

Abstract
Accumulating evidence demonstrates increasing bone turnover and bone loss in women prior to menopause and decreases in serum estradiol levels. Increased follicle-stimulating hormone levels have been correlated with some of these peri-menopausal changes. However, decreases in gonadal inhibins of the transforming growth factor (TGF)-β superfamily strongly correlate with increases in bone formation and resorption markers across the menopause transition and predict lumbar bone mass in peri-menopausal women, likely as a result of direct inhibin suppression of osteoblastogenesis and osteoclastogenesis. Inhibins bind specifically to cells during osteoblastogenesis and osteoclastogenesis. They can block bone morphogenetic protein (BMP)-stimulated osteoblast and osteoclast development as well as BMP-stimulated SMAD1 phosphorylation, likely via inhibin–β-glycan sequestration of BMP Type II receptor (BMPRII). Interestingly, continuous in vivo exposure to inhibin A is anabolic and protective against gonadectomyinduced bone loss in mice, suggesting that inhibins contribute to the endocrine regulation of bone metabolism via a bimodalmechanism of action whereby cycling inhibin exposure suppresses bone turnover and continuous exposure to inhibins is anabolic.
Keywords: inhibin, menopause, bone turnover, BMP, bone morphogenetic protein, osteoblast, osteoclast
Bone loss across the menopause transition
For over 50 years it has been suspected that osteoporosis is a result of hormone deficiency at menopause. Simple sex steroid deficiency has long been considered the cause of this menopausal bone loss and has provided the foundation for many approaches to the prevention and treatment of osteopenia and osteoporosis.1 Indeed, it is now widely accepted that estrogen plays a critical role in the maintenance of bone homeostasis and that the cellular basis of bone loss in postmenopausal women results from derepression of both osteoblast (OB) and osteoclast (OCL) development.2 The pathophysiology of postmenopausal osteoporosis involves the overproduction of OCLs relative to the integrally coupled increase in osteoblastogenesis, a process that also facilitates OCL development.3
Estrogen deficiency has been identified as a major risk factor for osteoporosis in women.4,5 Recent evidence suggests that estrogen deficiency may be responsible for the rapid bone loss of the early postmenopausal phase as well as be involved in the later slower phase of bone loss associated with aging. 6-8 However, in late premenopausal women with normal circulating estrogen levels, clinical indices of increased bone turnover are already elevated.9 More recently, more sensitive and precise computed tomography (CT) technology that allows for the independent assessment of cortical and trabecular volumetric bone mineral density (BMD) has been used. Cross-sectional10 and longitudinal11 population-based CT studies demonstrated that young women (and men) begin to lose both trabecular and cortical bone volume long before the onset of menopause in women or before aging in men.
The endocrine parameter that was identified to be best correlated with increases in bone turnover in late pre- or peri-menopausal women was elevated serum follicle-stimulating hormone (FSH) levels.9 However, early losses in trabecular bone volume in women or young men measured by CT did not consistently significantly correlate with either FSH or sex steroids, although the later postmenopausal loss in trabecular bone volume was correlated with the expected increases in FSH and decreases in bioactive steroids.11 Correlations of FSH with increasing bone turnover are consistent with recent demonstrations that FSH can enhance receptor activator of nuclear factor (NF)-κB ligand (RANKL)-stimulated OCL development and activity without effects on osteoblastogenesis. 12 However, lack of consistent clinical correlation in longitudinal studies demonstrates that the early onset pre- and peri-menopausal trabecular bone loss is neither explained by direct effects of FSH on osteoclastogenesis nor changes in sex steroids. Indeed, these findings highlight the importance for identifying additional endocrine markers that may be altered across the menopause transition in women and across age in men, both of which are causal in the pathogenesis of bone loss.
FSH production in premenopausal women is under ovarian negative feedback not only by estradiol but also by the production of the ovarian peptides inhibin A (InhA) and inhibin B (InhB), which are related heterodimeric members of the transforming growth factor (TGF)-β superfamily.13 These heterodimers are comprised of either αβA (InhA) or αβB (InhB) subunits. The α subunit is unique to inhibins, whereas the β subunits are derived from two genes βA and βB, which when combined form homodimeric βAβA or βBβB peptides comprising activin A (ActA) or activin B, respectively, and exert effects that oppose those of inhibins in a variety of cell types.14
Prior to the development of specific assays for InhA and InhB,15 the assay used to measure total inhibins was one based upon measurement of the inhibin-specific α-subunit. The assay did not distinguish between InhA or InhB and also detected a secreted nonbioactive free α-subunit monomer. Thus, it may not be surprising that the measurement of total inhibins using the α-subunit assay in the Ebeling study9 did not demonstrate significant correlations with bone turnover markers (see below).
