen signature and ‘other’.26 This classification has been validated in three separate cohorts of patients, and in one tested it discriminates patients with a shorter survival.
t(11;14)(q13;q32) and CCND3 translocations
Of all MM, 15% harbor t(11;14)(q13;q32), with consequent upregulation of cyclin D1. MM with t(11;14) is associated with CD20 expression, lymphoplasmacytic morphology, hyposecretory disease and λ-light chain usage.27,28 The majority of all cases of IgM MM have been reported by one series to have t(11;14)(q13;q32) and one half of all cases of light chain amyloidosis harbor this same translocation. The translocation can be observed in MGUS, in which it results in nuclear cyclin D1 expression, and yet patients may remain stable without disease progression for decades. Although these clinicopathological features are not unique to t(11;14)(q13;q32) MM, the entity seems unique, even when it is neutral with regard to prognosis.6,29 In most series tested, t(11;14)(q13;q32) seem to be associated with a favorable outcome, but this effect is not strong enough to be statistically significant. The difficulty in establishing a favorable outcome for patients with t(11;14)(q13;q32) may relate to heterogeneity within patients with this genetic aberration. For instance, some cases of MM with t(11;14)(q13;q32) manifest with an aggressive phenotype such as plasma cell leukemia. The group from the University of Arkansas for Medical Science (UAMS) has identified two distinct subsets of t(11;14)(q13;q32) with different outcomes reported such that the global effect of t(11;14)(q13;q32) on prognosis remains neutral.
t(4;14)(p16;q32)
Several groups have shown that t(4;14)(p16;q32) and t(14;16)(q32;q23) are associated with poor survival, irrespective of the treatment modality.6,7 The t(4;14)(p16;q32) affects the most telomeric portion of chromosome 4 and is detectable only by FISH or reverse transcriptase–PCR11,30–32. The consequence of the translocation is increased expression of FGFR3 and multiple myelom SET domain (MMSET).31,32 The translocation can be imbalanced with up to 25% losing the derivative chromosome 14 associated with the loss of FGFR3 expression11. Of immediate clinical application was the observation that patients with t(4;14)(p16;q32) have short-remission duration after high-dose chemotherapy with stem cell support.6,12,33,34 In some series, the median time to relapse is as short as 8 months after one high-dose therapy treatment, with patients becoming refractory to alkylators and steroids.6,12,33,34 Owing to this unmet medical need, new treatments that specifically target the translocations have been developed. One such compound is TKI-258, a small molecule inhibitor of FGFR3 with in vitro and xenograft animal model activity against t(4;14)(p16;q32) HMCLs.16,35 Studies are ongoing to evaluate the clinical worth of this compound. More recently, the prognostic value of the translocation has been challenged in series of patients treated with bortezomib.14 In one study of patients treated with bortezomib, melphalan and prednisone, the translocation did not discriminate outcome among newly diagnosed cases, although the numbers are still small to reach definitive conclusions.14 Some of the clincopathological features associated with the translocation include association with the use of IgA heavy chains, λ-light chain usage and a very high prevalence of chromosome 13 abnormalities (deletions/monosomy).6,7,11,36,37 Although the t(4;14)(p16;q32) is also observed in the premalignant stages of the disease, it seems to be less common in patients with MGUS and present more frequently in patients with smoldering multiple myeloma (SMM).38,39
t(14;16)(q32;q23), other maf translocations, and 16q abnormalities
Translocations involving maf genes have been described in 5–7% of all MM cases. These translocations arise from IgH rearrangements involving a fragile site in chromosome 16. These translocations have also been associated with a higher frequency of chromosome 13 deletion, IgA isotype, and in at least two series, a more aggressive clinical outcome associated with C-maf translocations.7 There are minimal data regarding the clinical implications of other maf translocations, but they would be predicted to have similar clinical outcomes as those of C-maf.
Secondary genetic events
Tumor clone development is believed to be a consequence of a multistep process that accumulates sequential genetic changes.19 It has not been well defined in MM what the specific steps associated with disease progression are and what steps associate with the different cytogenetic subtypes. Some of the genetic abnormalities that seem to reflect progression include deletions at 17p13, chromosome 1 abnormalities (1p deletion and 1q amplification) and C-myc translocations. It is also likely that some of the best prognostic markers will come from the complete understanding of secondary (progression) events.
Chromosome 13 deletion and monosomy
We have recently reexamined the role of chromosome 13 as a biological factor versus a surrogate marker of aggressive disease. The field of MM genetics was invigorated first by the observation that cases with abnormal metaphase cytogenetics were associated with a shortened survival, and later by the observation that chromosome 13 deletions were also associated with a shorter survival.7,40–44 In the case of chromosome 13 abnormalities, they are detected in 50% of cases.45–48 Of all cases with chromosome 13 abnormalities, 85% constitute monosomy, whereas the remaining 15% are interstitial deletions.45–48 There is no known difference in effect for prognosis for deletions versus monosomy. The prognostic significance for chromosome 13 likely emanates because of its close association with high-risk genetic features (for example, t(4;14)(p16;q32)).6,7,49,50 Chromosome 13 deletion is not significant in discriminating prognosis for non-hyperdiploid patients50 and is not capable of distinguishing prognosis for hyperdiploid patients.51 Its prognostic significance is thus now thought to be a surrogate of its association with nh-MM. Does that mean that chromosome 13 has no biological importance?50 More and more data suggest a crucial role for chromosome 13 as prerequisite for clonal expansion for tumors; nearly 90% of cases with t(4;14)(p16;q32) will harbor chromosome 13 deletion.45–48
Deletion of 17p13
The most important molecular cytogenetic factor for prognostication is the deletion of 17p13 (the locus for the tumor-suppressor gene, p53).6,7,52 In all series tested, 17p13 deletions confer a very negative effect on survival. We and others have shown that patients with 17p13 deletions (mostly monoallelic) have an overall shorter survival, more aggressive disease, higher prevalence of extramedullary disease (such as plasmacytomas) and hypercalcemia.6,7,52 Deletions of 17p13 predict for a short duration of response after HDT and involvement of the central nervous system.53,54 We have recently shown that extramedullary disease is likely a consequence of defective p53, as most cases of plasma cell leukemia (primary and secondary) have abnormalities in the p53.55 In support of this we know that most, if not all, HMCLs have p53 functional deficiency (M Kuehl, personal communication). Our hypothesis is that PCs are indeed capable of surviving at extramedullary locations, but that they will usually undergo apoptosis in the presence of an intact p53 response. In support of this hypothesis a confirmatory study recently showed that p53 levels of gene expression are lower in cases with monoallelic deletions and that introduction of p53 back into the HMCLs induces apoptosis at high levels.56 Most studies have shown a negative effect on the prognosis for patients with 17p13, something not even resolved by allogeneic stem cell transplant.57 The overall complete remission (CR) rate was 50%, with no differences between the genetic abnormalities except for patients with del(17p13) who achieved less CR (7 versus 56%; P = 0.001). For event-free survival, only age (hazard ratio 2.8; P = 0.01) and del(17p13) (hazard ratio: 2.05; P = 0.03) retained their negative prognostic value. It is worth commenting that chromosome 17 deletion is uncommon in MGUS.
