

Abstract
Mutations in genes encoding the intermediate filament nuclear lamins and associated proteins cause a wide spectrum of diseases sometimes called “laminopathies.” Diseases caused by mutations in LMNA encoding A-type lamins include autosomal dominant Emery-Dreifuss muscular dystrophy and related myopathies, Dunnigan-type familial partial lipodystrophy, Charcot-Marie-Tooth disease type 2B1 and developmental and accelerated aging disorders. Duplication in LMNB1 encoding lamin B1 causes autosomal dominant leukodystrophy and mutations in LMNB2 encoding lamin B2 are associated with acquired partial lipodystrophy. Disorders caused by mutations in genes encoding lamin-associated integral inner nuclear membrane proteins include X-linked Emery-Dreifuss muscular dystrophy, sclerosing bone dysplasias, HEM/Greenberg skeletal dysplasia and Pelger-Huet anomaly. While mutations and clinical phenotypes of “laminopathies” have been carefully described, data explaining pathogenic mechanisms are only emerging. Future investigations will likely identify new “laminopathies” and a combination of basic and clinical research will lead to a better understanding of pathophysiology and the development of therapies.
Keywords: lamin, nuclear envelope, intermediate filaments, muscular dystrophy, lipodystrophy, progeria
Introduction
Nuclear lamins are intermediate filament proteins primarily located at the inner aspect of the inner nuclear membrane [1–4]. Lamins interact with a variety of proteins in the nucleus, including several integral proteins of the inner nuclear membrane [5,6]. Lamins found in several different species were originally grouped into two types based on their isoelectric points determined by two-dimensional gel electrophoresis: A-type lamins, with near-neutral isoelectric points and B-type lamins, with acidic isoelectric points [7,8]. It is now known that in humans, one gene, LMNA, encodes A-type lamins, and two genes, LMNB1 and LMNB2, encode B-type lamins.
LMNA is localized to chromosome 1q21.2 [9]. It encodes two major somatic cell variants that arise by alternative RNA splicing, prelamin A and lamin C [10]. Prelamin A and lamin C are identical for their first 566 amino acids; lamin C has 6 unique carboxyl-terminal amino acids and prelamin A 98 unique carboxyl-terminal amino acids. Prelamin A, which contains a CAAX motif at its carboxyl-terminus, is farnesylated and carboxymethylated and undergoes endoproteolyic processing catalyzed by ZMPSTE24 to yield lamin A, which lacks the last 18 amino acids, including the farnesylated and carboxymethylated cysteine, of prelamin A [11,12]. Lamin A and lamin C are expressed in most somatic cells but absent from or expressed in significantly reduced quantities in early embryos, some hematopoietic cells, certain neurons, undifferentiated epithelial and mesenchymal cells and several types of cancer [13–17]. LMNA also encodes a germ cell-specific protein, lamin C2, which arises from alternative splicing of RNA encoded by an alternative first exon [18].
LMNB1 is localized to chromosome 5q23.3-q31.1 and encodes lamin B1 [9,19]. LMNB2, localized to chromosome 19p13.3, encodes lamin B2 and a germ cell isoform lamin B3 [20–22]. The B-type lamins have CAAX motifs at their carboxyl-termini and are fanesylated and carboxymethylated [11,12]. At least one of the B-type lamins appears to be expressed in all human cells [13–17,20]. This may be because of their requirement for spindle assembly in mitosis [23].
Approximately 80 proteins appear to be localized to the inner nuclear membrane in interphase mammalian cells [24]. Several of these proteins interact with lamins, likely playing a role in attaching the lamina to the inner nuclear membrane [5,6]. Integral membrane proteins reach the inner nuclear membrane after synthesis on the rough endoplasmic reticulum by lateral diffusion in the interconnected membranes of the endoplasmic reticulum, outer nuclear membrane, pore membranes and inner nuclear membrane [25–27]. Energy appears to be necessary for active restructuring of the nuclear pore complexes [28]. In some instances as shown in yeast, karyopherins may mediate transport of integral proteins to the inner nuclear membrane [29]. During mitosis, when the nuclear lamina depolymerizes, the integral proteins of the inner nuclear membrane are disbursed in the endoplasmic reticulum and then recruited to the decondensing chromatin when the daughter nuclei reform [27,30].
Up until the 1990’s, studies of the nuclear lamins and nuclear envelope were primarily in the domain of basic cell biologists. However, in 1994, Bione et al. [31] identified the gene responsible for X-linked Emery-Dreifuss muscular dystrophy and called the encoded integral membrane protein emerin. Soon afterwards, emerin was localized to the nuclear envelope [32,33]. Then, in 1999, Bonne et al. [34] reported that mutations in LMNA encoding A-type lamins cause autosomal dominant Emery-Dreifuss muscular dystrophy, a disease with the same phenotype as the X-linked inherited form. Since then, several different human diseases have been linked to mutations in LMNA and others to mutations in the genes encoding B-type lamins, lamin-associated proteins and other proteins of the nuclear envelope (Figures 1). As a result, the nuclear lamina is now positioned on the interface between cell biology and medicine. This review will summarize the wide range of diseases now sometimes referred to as “laminopathies.”
Figure 1.
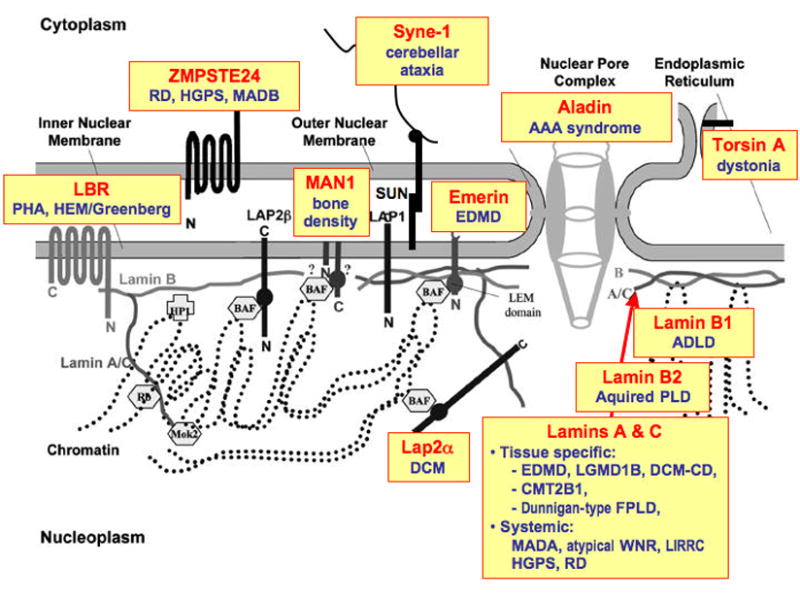
Schematic diagram of the nuclear envelope showing localizations and interactions of various proteins. Boxes highlight diseases caused by mutations in genes encoding the proteins indicated. RD: restrictive dermopathy, HGPS: Hutchinson-Gilford progeria syndrome, PHA: Pelger-Huet anaomaly, EDMD: Emery-Dreifuss muscular dystrophy, ADLD: Adult onset autosomal dominant leukodystrophy, BSS: Barraquer-Simons syndrome, LGMD: limb-girdle muscular dystrophy; DCM: dilated cardiomyopathy; CMT: Charcot-Marie-Tooth, FPLD: familial partial lipodystrophy, MADA / MADB: mandibuloacral dysplasia, WNR: Werner syndrome. For more specific disease nomenclature, see Table 1, Table 2 and Table 3.
