

Abstract
Background
This study was aimed to identify novel susceptibility variants for second primary tumor (SPT) or recurrence in curatively treated early stage head and neck squamous cell carcinoma (HNSCC) patients.
Methods
We constructed a custom chip containing a comprehensive panel of 9645 chromosomal and mitochondrial single nucleotide polymorphisms (SNPs) representing 998 cancer-related genes selected by a systematic prioritization schema. Using this chip, we genotyped 150 early-stage HNSCC patients with and 300 matched patients without SPT/recurrence from a prospectively conducted randomized trial and assessed the association of these SNPs with risk of SPT/recurrence.
Results
Individually, six chromosomal SNPs and seven mitochondrial SNPs (mtSNPs) were significantly associated with risk of SPT/recurrence after adjustment for multiple comparisons. A strong gene-dosage effect was observed these SNPs were combined, as evidenced by a progressively increasing SPT/recurrence risk as the number of unfavorable genotypes increased (P for trend < 1.00×10−20). Several polygenic analyses suggest an important role of interconnected functional network and gene-gene interaction in modulating SPT/recurrence. Furthermore, incorporation of these genetic markers into a multivariate model improved significantly the discriminatory ability over the models containing only clinical and epidemiologic variables.
Conclusions
This is the first large scale systematic evaluation of germline genetic variants for their roles in HNSCC SPT/recurrence. The study identified several promising susceptibility loci and demonstrated the cumulative effect of multiple risk loci in HNSCC SPT/recurrence. Furthermore, this study underscores the importance of incorporating germline genetic variation data with clinical and risk factor data in constructing prediction models for clinical outcomes.
Keywords: iSelect Infinium, Single nucleotide polymorphisms, Head and neck cancer, Secondary primary tumor, recurrence
INTRODUCTION
Approximately 10% of early-stage head and neck squamous cell carcinoma (HNSCC) patients develop loco-regional recurrence and 15–25% develop second primary tumors (SPT) within 5 years of initial diagnosis. (1, 2) As diagnostic and therapeutic approaches continue to improve, the ability to accurately predict SPT/recurrence in early-stage HNSCC patients would facilitate intensive surveillance or targeted interventions for high-risk patients and thereby reduce mortality and morbidity.
Clinical (index tumor site and disease stage) and lifestyle (continued smoking and alcohol drinking) factors contribute to the risk of SPT and recurrence. (3, 4) HNSCC tumorigenesis is a multistep process involving an accumulation of progressive genetic alterations, (5) including genomic alterations of multiple chromosomes (3p, 9p, 13q, and 17p), (6, 7) and mutations of essential oncogenes and tumor suppressor genes ( p53, p16, cyclin D1, KRAS, and FHIT ). (8, 9) Many of these somatic alterations have also been linked to SPT/recurrence development.
We previously reported that high mutagen sensitivity measured by an in vitro lymphocytic assay, reflecting constitutional genetic instability, was associated with increased risk of SPT/recurrence. (10, 11) While the association between single nucleotide polymorphisms (SNPs) and risk of HNSCC (12, 13) has been extensively investigated, no studies have investigated their association with SPT/recurrence. To address this issue, we conducted this nested case-control analysis to test the hypothesis that common sequence variants affect the risk of SPT/recurrence in curatively treated HNSCC patients. Because genome-wide scanning approach was not an option due to the limited sample availability of HNSCC patients who developed SPT/recurrence, we therefore constructed a comprehensive panel of 998 cancer-related genes and 9645 SNPs to assess both their individual and combined effects on SPT/recurrence. We also constructed risk prediction models of SPT/recurrence based on known clinical and epidemiologic risk factors, and SNPs identified from this study.
SUBJECTS AND METHODS
Study population and epidemiologic data
The subjects included in this study were participants enrolled (1991–1999) in the Retinoid Head and Neck Second Primary Trial (RHNSPT) designed to evaluate whetherdaily low dose 13-cis-retinoic acid (13-cRA) prevents SPT or tumor recurrence in early-stage HNSCC patients.(1) Briefly, patients with histologically confirmed stage Ior II HNSCC who were cancer-free for at least 16 weeks after the end of treatment were eligible forrandomization to either low-dose (30 mg/day) 13-cRA treatment or placebo for 3 years with a minimum of planned 4 years of follow-up. The stratification criteria for randomization included the primary tumor site (larynx, oral cavity, and pharynx), tumor stage (stage I or II), and smoking status (current, former, or never smoker). Never smokers were individuals who had smoked less than 20 total cigarettes during their lifetime. Former smokers were individuals who had stopped smoking for at least 1 year at the time of enrollment. (14) Patients were evaluated at 3, 6, 9, 12, 16, 20, 24, 28, 32, and 36 months after randomization. After completing treatment, patients were follow-up at 6-month intervals for an additional 4 years. Standard criteria for diagnosis of an SPT were applied. (15) The major sites of SPT in this population were lung (29.8%), head and neck (28.0%), prostate (14.2%), and bladder (5.1%). Local recurrence was defined as any tumor of similar histology appearing within 2 cm or within 3 years of the primary tumor. Among approximately 1190 patients enrolled, 354 developed SPT/recurrence. However, only 150 patients have blood DNA samples available. Therefore, we designed a nested case control study to evaluate these 150 patients with SPT/recurrence designated as cases and 300 patients without SPT/recurrence as controls. We performed analyses on these 150 cases and those not included in this study, and did not find significant differences in terms of age, sex, smoking, alcohol, tumor site, stage, radiotherapy, surgery or 13 cis retinoic acid treatment. Patients included in the study had higher percentage of Caucasians (95%) than patients not included (89%) (P=0.001). We are confident that there is minimal patient selection bias. The study was approved by the Institutional Review Board of The University of Texas M. D. Anderson Cancer Center. Informed consent was obtained from all participants.