Chromosome 1 abnormalities
Chromosome 1 abnormalities have long been known to be highly prevalent in MM. The majority of these abnormalities involve rearrangements located in the pericentromeric regions and frequently in the form of jumping translocations. Chromosome 1 abnormalities have been recently proposed as major prognostic factors for MM.58 Regarding chromosome 1, it should be noted that 1q gain and 1p loss are so closely related that it is hard to provide differentiation.8,10,59 We and others had previously reported that abnormalities of both the short and long arm of chromosome 1 were associated with shorter survival.8,60 In one study by Shaughnessy et al.58, they found and validated a gene expression signature for high-risk disease. This signature is enriched disproportionately for genes located in chromosome 1. This also builds on previous studies showing that chromosome 1 abnormalities are associated with an adverse outcome. Although an initial search suggested that CKS1B might be the responsible gene for this association, other studies have failed to validate this notion.61 Two recent series have failed to confirm the overriding negative prognostic association with chromosome 1 amplification detected by FISH.6,61 Although it is thus still unclear how chromosome 1 participates biologically in generating more aggressive clones, chromosome 1 abnormalities continue to emerge as regions important in establishing the prognosis of patients.
NF-κB activation
We and others have recently shown that, through multiple primary genetic mechanisms, there is constitutive activation of the NF-κB pathway in at least 50% of MM cases.18,62 This activation is a consequence of inactivation of suppressors (by either biallelic deletion of deletion/mutation combinations) or by hyperactivity as a consequence of amplification or chromosome translocations. The summary of the effects of this (epistatic mutations) is readily detectable by gene expression profiling. All of these aberrations ultimately result in increased processing of substrate and consequent NFB nuclear hyperactivity. These genetic events have not been fully positioned in the process of disease progression but likely are secondary genetic events as they transcend the primary genetic categories. One example of prediction is our aforementioned recent observation of non-canonical NF-κB activation in a subset of patients. We have found that the likelihood of responsiveness to bortezomib and sustainability of responses seem higher among patients who have intrinsic activation of this pathway.18 In our study, those with low level of TRAF3 gene expression had a much higher likelihood of response to bortezomib (90%) as opposed to all others (30%).
Ras mutation
Other studies have shown an adverse outcome for patients with K-ras mutations, but not with N-ras mutations.49,63–66 This is interesting as ras mutations cluster more among patients with t(11;14)(q13;q32), and are likely important factors for disease progression for this subtype.65,66
12p deletions
In a recent single nucleotide polymorphism array study, the IFM did show that deletions of the short arm of chromosome 12 occurred in about 12% of the patients and was associated with both a short event-free survival and short overall survival. The size of the deletions was variable, but the minimal deleted region was centered on the CD27 gene. Other studies have suggested that the low expression of CD27 was associated with a poor prognosis.
p16 methylation and inactivation of p18
The issue is less clear for p16 methylation. Although some original studies had suggested negative associations with prognosis,67–72 recent data on large data sets suggest that p16 methylation is prognostically neutral.73 However, it has been recently found that a low transcription of the p16 gene measured by quantitative real-time PCR is associated with short survival, which suggests a possible impact of this gene in the MM pathogenesis, but with limited prognostic value.74
miRNA
Recent studies of microRNAs (miRNAs) show that they may also be important in the pathogenesis of MM. Pichiorri et al.75 used miRNA microarrays and quantitative real-time PCR to profile miRNA expression in HMCLs (n = 49), PCs from MM (n = 16), MGUS (n = 6) and normal donors (n = 6). They identified expression of miR-21 (possibly blocking apoptosis in the early phases of clonal PC expansion), miR-106b~25 cluster, miR-181a and b in MM and MGUS (otherwise not expressed in normal PCs). Two sets of mir genes were found as upregulated in MM but not in MGUS, miR-32 and miR-17–92. In another study, ectopic expression of mir-21 made a cell line become interleukin-6 independent.76 Functional in vitro and subsequent xenograft experiments show that the postulated miRNA genes may have a significant role in the pathogenesis of MM through the reduction of P300/CBP-associates factor (PCAF) (a positive p53 regulator), by downregulation of suppression of cytokine signaling 1 (SOCS-1) and BCLZ like 11 (BIM).75
16q abnormalities
A recent paper using single nucleotide polymorphism arrays lead to the conclusion that 16q abnormalities are also a recurrent and important genetic aberration in MM (20% of cases).77 The region 16q is unique in that it frequently harbors both deletions, and leads to the postulation of WWOX as a putative tumor-suppressor gene in MM.77 One study reported a 40% loss of hetreozygocity (LOH) of 16q, including deletion of the entire chromosome or the whole arm in 12 of 55 cases (22%), interstitial deletions in 7 of 55 cases (13%) and uniparental disomy (UPD) of the entire chromosome 16 or 16q in 4 of 55 cases (7%).78 These studies need confirmation.
Prognostic classifications
Prognosis by specific genetic aberrations
Although it is now clear that much of the major prognostic variation of MM is dictated by primary genetic categories, secondary changes can also have a profound influence in outcome by providing clonal survival/proliferation advantages. Some of the basic genetic categories have not resulted (yet) in specific clinical outcome difference, yet define unique subtypes (for example, t(11;14)(q13;q32)).6,7 The clinical consequences of secondary genetic changes tend not to be related to the therapy administered. One possible way to define prognostic markers is that they associate with baseline features of aggressiveness (pathobiology) and they should exert their influence if patients are not treated (natural history) (Table 1). It is possible that these markers will also identify patients who are more likely to progress from the premalignant stages of the disease. In general, the effect on overall outcome for validated prognostic markers will be evident irrespective of treatment modality, even when the hazard ratios for their influence may vary. As one example, we cite the negative implications for outcome for the t(4;14)(p16;q32) as it identifies patients with shorter clinical benefit from standard and HDT (Table 1). Although it seems that some of the prognostic ability can be challenged with novel agents, it is still too early to negate prognostication ability for t(4;14)(p16;q32) for patients receiving agents such as bortezomib.79 The same negative effect for prognosis is evident for the t(14;16)(q32;q23), as two series using conventional therapy (the Eastern Cooperative Oncology Group) and HDT (UAMS) have shown the deleterious effect on survival.3,7 Minimal data are available regarding the other musculoaponeurotic fibrosarcoma (MAF) variants. Other prognostic markers exert effects across the major biological subtypes of MM. Some are well established, including the aforementioned effect of 17p13 in prognosis.6,7,52
Gene expression profiling
Using high-throughput genomic tools is likely to unravel novel means of predicting patient outcome. A major effort at the University of Arkansas has identified a set of 70 genes (signature) capable of predicting high-risk MM.58 The team further shows that a simplified list of 17 genes is capable of providing the same prognostic discrimination.58 This last model discriminates with unprecedented ability ‘high-risk’ disease. This high-risk profile was indeed enriched for genes located in chromosome 1. The IFM also demonstrated in an independent series of 250 patients that a set of 15 genes was able to identify the patients with the poorest prognosis. It is possible that reverse transcriptase–PCR or immunohistochemistry-based strategies can be used to derive clinically applicable prognostication models for the disease. Other markers could include proliferation index by GEP, centrosome index by GEP and cancer testis antigens.5,51,80 It is important to note that there is minimal overlap between the different proposed signatures. The ability of each one of these signatures to be used in different contexts of treatments and stages is still being validated. Furthermore, it is conceivable that novel GEP derived signatures could be developed in the future and that will better predict patient outcomes. This will also be important and relevant with further developments of anti-MM therapies.