Diseases caused by mutations in LMNA encoding A-type lamins
Over the past 10 years, there has been a rapid pace in the discovery of diseases caused by mutations in genes encoding nuclear lamins and associated nuclear envelope proteins (Figure 2). Mutations in LMNA encoding A-type lamins cause a wide spectrum of human diseases and more than 10 different clinical syndromes have been attributed to LMNA mutations (Table 1). However, careful analysis reveals that there is considerable similarity between some of these clinically distinct diseases. Broadly, mutations in LMNA can be considered to cause four different groups of disorders with overlap between them: 1) diseases of striated muscle, 2) lipodystrophy syndromes, 3) a peripheral neuropathy and 4) accelerated aging disorders.
Figure 2.
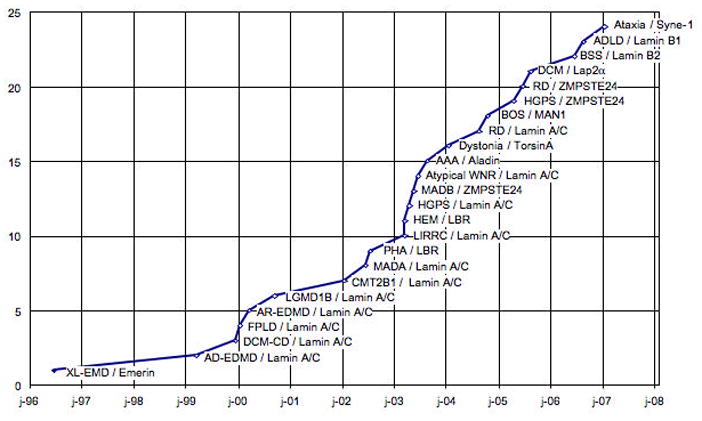
Graph showing number of diseases caused by mutations in genes encoding nuclear lamins or associated proteins reported (y-axis) versus year (x-axis). Emerin, which is mutated in X-linked EDMD, was localized to the nuclear envelope in 1996. LMNA mutations were reported to cause autosomal dominant EDMD in 1999. In 2000, LMNA mutations were reported to cause autosomal recessive EDMD, cardiomyopathy dilated 1A, limb girdle muscular dystrophy type 1B and Dunnigan-type familial partial lipodystrophy. In 2002, LMNA mutations were reported to cause mandibulacral dysplasia and Charcot-Marie-Tooth disorder type 2B1 and mutations in LBR were reported to cause Pelger-Huet anomaly. In 2003, LMNA mutations were reported to cause Hutchinson-Gilford progeria syndrome, atypical Werner syndrome and lipoatrophy with diabetes, hepatic steatosis, hypertrophic cardiomyopathy and leukomelanodermic papules; LBR mutations were reported in HEM/Greenberg dysplasia; ZMPSTE24 reported in mandibulacral dysplasia and mutation in the gene encoding Aladin reported in triple A syndrome. In 2004, mutations in torsinA that cause DYT1 dystonia were reported to lead to abnormal protein localization in the perinuclear space, LMNA mutations were reported to cause restrictive dermopathy and LEMD3 mutations to cause Buschke-Ollendorff Syndrome and related sclerosing bone dysplasias. In 2005, ZMPSTE24 mutations were reported to cause a progeria syndrome and restrictive dermopathy; Lap2α polymorphisms were reported in two siblings with dilated cardiomyopathy. In 2006, duplication of LMNB1 was reported to cause adult- onset, autosomal dominant leukodystrophy and mutations in LMNB2 Barraquer-Simons syndrome. In 2007, mutations in SYNE1 were reported to cause a form of cerebellar ataxia.
Table 1.
Diseases caused by mutations in LMNA. On-line Mendelian Inheritance in Man (OMIM; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM) entry numbers are given in parentheses.
| Diseases of Striated Muscle |
| Autosomal Dominant Emery-Dreifuss Muscular Dystrophy (#181350) |
| Autosomal Recessive Emery-Dreifuss Muscular Dystrophy (#604929) |
| Autosomal Dominant Cardiomyopathy Dilated 1A (#115200) |
| Autosomal Dominant Limb Girdle Muscular Dystrophy Type 1B (#159001) |
| Peripheral Neuropathy |
| Autosomal Recessive Charcot-Marie-Tooth Disorder Type 2B1 (#605588) |
| Lipodystrophy Syndromes |
| Autosomal Dominant Dunnigan-type Familial Partial Lipodystrophy (#151660) |
| Autosomal Dominant Lipoatrophy with Diabetes, Hepatic Steatosis, Hypertrophic Cardiomyopathy and Leukomelanodermic Papules (#608056) |
| Autosomal Recessive Mandibuloacral Dysplasia (#248370)* |
| Accelerated Aging Disorders |
| Autosomal Dominant Atypical Werner Syndrome (#277700 for Werner syndrome) |
| Autosmal Dominant Hutchinson-Gilford Progeria Syndrome (#176670) |
| Autosomal Dominant Restrictive Dermopathy Lethal (#275210) |
Mandibuloacral dysplasia also has features of accelerated aging.
Mutations in LMNA were first shown to cause autosomal dominant Emery-Dreifuss muscular dystrophy [34]. LMNA mutations also cause rare cases of Emery-Dreifuss muscular dystrophy demonstrating autosomal recessive inheritance [35]. Emery-Dreifuss muscular dystrophy has distinctive clinical features of early contractures, humero-peroneal weakness and dilated cardiomyopathy with conduction defects [36]. Subsequently, mutations in LMNA were shown to cause dilated cardiomyopathy 1A [37] and limb-girdle muscular dystrophy 1B [38], both with autosomal dominant inheritance. Both of these conditions have dilated cardiomyopathy with conduction abnormalities similar to those in Emery-Dreifuss muscular dystrophy but with either minimal to no skeletal muscle involvement or skeletal muscle affected in a different pattern. Subsequent molecular genetic and phenotypic analyses of additional subjects showed that the same LMNA mutation can cause what is clinically diagnosed as Emery-Dreifuss muscular dystrophy, limb-girdle muscular dystrophy 1B and dilated cardiomyopathy 1A, even within the same family [39,40]. Hence, one group of mutations in LMNA cause disorders of striated muscle with predominant cardiomyopathy, which based on clinical features and inheritance pattern are classified as four different diseases.