With the development of specific assays to selectively measure InhB,15 the rise in FSH at the onset of menopause was determined to be the result of declining levels of gonadal InhB, providing the first indication of loss of ovarian function and diminished ovarian follicle number.16-18 As the loss of gonadal function proceeds in late peri- and postmenopausal women, the well-established decreases in estrogen accompany the already declining levels of both InhB and InhA, further increasing serum FSH16-18 and markedly increasing bone loss.9,19 Thus, during the menopause transition, the importance of inhibins in the negative feedback control of FSH secretion is now well established. These independent findings suggested the possibility that menopausal changes in gonadal inhibins in the presence of normal (or elevated) estradiol may contribute to the initial increases in bone turnover observed in peri-menopausal women and may in fact be the endocrine hormone that plays a causal role in trabecular bone loss observed by Riggs et al.11 prior to the onset of menopause.
Inhibin suppresses osteoblast and osteoclast differentiation in vitro
In support of this hypothesis, initial studies in murine bone marrow cell cultures demonstrated that both InhA and InhB suppress both OB and OCL differentiation.20 Inhibins suppressed the recruitment of cells into the osteoblastic lineage (formation of alkaline positive [AP+] colony-forming units-fibroblasts [CFU-F]) as well as the later osteoblastic differentiation into bone nodules (formation of CFU-OB).20 Formation of multinucleated tartrate-resistant acid phosphatase positive (TRAP+) cells, an indicator of osteoclastogenesis, was also suppressed by inhibins.20 As has been demonstrated with many activin target tissues, inhibins exert dominant and opposing effects to activin on both OB and OCL development.13,20 Surprisingly, however, inhibins also blocked the well-described stimulatory effects of bone morphogenetic proteins (BMPs) on osteoblastogenesis and osteoclastogenesis, regardless of whether the BMP was generated endogenously in the culture or provided by exogenous administration.20 Collectively, these data were the first to demonstrate 1) direct effects of inhibins on OB and OCL differentiation and 2) antagonism/blockade of the stimulatory effects of BMPs by inhibins in any tissue, a finding that was later confirmed in pituitary and other cell types.21 The demonstration that inhibins block the stimulatory effects of both BMP and activin support the concept that bone is an endocrine target tissue of inhibin action. Indeed, 125I-InhA rapidly accumulates in the bone marrow of 25-day-old rats (within 10 min of i.v. injection) and is retained for at least an hour.22 Moreover, it suggests that regulation of OB and OCL differentiation by gonadal inhibins can be dominant over the local paracrine/autocrine effects of BMPs, activin A, and their soluble antagonists (chordin, follistatin, and gremlin) also expressed in bone (reviewed in Ref. 23).
The suppressive effects of inhibins on murine OB and OCL differentiation were subsequently confirmed in human cultures of isolated mesenchymal stem cells and peripheral blood mononuclear cells, respectively.24 Inhibin treatment blocked recruitment of cells into the osteoblastic lineage (AP expression) as well as later differentiation (mineralized matrix formation). Interestingly, inhibins suppressed RANKL-induced osteoclastogenesis to the same extent as receptor activator of NF-κB (RANK)-Fc, indicating that the mechanism of suppression in heterogeneous murine bone marrow cultures was not merely occurring through inhibition of RANKL production. These data provided further evidence in human cells that changes in ovarian inhibins might directly influence bone turnover via changes in cell differentiation. Moreover, the demonstrated effects of inhibins on bone cell differentiation suggested that they may be a component in the normal endocrine repertoire that regulates bone homeostasis such that changes in serum inhibins may regulate OB and OCL differentiation and thereby bone turnover, independent of changes in sex steroids.