aCGH prognostication
The availability of aCGH has provided new opportunities for the identification of new biomarkers capable of discerning prognosis. Two aforementioned studies have identified recurrent genetic aberrations associated with prognosis. A study by Carrasco et al.10 found that patients could be divided into four subgroups (the two major branches being h-MM and nh-MM). Furthermore the h-MM could be divided into cases with chromosome 13 deletions and 1 abnormality, and they had a shorter survival. This study was small and needs confirmation. In another study, Avet-Loiseau et al. identified three recurrent abnormalities as predictive of outcome, gain of 1q, loss of 12p and gain of chromosome 5. Identification of biomarkers through aCGH is promising, as they can be easily converted to other diagnostic tools such as FISH.25
Diagnostic tests needed
Summary and technical aspects
We believe that a comprehensive cytogenetic evaluation should be carried out in all cases at the time of diagnosis in both the clinical and research trials (Table 2). This paper has not focused as much on the technical aspects of detecting genetic aberrations and advantages or pitfalls of each technology, but rather on the importance of detecting each one of these categories. We believe that clinical testing at this point should include at a minimum interphase FISH in purified PCs or in combination with immunofluorescent detection of light chain-restricted PC cytoplasmic immunoglobulin enhanced FISH (cIg-FISH).81 It is imperative that all FISH testing in MM incorporates one of these two strategies to improve on the rate of abnormality detection. It is common for the cytogenetics portion of a bone marrow aspirate draw to be the last tube collected. Owing to this, hemodilution of the bone marrow aspirate will result in a lower concentration of PCs and this will reduce the yield even further. The two aforementioned methods are equally effective at selecting the cells to be studied and could be implemented more on the basis of laboratory experience. Selection of cells has been more commonly used in Europe, whereas cIg-FISH is now performed by at least two reference laboratories in the United States.
Clinical testing
At a bare minimum, a FISH panel for MM should include testing for t(4;14)(p16;q32), t(14;16)(q32;q23) and −17p13 (Table 2). These three probes have been proposed by one group and have been used by others in the stratification of cases into high- and standard-risk disease.
The expansion of this panel to other probes may be desirable as it provides a more comprehensive assessment of the disease biology, clinical features and likely outcome. Among these are those that identify 1q amplification, 1p deletions and others. Those additional markers emanating from the aCGH analysis, such as loss of 12p and gain of chromosome 5, could also be converted to FISH strategies(Table 2). Other markers include t(11;14)(q13;q32) (see below), hyperdiploidy and IgH translocations, not otherwise specified. Although chromosome 13 alone is no longer considered a strong prognostic indicator, and hence could be abandoned, its use in combination with other variables (such as the β2-microglobulin or others) results in effective segregation of cases into the high-risk category.
The t(11;14)(q13;q32) deserves special attention as it is at a very high frequency in cases of light chain amyloidosis (~35–50%)82,83 and almost universal in cases of IgM MM (>90%).84 Detection of this translocation in a patient with monoclonal IgM strongly supports the diagnosis of IgM myeloma, as the clonal cells in patients with wladenstrom macroglobulinemia (WM) almost never harbor IgH translocations.85 Patients with t(11;14)(q13;q32) MM can have lymphoplasmacytic morphology, occasionally creating diagnostic confusion and also have a higher rate of CD20 expression.28
The frequency of testing is not well defined (Tables 1 and 2). It is now accepted that the major genetic subtypes of the disease will not change over time. However, genetic progression events are likely to arise with additional follow-up of cases and repeat testing may be desirable. Two examples are testing for −17p13 and chromosome 1 amplification, and probably chromosome 13 too. The repeat testing for other markers is not defined yet. The utilization of FISH strategies for determination of minimal residual disease is not validated yet at the clinical level.
Clinical trial testing
The aforementioned testing should be considered in all ongoing and future clinical trials(Tables 1 and 2). Trials that will be based on the targeting of specific genetic abnormalities need accurate determination of such subgroups and adequate clinical testing is needed. It should be considered mandatory that for demographic description of patients in future clinical trials, the table describing the cohort contains descriptors of the genetic subtypes of patients studied.
It is highly desirable to incorporate gene expression profiling into the correlative science of ongoing clinical trials. Likewise, as new technology emerges, the incorporation of other high-throughput platforms such as aCGH and exon gene expression profiling should be considered. The power of gene expression profiling has already been exploited at some centers for the selection of therapy for patients for different treatment algorithms. The introduction of new predictive markers (such as those for bortezomib sensitivity) is also highly desirable for all future clinical trials.
Summary and consensus recommendations
International MM working genetic classification
We recommend that the following working genetic classification (Table 5) be adopted until further elucidation of the pathogenesis of other subtypes of MM is provided.
Table 5.
New proposed International Myeloma Working Group molecular cytogenetic classification
| Percentage of patients |
Clinical and laboratory features | |
|---|---|---|
| Hyperdiploid | 45 | More favorable, IgG-κ, older patients. |
| Non-hyperdiploid | 40 | Aggressive, IgA-λ, younger individuals |
| Cyclin D translocation |
18 | |
| t(11;14)(q13;q32) | 16 | Upregulation of CCND1; favorable prognosis; bone lesions. Two subtypes by GEP |
| t(6;14q)(p21;32) | 2 | Probably same as CCND1 |
| t(12;14)(p13;q32) | <1 | Rare |
| MMSET translocation |
15 | |
| t(4;14)(p16;q32) | 15 | Upregulation of MMSET; upregulation of FGFR3 in 75% unfavorable prognosis with conventional therapy; bone lesions less frequent |
| MAF translocation | 8 | Aggressive |
| t(14;16)(q32;q23) | 5 | Confirmed as aggressive by at least two series |
| t(14;20)(q32;q11) | 2 | One series shows more aggressive disease. |
| t(8;14)(q24;q32) | 1 | Unknown effect on outcome but presumed aggressive. |
| Unclassified (other) | 15 | Various subtypes and some with overlap |
If clinical testing for GEP existed, all (most) of these categories could be also identified. There is significant overlap between the translocations and cyclin d (TC) classification and the UAMS molecular classification (Table 4), but there is excellent concordance for the MS and MF group corresponding to the 4p16 and Maf groups, respectively, with 100% concordance. There is still good but imperfect concordance (88%) for the CD-1 and CD-2 groups together corresponding to the 11q13 and 6p21 groups. If one joins the TC D1 and D1 + D2 groups, there is 96% concordance with the HY group. Significantly, overlap is also seen between the D2 cases and LB from UAMS. As mentioned before, the PR group seems to encompass many of the other genetic subtypes that have acquired secondary genetic events.