Approximately one year after the first report that mutations in LMNA cause Emery-Dreifuss muscular dystrophy, three groups reported that mutations in LMNA also cause Dunnigan-type familial partial lipodystrophy [41–43]. Individuals with this autosomal dominant disorder have a normal fat distribution in early childhood but subcutaneous adipose tissue gradually disappears from the upper and lower extremities at the onset of puberty with adipose accumulation on the face and neck. Virtually all affected individuals have insulin resistance and many develop diabetes mellitus. A heterozygous LMNA mutation has also been reported in a subject with a lipodystrophy syndrome characterized by generalized lipoatrophy, diabetes mellitus, hepatic steatosis, hypertrophic cardiomyopathy and cutaneous leukomelanodermic papules [44]. More recently, a heterozygous missense mutation in LMNA has been reported in a subject with a type A insulin resistance syndrome characterized by glucose tolerance, insulin resistance and acanthosis nigricans in the absence of clinical lipodystrophy [45]. While the vast majority of subjects with muscular dystrophies and cardiomyopathy caused by LMNA mutations do not have lipodystrophy, a few case reports suggest that there may be overlap between striated muscle disease and lipodystrophy in rare subjects [46,47].
Mandibuloacral dysplasia is an autosomal recessive disorder, characterized by postnatal growth retardation, craniofacial anomalies, skeletal malformations and mottled cutaneous pigmentation frequently associated with partial lipodystrophy and insulin resistance. Novelli et al. [48] first described a homozygous missense LMNA mutation (R527H) that was shared by all affected subjects with mandibuloacral dysplasia in five consanguineous Italian families. The same mutation was subsequently described in affected individuals in other kindreds [49]. In one reported subject with mandibuloacral dysplasia associated with progeroid appearance and more generalized lipodystrophy, compound heterozygous mutations in ZMPSTE24 encoding the protease that processes prelamin A to lamin A have been described [50].
A homozygous LMNA missense mutation has been reported to cause autosomal recessive Charcot-Marie-Tooth type 2B1, a peripheral neuropathy [51]. Combined peripheral neuropathy and muscular dystrophy have also been reported in subjects some dominantly inherited LMNA mutations, including a mutation leading to haploinsufficiency of A-type lamins [52–54]. However, peripheral neuropathy has not been reported in most subjects with Emery-Dreifuss muscular dystrophy [31,34–36].
Arguably the most dramatic disease phenotype caused by mutation in LMNA is Hutchinson-Gilford progeria syndrome [55,56]. This is a rare, sporadic, multi-system disorder characterized by features of premature aging, with most affected subjects dying in the second decade from cardiovascular disease [57]. The LMNA mutations that cause Hutchinson-Gilford progeria syndrome, by far the most common being G608G, create an abnormal splice donor site in exon 11 leading to an mRNA that encodes a protein with 50 amino acids deleted from its carboxyl-terminal domain [55,56]. This leads to abnormal processing of prelamin A because the ZMPSTE24 endoprotease responsible for cleavage “upstream” of the farnesylated cysteine of prelamin A is lacking from the truncated protein. This truncated prelamin A retains at its carboxyl-terminus the farnesylated and carboxymethylated cysteine residue [11,12]. Lamin C, which is encoded by exons 1 through 10 of LMNA [10], is unaffected. Restrictive dermopathy, a neonatal, lethal syndrome with severe intrauterine growth retardation, congenital contractures and tense skin, is also caused by heterozygous splicing mutations in LMNA resulting in complete or partial loss of exon 11 as well as by mutations in the gene encoding ZMPSTE24 protease [58]. LMNA missense mutations have also been reported in subjects with variable feature of accelerated aging and a diagnosis of atypical Werner syndrome [59]. However, atypical Werner syndrome may not be a specific diagnosis but rather subjects with LMNA mutations that cause overlapping phenotypes or a multi-system phenotype with progeroid features, such as mandibuloacral dysplasia [60].
How do different mutations in LMNA cause several different diseases?
One of the key questions facing scientists who study diseases caused by LMNA mutations is: how do different mutations in this single gene encoding proteins expressed in most differentiated somatic cells cause different, often system-specific, disease phenotypes (Figure 3)? Despite intensive investigation over the past several years, there are no definitive answers. However, relatively recent research has provided some clues.
Figure 3.
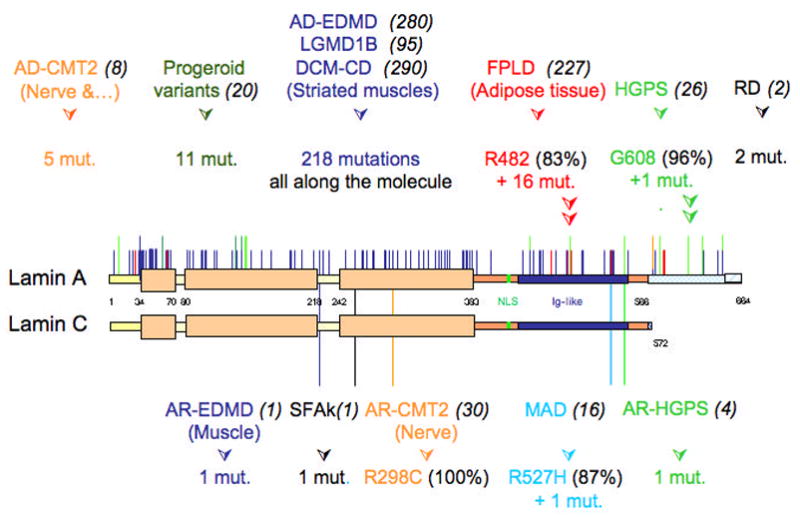
Spectrum of disease-causing LMNA mutations. Schematic representations of lamin A and lamin C are shown. Portions of the proteins affected by LMNA mutations identified causing different diseases are indicated. Dominant disorders due to heterozygous LMNA mutations are depicted on the top and recessive disorders due to homozygous mutations are presented below (for more details, see UMD-LMNA mutation database at http://www.umd.be). Numbers in parentheses in black next to each disease acronym indicate the numbers of reported individuals carrying that LMNA mutation and presenting the corresponding phenotype. Figure is updated from Broers J, Ramaekers F, Bonne G, Hutchison C. The nuclear lamins: laminopathies and their role in premature ageing. Physiological Reviews 2006;86:967–1008 (used with permission).