Serum levels predict changes in markers of bone turnover
Subsequent to the demonstration that inhibins exert direct effects on osteoblastogenesis and osteoclastogenesis, clinical correlation studies of InhAand InhB with markers of bone turnover and bone mass were performed. The first demonstration that InhB levels are correlated with bone mass investigated the relationships between the levels of gonadotrophins, estradiol, and InhB with bone mass and bone turnover in regularly menstruating women aged 35–50 years.25 Increases in bone resorption were correlated with increased FSH, confirming the initial peri-menopausal bone turnover study.9 Interestingly, increased bone resorption was also correlated with decreases in InhB levels.25 Multivariate analysis showed an independent contribution of InhB in the prediction of lumbar bone mass, whereas neither FSH nor estradiol levels in these 35–50-year-old women were significantly associated with bone mass.
These findings were confirmed and expanded in a more recent cross-sectional study of women across the menopause transition from 21–85 years of age in which both InhA and InhB were measured in addition to FSH and estradiol. These hormones were correlated with markers of bone formation and bone resorption in univariate and multivariate analyses.24 Serum InhA and InhB levels significantly correlated inversely with markers of bone formation and bone resorption in both premenopausal and peri-menopausal women. InhA, but not InhB, was also inversely correlated with markers of bone formation in postmenopausal women. FSH was not significantly correlated with bone turnover in either pre- or postmenopausalwomen. However, confirming the studies described above, FSH was significantly correlated with bone resorption (C-terminal collagen I cross-link) in a subgroup of women of peri-menopausal age (45–54 years). In multivariate analyses serum InhA better predicted bone formation and resorption markers in premenopausal women than either FSH or bioavailable estradiol.
Collectively, these clinical findings demonstrate that decreases in inhibin levels across the menopausal transition are associated with increasing bone turnover24,25 as well as with bone mass in peri-menopausal women,24,25 regardless of changes in sex steroids or FSH.
Furthermore, it is likely that decreases in in-hibin levels, in the absence of changes in estradiol, that occur throughout reproductive life are associated with the reported increases in bone turnover and observed decreases in volumetric BMD in these women.11 High-resolution peripheral quantitative CT assessment of women before and after menopause demonstrated that young adult women (and men) already have marked diminished volumetric BMD in the trabecular bone of the distal radius, distal tibia, and lumbar spine, whereas cortical bone loss begins in midlife.11 As expected, the cortical bone loss in postmenopausal women (and men over age 75) was associated with lower levels of bioavailable estradiol and testosterone and with higher levels of FSH and bone turnover markers.11 However, the early trabecular bone loss did not consistently correlate with putative causal factors except for a trend with insulin-like growth factor-related variables at the distal tibia in women. Indeed, this was identified in men in an earlier cross-sectional study by the same group.26 The authors concluded that the early onset substantial trabecular bone loss in both sexes during sex steroid sufficiency is incompletely explained and indicates that current paradigms on the pathogenesis of osteoporosis are incomplete.11 Unfortunately, inhibins were not assessed in this study. However, it is interesting to speculate that declines in cycling gonadally derived serum inhibins may lead to subtle increases in activin/BMP tone inthe local bone microenvironment, enhancing bone cell differentiation and bone turnover, resulting in bone loss in these younger subjects.
Mechanisms of inhibin action
The mechanisms of inhibin action in any tissue are only recently becoming clear. InhA and InhB both bind with high affinity to their specific receptor, β-glycan (BG), also known as type III TGF-β receptor.27 Inhibin–BG binding facilitates interaction with other members of the type II TGF-β receptor superfamily, namely type II receptors for activin (ActRII and ActRIIB), BMPs (BMPRII), and TGFβ (TβRII).28-31 The stable formation of the inhibin–BG type II receptor complex sequesters the type II receptor and prevents its availability for the productive formation of ligand type II–type I receptor complexes, thereby providing a mechanism by which inhibin can exert its known antagonistic effects of activin,29 BMP,20,28,30 and TGF-β action,31,32 such as that shown for BMP2 antagonism in Figure 1.
Figure 1.