Development of the iSelect Infinium II cancer gene/SNP Beadchip
We developed a customized and comprehensive panel of cancer-related genes involved in 12 major cellular pathways (Supplementary Table 1). For each specific pathway, genes were subcategorized according to their major reported functions. To generate an unbiased relevant gene list, we utilized the Gene Ontology (GO) (http://www.geneontology.org), a comprehensive database of gene annotation. We further used the Cancer Genome Anatomy Project (CGAP) GO Browser (http://cgap.nci.nih.gov/Genes/GOBrowser) to pinpoint all relevant ontology terms for probing the GO database. We performed an extensive literature review on the genes returned by the GO database, using the HUGO name and the common aliases (http://www.gene.ucl.ac.uk/nomenclature) and “cancer” as keywords to interrogate the PubMed to further scrutinize for cancer relevance. We then assigned a priority score to each gene based on the gene’s importance and relevance to the specific cancer pathway. For each gene with a high priority score, we identified the tagSNPs ranging from 10 kb upstream of the 5’ untranslated region (UTR) to 10 kb downstream of the 3’ UTR of the gene.(16) We also included potentially functional SNPs which located in the functional regions of the genes, including coding (synonymous SNPs and nsSNPs), and regulatory (promoter, splicing site, 5’ UTR, and 3’ UTR) regions. Each gene was then analyzed using the LDSelect program (http://droog.gs.washington.edu/ldSelect.html) to divide SNPs into bins based on the r2 threshold of 0.8 and minor allele frequency (MAF) ≥ 0.05 in Caucasians. For genes with a medium priority score, only potentially functional SNPs were identified. For tagSNP selection, we selected one SNP from each bin according to pre-set criteria considering the validation status, designability score, position, and beadtype number of specific SNPs. For potentially functional SNP selection, we included all two-hit or HapMap validated SNPs with a designability score ≥ 0.6 and a MAF ≥ 0.01 in Caucasians. Overall, 9645 SNPs were included on the BeadChip (Supplementary Table 1). The complete set of selected SNPs was submitted to Illumina technical support for the Infinium II chemistry designability and beadtype analyses using a proprietary program developed by Illumina. (17)
Genotyping
Genomic DNA was extracted from peripheral blood lymphocytes. Genotyping was carried out according to the standard 3-day protocol provided by Illumina. The genotypes were auto-called using the BeadStudio software.
Statistical analysis
Statistical analyses were performed using Intercooled STATA software (STATA Corp., College Station, TX) and SAS/Genetics, version 9.0 (SAS Institute). Chi-square analysis was used to assess the differences between subject groups with regard to categorical variables and Student’s t test for continuous variables For each chromosomal SNP, the risks of SPT/recurrence were estimated as hazard ratios (HRs) and 95% confidence intervals (CIs) using multivariable Cox proportional hazard regression models adjusted for age, gender, ethnicity, smoking status, tumor site, stage, and treatment, where appropriate. Three genetic models (dominant, recessive, and additive) were tested for each SNP and the model with the highest significance was considered the best-fitting model and used to measure the statistical significance of each SNP. (18) For mitochondrial SNPs (mtSNPs), the heterozygous genotypes were treated as missing data since these calls typically result either from DNA contamination or heteroplasmy. (19) The wild-type and variant genotypes of mtSNPs were then analyzed in the same way as chromosomal SNPs. Multiple hypothesis testing was performed using the q value, a measure of significance in terms of the false discovery rate and implemented in the R package. (20) The multiple comparison adjustment was carried out for the best-fitting model representing the significance of the association for each SNP. We applied a bootstrap resampling method to internally validate the results. We generated bootstrap 100 samples. Each time a bootstrap sample was drawn from the original dataset and the p value was obtained for each SNP among the dominant, recessive, and additive models. The cumulative effects of unfavorable genotypes on SPT/recurrence were tested for the combined top SNPs that showed a significant q value (<0.05) and also had a bootstrap p value below 0.01 at least 80% times. Based on the percentage of patients developing SPT/recurrence, subjects were categorized into low-risk (< 25%), medium low-risk (25–50%), medium high-risk (51–75%), and high-risk (> 75%) groups by number of unfavorable genotypes. We calculated the HRs and 95% CIs for all other groups compared to the low-risk reference group, using a multivariable Cox proportional hazard regression model. Kaplan-Meier estimates were calculated to plot the event-free curve for each group and the log-rank test was used to compare survival between these groups. We also constructed receiver operating characteristic (ROC) curves and calculated the area under the curve (AUC) to evaluate the specificity and sensitivity of predicting SPT/recurrence by incorporating different combinations of epidemiological, clinical, and genetic predictor variables. We only included SNPs internal validated by bootstrapping in these analyses. A two-sided P ≤ 0.05 was considered the threshold of statistical significance.
RESULTS
Characteristics of the study population
One hundred and fifty patients with SPT/recurrence (cases) were 1:2 matched to 300 patients without SPT/recurrence (controls) by age (±5 years), gender, and ethnicity (Supplementary Table 2). There were no significant differences between these two groups in radiotherapy (P=0.71), surgery (P=0.34), or 13 cis retinoic acid treatment arm (P=0.42). There appeared to be more current smokers (42%) in SPT/recurrence group than in no event group (34%), and more high stage (stage II) patients in the former group (41%) than the later group (34%), although these two comparisons did not reach statistical significance (P=0.22 and 0.13, respectively). However, significant differences were observed between the two groups in pack-years (P=0.007), and tumor site (P=6.0 × 10−5).
iSelect Infiniumm II Beadchip content and genotyping quality controls
There were 998 genes represented by 9645 SNPs on the Beadchip (Supplementary Table 3). 78% were tagging SNPs and 22% were potentially functional SNPs. The initial conversion rate of the Beadchip synthesis was 90.61%, leaving 8739 SNPs (8583 chromosomal SNPs and 156 mtSNPs) with reliable genotyping data. Individuals with > 5% missing genotypes, SNPs with > 5% missing calls, chromosomal SNP with < 1% MAF or mtSNPs with < 5% MAF were excluded. After applying these filters, 8370 SNPs and 440 study subjects (147 cases and 293 controls) were included in the following analyses.