Figure 4 summarizes some of the proposed pathological mechanisms as to how mutations in LMNA and the processing of prelamin A may cause disease. In Hutchinson-Gilford progeria syndrome and some cases of restrictive dermopathy, LMNA mutations lead to expression of truncated forms of prelamin A that remain farnesylated in cells because of deletion of the ZMPSTE24 protease site. Similarly, unprocessed, farnesylated prelamin A is expressed in related conditions resulting from mutations in the gene encoding ZMPSTE24. Zmpste24 null mice develop multiple abnormalities and progeroid features [61,62]. When Zmpste24 null mice are crossed to Lmna +/− mice, reducing the prelamin A content by half the offspring are apparently normal, suggesting that farnesylated prelamin A is toxic and that reducing its level provides protection from disease [63]. “Knockdown” of ZMPSTE24 using RNA interference in cultured cells leads to dramatic changes in nuclear morphology and premature cell death and this is prevented by “knockdown” of prelamin A 24 hours earlier [64]. In fibroblasts from human subjects with Hutchinson-Gilford progeria syndrome, introduction of wild type lamin A does not rescue cellular morphological abnormalities; however, correction of the aberrant splicing event using a modified oligonucleotide has been reported to reverse abnormalities in nuclear morphology and induce proper expression of misregulated genes [65]. Additional data suggest that the farnesyl group of unprocessed lamin A and truncated prelamin A is responsible for “toxicity.” Treatment of cells from mice with Zmpste24 deficiency, mice with a targeted Hutchinson-Gilford progeria syndrome mutation, human subjects with Hutchinson-Gilford progeria syndrome and cultured cells overexpressing the truncated prelamin A in this disorder with farnesyltransferase inhibitors reverses the nuclear morphological abnormalities normally present [66–70]. Furthermore, treatment with farnesyltransferase inhibitors improves the whole animal abnormal phenotypes of mice with Zmpste24 deficiency and mice with a targeted Hutchinson-Gilford progeria syndrome mutation [71,72]. These results suggests that farnesylated prelamin A that accumulates with ZMPSTE24 mutations in some cases of restrictive dermopathy and farnesylated truncated prelamin A resulting from LMNA mutations in Hutchinson-Gilford progeria syndrome are involved in the pathogenesis of these disorders.
Figure 4.
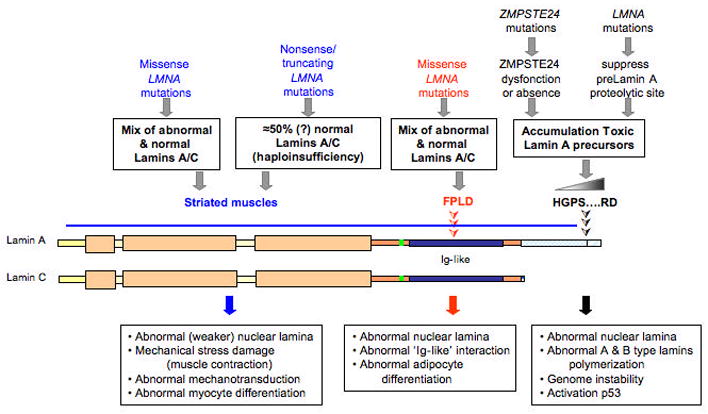
Summary of potential pathogenic mechanisms resulting from mutations in LMNA and ZMPSTE24. See text for details.
It is still not known how the abnormal farnesylated polypeptides resulting from LMNA and ZMPSTE24 mutations contribute to an accelerated aging phenotype. Some data suggest that these proteins perturb DNA damage response and repair, resulting in genomic instability [73]. However, blocking protein farnesylation does not appear to reverse these DNA damage responses [74]. Other studies suggest that farnesylated prelamin A activates p53, which may somehow be linked to accelerated aging [75]. The truncated prelamin A in Hutchinson-Gilford progeria syndrome also alters the mechanism of A-type and B-type lamin polymerization into the lamina and cells expressing this polypeptide have abnormal nuclear mechanics and an abnormal response to heat stress [76–78].
Virtually all LMNA mutations causing Dunnigan-type familial partial lipodystrophy lead to amino acid substitutions on the surface of an immunoglobulin-like fold in the tails of lamin A and lamin C, altering the surface charge but not overall fold structure [79,80] (see Figure 3). As the lipodystrophy-causing amino acid substitutions change the charge of a very precise domain of the proteins, they may lead to alteration of an adipocyte-specific function of A-type lamins. One study suggests that these amino acid changes may alter the binding of A-type lamins to the adipogenic differentiation factor, sterol responsive element binding protein 1 [81].
The majority of LMNA mutations in subjects with striated muscle diseases are missense and lead to amino acid substitutions almost anywhere in lamins A and C and not in a specific domain of the proteins [34,39,82,83; see also UMD-LMNA mutation database at http://www.UMD.be:2000]. The mutations that cause amino acid substitutions in the immunoglobulin-like fold of lamins A and C lead to overall disruption of fold structure in contrast to the less dramatic alterations occurring with mutations causing Dunnigan-type familial partial lipodystrophy [79,80]. Nonsense or other truncating mutations leading to haploinsufficiency of A-type lamins, which comprise approximately 15% of all LMNA mutations, have only been reported in subjects with striated muscle disease (see UMD-LMNA mutation database at http://www.UMD.be:2000). Thus, striated muscle appears to be sensitive to both the expression of A-type lamins with certain amino acid changes as well as to reduced expression of A-type lamins. Similarly, both Lmna “knockout” mice and “knock-in” mice with Lmna mutations corresponding to those that cause striated muscle abnormalities in humans develop skeletal muscle and cardiac disease [84–87]. Lmna “knockout” mice to do not have lipodystrophy [88].
LMNA missense mutations that cause striated muscle diseases appear to encode polypeptides that disrupt the structure of the nuclear lamina and nuclear envelope in a “dominant” manner. Expression of several such mutant lamin polypeptides in transfected cells produces visible alterations in the lamina and envelope [89,90]. Fibroblasts from human subjects with LMNA mutations that cause muscle disorders have similar abnormalities in nuclear structure [91]. Cardiac-selective transgenic overexpression of human lamin A with an amino acid substitution encoded by a LMNA mutation causing Emery-Dreifuss muscular dystrophy leads to profound nuclear shape abnormalities and severe heart damage while overexpression of wild type human lamin A has minimal effects [92]. However, various nuclear structural abnormalities have been reported in cells expressing A-type lamins causing other “laminopathies” and are not specific for striated muscle diseases [55,56,58,59,65,66–70,74,76–78,91,93–96].