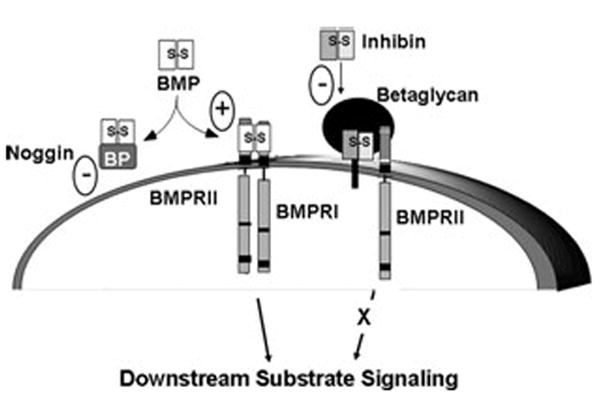
Inhibin antagonism of receptor serine kinase signaling via β-glycan (transforming growth factor [TGF]-β RIII). Bone morphogenetic protein (BMP) signals through the heteromeric type II and type I receptor serine kinases. BMP binds BMPRII, which in turn recruits and phosphorylates BMPRI to stimulate cytoplasmic downstream substrate signaling, primarily through phosphorylation of SMADs 1, 5, and 8. This action can be blocked by soluble BMP antagonists, such as noggin, and enhanced by BMP–β-glycan interaction. Inhibins can also antagonize receptor serine kinase signaling. Inhibin binds with high affinity to β-glycan, allowing for a complex formation with BMPRII. Inhibin–β-glycan–BMPRII interactions sequester BMPRII, preventing its subsequent recruitment of BMPRI and downstream substrate signaling, such as SMAD phosphorylation.
BG expression in cells of the osteoblastic lineage and in bone marrow cells has been well characterized. 33-35 BG expression on the surface of OB precursors has been shown to limit TGF-β signaling via TβRII and in this way delay TGF-β-dependent activation of OBs.33-35 Inhibin suppression of osteoblastogenesis is likely similar, as inhibin has been shown to use a similar mechanism via BG binding in other cell types.31 Support of specific binding of inhibins to BG and type II receptor serine kinase complexes in bone cells is provided in Figure 2. Murine bone marrow cells were cultured toward osteoblastogenesis and osteoclastogenesis for 20 and 10 days, respectively, prior to affinity labeling with 125I-InhA binding and autoradiography of cells (Fig. 2A) as well as by SDS-PAGE of cross-linked receptor complexes (Fig. 2B). In both osetoblastic and osteoclastic cultures, InhA bound with high affinity to receptors corresponding to BG, type II, and type I. InhA binding was competed with 50-fold molar excess of InhA but not ActA.
Figure 2.
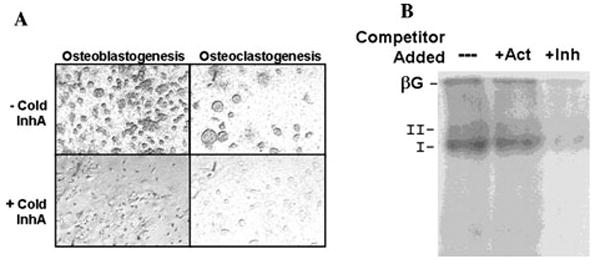
125I inhibin-A (InhA) binds specifically to murine bone marrow cells during cell differentiation. (A) Adult murine bone marrow cells were cultured for 20 days to stimulate osteoblastogenesis in α modified Eagle’s medium (αMEM), 15% fetal bovine serum(FBS), 50 mmol/L ascorbate, and 10 nmol/L β-glycerolphosphate (left) or cultured for 10 days to stimulate osteoclastogenesis in αMEM, 15% FBS, and 10 nmol/L 1.25 (OH)2 Vitamin D3 (right). Subsequently, cells were incubated for 3 h at room temperature in blocking buffer: Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (1:1), 20 mmol/L HEPES, 0.05% cytochrome c, 0.3% bovine serum albumin, 0.01 mg/mL phenylmethylsulfonyl fluoride, 0.01% bacitracin, and 0.4 mg/mL leupeptin. Slides were then incubated at room temperature overnight in the same buffer containing 40 pmol/L lactoperoxidase-labeled 125I-rh (recombinant human) InhA, alone (top) or in the presence of 40 nmol/L InhA, to define a nonspecific background (bottom). Cells were then washed with buffer to remove unbound label, and slides were dipped in emulsion and left to expose in the dark for 1 week. Following development, photographs were taken using the 20× phase objective without counterstaining. (B) Adult murine bone marrow cells were cultured for 20 days to stimulate osteoblastogenesis and then exposed to 125I-InhA prepared as in (A) for 30 min on ice in the absence or presence of 50-fold molar excess of activin A (Act) or InhA (Inh). Cells were cross-linked with disuccidimidyl suberate, lysed in radioimmunoprecipitation assay buffer, and the affinity-labeled complexes analyzed by SDS-PAGE and autoradiography. 125I-InhA formed high-affinity complexes with proteins corresponding to the sizes for β-glycan (βG; type III TGF-β receptor), type II, and type I receptors that were competed for by Inh but not Act.