Significant individual SNPs associated with SPT/recurrence in the main effect analysis
Since the genetic background and replication patterns are significantly different for chromosomal and mtSNPs, we performed analyses separately for these two groups. Table 1 lists the top 20 chromosomal SNPs sorted by p values. Six SNPs remained statistically significant after multiple comparison adjustment using q value (Table 1). The most significant SNP (rs12359892) was located in the 3’ region of the MKI67 gene. The homozygous variant genotype was associated with a 2.65-fold (95% CI 1.72–4.11, P=1.25 × 10−5, q=0.042) increased risk of SPT/recurrence under the recessive genetic model. Seven mtSNPs had significant q values after multiple comparison adjustment (Table 2). MitoA11813G located in the NADH dehydrogenase subunit 4 (ND4) gene was the most significant mtSNP. The HR of the variant allele was 0.06 (95% CI 0.01-0.44, P=1.24 × 10−6, q=1.98 × 10−5) compared to the wild-type allele. We then performed bootstrap 100 times for internal validation and listed the number of times that the bootstrap p value was less than 0.01 for each SNP (Tables 1 and 2). For the top 20 chromosomal SNPs, 12 had a bootstrap p value <0.01 at least 80% times (Table 1, shaded SNPs). The top SNP, MKI67 rs12359892, exhibited a highly consistent result with a p value < 0.01 96 times in 100 bootstrap samples (Table 1). The top 3 mitochondrial SNPs had a bootstrap p value below 0.01 at least 80% times (Table 2). The top mitochondrial SNP, mitoA11813G, exhibited a highly consistent result with a bootstrap p value < 0.01 for 98 times.
Table 1.
Associations of SPT/Recurrence with the Top 20 Chromosomal SNPs
| SNP | Host gene | Pathway | Position in host gene | Chromosome region | Allelic change | Genotype Counts* | Best-fitting model | Number of times in Bootstrap Sample P<0.01 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SPT | No SPT | Model | HR (95% CI)** | P Value | q value | |||||||
| rs12359892 | MKI67 | Cell cycle | 3’ region | 10q26 | T>C | 101/14/27 | 221/38/18 | Recessive | 2.65 (1.72–4.11) | 1.25×10−5 | 0.042 | 96 |
| rs359974 | NHEJ1 | DNA repair | 3’ region | 2q35 | T>C | 93/44/10 | 213/76/4 | Recessive | 4.26 (2.18–8.32) | 2.15×10−5 | 0.042 | 81 |
| rs7781436 | CDK6 | Cell cycle | Intron 4 | 7q21–q22 | T>C | 97/41/9 | 186/104/3 | Recessive | 4.51 (2.22–9.17) | 3.24×10−5 | 0.042 | 68 |
| rs876435 | TNFRSF10B | Apoptosis | 3’ region | 8p22–p21 | A>G | 51/63/33 | 114/148/31 | Recessive | 2.26 (1.52–3.36) | 6.04×10−5 | 0.042 | 90 |
| rs12888332 | MNAT1 | DNA repair | Intron 7 | 14q23 | T>G | 125/19/3 | 280/10/3 | Dominant | 2.57 (1.62–4.09) | 6.27×10−5 | 0.042 | 88 |
| rs506008 | GSTM4 | Carcinogen metabolism | sSNP in exon 7 § | 1p13 | G>A | 94/46/3 | 232/54/1 | Dominant | 2.09 (1.46–3.00) | 6.30×10−5 | 0.042 | 91 |
| rs7561607 | GLI2 | SHH stem cell | Intron 2 | 2q14 | C>T | 26/82/39 | 103/128/62 | Dominant | 2.30 (1.49–3.55) | 1.63×10−4 | 0.094 | 94 |
| rs6684195 | RNF2 | Epigentic regulation | Intron 1 | 1q25 | A>G | 50/64/33 | 111/150/32 | Recessive | 2.12 (1.43–3.14) | 1.93×10−4 | 0.097 | 87 |
| rs17387169 | PROM1 | EMT, stem cell | Intron 13 | 4p15 | G>A | 129/19/2 | 202/85/6 | Dominant | 0.42 (0.26–0.67) | 2.71×10−4 | 0.121 | 92 |
| rs7168671 | IGF1R | Growth factor signaling | Intron 7 | 15q26 | C>T | 94/44/9 | 194/93/6 | Recessive | 3.98 (1.87–8.44) | 3.22×10−4 | 0.13 | 61 |
| rs7591 | AXIN1 | WNT stem cell | 3’ UTR | 16p13 | A>T | 33/93/21 | 110/124/59 | Dominant | 2.03 (1.37–3.01) | 4.10×10−4 | 0.15 | 89 |
| rs2306536 | CHFR | Cell cycle | nsSNP §§ in exon 15 | 12q24 | C>T | 87/49/11 | 88/97/8 | Recessive | 3.14 (1.65–5.99) | 5.13×10−4 | 0.172 | 70 |
| rs7118388 | CAT | Carcinogen metabolism | 5’ region | 11p13 | G>A | 51/66/30 | 70/139/84 | Additive | 0.67 (0.53–0.84) | 5.64×10−4 | 0.175 | 85 |
| rs2237724 | CFTR | Carcinogen metabolism | Intron 3 | 7q31-q32 | G>A | 92/45/10 | 198/89/6 | Recessive | 3.12 (1.62–5.98) | 6.30×10−4 | 0.181 | 68 |
| rs604337 | GSTM4 | Carcinogen metabolism | 5’ region | 1p13 | C>T | 94/50/3 | 223/64/6 | Dominant | 1.84 (1.29–2.61) | 6.80×10−4 | 0.183 | 71 |
| rs3826537 | MRC2 | Growth factor signaling | sSNP in exon 30 | 17q23 | A>G | 67/63/17 | 88/150/55 | Dominant | 0.57 (0.41–0.79) | 8.41×10−4 | 0.202 | 84 |
| rs9562605 | BRCA2 | DNA repair | Intron 1 | 13q12–q13 | C>T | 107/36/4 | 168/114/11 | Dominant | 0.54 (0.37–0.78) | 9.34×10−4 | 0.202 | 80 |
| rs2300181 | CAT | Carcinogen metabolism | Intron 6 | 11p13 | G>A | 76/58/13 | 163/124/6 | Recessive | 2.67 (1.49–4.78) | 9.38×10−4 | 0.202 | 79 |
| rs11047917 | KRAS | RAS signaling | Intron 2 | 12p12 | C>T | 123/24/0 | 271/22/0 | Dominant | 2.12 (1.36–3.31) | 9.51×10−4 | 0.202 | 69 |
| rs9622978¶ | PDGFB | Growth factor signaling | Intron 5 | 22q12–q13 | G>T | 49/63/35 | 98/160/35 | Recessive | 1.92 (1.30–2.85) | 1.08×10−3 | 0.213 | 76 |
Genotype counts: common homozygous/ heterozygous/rare homozygous genotype
Adjusted for age, gender, ethnicity, smoking status, tumor site, tumor stage, and treatment.