The observed morphological abnormalities in cells expressing A-type lamin variants causing striated muscle diseases, along with similar abnormalities in cells lacking A-type lamins [84], has led to the hypothesis that either expression of lamin polypeptides with certain amino acid changes or partial loss of A-type lamins make the lamina “weaker.” As maintenance of the structural integrity of the cell nucleus was one of the first proposed functions of the nuclear lamina, a “weakened” lamina may lead to overall loss of a cell’s ability to withstand stress-induced damage, which may be of critical significance in contracting skeletal and cardiac muscle. Further support for this as a possible pathogenic mechanism comes from the observations that fibroblasts from Lmna null mice have defects in nuclear mechanics and mechanotransduction [97,98].
Some studies suggest that A-type lamins with amino acid substitutions causing striated muscle disease and lipodystrophy inhibit differentiation of precursor cells into myocytes and adipocytes. Expression of a mutant lamin A that causes Emery-Dreifuss muscular dystrophy inhibits in vitro differentiation of C2C12 myoblasts [99,100]. Overexpression of both wild type lamin A and a mutant lamin A that causes Dunnigan-type familial partial lipodystrophy inhibits differentiation of 3T3-L1 preadipocytes into adipocytes, suggesting that lamin A may negatively regulate adipocyte differentiation and that amino acid changes on the surface of the immunoglobulin-like fold of A-type lamins somehow lead to a “gain” of this function [101]. Another study has shown that accumulation of unprocessed prelamin A blocks adipocyte differentiation [102]. It remains to be determined how these in vitro observations relate to the maintenance of normally differentiated tissues in whole animals.
Diseases caused by genes encoding B-type lamins
Abnormalities in genes encoding B-type lamins have been linked to two very different disorders (Table 2). Adult-onset autosomal dominant leukodystrophy is a slowly progressive disorder characterized by symmetrical widespread myelin loss in the central nervous system with a phenotype is similar chronic progressive multiple sclerosis [103]. In 2000, Coffeen et al. [104] reported that the responsible genetic locus was localized to chromosome 5q31. In 2006, the same group reported that duplication of LMNB1 was the cause of adult-onset autosomal dominant leukodystrophy and that overexpression of lamin B1 was detected in brains of affected individuals [105]. A very different disease, Barraquer-Simons syndrome, is a mostly sporadic acquired form of progressive lipodystrophy [106]. In 2006, based on the fact the LMNA mutations cause a type of lipodystrophy, Hegele et al. [107] sequenced LMNB2 and found several nucleotide variants with a higher frequency in individuals with acquired Barraquer-Simons syndrome than in the control population.
Table 2.
Diseases caused by mutations in LMNB1 and LMNB2 encoding B-type lamins. On-line Mendelian Inheritance in Man entry numbers are given in parentheses.
| LMNB1 |
| Adult-onset, Autosomal dominant Leukodystrophy (#169500) |
| LMNB2 |
| Barraquer-Simons Syndrome, Heterozygous Mutations (#608709) |
There are little data as to how mutations in genes encoding B-type lamins cause disease. Homozygous mice with Lmnb1 deleted survive embryonic development but die at birth with defects in lung and bone [108]. Embryonic fibroblasts from these mice have severe abnormalities in nuclear structure but normal mechanical properties [108,109]. Overexpression of lamin B1 in Drosophila eye leads to a degenerative phenotype and overexpression in cultured cells induced morphological abnormalities similar to those in cell with abnormal A-type lamins [105].
Diseases caused by genes encoding lamin-associated integral proteins of the inner nuclear membrane
Abnormalities in genes encoding lamin-associated integral proteins of the inner nuclear membrane have been linked todisorders primarily affecting nuclear structure, striated muscle or bone (Table 3). As noted above, X-linked Emery-Dreifuss muscular dystrophy is due to mutation in the EMD gene encoding emerin, an integral protein of the inner nuclear membrane [31–33]. The clinical features of X-linked and autosomal dominant Emery-Dreifuss muscular dystrophy are very similar, with the only reported difference being no significant increase in cardiac sudden death in individuals with the X-linked disorder (82,110). In contrast, implantation of a cardiac defibrillator has been recommended for individuals with cardiomyopathy and LMNA mutations [111].
Table 3.
Diseases caused by mutations in genes encoding lamin-associated proteins of the inner nuclear. On-line Mendelian Inheritance in Man entry numbers are given in parentheses.
| EMD |
| X-linked Emery-Dreifuss Muscular Dystrophy (#310300) |
| LBR |
| Pelger-Huet Anomaly, Autosomal Recessive (#169400) |
| Hydrops-Ectopic Calcification-Moth-Eaten Skeletal Dysplasia (HEM)/Greenberg Skeletal Dysplasia, Autosomal Dominant (#215140) |
| LEMD3 (MAN1) |
| Buschke-Ollendorff Syndrome, Autosomal Recessive (#166700) |
| Melorheostosis, Familial and Melorheostosis with Osteopoikilosis, Autosomal Recessive (#155950) |
As with LMNA mutations, it is not understood how EMD mutations cause striated muscle disease. However, emerin interacts with A-type lamins [84,112–115], providing a rationale as to why certain mutations in LMNA encoding A-type lamins and mutations in EMD encoding emerin cause disorders with very similar phenotypes. The vast majority of EMD mutations causing Emery-Dreifuss muscular dystrophy lead to the absence of emerin from cells [32,33; see also UMD-EMD mutation database at http://www.umd.be:2010]. Similarly, many LMNA mutations causing striated muscle disease or loss of A-type lamins from cells leads to a redistribution of emerin from the nuclear envelope to the bulk endoplasmic reticulum [84,89,90]. Emerin that has “escaped” from the nuclear envelope in cells lacking A-type lamins may be degraded by proteasomes [116]. Hence, both EMD mutations and LMNA mutations that cause Emery-Dreifuss muscular dystrophy may lead to some loss of an emerin at the inner nuclear membrane. Emerin may also regulate the activities of several other proteins involved in muscle cell differentiation and function [117]. In rare human subjects with mutations in both LMNA and EMD, the presentation severity of Emery-Dreifuss muscular dystrophy is increased, provide genetic evidence for the interaction between emerin and A-type lamins [118].
Deletion of Emd from mice does not lead to significant pathology [119,120]. However, fragility of myocyte nuclei and impaired expression of mechanosensitive genes in embryonic fibroblasts in response to strain have been reported in these emerin-deficient mice [120,121]. Furthermore, regenerating muscle from Emd null mice have cell cycle abnormalities and delayed myogenic differentiation, which are associated with perturbations to transcriptional pathways regulated by the retinoblastoma protein and MyoD [119]. Analysis of transcriptomes from muscles of human subjects with X-linked and autosomal dominant Emery-Dreifuss muscular dystrophy similarly shows disruption of retinoblastoma protein and MyoD pathways in muscle regeneration [122].