More recently BG was shown to also serve as a co-receptor for BMPs, including BMP2, BMP4, and BMP7,30 which stimulate bone cell differentiation and bone formation. Binding of BG to BMP ligands occurs with similar kinetics and ligand-binding domains, as previously identified forTGF-β and inhibin.21 BMP binding to BG enhances ligand binding to the BMP type I receptors and amplifies downstream BMP signaling,30 whereas inhibin–BG complexes bind to type II receptors to block BMP as well as activin signaling.27
To provide evidence supporting the mechanism of inhibin suppression of rapid BMP signaling (Fig. 1), we tested whether the well-known stimulation of SMAD1 phosphorylation that is required for BMP-stimulated osteoblastogenesis could be inhibited by inhibin treatment. Human mesenchymal stem cells were cultured toward osteoblastogenesis24 and stimulated with BMP2 in the presence or absence of InhA. BMP2 stimulated rapid phosphorylation of SMAD1, which was completely blocked by treatment with InhA (Fig. 3), although total SMAD1 levels were unchanged (data not shown).
Figure 3.
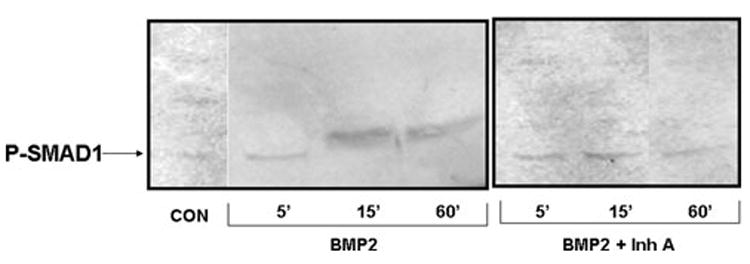
InhA blocks BMP2-induced SMAD1 phosphorylation during osteoblastogenesis of human mesenchymal cells. Adult human mesenchymal stem cells (Lonza, Walkersville, MD) were used to initiate osteoblast cultures according to themanufacturer’s protocol. Cultures were treated with BMP2 or BMP2 + InhA to determine the ability of InhA to antagonize BMP signaling. Cells were harvested at 5, 15, and 60 min following treatment and protein isolated and analyzed by immunoblot. The presence of SMAD1 phosphorylation was determined using a phospho-specific antibody (R&D Systems, Inc., Minneapolis, MN) as a measure of the initiation of BMP signaling. BMP2 treatment of osteoblast cultures led to increased SMAD1 phosphorylation by 15 min that could still be observed with 60 min of treatment, which was blocked in the presence of 50 ng/mL InhA as early as 15 min of treatment. Neither BMP2 nor InhA had any effect on total SMAD1 protein content (data not shown).
Collectively, the competition for ligand binding to BG between inhibins, BMPs, and TGF-β, as well as the competition between inhibin and activin/BMP for binding to the type II activin/BMP receptors provides potential mechanisms by which inhibins exert their dominant suppressive effects over the stimulatory actions of BMPs or activin in both mouse20 and human24 cells.
Inhibin effects in vivo
Although clear evidence inmultiple species demonstrates the in vitro suppressive effects of inhibins on both OB and OCL, the effects of inhibins on the regulation of bone mass are not as well documented. Transgenic mouse models have been used to inducibly express recombinant human InhA in intact and gonadectomized mice.36 Continuous systemic exposure to InhA for 4 weeks was strongly anabolic in intact adult mice at multiple skeletal sites, including the tibia, spine, and humerus in both sexes.36 Moreover, long-term (4 weeks) InhA overexpression prevented orchidectomy-induced bone loss at the tibia and spine and prevented loss of vertebral bone strength from gonadectomy; these anabolic effects were the result of increased OB formation and activity rather than decreases in OCL parameters.36
However, shorter times of continuous InhA exposure in vivo (1 week) had the same suppressive effects on ex vivo osteoblastogenesis potential as those observed after direct InhA stimulation exposure in vitro.36 These data suggest that the length of inhibin exposure and/or the mode of exposure (continuous versus intermittent) determines whether the effect of InhA is either to suppress cell differentiation or stimulate OB activity and bone formation. This is a situation reminiscent of the opposing effects of continuous/catabolic and intermittent/anabolic exposure to parathyroid hormone.