sSNP, synonymous SNP;
nsSNP, nonsynonous SNP.
Shaded SNPs were those with a p<0.01 at least 80% of the times in bootstrapping.
Table 2.
Associations of SPT/Recurrence with Mitochondrial SNPs Remaining Significant after Multiple Comparison Adjustment
| SNP | Host gene/region * | SNP type** | Mitochondrial Position | Allelic change | Genotype Counts§ | Cox model | Number of times in Bootstrap Sample | |||
|---|---|---|---|---|---|---|---|---|---|---|
| SPT | No SPT | HR (95% CI) ¶ | P Value | q value | P<0.01 | |||||
| mitoA11813G | Mt-ND4 | sSNP | 11812 | A>G | 146/1 | 259/34 | 0.06 (0.01–0.44) | 1.24×10−6 | 1.98×10-5 | 98 |
| mitoG15929A | Mt-TT | Non-coding | 15928 | G>A | 143/4 | 252/39 | 0.20 (0.08–0.56) | 5.47×10−5 | 4.37×10−4 | 94 |
| mitoA14906G | Mt-CYB | sSNP | 14905 | A>G | 141/6 | 248/41 | 0.28 (0.12−0.63) | 1.95×10−4 | 1.04×10−3 | 83 |
| mitoT10464C | Mt-TR | Non-coding | 10463 | T>C | 140/7 | 253/39 | 0.34 (0.16–0.73) | 1.04×10−3 | 4.17×10−3 | 73 |
| mitoA11252G | Mt-ND4 | sSNP | 11251 | A>G | 132/14 | 230/60 | 0.47 (0.27–0.82) | 3.21×10−3 | 1.03×10−2 | 68 |
| mitoG3012A | Mt-RNR2 | Non-coding | 3010 | G>A | 101/46 | 237/56 | 1.73 (1.21–2.49) | 3.87×10−3 | 1.03×10−2 | 70 |
| mitoT14767C | Mt-CYB | Thr > Ile | 14766 | T>C | 60/87 | 166/127 | 1.60 (1.14–2.25) | 5.90×10−3 | 1.35×10−2 | 67 |
ND4, NADH dehydrogenase subunit 4; TT, tRNA threonine; CYB, Cytochrome b; TR, tRNA arginine; RNR2, 16s ribosomal RNA.
sSNP, synonymous SNP; nsSNP, nonsynonymous SNP.
Genotype counts: wild genotype/variant genotype
Adjusted for age, gender, ethnicity, smoking status, tumor site, tumor stage, and treatment.
To increase sample size and statistical power, we grouped all SPT cases in our analysis. Since the relevance of prostate cancer and other non-smoking related or non-aerodigestive tract cancer as SPT may not be clear, we also performed separate analyses of smoking-related and aerodigestive SPT and compared the results to the entire SPT group. Of the top 20 chromosomal SNPs that were significant in the entire SPT cases (Table 1), 19 remained significant at significance level 0.05 in both smoking-related and aerodigestive tract SPT subgroup analyses and the remaining SNP had a p value of 0.11 when considering smoking-related SPT cases and p value of 0.15 when considering aerodigestive tract SPT cases. The HRs estimates were similar and the best fitting models were the same for the top 20 chromosomal SNPs (Supplementary Table 4). A similar pattern was observed for the top mtSNPs (Supplementary Table 5). We chose to present data from entire SPT cases to reflect general risk for developing any new tumors.
Cumulative effects of the unfavorable genotypes
We further evaluated the cumulative effects of the high-risk genotypes on SPT/recurrence by summing the unfavorable genotypes of the above described top risk-conferring chromosomal SNPs and mtSNPs that had bootstrap p values < 0.01 at least 80% times. Twelve chromosome SNPs and one mtSNP (mitoG15929A and mitoA14906G were excluded because of high LD with mitoA11813G) were included in this analysis. As shown in Table 3, there was a significant gene-dosage effect. Compared with those in the low-risk reference group ( 4 unfavorable genotypes), subjects with medium low- (5 ~ 6 unfavorable genotypes), medium high- (7), and high-risk ( 8) had 4.29-fold (95% CI 2.52–7.29, P=1.58×10−8), 9.16-fold (95% CI 5.52–17.83, P=3.68×10−14), and 26.72-fold (95% CI 14.00–50.99, P<1×10−20) increased SPT/recurrence risks, respectively (P for trend < 1×10−20). The event-free median survival times (MST) were 14.6 months, 49.2 months, and 79.4 months for these three risk groups, respectively, compared with > 93.0 months for the low-risk groups (log-rank P=9.92 × 10−38) (Fig. 1).