LBR is an integral inner nuclear membrane with a basic, amino-terminal domain that faces the nucleoplasm and a hydrophobic domain with multiple transmembrane segments [123,124]. LBR binds to B-type lamins, DNA and chromatin proteins [124–126]. Its hydrophobic domain is highly homologous to sterol reductases [127]. Heterozygous mutations in LBR cause Pelger-Huet anomaly [128]. Homozygous mutations lead to HEM/Greenberg skeletal dysplasia, which is associated with 3 beta-hydroxysterol delta 14-reductase deficiency [129]. Whereas Pelger-Huet anomaly is a benign disorder characterized by abnormal nuclear shape and chromatin organization in blood granulocytes, HEM/Greenberg skeletal dysplasia is generally lethal in utero and characterized by chondrodystrophy, fetal hydrops, short limbs and abnormal chondro-osseous calcification. Loss of function of one LBR allele therefore leads to subtle abnormalities of a highly structured nucleus whereas mutation in both alleles leads to a severe phenotype, which may result both from loss of the protein’s role in maintaining nuclear structure as well as its sterol reductase activity. Mutations in Lbr are responsible for the ichthyosis mouse, which have misshapen granulocyte nuclei similar to humans with Pelger-Huet anomaly and develop other abnormalities including alopecia, variable expression of syndactyly and hydrocephalus [130].
MAN1, encoded by the gene now called LEMD3, is an integral inner nuclear membrane protein with an amino-terminal nucleoplasmic domain that contained a LEM motif, two transmembrane segments and a nucleoplasmic carboxyl-terminal domain [131]. The amino-terminal domain of MAN1 interacts directly with A-type lamins and the protein barrier-to-autointegration factor [132]. The carboxyl-terminal nucleoplasmic domain binds to regulatory-Smads and inhibits signaling by transforming growth factor-beta and bone morphogenic protein [133–137]. This explains why heterozygous loss-of-function mutations cause the sclerosing bone dysplasias osteopoikilosis, Buschke-Ollendorff syndrome, and familial melorheostosis, which are sometimes associated with hyperproliferative skin and tendon lesions [135]. LEMD3 germline mutations are not found in most subjects with sporadic, non-familial melorheostosis; however, somatic mutations have not been excluded [138,139]. The portion of MAN1 involved in Smad-binding also exhibits a DNA-binding winged helix domain [140]. Genetically modified mice that do not express MAN1 have abnormal remodeling of the embryonic vasculature, a process mediated by transforming growth factor-beta [141]. Mice with one copy of the allele encoding MAN1 are apparently normal [141].
Concluding comments and future directions
The nuclear lamina and nuclear envelope have become focal points of investigation for scientists and clinicians interested in a wide spectrum of human diseases. In the future, it is likely that more “laminopathies” will be identified as mutations are discovered in other genes encoding lamin-associated proteins and perhaps other mutations in genes encoding lamins are shown to cause additional disorders. Taylor et al. [142] recently reported a polymorphism in the TMPO gene encoding LAP2-alpha in two family members with cardiomyopathy. The polymorphism affected an amino acid located in the domain of the LAP2-alpha known to interact with A-type lamins, compromising the interaction. Whether the reported polymorphism is a mutation causing dilated cardiomyopathy still requires further confirmation with the identification of additional cases. More recently, Gros-Louis [143] reported that mutations in SYNE1 cause a rare form of autosomal recessive cerebellar ataxia. SYNE1 is one of two mammalian genes encoding multiple isoforms of proteins that have been given several different names but are most commonly referred to as nesprins. Depending on their size, nesprins are localized to either the inner nuclear membrane, where they may interact with lamins, or to the outer nuclear membrane, where they interact with transmembrane SUN proteins in the perinuclear space, which in turn bind to lamins inside the nucleus [144].
Studies of other inherited diseases involving proteins localized to the nuclear envelope may provide insights into the pathophysiology of “laminopathies.” Mutations in a gene encoding a nuclear pore complex protein called ALADIN cause triple A syndrome, an autosomal recessive disorder characterized by adrenal insufficiency, achalasia and alacrima [145]. Notably, little attention has been given to examining nucleocytoplasmic transport in “laminopathies” but selective defects in this process could potentially be involved in pathogenesis. Mutations in the gene DYT1 encoding torsinA cause the movement disorder DYT1 dystonia [146]. TorsinA is an ATPase normally localized to the bulk endoplasmic reticulum but the mutations cause the protein to be concentration in the perinuclear space, between the inner and outer nuclear membrane [147–149]. In the perinuclear space, torsinA interacts with the luminal portion of the transmembrane protein lamina-associated polypeptide 1, which binds to lamins on the other side of the inner nuclear membrane [150].
Future clinical and genetic studies of “laminopathies” need to consider newly described functions of lamins, such as their role connecting the nucleus to the cytoskeleton via SUNs and nesprins [144]. At the same time, scientists studying the properties of nuclear lamins and associated proteins in vitro, in cell culture and in animal models must consider what is learned from genetics and clinical investigations involving human subjects. Only a combination of basic and clinical research will provide novel insights into pathophysiology and the development of novel treatments.