Decreases in gonadal inhibin contribute to increased bone turnover in women
Our in vitro, short-term (1 week) in vivo, and clinical findings provide a cellular basis by which inhibins can exert their suppressive effects on bone turnover through their suppression of bone marrow cell differentiation and enable us to suggest a model, summarized in Figure 4, by which changes in the inhibin/activin to BMP ratio can regulate bone turnover. Both activin and its antagonist follistatin as well as BMPs and noggin have been previously localized in bone marrow and shown to exert opposing effects on both osteoblast and osteoclast differentiation. 20,37 Superimposed on the balance of these locally produced ligands and antagonists is the suppressive effect of endocrine-derived inhibins on both osteoblastogenesis and osteoclastogenesis. These data are consistent with circulating inhibins in cycling women (Fig. 4, left panel) serving to suppress the overall rate of bone turnover and thus modulate the basal rate driven by the balance of local BMPs and noggin. In cycling pre-menopausal women, monthly episodes of increased bone resorption have been reported.38 Cyclic fluctuations in circulating inhibins as well as gonadal steroids38 may serve to suppress basal bone turnover driven by the balance of local activin, BMPs, and noggin and thus maintain normal bone turnover.
Figure 4.
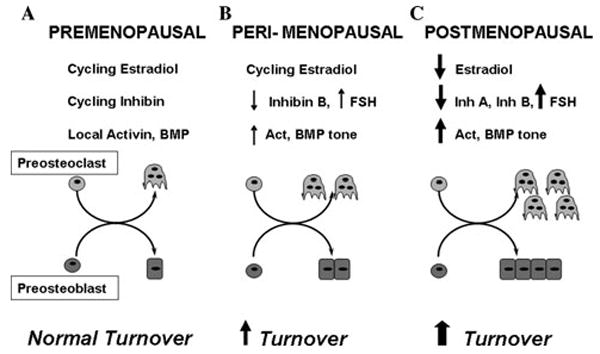
Premenopausal women (A) demonstrate normal cycling hormones and autocrine-paracine levels of activin/BMP, resulting in normal adult bone cell differentiation and bone turnover. (B) Peri-menopausal decreases in inhibin B (InhB) increase circulating FSH and local activin/BMP tone, increasing cell differentiation and turnover. (C) Postmenopausal decreases in InhA and estradiol amplify increases in cell differentiation and dramatically increase bone turnover. FSH, follicle-stimulating hormone.
At menopause (Fig. 4, middle panel), even prior to changes in serum estrogen levels, the selective decrease in InhB levels16,17 may cause a local increase in marrow activin and BMP tone. Increases in activin, BMP tone, and modest increases in FSH would promote osteoblast recruitment and osteoclast differentiation, resulting in a net increase in the rate of bone turnover and subsequent loss of bone.
In postmenopausal women (Fig. 4, right panel), concurrent with the well-established loss of estrogen and elevated FSH, total inhibin levels are dramatically diminished,9 resulting in a further increase in activin/BMP tone and increases in marrow cell differentiation. The increased activin and BMP tone may enhance bone remodeling that is already significantly upregulated as a result of the loss of estrogen.
Finally, although the bone loss and eventual osteoporosis following loss of gonadal function has been previously attributed solely to estrogen deficiency, these data suggest that the contributions of endocrine-derived inhibins may also play an important role in regulation of bone turnover throughout the reproductive life span.
Footnotes
Conflict of interest The authors declare no conflicts of interest.