Testing recommendation
We recommend that at a minimum baseline genetic information should be obtained in all MM cases. Although many centers continue to collect karyotypes as an important predictor of outcome, further clarification using molecular cytogenetics is warranted. This testing must be done with either cytoplasmic immunoglobulin-enhanced FISH or FISH carried out on the nuclei from purified PCs. Performing FISH in unsorted samples carries a significant risk of low sensitivity for detection of chromosome abnormalities. The minimum panel required for prognostic estimation should include t(4;14)(p16;q32), t(14;16)(q32;q23) and 17p13 deletions. A more comprehensive panel should include testing for t(11;14)(q13;q32), chromosome 13 deletion, ploidy category and chromosome 1 abnormalities. The utility of this information is both at the biological subtype determination, as well as prognostic recommendations based on the minimal panel for testing. As new prognostic markers emerge from ongoing aCGH studies, new markers may be added to these screening panels.
GEP profiling: classification and prognosis
Our group also recognizes that the greatest prognostic ability for MM resides in the comprehensive analysis of GEP. At a minimum, all clinical trials should consider incorporation of GEP into the correlative science studies to identify subgroups of high-risk disease. We also propose that methodology to include GEP into the clinical testing is urgently needed and methods for implementation should be identified. Alternatively, conversion of GEP signature profiles into other routine clinical diagnostic tools is also likely to lead to rapid conversion of these prognostic signatures into widely available clinical tests.
Appendix
Rafat Abonour, Indiana University School of Medicine, Indianapolis, IN, USA
Ray Alexanian, MD Anderson, Houston, TX, USA
Kenneth Anderson, DFCI, Boston, MA, USA
Michael Attal, Purpan Hospital, Toulouse, France
Herve Avet-Loiseau, Institute de Biologie, Nantes, France
Ashraf Badros, University of Maryland, Baltimore, MD, USA
Leif Bergsagel, Mayo Clinic Scottsdale, Scottsdale, AZ, USA
Joan Bladé, Hospital Clinica, Barcelona, Spain
Bart Barlogie, MIRT UAMS Little Rock, AR, USA
Regis Batille, Institute de Biologie, Nantes, France
Meral Beksac, Ankara University, Ankara, Turkey
Andrew Belch, Cross Cancer Institute, Alberta, Canada
Bill Bensinger, Fred Hutchinson Cancer Center, Seattle, WA, USA
Mario Boccadoro, University of Torino, Torino, Italy
Michele Cavo, Universita di Bologna, Bologna, Italy
Wen Ming Chen, MM Research Center of Beijing, Beijing, China
Tony Child, Leeds General Hospital, Leeds, UK
James Chim, Department of Medicine, Queen Mary Hospital, Hong Kong
Ray Comenzo, Tufts Medical Center, Boston, MA, USA
John Crowley, Cancer Research and Biostatistics, Seattle, WA, USA
William Dalton, H Lee Moffitt, Tampa, FL, USA
Faith Davies, Royal Marsden Hospital, London, England
Cármino de Souza, Univeridade de Campinas, Caminas, Brazil
Michel Delforge, University Hospital Gasthuisberg, Leuven, Belgium
Meletios Dimopoulos, Alexandra Hospital, Athens, Greece
Angela Dispenzieri, Mayo Clinic, Rochester, MN, USA
Brian GM Durie, Cedars-Sinai Outpatient Cancer Center, Los Angeles, CA, USA
Hermann Einsele, Universitätsklinik Würzburg, Würzburg, Germany
Theirry Facon, Centre Hospitalier Regional Universitaire de Lille, Lille, France
Dorotea Fantl, Socieded Argentinade Hematolgia, Buenos Aires, Argentina
Jean-Paul Fermand, Hopitaux de Paris, Paris, France
Rafael Fonseca, Mayo Clinic Arizona, Scottsdale, AZ, USA
Gosta Gahrton, Karolinska Institute for Medicine, Huddinge, Sweden
Morie Gertz, Mayo Clinic, Rochester, MN, USA
John Gibson, Royal Prince Alfred Hospital, Sydney, Australia
Sergio Giralt, MD Anderson Cancer Center, Houston, TX, USA
Hartmut Goldschmidt, University Hospital Heidelberg, Heidelberg, Germany
Philip Greipp, Mayo Clinic, Rochester, MN, USA
Roman Hajek, Brno University, Brno, Czech Republic
Izhar Hardan, Tel Aviv University, Tel Aviv, Israel
Jean-Luc Harousseau, Institute de Biologie, Nantes, France
Hiroyuki Hata, Kumamoto University Hospital, Kumamoto, Japan
Yutaka Hattori, Keio University School of Medicine, Tokyo, Japan
Joy Ho, Royal Prince Alfred Hospital, Sydney, Australia
Vania Hungria, Clinica San Germano, Sao Paolo, Brazil
Shinsuke Ida, Nagoya City University Medical School, Nagoya, Japan
Peter Jacobs, Constantiaberg Medi-Clinic, Plumstead, South Africa
Sundar Jagannath, St Vincent’s Comprehensive Cancer Center, New York, NY, USA
Hou Jian, Shanghai Chang Zheng Hospital, Shanghai, China
Douglas Joshua, Royal Prince Alfred Hospital, Sydney, Australia
Michio Kawano, Yamaguchi University, Ube, Japan
Nicolaus Kröger, University Hospital Hamburg, Hamburg, Germany
Shaji Kumar, Department of Hematology, Mayo Clinic, MN, USA
Robert Kyle, Department of Laboratory Med. and Pathology, Mayo Clinic, MN, USA
Juan Lahuerta, Grupo Espanol di Mieloma, Hospital Universitario, Madrid, Spain
Jae Hoon Lee, Gachon University Gil Hospital, Incheon, Korea
Xavier LeLeu, Hospital Huriez, CHRU Lille, France
Suzanne Lentzsch, University of Pittsburgh, Pittsburgh, PA, USA
Henk Lokhorst, University Medical CenterUtrecht, Utrecht, The Netherlands
Sagar Lonial, Emory University Medical School, Atlanta, GA, USA
Heinz Ludwig, Wilhelminenspital Der Stat Wien, Vienna, Austria
Angelo Maiolino, Rua fonte da Saudade, Rio de Janeiro, Brazil
Maria Mateos, University of Salamanca, Salamanca, Spain
Jayesh Mehta, Northwestern University, Chicago, IL, USA
GianPaolo Merlini, University of Pavia, Pavia, Italy
Joseph Mikhael, Mayo Clinic Arizona, Scottsdale, AZ, USA
Philippe Moreau, University Hospital, Nantes, France
Gareth Morgan, Royal Marsden Hospital, London, England
Nikhil Munshi, Diane Farber Cancer Institute, Boston, MA, USA
Ruben Niesvizky, Weill Medical College of Cornell University, New York, NY, USA
Yana Novis, Hospital SírioLibanês, Bela Vista, Brazil
Amara Nouel, Hospital Rutz y Paez, Bolivar, Venezuela
Robert Orlowski, MD Anderson Cancer Center, Houston, TX, USA
Antonio Palumbo, Cathedra Ematologia, Torino, Italy
Santiago Pavlovsky, Fundaleu, Buenos Aires, Argentina