Acknowledgments
HJW is supported by grants from the United States National Institutes of Health (AR048997 and AG025240) and the Muscular Dystrophy Association (MDA4287). GB is supported by grants from Association Française contre les Myopathies (#11057 and #10722) and from EU-FP6 Euro-laminopathies (# 018690).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McKeon FD, Kirschner MW, Caput D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature. 1986;319:463–468. doi: 10.1038/319463a0. [DOI] [PubMed] [Google Scholar]
- 2.Aebi U, Cohn J, Buhle L, Gerace L. The nuclear lamina is a meshwork of intermediate-type filaments. Nature. 1986;323:560–564. doi: 10.1038/323560a0. [DOI] [PubMed] [Google Scholar]
- 3.Goldman AE, Maul G, Steinert PM, Yang HY, Goldman RD. Keratin-like proteins that coisolate with intermediate filaments of BHK-21 cells are nuclear lamins. Proc Natl Acad Sci U S A. 1986;83:3839–3843. doi: 10.1073/pnas.83.11.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher DZ, Chaudhary N, Blobel G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc Natl Acad Sci U S A. 1986;83:6450–6454. doi: 10.1073/pnas.83.17.6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Worman HJ, Courvalin JC. The inner nuclear membrane. J Membr Biol. 2000;177:1–11. doi: 10.1007/s002320001096. [DOI] [PubMed] [Google Scholar]
- 6.Zastrow MS, Vlcek S, Wilson KL. Proteins that bind A-type lamins: integrating isolated clues. J Cell Sci. 2004;117:979–987. doi: 10.1242/jcs.01102. [DOI] [PubMed] [Google Scholar]
- 7.Gerace L, Blobel G. The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell. 1980;19:277–287. doi: 10.1016/0092-8674(80)90409-2. [DOI] [PubMed] [Google Scholar]
- 8.Krohne G, Benavente R. The nuclear lamins. A multigene family of proteins in evolution and differentiation. Exp Cell Res. 1986;162:1–10. doi: 10.1016/0014-4827(86)90421-0. [DOI] [PubMed] [Google Scholar]
- 9.Wydner KL, McNeil JA, Lin F, Worman HJ, Lawrence JB. Chromosomal assignment of human nuclear envelope protein genes LMNA, LMNB1, and LBR by fluorescence in situ hybridization. Genomics. 1996;15:474–478. doi: 10.1006/geno.1996.0146. [DOI] [PubMed] [Google Scholar]
- 10.Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993;268:16321–16326. [PubMed] [Google Scholar]
- 11.Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria--new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res. 2005;46:2531–2558. doi: 10.1194/jlr.R500011-JLR200. [DOI] [PubMed] [Google Scholar]
- 12.Rusinol AE, Sinensky MS. Farnesylated lamins, progeroid syndromes and farnesyl transferase inhibitors. J Cell Sci. 2006;119:3265–3272. doi: 10.1242/jcs.03156. [DOI] [PubMed] [Google Scholar]
- 13.Stewart C, Burke B. Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell. 1987;51:383–392. doi: 10.1016/0092-8674(87)90634-9. [DOI] [PubMed] [Google Scholar]
- 14.Guilly MN, Bensussan A, Bourge JF, Bornens M, Courvalin JC. A human T lymphoblastic cell line lacks lamins A and C. EMBO J. 1987;6:3795–3799. doi: 10.1002/j.1460-2075.1987.tb02715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Worman HJ, Lazaridis I, Georgatos SD. Nuclear lamina heterogeneity in mammalian cells. Differential expression of the major lamins and variations in lamin B phosphorylation. J Biol Chem. 1988;263:12135–12141. [PubMed] [Google Scholar]
- 16.Rober RA, Weber K, Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development. 1989;105:365–378. doi: 10.1242/dev.105.2.365. [DOI] [PubMed] [Google Scholar]
- 17.Cance WG, Chaudhary N, Worman HJ, Blobel G, Cordon-Cardo C. Expression of the nuclear lamins in normal and neoplastic human tissues. J Exp Clin Cancer Res. 1992;11:233–246. [Google Scholar]
- 18.Furukawa K, Inagaki H, Hotta Y. Identification and cloning of an mRNA coding for a germ cell-specific A-type lamin in mice. Exp Cell Res. 1994;212:426–430. doi: 10.1006/excr.1994.1164. [DOI] [PubMed] [Google Scholar]
- 19.Lin F, Worman HJ. Structural organization of the human gene (LMNB1) encoding nuclear lamin B1. Genomics. 1995;27:230–236. doi: 10.1006/geno.1995.1036. [DOI] [PubMed] [Google Scholar]
- 20.Höger TH, Zatloukal K, Waizenegger I, Krohne G. Characterization of a second highly conserved B-type lamin present in cells previously thought to contain only a single B-type lamin. Chromosoma. 1990;99:379–390. doi: 10.1007/BF01726689. [DOI] [PubMed] [Google Scholar]
- 21.Biamonti G, Giacca M, Perini G, Contreas G, Zentilin L, Weighardt F, Guerra M, Della Valle G, Saccone S, Riva S, Falaschi A. The gene for a novel human lamin maps at a highly transcribed locus of chromosome 19 which replicates at the onset of S-phase. Mol Cell Biol. 1992;12:3499–3506. doi: 10.1128/mcb.12.8.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furukawa K, Hotta Y. cDNA cloning of a germ cell specific lamin B3 from mouse spermatocytes and analysis of its function by ectopic expression in somatic cells. EMBO J. 1993;12:97–106. doi: 10.1002/j.1460-2075.1993.tb05635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai MY, Wang S, Heidinger JM, Shumaker DK, Adam SA, Goldman RD, Zheng Y. A mitotic lamin B matrix induced by RanGTP required for spindle assembly. Science. 2006;311:1887–1893. doi: 10.1126/science.1122771. [DOI] [PubMed] [Google Scholar]
- 24.Schirmer EC, Florens L, Guan T, Yates JR, 3rd, Gerace L. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science. 2003;301:1380–1382. doi: 10.1126/science.1088176. [DOI] [PubMed] [Google Scholar]
- 25.Soullam B, Worman HJ. The amino-terminal domain of the lamin B receptor is a nuclear envelope targeting signal. J Cell Biol. 1993;120:1093–1100. doi: 10.1083/jcb.120.5.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soullam B, Worman HJ. Signals and structural features involved in integral membrane protein targeting to the inner nuclear membrane. J Cell Biol. 1995;130:15–27. doi: 10.1083/jcb.130.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellenberg J, Siggia ED, Moreira JE, Smith CL, Presley JF, Worman HJ, Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohba T, Schirmer EC, Nishimoto T, Gerace L. Energy- and temperature-dependent transport of integral proteins to the inner nuclear membrane via the nuclear pore. J Cell Biol. 2004;167:1051–1062. doi: 10.1083/jcb.200409149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King MC, Lusk CP, Blobel G. Karyopherin-mediated import of integral inner nuclear membrane proteins. Nature. 2006;442:1003–1007. doi: 10.1038/nature05075. [DOI] [PubMed] [Google Scholar]
- 30.Yang L, Guan T, Gerace L. Integral membrane proteins of the nuclear envelope are dispersed throughout the endoplasmic reticulum during mitosis. J Cell Biol. 1997;137:1199–1210. doi: 10.1083/jcb.137.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–327. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 32.Nagano A, Koga R, Ogawa M, Kurano Y, Kawada J, Okada R, Hayashi YK, Tsukahara T, Arahata K. Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nat Genet. 1996;12:254–259. doi: 10.1038/ng0396-254. [DOI] [PubMed] [Google Scholar]