Table 3.
The Cumulative Effects of Unfavorable Genotypes on SPT/Recurrence
| Number of unfavorable genotypes * | SPT/Recurrence N (%) | No SPT/Recurrence N (%) | HR (95% CI) ** | P value |
|---|---|---|---|---|
| Reference group ≤4 | 18(10.91) | 147(89.09) | 1 | Reference |
| 5~6 | 62(37.58) | 103(62.42) | 4.29(2.52–7.29) | 7.59×10−8 |
| 7 | 34(61.82) | 21(38.18) | 9.16(5.52–17.83) | 1.80×10−14 |
| ≥8 | 25(96.15) | 1(3.85) | 26.72(14.00–50.99) | <1.00×10−20 |
| P for trend | <1.00×10−20 |
Unfavorable genotype was based on the 12 chromosomal SNPs and one mitochondrial SNP as described in text.
Adjusted for age, gender, smoking status, ethnicity, tumor site, tumor stage, and treatment.
Figure 1.

Kaplan-Meier event-free survival curve on SPT/recurrence by the unfavorable genotypes of 18 chromosomal and three mitochondrial SNPs.
Model discrimination ability
We next constructed prediction models by incorporating established prognostic clinical variables (tumor site, stage, treatment), epidemiological variables (smoking pack-years), and genetic variables (12 chromosomal SNPs and one mtSNP identified in this study) (Figure 2). The AUC increased from 0.61 (clinical variables only), to 0.64 (clinical-smoking variables), and to 0.84 (clinical, smoking, and genetic variables). The observed difference in AUC between the third and second models was 0.23, and the bias corrected 95% confidence intervals based on 10,000 bootstrap samples were 0.18–0.29, suggesting significant differences between these two models.
Figure 2.
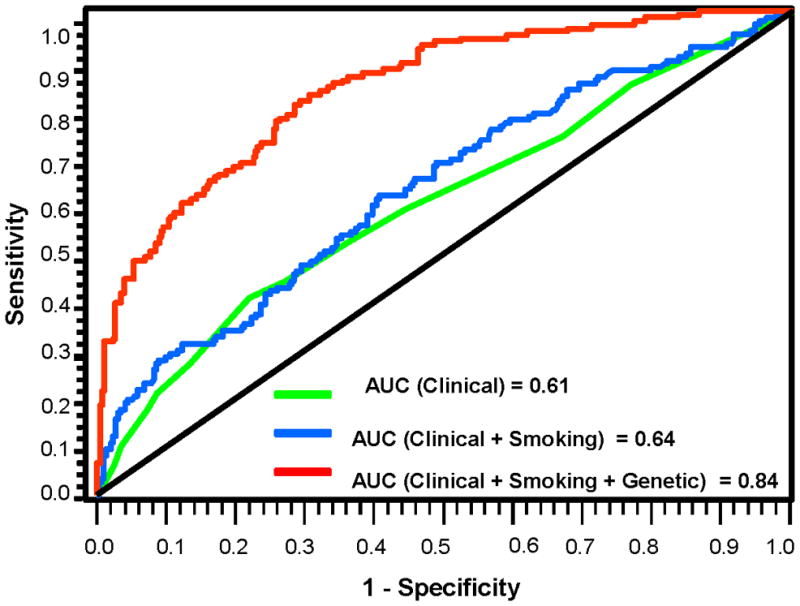
ROC curves from various models showing improvement of discrimination ability.
Because age, gender and ethnicity were matched by study design, the above models may be weak in terms of epidemiological risk factors. However, we analyzed the entire cohort data to explore the main effects of age, gender, and ethnicity on SPT/recurrence and constructed ROC curve based on these data. We found a significant effect of age on SPT/recurrence, but neither sex nor ethnicity was significantly associated with SPT/recurrence. However, adding age to the clinical-smoking model did not significantly change the AUC of the clinical-smoking model (data not shown). .
DISCUSSION
In this large scale systematic evaluation of 9645 SNPs in 998 cancer-related genes, we identified six chromosomal and seven mitochondrial SNPs significantly associated with risk of SPT/recurrence after correction for type I errors, with evidence of a significant gene-dosage effect. These results support the notion that SPT and tumor recurrences are polygenic traits determined by multiple low penetrance loci.
We developed a customized SNP chip encompassing well-established pathways through comprehensive and exhaustive database interrogation and literature review. The associations identified are biologically plausible. Among the six significant chromosomal variants, the most significant is localized in the MKI67 gene, an important cell cycle proliferation marker whose expression is correlated with the development and progression of various malignancies including HNSCC. (21) CDK6 mostly functions in the progression of G1 phase through interacting with multiple cyclins and inhibiting tumor suppressor protein RB. (22) Both CDK6 and MKI67 are reported to promote HNSCC progression through enhancing expression of protein kinases to phosphorylate and activate proliferative transcription factors. (23) MNAT1 is a key component of the protein complex CAK (CDK-activating kinase), which phosphorylates CDKs to activate cell cycle progression and also interacts with transcription factor TFIIH to stimulate nucleotide excision repair. (24) NHEJ1 gene product interacts with both XRCC4 and LIG4 as a core component of the protein complex responsible for non-homologous end joining pathway of double-stranded DNA break repair. (25) Suboptimal DNA repair capacity have been shown to increase the risk of HNSCC and SPT/recurrence. (10, 11) TNFRSF10B encodes a member of the tumor necrosis factor (TNF)-receptor superfamily involved in extrinsic apoptosis pathway. (26) Mutations in TNFRSF10B have been identified in multiple cancers including HNSCC. (10) GSTM4 belongs to the Mu subclass of the GST family, essential in the detoxification of electrophilic compounds and polymorphisms of this gene family have been extensively associated with the risk and outcomes of HNSCC. (27, 28) Taken together, there is strong biological plausibility for the associations between the six identified chromosomal genes and HNSCC.