Linda Pilarski, University of Alberta, Alberta, Canada
Raymond Powles, Leukaemia & Myeloma, Wimbledon, England
S. Vincent Rajkumar, Mayo Clinic, Rochester, MN, USA
Donna Reece, Princess Margaret Hospital, Toronto, Canada
Tony Reiman, Cross Cancer Institute, Alberta, Canada
Paul Richardson, Dana Farber Cancer Institute, Boston, MA, USA
Angelina Rodriquez Morales, Bonco Metro Politano de Sangre, Caracas, Venezuela
Orhan Sezer, Department of Hem/Onc, Universitatsklinikum
Charite, Berlin, Germany
John Shaughnessy, MIRT UAMS, Little Rock, AR, USA
Kazuyuki Shimizu, Nagoya City Midori General Hospital, Nagoya, Japan
David Siegel, Hackensack, Cancer Center, Hackensack, NJ, USA
Jesus San Miguel, University of Salamanca, Salamanca, Spain
Chaim Shustik, McGill University, Montreal, Canada
Seema Singhal, Northwestern University, Chicago, IL, USA
Pieter Sonneveld, Erasmus MC, Rotterdam, The Netherlands
Andrew Spencer, The Alfred Hospital, Melbourne, Australia
Edward Stadtmauer, University of Pennsylvania, Philadelphia, PA, USA
A Keith Stewart, Mayo Clinic Arizona, Scottsdale, AZ, USA
Patrizia Tosi, Italian Cooperative Group, Istituto di Ematologia Seragnoli, Bologna, Italy
Guido Tricot, Huntsman Cancer Institute, Salt Lake City, UT, USA
Ingemar Turesson, Department of Hematology, Malmo University, Malmo, Sweden
Brian Van Ness, University of Minnesota, Minneapolis, MN, USA
Ivan Van Riet, Brussels Vrija University, Brussels, Belgium
Robert Vescio, Cedars-Sinai Cancer Center, Los Angeles, CA, USA
David Vesole, Loyola University Chicago, IL, USA
Anders Waage, University Hospital, Trondheim, Norway NSMG
Michael Wang, MD Anderson, Houston, TX, USA
Donna Weber, MD Anderson, Houston, TX, USA
Jan Westin, Sahlgrenska University Hospital, Gothenburg, Sweden
Keith Wheatley, University of Birmingham, Birmingham, UK
Dina B Yehuda, Department of Hematology, Hadassah University Hospital, Hadassah, Israel
Jeffrey Zonder, SWOG, Department of Hem/Onc., Karmanos Cancer Institute, MI, USA
Footnotes
Conflict of interest R Fonseca is a consultant for Genzyme, Celgene, BMS, Otsuka, Halozyme and Medtronic. His research was funded by Cylene and Proteolix. PL Bergsagel is on the advisory board of Amgen, Genentech, Celgene. Hid research was funded by Merck. B Van Ness serves on the IMF Scientific Advisory Board. The rest of the authors declare no conflict of interest. Shaughnessy’s work was patented by Myelogix, Genzyme and Novartis. He is a scientific advisor to Myelogix, Genzyme, Novartis and Celgene. He receives royalties from Myelogix, Genzyme, Novartis and Celgene, and is the owner of Myelogix. Bart Barlogie’s research was funded by NCI, Millennium, Celgene and Novartis. He has received honoraria from Millennium, Celgene and IMF. He is on the speakers Bureau of Millennium, Celgene and SWOG, and is a consultant for Celegene and Genzyme. He also has membership on an entity’s Board of Directors or advisory committees of IMF, MMRF and SWOG.
Members of the IMWG are listed in the appendix.
References
- 1.Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351:1860–1873. doi: 10.1056/NEJMra041875. [see comment]. [published erratum appears in N Engl J Med 2005; 352:1163] [DOI] [PubMed] [Google Scholar]
- 2.Fonseca R. Many and multiple myeloma(s) Leukemia. 2003;17:1943–1944. doi: 10.1038/sj.leu.2403090. [DOI] [PubMed] [Google Scholar]
- 3.Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood. 2006;108:2020–2028. doi: 10.1182/blood-2005-11-013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ludwig H, Durie BG, Bolejack V, Turesson I, Kyle RA, Blade J, et al. Myeloma in patients younger than age 50 years presents with more favorable features and shows better survival: an analysis of 10 549 patients from the International Myeloma Working Group. Blood. 2008;111:4039–4047. doi: 10.1182/blood-2007-03-081018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J, et al. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106:296–303. doi: 10.1182/blood-2005-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood. 2007;109:3489–3495. doi: 10.1182/blood-2006-08-040410. [DOI] [PubMed] [Google Scholar]
- 7.Fonseca R, Blood E, Rue M, Harrington D, Oken MM, Kyle RA, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101:4569–4575. doi: 10.1182/blood-2002-10-3017. [DOI] [PubMed] [Google Scholar]
- 8.Debes-Marun C, Dewald G, Bryant S, Picken E, Santana-Dávila S, González-Paz N, et al. Chromosome abnormalities clustering and its implications for pathogenesis and prognosis in myeloma. Leukemia. 2003;17:427–436. doi: 10.1038/sj.leu.2402797. [DOI] [PubMed] [Google Scholar]
- 9.Smadja NV, Fruchart C, Isnard F, Louvet C, Dutel JL, Cheron N, et al. Chromosomal analysis in multiple myeloma: cytogenetic evidence of two different diseases. Leukemia. 1998;12:960–969. doi: 10.1038/sj.leu.2401041. [DOI] [PubMed] [Google Scholar]
- 10.Carrasco DR, Tonon G, Huang Y, Zhang Y, Sinha R, Feng B, et al. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell. 2006;9:313–325. doi: 10.1016/j.ccr.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 11.Keats JJ, Reiman T, Maxwell CA, Taylor BJ, Larratt LM, Mant MJ, et al. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood. 2003;101:1520–1529. doi: 10.1182/blood-2002-06-1675. [DOI] [PubMed] [Google Scholar]
- 12.Gertz MA, Lacy MQ, Dispenzieri A, Greipp PR, Litzow MR, Henderson KJ, et al. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16 3 q32) and −17p13 in myeloma patients treated with high-dose therapy. Blood. 2005;106:2837–2840. doi: 10.1182/blood-2005-04-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jagannath S, Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, et al. Bortezomib appears to overcome the poor prognosis conferred by chromosome 13 deletion in phase 2 and 3 trials. Leukemia. 2007;21:151–157. doi: 10.1038/sj.leu.2404442. [DOI] [PubMed] [Google Scholar]
- 14.San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359:906–917. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 15.Chng WJ, Kuehl WM, Bergsagel PL, Fonseca R. Translocation t(4;14) retains prognostic significance even in the setting of high-risk molecular signature. Leukemia. 2008;22:459–461. doi: 10.1038/sj.leu.2404934. [published erratum appears in Leukemia 2008; 22: 462] e-pub ahead of print 6 September 2007. No abstract available. [DOI] [PubMed] [Google Scholar]