References
- 1.Albright F, Bloomberg E, Smith PH. Postmenopausal osteoporosis. Trans Assoc Am Phys. 1940;55:298–305. [Google Scholar]
- 2.Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–137. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- 3.Martin TJ, Sims NA. Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med. 2005;11:76–81. doi: 10.1016/j.molmed.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Manolagas SC, Kousteni S, Jilka RL. Sex steroids and bone. Recent Prog Horm Res. 2002;57:385–409. doi: 10.1210/rp.57.1.385. [DOI] [PubMed] [Google Scholar]
- 5.Riggs BL. Endocrine causes of age-related bone loss and osteoporosis. Novartis Found Symp. 2002;242:247–259. discussion 260–4. [PubMed] [Google Scholar]
- 6.Khosla S, et al. Effects of age and estrogen status on serum parathyroid hormone levels and biochemical markers of bone turnover in women: a population-based study. J Clin Endocrinol Metab. 1997;82:1522–1527. doi: 10.1210/jcem.82.5.3946. [DOI] [PubMed] [Google Scholar]
- 7.Khosla S, et al. Relationship of serum sex steroid levels and bone turnover markers with bone mineral density in men and women: a key role for bioavailable estrogen. J Clin Endocrinol Metab. 1998;83:2266–2274. doi: 10.1210/jcem.83.7.4924. [DOI] [PubMed] [Google Scholar]
- 8.Riggs BL, Khosla S, Melton LJ., 3rd Primary osteoporosis in men: role of sex steroid deficiency. Mayo Clin Proc. 2000;75(Suppl):S46–S50. [PubMed] [Google Scholar]
- 9.Ebeling PR, et al. Bone turnover markers and bone density across the menopausal transition. J Clin Endocrinol Metab. 1996;81:3366–3371. doi: 10.1210/jcem.81.9.8784098. [DOI] [PubMed] [Google Scholar]
- 10.Riggs BL, et al. Population-based study of age and sex differences in bone volumetric density, size, geometry, and structure at different skeletal sites. J Bone Miner Res. 2004;19:1945–1954. doi: 10.1359/JBMR.040916. [DOI] [PubMed] [Google Scholar]
- 11.Riggs BL, et al. A population-based assessment of rates of bone loss at multiple skeletal sites: evidence for substantial trabecular bone loss in young adult women and men. J Bone Miner Res. 2008;23:205–214. doi: 10.1359/JBMR.071020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L, et al. FSH directly regulates bone mass. Cell. 2006;125:247–260. doi: 10.1016/j.cell.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 13.Bilezikjian LM, et al. Autocrine/paracrine regulation of pituitary function by activin, inhibin and follistatin. Mol Cell Endocrinol. 2004;225:29–36. doi: 10.1016/j.mce.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 14.Cook RW, et al. Molecular biology of inhibin action. Semin Reprod Med. 2004;22:269–276. doi: 10.1055/s-2004-831902. [DOI] [PubMed] [Google Scholar]
- 15.Groome NP, et al. Measurement of dimeric inhibin B throughout the human menstrual cycle. J Clin Endocrinol Metab. 1996;81:1401–1405. doi: 10.1210/jcem.81.4.8636341. [DOI] [PubMed] [Google Scholar]
- 16.Klein NA, et al. Decreased inhibin B secretion is associated with themonotropic FSH rise in older, ovulatory women: a study of serum and follicular fluid levels of dimeric inhibin A and B in spontaneous menstrual cycles. J Clin Endocrinol Metab. 1996;81:2742–2745. doi: 10.1210/jcem.81.7.8675606. [DOI] [PubMed] [Google Scholar]
- 17.Welt CK, et al. Female reproductive aging is marked by decreased secretion of dimeric inhibin. J Clin Endocrinol Metab. 1999;84:105–111. doi: 10.1210/jcem.84.1.5381. [DOI] [PubMed] [Google Scholar]
- 18.Burger HG, et al. Hormonal changes in the menopause transition. Recent Prog Horm Res. 2002;57:257–275. doi: 10.1210/rp.57.1.257. [DOI] [PubMed] [Google Scholar]
- 19.Guthrie JR, et al. A prospective study of bone loss in menopausal Australian-born women. Osteoporos Int. 1998;8:282–290. doi: 10.1007/s001980050066. [DOI] [PubMed] [Google Scholar]
- 20.Gaddy-Kurten D, et al. Inhibin suppresses and activin stimulates osteoblastogenesis and osteoclastogenesis in murine bone marrow cultures. Endocrinology. 2002;143:74–83. doi: 10.1210/endo.143.1.8580. [DOI] [PubMed] [Google Scholar]