We also identified several mtSNPs as predictors of HNSCC SPT/recurrence. Mitochondrial dysfunction may lead to tumorigenesis through apoptotic regulation, reactive oxygen species (ROS) generation, metabolic regulation, and nucleus-mitochondria communications. (29) Altered mitochondrial function with increased aerobic glycolysis, the Warburg effect, is a common feature in many tumors. (30) Aberrations of mtDNA have been observed in almost all types of solid cancers including HNSCC. (31) Polymorphisms in the mitochondrial genome have also been associated with many common diseases, including diabetes and cancer. (32) The most significant mtSNP, mitoA11813G, is located in the ND4 gene, which has been implicated in head and neck cancer by multiple independent studies. (33, 34) Mutations of cytochrome b (CYB) and 16s ribosomal RNA (RNR2) were also identified in HNSCC. (31) mtSNPs may be involved in the initiation and progression of both index tumors and SPT/recurrence due to possible disruptive effects on mitochondria genes and energy metabolism, (35) or related to the central role of mitochondria in apoptosis and ROS production.
We further used Ingenuity Pathway Analysis to explore whether certain canonical pathways were overrepresented for significant associations, by inputting chromosomal genes containing SNPs with P value<0.01 (a total of 170 genes). (36) The top pre-defined canonical pathways to which these genes belong include aryl hydrocarbon receptor signaling, PTEN signaling, LPS/IL-1 mediated inhibition of RXR function, xenobiotic metabolism signaling, and cell cycle (Supplementary Table 6), most of which are implicated in carcinogen or drug metabolism and treatment-related cellular response. Because of the etiologic role of tobacco and alcohol in HNSCC carcinogenesis, these results are not surprising. Most genetic markers of clinical outcome have only modest effects, and there is likely to be an enhanced predictive power when SNPs are analyzed jointly (18, 37, 38), as we noted. Another data-mining tool we explored is the survival tree analysis, which uses a binary recursive partitioning to produce a tree structure with many binary splits. Our survival tree analysis produced a decision tree with 14 terminal nodes, each with a different SPT/recurrence risk based on distinct combination of genotypes (Supplementary Fig. 1). The terminal nodes from the final tree were grouped into four risk groups based on the percentage of patients developing SPT/recurrence in each terminal node, low-risk (< 25%), medium low-risk ( 25 to 50%), medium high-risk (51 to 75%), and high-risk (> 75%). Compared to the low-risk group, the risk increased from 3.48 to 17.04 fold for medium low to high-risk groups (Supplementary Fig. 1). We validated the risk groups by bootstrapping the samples 10,000 times. These data support an important role of gene-gene interactions in modulating SPT/recurrence. Furthermore, when we incorporated the genetic variables into a multivariate model, we obtained a significant improvement of discriminatory ability (Fig. 2), underscoring the importance of incorporating germline genetic variation data with clinical and risk factor data into prediction models for clinical outcomes.
There are also a few limitations of this study. First, the sample size is limited due to the rarity of events and availability of germline DNA. We calculated statistical power based on the minor allele frequency (MAF) and genetic models (supplemental Table 7). Power is adequate for additive and dominant models to detect an OR 2.5 or higher when MAF >0.05. At an MAF of 0.05, we have more than 91% power and 94% power to detect an increased OR of 2.5 in dominant and additive models, respectively. The power to detect OR of 2.5 is close to 100% for larger MAFs. For a recessive model, we have more than 80% power to detect an increased OR of 3.0 when MAF is 0.20 or higher. However, power is limited when MAF is lower in recessive model. We calculated power to detect ORs instead of HRs. In cohort studies with long follow-up time, the HR approach based on survival analysis for time to event endpoint is even more efficient than the OR approach based on logistic regression for binary endpoint. Second, due to the sample size, we could not perform stratified analyses, for example, on smoking and tumor site. Hence we adjusted these variables in all our analyses. We also do not have information on HPV-16 status. Third, due to the difficulty in identifying an external validation population, we are unable to validate the significant SNPs in an independent population. Such external validation would be a critical next step. Finally, we used a nested 1:2 case-control study design, which may not reflect the population of early stage HNSCC, although the 1:2 case-control ratio is comparable to the roughly 30% of SPT/recurrence incidence in the original population.
There are many strengths of this study. This is the first large scale study to systematically evaluate germline genetic variants in HNSCC SPT/recurrence. Because a genome-wide scanning approach was not possible due to the limited numbers of HNSCC patients who developed SPT/recurrence, our pathway-based custom SNP array is the best option. There is minimal selection bias since the cases and controls were well matched and were all early stage HNSCC patients enrolled in a prospectively conducted randomized chemoprevention trial. The significant SNPs identified may be useful for clinicians in assessing the risk for SPT/recurrence in early stage HNSCC patients. The genotyping technology is robust and consistent. Obtaining DNA from peripheral blood is non-invasive and inexpensive. We can generate thousands of genotypes from one drop of blood and get the patients’ genetic profile predictive of SPT/recurrence, which can be incorporated into a risk prediction model to identify high-risk patients to undergo intensive screening, smoking cessation, or dietary modification. Chemoprevention trials have been mostly negative in head and neck cancer. Although the main reason for these negative results probably is that the tested chemoprevention agents are not the best, we also think that patients are heterogeneous and these agents may not work in all patients. Not considering patients’ genetic background in patient stratification may at least partially contribute to the negative results. Patients with a specific genetic background may respond better to certain chemoprevention agents.