- 16.Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, et al. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105:2941–2948. doi: 10.1182/blood-2004-10-3913. [DOI] [PubMed] [Google Scholar]
- 17.Shi J, Tricot GJ, Garg TK, Malaviarachchi PA, Szmania SM, Kellum RE, et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood. 2008;111:1309–1317. doi: 10.1182/blood-2007-03-078535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nature Rev Cancer. 2002;2:175–187. doi: 10.1038/nrc746. [DOI] [PubMed] [Google Scholar]
- 20.Fonseca R, Debes-Marun CS, Picken EB, Dewald GW, Bryant SC, Winkler JM, et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood. 2003;102:2562–2567. doi: 10.1182/blood-2003-02-0493. [DOI] [PubMed] [Google Scholar]
- 21.Chng WJ, Winkler JM, Greipp PR, Jalal SM, Bergsagel PL, Chesi M, et al. Ploidy status rarely changes in myeloma patients at disease progression. Leuk Res. 2006;30:266–271. doi: 10.1016/j.leukres.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Chng WJ, Van Wier SA, Ahmann GJ, Winkler JM, Jalal SM, Bergsagel PL, et al. A validated FISH trisomy index demonstrates the hyperdiploid and nonhyperdiploid dichotomy in MGUS. Blood. 2005;106:2156–2161. doi: 10.1182/blood-2005-02-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brousseau M, Leleu X, Gerard J, Gastinne T, Godon A, Genevieve F, et al. Hyperdiploidy is a common finding in monoclonal gammopathy of undetermined significance and monosomy 13 is restricted to these hyperdiploid patients. Clin Cancer Res. 2007;13:6026–6031. doi: 10.1158/1078-0432.CCR-07-0031. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Pennisi A, Zhan F, Sawyer JR, Shaughnessy JD, Yaccoby S. Establishment and exploitation of hyperdiploid and non-hyperdiploid human myeloma cell lines. Br J Haematol. 2007;138:802–811. doi: 10.1111/j.1365-2141.2007.06742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Avet-Loiseau H, Li C, Magrangeas F, Gouraud W, Charbonnel C, Harousseau J-L, et al. J Clin Oncol. Published online 17 August 2009, doi:10.1200/JCO.2008.20.6136. [Google Scholar]
- 26.Chng WJ, Kumar S, Vanwier S, Ahmann G, Price-Troska T, Henderson K, et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007;67:2982–2989. doi: 10.1158/0008-5472.CAN-06-4046. [DOI] [PubMed] [Google Scholar]
- 27.Hoyer JD, Hanson CA, Fonseca R, Greipp PR, Dewald GW, Kurtin PJ. The (11;14)(q13;q32) translocation in multiple myeloma A morphologic and immunohistochemical study. Am J Clin Pathol. 2000;113:831–837. doi: 10.1309/4W8E-8F4K-BHUP-UBE7. [DOI] [PubMed] [Google Scholar]
- 28.Garand R, Avet-Loiseau H, Accard F, Moreau P, Harousseau J, Bataille R. t(11;14) and t(4;14) translocations correlated with mature lymphoplasmocytoid and immature morphology, respectively, in multiple myeloma. Leukemia. 2003;17:2032–2035. doi: 10.1038/sj.leu.2403091. [DOI] [PubMed] [Google Scholar]
- 29.Fonseca R, Harrington D, Oken M, Kyle R, Dewald G, Bailey R, et al. Myeloma and the t(11;14)(q13;q32) represents a uniquely defined biological subset of patients. Blood. 2002;99:3735–3741. doi: 10.1182/blood.v99.10.3735. [DOI] [PubMed] [Google Scholar]
- 30.Sawyer JR, Lukacs JL, Thomas EL, Swanson CM, Goosen LS, Sammartino G, et al. Multicolour spectral karyotyping identifies new translocations and a recurring pathway for chromosome loss in multiple myeloma. Br J Haematol. 2001;112:167–174. doi: 10.1046/j.1365-2141.2001.02546.x. [DOI] [PubMed] [Google Scholar]
- 31.Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16 3 q32 3 ) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chesi M, Nardini E, Lim R, Smith K, Kuehl W, Bergsagel P. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998;92:3025–3034. [PubMed] [Google Scholar]
- 33.Chang H, Sloan S, Li D, Zhuang L, Yi QL, Chen CI, et al. The t(4;14) is associated with poor prognosis in myeloma patients undergoing autologous stem cell transplant. Br J Haematol. 2004;125:64–68. doi: 10.1111/j.1365-2141.2004.04867.x. [DOI] [PubMed] [Google Scholar]
- 34.Chang H, Qi XY, Samiee S, Yi QL, Chen C, Trudel S, et al. Genetic risk identifies multiple myeloma patients who do not benefit from autologous stem cell transplantation. Bone Marrow Transplant. 2005;36:793–796. doi: 10.1038/sj.bmt.1705131. [DOI] [PubMed] [Google Scholar]
- 35.Trudel S, Ely S, Farooqi Y, Affer M, Robbiani DF, Chesi M, et al. Inhibition of fibroblast growth factor receptor 3 induces differentiation and apoptosis in t(4;14) myeloma. Blood. 2004;103:3521–3528. doi: 10.1182/blood-2003-10-3650. [DOI] [PubMed] [Google Scholar]
- 36.Avet-Loiseau H, Facon T, Grosbois B, Magrangeas F, Rapp MJ, Harousseau JL, et al. Oncogenesis of multiple myeloma: 14q32 and 13q chromosomal abnormalities are not randomly distributed, but correlate with natural history, immunological features, and clinical presentation. Blood. 2002;99:2185–2191. doi: 10.1182/blood.v99.6.2185. [DOI] [PubMed] [Google Scholar]
- 37.Fonseca R, Oken MM, Greipp PR. The t(4;14)(p16.3;q32) is strongly associated with chromosome 13 abnormalities in both multiple myeloma and monoclonal gammopathy of undetermined significance. Blood. 2001;98:1271–1272. doi: 10.1182/blood.v98.4.1271. [DOI] [PubMed] [Google Scholar]
- 38.Avet-Loiseau H, Facon T, Daviet A, Godon C, Rapp MJ, Harousseau JL, et al. 14q32 translocations and monosomy 13 observed in monoclonal gammopathy of undetermined significance delineate a multistep process for the oncogenesis of multiple myeloma. Intergroupe Francophone du Myelome. Cancer Res. 1999;59:4546–4550. [PubMed] [Google Scholar]
- 39.Fonseca R, Bailey RJ, Ahmann GJ, Rajkumar SV, Hoyer JD, Lust JA, et al. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood. 2002;100:1417–1424. [see comment] [PubMed] [Google Scholar]
- 40.Tricot G, Barlogie B, Jagannath S, Bracy D, Mattox S, Vesole DH, et al. Poor prognosis in multiple myeloma is associated only with partial or complete deletions of chromosome 13 or abnormalities involving 11q and not with other karyotype abnormalities. Blood. 1995;86:4250–4256. [PubMed] [Google Scholar]