- 21.Wiater E, Vale W. Inhibin is an antagonist of bone morphogenetic protein signaling. J Biol Chem. 2003;278:7934–7941. doi: 10.1074/jbc.M209710200. [DOI] [PubMed] [Google Scholar]
- 22.Woodruff TK, et al. Pharmacokinetic profile of recombinant human (rh) inhibin A and activin A in the immature rat. II. Tissue distribution of 125Irh-inhibin A and 125Irh-activin A in immature female and male rats. Endocrinology. 1993;132:725–734. doi: 10.1210/endo.132.2.8425491. [DOI] [PubMed] [Google Scholar]
- 23.Rosen V. BMP and BMP inhibitors in bone. Ann N Y Acad Sci. 2006;1068:19–25. doi: 10.1196/annals.1346.005. [DOI] [PubMed] [Google Scholar]
- 24.Perrien DS, et al. Bone turnover across the menopause transition: correlations with inhibins and follicle-stimulating hormone. J Clin Endocrinol Metab. 2006;91:1848–1854. doi: 10.1210/jc.2005-2423. [DOI] [PubMed] [Google Scholar]
- 25.Vural F, et al. Ovarian aging and bone metabolism in menstruating women aged 35–50 years. Maturitas. 2005;52:147–153. doi: 10.1016/j.maturitas.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Khosla S, et al. Hormonal and biochemical determinants of trabecular microstructure at the ultradistal radius in women and men. J Clin Endocrinol Metab. 2006;91:885–891. doi: 10.1210/jc.2005-2065. [DOI] [PubMed] [Google Scholar]
- 27.Lewis KA, et al. Betaglycan binds inhibin and canmediate functional antagonism of activin signalling. Nature. 2000;404:411–414. doi: 10.1038/35006129. [DOI] [PubMed] [Google Scholar]
- 28.Farnworth PG, et al. Inhibins differentially antagonize activin and bone morphogenetic protein action in a mouse adrenocortical cell line. Endocrinology. 2006;147:3462–3471. doi: 10.1210/en.2006-0023. [DOI] [PubMed] [Google Scholar]
- 29.Vale W, et al. Activins and inhibins and their signaling. Ann NY Acad Sci. 2004;1038:142–147. doi: 10.1196/annals.1315.023. [DOI] [PubMed] [Google Scholar]
- 30.Kirkbride KC, et al. Bonemorphogenetic proteins signal through the transforming growth factor-beta type III receptor. J Biol Chem. 2008;283:7628.37. doi: 10.1074/jbc.M704883200. [DOI] [PubMed] [Google Scholar]
- 31.Esparza-Lopez J, et al. Ligand binding and functional properties of betaglycan, a co-receptor of the transforming growth factor-beta superfamily. Specialized binding regions for transforming growth factor-beta and inhibin. A J Biol Chem. 2001;276:14588–14596. doi: 10.1074/jbc.M008866200. [DOI] [PubMed] [Google Scholar]
- 32.Cheifetz S, Andres JL, Massague J. The transforming growth factor-beta receptor type III is a membrane proteoglycan. Domain structure of the receptor. J Biol Chem. 1988;263:16984–16991. [PubMed] [Google Scholar]
- 33.McCarthy TL, et al. Prostaglandin E2 increases transforming growth factor-beta type III receptor expression through CCAAT enhancer-binding protein delta in osteoblasts. Mol Endocrinol. 2007;21:2713–2724. doi: 10.1210/me.2007-0210. [DOI] [PubMed] [Google Scholar]
- 34.Centrella M, et al. Transforming growth factorbeta gene family members and bone. Endocr Rev. 1994;15:27–39. doi: 10.1210/edrv-15-1-27. [DOI] [PubMed] [Google Scholar]
- 35.Centrella M, McCarthy TL, Canalis E. Transforming growth factor-beta and remodeling of bone. J Bone Joint Surg Am. 1991;73:1418–1428. [PubMed] [Google Scholar]
- 36.Perrien DS, et al. Inhibin A is an endocrine stimulator of bone mass and strength. Endocrinology. 2007;148:1654–1665. doi: 10.1210/en.2006-0848. [DOI] [PubMed] [Google Scholar]
- 37.Abe M, et al. Potent induction of activin A secretion from monocytes and bone marrow stromal fibroblasts by cognate interaction with activated T cells. J Leukoc Biol. 2002;72:347–352. [PubMed] [Google Scholar]
- 38.Chiu KM, et al. Changes in bone resorption during the menstrual cycle. J Bone Miner Res. 1999;14:609–615. doi: 10.1359/jbmr.1999.14.4.609. [DOI] [PubMed] [Google Scholar]