The present study focused on comprehensive risk-modeling analyses of SNPs to identify early-stage head and neck HNSCC cancer patients at the highest risk of SPT/recurrence and conducted within a large-scale randomized trial of 13 cis retinoic acid. Ongoing work that is beyond the scope of this paper is examining pharmacogenetic interactions to see if there are certain germline alterations associated with a better outcome of 13 cis retinoic acid treatment. This treatment was a covariate in the risk modeling analysis, which was adjusted for this factor. We identified the top 20 chromosomal SNPs associated with a high risk of SPT/recurrence (Table 1); of these 20 SNPs, only one, which is in MK167, a cell-cycle gene, was associated with the retinoid effect of a significantly reduced SPT/recurrence risk (62%), making this SNP both highly prognostic and predictive (data not shown). This preliminary observation is advantageous in that it appears to mark high-risk patients with the greatest need and their sensitivity to an agent; it is being examined further in the broader pharmacogenomic studies mentioned above. If these studies identify a predictive marker or signature based on individual patients’ germline genetic variations, we can design a better patient stratification plan in future chemoprevention trials, targeting chemoprevention agents to patients with a high risk of SPT/recurrent and more likely to benefit from treatment. Through this personalized chemoprevention, we may have better success in chemoprevention trials.
Supplementary Material
Acknowledgments
The study was supported in part by the NIH grants CA52051 (W.K.H.), CA97007 (W.K.H. and S.M.L.), and CA86390 (M.R.S.). Dr. Waun Ki Hong is an American Cancer Society Clinical Research Professor
References
- 1.Khuri FR, Kim ES, Lee JJ, Winn RJ, Benner SE, Lippman SM, et al. The impact of smoking status, disease stage, and index tumor site on second primary tumor incidence and tumor recurrence in the head and neck retinoid chemoprevention trial. Cancer Epidemiol Biomarkers Prev. 2001;10(8):823–9. [PubMed] [Google Scholar]
- 2.Khuri FR, Lee JJ, Lippman SM, Kim ES, Cooper JS, Benner SE, et al. Randomized phase III trial of low-dose isotretinoin for prevention of second primary tumors in stage I and II head and neck cancer patients. J Natl Cancer Inst. 2006;98(7):441–50. doi: 10.1093/jnci/djj091. [DOI] [PubMed] [Google Scholar]
- 3.Perez-Ordonez B, Beauchemin M, Jordan RC. Molecular biology of squamous cell carcinoma of the head and neck. J Clin Pathol. 2006;59(5):445–53. doi: 10.1136/jcp.2003.007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedi GC, Westra WH, Gabrielson E, Koch W, Sidransky D. Multiple head and neck tumors: evidence for a common clonal origin. Cancer Res. 1996;56(11):2484–7. [PubMed] [Google Scholar]
- 5.Mao L, Hong WK, Papadimitrakopoulou VA. Focus on head and neck cancer. Cancer Cell. 2004;5(4):311–6. doi: 10.1016/s1535-6108(04)00090-x. [DOI] [PubMed] [Google Scholar]
- 6.Maestro R, Gasparotto D, Vukosavljevic T, Barzan L, Sulfaro S, Boiocchi M. Three discrete regions of deletion at 3p in head and neck cancers. Cancer Res. 1993;53(23):5775–9. [PubMed] [Google Scholar]
- 7.Bockmuhl U, Wolf G, Schmidt S, Schwendel A, Jahnke V, Dietel M, et al. Genomic alterations associated with malignancy in head and neck cancer. Head Neck. 1998;20(2):145–51. doi: 10.1002/(sici)1097-0347(199803)20:2<145::aid-hed8>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 8.Izzo JG, Papadimitrakopoulou VA, Li XQ, Ibarguen H, Lee JS, Ro JY, et al. Dysregulated cyclin D1 expression early in head and neck tumorigenesis: in vivo evidence for an association with subsequent gene amplification. Oncogene. 1998;17(18):2313–22. doi: 10.1038/sj.onc.1202153. [DOI] [PubMed] [Google Scholar]
- 9.El-Naggar AK, Lai S, Clayman G, Lee JK, Luna MA, Goepfert H, et al. Methylation, a major mechanism of p16/CDKN2 gene inactivation in head and neck squamous carcinoma. Am J Pathol. 1997;151(6):1767–74. [PMC free article] [PubMed] [Google Scholar]
- 10.Pai SI, Wu GS, Ozoren N, Wu L, Jen J, Sidransky D, et al. Rare loss-of-function mutation of a death receptor gene in head and neck cancer. Cancer Res. 1998;58(16):3513–8. [PubMed] [Google Scholar]
- 11.Spitz MR, Lippman SM, Jiang H, Lee JJ, Khuri F, Hsu TC, et al. Mutagen sensitivity as a predictor of tumor recurrence in patients with cancer of the upper aerodigestive tract. J Natl Cancer Inst. 1998;90(3):243–5. doi: 10.1093/jnci/90.3.243. [DOI] [PubMed] [Google Scholar]
- 12.Sturgis EM, Castillo EJ, Li L, Zheng R, Eicher SA, Clayman GL, et al. Polymorphisms of DNA repair gene XRCC1 in squamous cell carcinoma of the head and neck. Carcinogenesis. 1999;20(11):2125–9. doi: 10.1093/carcin/20.11.2125. [DOI] [PubMed] [Google Scholar]
- 13.Cheng L, Sturgis EM, Eicher SA, Char D, Spitz MR, Wei Q. Glutathione-S-transferase polymorphisms and risk of squamous-cell carcinoma of the head and neck. Int J Cancer. 1999;84(3):220–4. doi: 10.1002/(sici)1097-0215(19990621)84:3<220::aid-ijc4>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 14.Leibovici D, Grossman HB, Dinney CP, Millikan RE, Lerner S, Wang Y, et al. Polymorphisms in inflammation genes and bladder cancer: from initiation to recurrence, progression, and survival. J Clin Oncol. 2005;23(24):5746–56. doi: 10.1200/JCO.2005.01.598. [DOI] [PubMed] [Google Scholar]