- 41.Tricot G, Sawyer JR, Jagannath S, Desikan KR, Siegel D, Naucke S, et al. Unique role of cytogenetics in the prognosis of patients with myeloma receiving high-dose therapy and autotransplants. J Clin Oncol. 1997;15:2659–2666. doi: 10.1200/JCO.1997.15.7.2659. [DOI] [PubMed] [Google Scholar]
- 42.Perez-Simon JA, Garcia-Sanz R, Tabernero MD, Almeida J, Gonzalez M, Fernandez-Calvo J, et al. Prognostic value of numerical chromosome aberrations in multiple myeloma: A FISH analysis of 15 different chromosomes. Blood. 1998;91:3366–3371. [PubMed] [Google Scholar]
- 43.Zojer N, Konigsberg R, Ackermann J, Fritz E, Dallinger S, Kromer E, et al. Deletion of 13q14 remains an independent adverse prognostic variable in multiple myeloma despite its frequent detection by interphase fluorescence in situ hybridization. Blood. 2000;95:1925–1930. [PubMed] [Google Scholar]
- 44.Dewald GW, Therneau T, Larson D, Lee YK, Fink S, Smoley S, et al. Relationship of patient survival and chromosome anomalies detected in metaphase and/or interphase cells at diagnosis of myeloma. Blood. 2005;106:3553–3558. doi: 10.1182/blood-2005-05-1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fonseca R, Oken MM, Harrington D, Bailey RJ, Van Wier SA, Henderson KJ, et al. Deletions of chromosome 13 in multiple myeloma identified by interphase FISH usually denote large deletions of the q arm or monosomy. Leukemia. 2001;15:981–986. doi: 10.1038/sj.leu.2402125. [DOI] [PubMed] [Google Scholar]
- 46.Fonseca R, Harrington D, Oken M, Dewald G, Bailey R, Van Wier S, et al. Biologic and prognostic significance of interphase FISH detection of chromosome 13 abnormalities (D13) in multiple myeloma: an Eastern Cooperative Oncology Group (ECOG) Study. Cancer Res. 2002;62:715–720. [PubMed] [Google Scholar]
- 47.Avet-Loiseau H, Li JY, Morineau N, Facon T, Brigaudeau C, Harousseau JL, et al. Monosomy 13 is associated with the transition of monoclonal gammopathy of undetermined significance to multiple myeloma. Intergroupe Francophone du Myelome. Blood. 1999;94:2583–2589. [PubMed] [Google Scholar]
- 48.Avet-Loiseau H, Daviet A, Saunier S, Bataille R. Chromosome 13 abnormalities in multiple myeloma are mostly monosomy 13. Br J Haematol. 2000;111:1116–1117. doi: 10.1046/j.1365-2141.2000.02488.x. [DOI] [PubMed] [Google Scholar]
- 49.Gutierrez NC, Castellanos MV, Martin ML, Mateos MV, Hernandez JM, Fernandez M, et al. Prognostic and biological implications of genetic abnormalities in multiple myeloma undergoing autologous stem cell transplantation: t(4;14) is the most relevant adverse prognostic factor, whereas RB deletion as a unique abnormality is not associated with adverse prognosis. Leukemia. 2007;21:143–150. doi: 10.1038/sj.leu.2404413. [DOI] [PubMed] [Google Scholar]
- 50.Chiecchio L, Protheroe RK, Ibrahim AH, Cheung KL, Rudduck C, Dagrada GP, et al. Deletion of chromosome 13 detected by conventional cytogenetics is a critical prognostic factor in myeloma. Leukemia. 2006;20:1610–1617. doi: 10.1038/sj.leu.2404304. [DOI] [PubMed] [Google Scholar]
- 51.Chng WJ, Santana-Davila R, Van Wier SA, Ahmann GJ, Jalal SM, Bergsagel PL, et al. Prognostic factors for hyperdiploid-myeloma: effects of chromosome 13 deletions and IgH translocations. Leukemia. 2006;20:807–813. doi: 10.1038/sj.leu.2404172. [DOI] [PubMed] [Google Scholar]
- 52.Drach J, Ackermann J, Fritz E, Kromer E, Schuster R, Gisslinger H, et al. Presence of a p53 gene deletion in patients with multiple myeloma predicts for short survival after conventional-dose chemotherapy. Blood. 1998;92:802–809. [PubMed] [Google Scholar]
- 53.Chang H, Qi C, Yi QL, Reece D, Stewart AK. p53 gene deletion detected by fluorescence in situ hybridization is an adverse prognostic factor for patients with multiple myeloma following autologous stem cell transplantation. Blood. 2005;105:358–360. doi: 10.1182/blood-2004-04-1363. [DOI] [PubMed] [Google Scholar]
- 54.Chang H, Sloan S, Li D, Stewart A Keith. Multiple myeloma involving central nervous system: high frequency of chromosome 17p13.1 (p53) deletions. Br J Haematol. 2004;127:280–284. doi: 10.1111/j.1365-2141.2004.05199.x. [DOI] [PubMed] [Google Scholar]
- 55.Tiedemann RE, Gonzalez-Paz N, Kyle RA, Santana-Davila R, Price-Troska T, Van Wier SA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22:1044–1052. doi: 10.1038/leu.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiong W, Wu X, Starnes S, Johnson SK, Haessler J, Wang S, et al. An analysis of the clinical and biologic significance of TP 53 loss and the identification of potential novel transcriptional targets of TP53 in multiple myeloma. Blood. 2008;112:4235–4246. doi: 10.1182/blood-2007-10-119123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schilling G, Hansen T, Shimoni A, Zabelina T, Perez-Simon JA, Gutierrez NC, et al. Impact of genetic abnormalities on survival after allogeneic hematopoietic stem cell transplantation in multiple myeloma. Leukemia. 2008;22:1250–1255. doi: 10.1038/leu.2008.88. [DOI] [PubMed] [Google Scholar]
- 58.Shaughnessy JD, Jr, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109:2276–2284. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 59.Sawyer JR, Tricot G, Mattox S, Jagannath S, Barlogie B. Jumping translocations of chromosome 1q in multiple myeloma: evidence for a mechanism involving decondensation of pericentromeric heterochromatin. Blood. 1998;91:1732–1741. [PubMed] [Google Scholar]
- 60.Hanamura I, Stewart JP, Huang Y, Zhan F, Santra M, Sawyer JR, et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood. 2006;108:1724–1732. doi: 10.1182/blood-2006-03-009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fonseca R, Van Wier SA, Chng WJ, Ketterling R, Lacy MQ, Dispenzieri A, et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia. 2006;20:2034–2040. doi: 10.1038/sj.leu.2404403. [DOI] [PubMed] [Google Scholar]
- 62.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu P, Leong T, Quam L, Billadeau D, Kay NE, Greipp P, et al. Activating mutations of N- and K-ras in multiple myeloma show different clinical associations: analysis of the Eastern Cooperative Oncology Group Phase III Trial. Blood. 1996;88:2699–2706. [PubMed] [Google Scholar]
- 64.Bezieau S, Devilder MC, Avet-Loiseau H, Mellerin MP, Puthier D, Pennarun E, et al. High incidence of N and K-Ras activating mutations in multiple myeloma and primary plasma cell leukemia at diagnosis. Human Mutat. 2001;18:212–224. doi: 10.1002/humu.1177. [DOI] [PubMed] [Google Scholar]
- 65.Rasmussen T, Kuehl M, Lodahl M, Johnsen HE, Dahl IM. Possible roles for activating RAS mutations in the MGUS to MM transition and in the intramedullary to extramedullary transition some plasma cell tumors. Blood. 2005;105:317–323. doi: 10.1182/blood-2004-03-0833. [DOI] [PubMed] [Google Scholar]
- 66.Chng WJ, Gonzalez-Paz N, Price-Troska T, Jacobus S, Rajkumar SV, Oken MM, et al. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia. 2008;22:2280–2284. doi: 10.1038/leu.2008.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Urashima M, Teoh G, Ogata A, Chauhan D, Treon SP, Sugimoto Y, et al. Characterization of p16(INK4A) expression in multiple myeloma and plasma cell leukemia. Clin Cancer Res. 1997;3:2173–2179. [PubMed] [Google Scholar]
- 68.Tasaka T, Asou H, Munker R, Said JW, Berenson J, Vescio RA, et al. Methylation of the p16INK4A gene in multiple myeloma. Br J Haematol. 1998;101:558–564. doi: 10.1046/j.1365-2141.1998.00724.x. [DOI] [PubMed] [Google Scholar]
- 69.Mateos MV, Garcia-Sanz R, Lopez-Perez R, Moro MJ, Ocio E, Hernandez J, et al. Methylation is an inactivating mechanism of the p16 gene in multiple myeloma associated with high plasma cell proliferation and short survival. Br J Haematol. 2002;118:1034–1040. doi: 10.1046/j.1365-2141.2002.03749.x. [DOI] [PubMed] [Google Scholar]
- 70.Chen W, Wu Y, Zhu J, Liu J, Tan S, Xia C. Methylation of p16 and p15 genes in multiple myeloma. Chi Med Sci J. 2002;17:101–105. [PubMed] [Google Scholar]
- 71.Guillerm G, Depil S, Wolowiec D, Quesnel B. Different prognostic values of p15(INK4b) and p16(INK4a) gene methylations in multiple myeloma. Haematologica. 2003;88:476–478. [PubMed] [Google Scholar]
- 72.Uchida T, Kinoshita T, Ohno T, Ohashi H, Nagai H, Saito H. Hypermethylation of p16 INK4A gene promoter during the progression of plasma cell dyscrasia. Leukemia. 2001;15:157–165. doi: 10.1038/sj.leu.2401991. [DOI] [PubMed] [Google Scholar]
- 73.Gonzalez-Paz N, Chng WJ, McClure RF, Blood E, Oken MM, Van Ness B, et al. Tumor suppressor p16 methylation in multiple myeloma: biological and clinical implications. Blood. 2007;109:1228–1232. doi: 10.1182/blood-2006-05-024661. [DOI] [PubMed] [Google Scholar]
- 74.Sarasquete ME, Garcia-Sanz R, Armellini A, Fuertes M, Martin-Jimenez P, Sierra M, et al. The association of increased p14ARF/p16INK4a and p15INK4a gene expression with proliferative activity and the clinical course of multiple myeloma. Haematologica. 2006;91:1551–1554. [PubMed] [Google Scholar]
- 75.Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci USA. 2008;105:12885–12890. doi: 10.1073/pnas.0806202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermuller J, Kretzschmar AK, et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of micro-RNA-21 through a highly conserved enhancer. Blood. 2007;110:1330–1333. doi: 10.1182/blood-2007-03-081133. [DOI] [PubMed] [Google Scholar]
- 77.Walker BA, Leone PE, Jenner MW, Li C, Gonzalez D, Johnson DC, et al. Integration of global SNP-based mapping and expression arrays reveals key regions, mechanisms, and genes important in the pathogenesis of multiple myeloma. Blood. 2006;108:1733–1743. doi: 10.1182/blood-2006-02-005496. [DOI] [PubMed] [Google Scholar]
- 78.Jenner MW, Leone PE, Walker BA, Ross FM, Johnson DC, Gonzalez D, et al. Gene mapping and expression analysis of 16q loss of heterozygosity identifies WWOX and CYLD as being important in determining clinical outcome in multiple myeloma. Blood. 2007;110:3291–3300. doi: 10.1182/blood-2007-02-075069. [DOI] [PubMed] [Google Scholar]
- 79.Mateos MV, Hernandez JM, Hernandez MT, Gutierrez NC, Palomera L, Fuertes M, et al. Bortezomib plus melphalan and prednisone in elderly untreated patients with multiple myeloma: results of a multicenter phase I/II study. Blood. 2006;108:2165–2172. doi: 10.1182/blood-2006-04-019778. [DOI] [PubMed] [Google Scholar]
- 80.Chng WJ, Ahmann GJ, Henderson K, Santana-Davila R, Greipp PR, Gertz MA, et al. Clinical implication of centrosome amplification in plasma cell neoplasm. Blood. 2006;107:3669–3675. doi: 10.1182/blood-2005-09-3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ahmann GJ, Jalal SM, Juneau AL, Christensen ER, Hanson CA, Dewald GW, et al. A novel three-color, clone-specific fluorescence in situ hybridization procedure for monoclonal gammopathies. Cancer Genet Cytogenet. 1998;101:7–11. doi: 10.1016/s0165-4608(97)00058-7. [DOI] [PubMed] [Google Scholar]
- 82.Harrison CJ, Mazzullo H, Ross FM, Cheung KL, Gerrard G, Harewood L, et al. Translocations of 14q32 and deletions of 13q14 are common chromosomal abnormalities in systemic amyloidosis. Br J Haematol. 2002;117:427–435. doi: 10.1046/j.1365-2141.2002.03438.x. [DOI] [PubMed] [Google Scholar]
- 83.Hayman SR, Bailey RJ, Jalal SM, Ahmann GJ, Dispenzieri A, Gertz MA, et al. Translocations involving heavy-chain locus are possible early genetic events in patients with primary systemic amyloidosis. Blood. 2001;98:2266–2268. doi: 10.1182/blood.v98.7.2266. [DOI] [PubMed] [Google Scholar]
- 84.Avet-Loiseau H, Garand R, Lode L, Harousseau J-L, Bataille R. Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE, and nonsecretory multiple myeloma variants. Blood. 2003;101:1570–1571. doi: 10.1182/blood-2002-08-2436. [DOI] [PubMed] [Google Scholar]
- 85.Schop RF, Van Wier SA, Xu R, Ghobrial I, Ahmann GJ, Greipp PR, et al. 6q deletion discriminates Waldenstrom macroglobulinemia from IgM monoclonal gammopathy of undetermined significance. Cancer Genet Cytogenet. 2006;169:150–153. doi: 10.1016/j.cancergencyto.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 86.Chng WJ, Glebov O, Bergsagel PL, Kuehl WL. Genetic events in the pathogenesis of multiple myeloma. Best Prac Res Clin Haematol. 2007;20:571–596. doi: 10.1016/j.beha.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]