- 15.Wu X, Gu J, Dong Q, Huang M, Do KA, Hong WK, et al. Joint effect of mutagen sensitivity and insulin-like growth factors in predicting the risk of developing secondary primary tumors and tumor recurrence in patients with head and neck cancer. Clin Cancer Res. 2006;12(23):7194–201. doi: 10.1158/1078-0432.CCR-06-0671. [DOI] [PubMed] [Google Scholar]
- 16.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74(1):106–20. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steemers FJ, Chang W, Lee G, Barker DL, Shen R, Gunderson KL. Whole-genome genotyping with the single-base extension assay. Nat Methods. 2006;3(1):31–3. doi: 10.1038/nmeth842. [DOI] [PubMed] [Google Scholar]
- 18.Zheng SL, Sun J, Wiklund F, Smith S, Stattin P, Li G, et al. Cumulative Association of Five Genetic Variants with Prostate Cancer. N Engl J Med. 2008 doi: 10.1056/NEJMoa075819. [DOI] [PubMed] [Google Scholar]
- 19.Saxena R, de Bakker PI, Singer K, Mootha V, Burtt N, Hirschhorn JN, et al. Comprehensive association testing of common mitochondrial DNA variation in metabolic disease. Am J Hum Genet. 2006;79(1):54–61. doi: 10.1086/504926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slootweg PJ, Koole R, Hordijk GJ. The presence of p53 protein in relation to Ki-67 as cellular proliferation marker in head and neck squamous cell carcinoma and adjacent dysplastic mucosa. Eur J Cancer B Oral Oncol. 1994;30B(2):138–41. doi: 10.1016/0964-1955(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 22.Wikman H, Kettunen E. Regulation of the G1/S phase of the cell cycle and alterations in the RB pathway in human lung cancer. Expert Rev Anticancer Ther. 2006;6(4):515–30. doi: 10.1586/14737140.6.4.515. [DOI] [PubMed] [Google Scholar]
- 23.Santos CR, Rodriguez-Pinilla M, Vega FM, Rodriguez-Peralto JL, Blanco S, Sevilla A, et al. VRK1 signaling pathway in the context of the proliferation phenotype in head and neck squamous cell carcinoma. Mol Cancer Res. 2006;4(3):177–85. doi: 10.1158/1541-7786.MCR-05-0212. [DOI] [PubMed] [Google Scholar]
- 24.Zhang S, He Q, Peng H, Tedeschi-Blok N, Triche TJ, Wu L. MAT1-modulated cyclin-dependent kinase-activating kinase activity cross-regulates neuroblastoma cell G1 arrest and neurite outgrowth. Cancer Res. 2004;64(9):2977–83. doi: 10.1158/0008-5472.can-03-4018. [DOI] [PubMed] [Google Scholar]
- 25.Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124(2):301–13. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 26.Takeda K, Stagg J, Yagita H, Okumura K, Smyth MJ. Targeting death-inducing receptors in cancer therapy. Oncogene. 2007;26(25):3745–57. doi: 10.1038/sj.onc.1210374. [DOI] [PubMed] [Google Scholar]
- 27.Singh M, Shah PP, Singh AP, Ruwali M, Mathur N, Pant MC, et al. Association of genetic polymorphisms in glutathione S-transferases and susceptibility to head and neck cancer. Mutat Res. 2008;638(1–2):184–94. doi: 10.1016/j.mrfmmm.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Cabelguenne A, Loriot MA, Stucker I, Blons H, Koum-Besson E, Brasnu D, et al. Glutathione-associated enzymes in head and neck squamous cell carcinoma and response to cisplatin-based neoadjuvant chemotherapy. Int J Cancer. 2001;93(5):725–30. doi: 10.1002/ijc.1392. [DOI] [PubMed] [Google Scholar]
- 29.Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer. 2002;1:9. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 31.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25(34):4663–74. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 32.Bai RK, Leal SM, Covarrubias D, Liu A, Wong LJ. Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 2007;67(10):4687–94. doi: 10.1158/0008-5472.CAN-06-3554. [DOI] [PubMed] [Google Scholar]
- 33.Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, et al. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 2000;287(5460):2017–9. doi: 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- 34.Allegra E, Garozzo A, Lombardo N, De Clemente M, Carey TE. Mutations and polymorphisms in mitochondrial DNA in head and neck cancer cell lines. Acta Otorhinolaryngol Ital. 2006;26(4):185–90. [PMC free article] [PubMed] [Google Scholar]
- 35.Wallace DC. Mitochondria and cancer: Warburg addressed. Cold Spring Harb Symp Quant Biol. 2005;70:363–74. doi: 10.1101/sqb.2005.70.035. [DOI] [PubMed] [Google Scholar]
- 36.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437(7061):1032–7. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 37.Gordon MA, Gil J, Lu B, Zhang W, Yang D, Yun J, et al. Genomic profiling associated with recurrence in patients with rectal cancer treated with chemoradiation. Pharmacogenomics. 2006;7(1):67–88. doi: 10.2217/14622416.7.1.67. [DOI] [PubMed] [Google Scholar]
- 38.Wu X, Gu J, Wu TT, Swisher SG, Liao Z, Correa AM, et al. Genetic variations in radiation and chemotherapy drug action pathways predict clinical outcomes in esophageal cancer. J Clin Oncol. 2006;24(23):3789–98. doi: 10.1200/JCO.2005.03.6640. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.