- 33.Manilal S, Nguyen TM, Sewry CA, Morris GE. The Emery-Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum Mol Genet. 1996;5:801–808. doi: 10.1093/hmg/5.6.801. [DOI] [PubMed] [Google Scholar]
- 34.Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–288. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 35.Raffaele Di Barletta M, Ricci E, Galluzzi G, Tonali P, Mora M, Morandi L, Romorini A, Voit T, Orstavik KH, Merlini L, Trevisan C, Biancalana V, Housmanowa-Petrusewicz I, Bione S, Ricotti R, Schwartz K, Bonne G, Toniolo D. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2000;66:1407–1412. doi: 10.1086/302869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emery AE. Emery-Dreifuss muscular dystrophy - a 40 year retrospective. Neuromuscul Disord. 2000;10:228–232. doi: 10.1016/s0960-8966(00)00105-x. [DOI] [PubMed] [Google Scholar]
- 37.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 38.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum Mol Genet. 2000;9:1453–1459. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 39.Bonne G, Mercuri E, Muchir A, Urtizberea A, Becane HM, Recan D, Merlini L, Wehnert M, Boor R, Reuner U, Vorgerd M, Wicklein EM, Eymard B, Duboc D, Penisson-Besnier I, Cuisset JM, Ferrer X, Desguerre I, Lacombe D, Bushby K, Pollitt C, Toniolo D, Fardeau M, Schwartz K, Muntoni F. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. 2000;48:170–180. [PubMed] [Google Scholar]
- 40.Brodsky GL, Muntoni F, Miocic S, Sinagra G, Sewry C, Mestroni L. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation. 2000;101:473–476. doi: 10.1161/01.cir.101.5.473. [DOI] [PubMed] [Google Scholar]
- 41.Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9:109–112. doi: 10.1093/hmg/9.1.109. [DOI] [PubMed] [Google Scholar]
- 42.Shackleton S, Lloyd DJ, Jackson SN, Evans R, Niermeijer MF, Singh BM, Schmidt H, Brabant G, Kumar S, Durrington PN, Gregory S, O'Rahilly S, Trembath RC. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24:153–166. doi: 10.1038/72807. [DOI] [PubMed] [Google Scholar]
- 43.Speckman RA, Garg A, Du F, Bennett L, Veile R, Arioglu E, Taylor SI, Lovett M, Bowcock AM. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am J Hum Genet. 2000;66:1192–1198. doi: 10.1086/302836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caux F, Dubosclard E, Lascols O, Buendia B, Chazouilleres O, Cohen A, Courvalin JC, Laroche L, Capeau J, Vigouroux C, Christin-Maitre S. A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. J Clin Endocr Metab. 2003;88:1006–1013. doi: 10.1210/jc.2002-021506. [DOI] [PubMed] [Google Scholar]
- 45.Young J, Morbois-Trabut L, Couzinet B, Lascols O, Dion E, Bereziat V, Feve B, Richard I, Capeau J, Chanson P, Vigouroux C. Type A insulin resistance syndrome revealing a novel lamin A mutation. Diabetes. 2005;54:1873–1878. doi: 10.2337/diabetes.54.6.1873. [DOI] [PubMed] [Google Scholar]
- 46.Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene. Am J Med. 2002;112:549–555. doi: 10.1016/s0002-9343(02)01070-7. [DOI] [PubMed] [Google Scholar]
- 47.van der Kooi AJ, Bonne G, Eymard B, Duboc D, Talim B, Van der Valk M, Reiss P, Richard P, Demay L, Merlini L, Schwartz K, Busch HF, de Visser M. Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy. Neurology. 2002;59:620–623. doi: 10.1212/wnl.59.4.620. [DOI] [PubMed] [Google Scholar]
- 48.Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D'Apice MR, Massart C, Capon F, Sbraccia P, Federici M, Lauro R, Tudisco C, Pallotta R, Scarano G, Dallapiccola B, Merlini L, Bonne G. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71:426–431. doi: 10.1086/341908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simha V, Agarwal AK, Oral EA, Fryns JP, Garg A. Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J Clin Endocr Metab. 2003;88:2821–2824. doi: 10.1210/jc.2002-021575. [DOI] [PubMed] [Google Scholar]
- 50.Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Molec Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- 51.De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, Grid D, Levy N. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70:726–736. doi: 10.1086/339274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goizet C, Yaou RB, Demay L, Richard P, Bouillot S, Rouanet M, Hermosilla E, Le Masson G, Lagueny A, Bonne G, Ferrer X. A new mutation of the lamin A/C gene leading to autosomal dominant axonal neuropathy, muscular dystrophy, cardiac disease, and leuconychia. J Med Genet. 2004;41:e29. doi: 10.1136/jmg.2003.013383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benedetti S, Bertini E, Iannaccone S, Angelini C, Trisciani M, Toniolo D, Sferrazza B, Carrera P, Comi G, Ferrari M, Quattrini A, Previtali SC. Dominant LMNA mutations can cause combined muscular dystrophy and peripheral neuropathy. J Neurol Neurosurg Psychiatry. 2005;76:1019–1021. doi: 10.1136/jnnp.2004.046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walter MC, Witt TN, Weigel BS, Reilich P, Richard P, Pongratz D, Bonne G, Wehnert MS, Lochmuller H. Deletion of the LMNA initiator codon leading to a neurogenic variant of autosomal dominant Emery-Dreifuss muscular dystrophy. Neuromuscul Disord. 2005;15:40–44. doi: 10.1016/j.nmd.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 55.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N. Lamin A truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 57.DeBusk FL. The Hutchinson-Gilford progeria syndrome. J Pediatr. 1972;80:697–724. doi: 10.1016/s0022-3476(72)80229-4. [DOI] [PubMed] [Google Scholar]
- 58.Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I, Boyer A, Genevieve D, Hadj-Rabia S, Gaudy-Marqueste C, Smitt HS, Vabres P, Faivre L, Verloes A, Van Essen T, Flori E, Hennekam R, Beemer FA, Laurent N, Le Merrer M, Cau P, Levy N. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Molec Genet. 2004;13:2493–2503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- 59.Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg E, Hanson NB, Martin GM, Mian IS, Kennedy BK, Oshima J. LMNA mutations in atypical Werner's syndrome. Lancet. 2003;362:440–445. doi: 10.1016/S0140-6736(03)14069-X. [DOI] [PubMed] [Google Scholar]
- 60.Bonne G, Levy N. LMNA mutations in atypical Werner's syndrome. Lancet. 2003;362:1585–1586. doi: 10.1016/S0140-6736(03)14761-7. [DOI] [PubMed] [Google Scholar]
- 61.Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodriguez F, Tryggvason K, Lopez-Otin C. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002;31:94–99. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- 62.Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RA, Michaelis S, Griffey SM, Young SG. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A. 2002;99:13049–13054. doi: 10.1073/pnas.192460799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fong LG, Ng JK, Meta M, Cote N, Yang SH, Stewart CL, Sullivan T, Burghardt A, Majumdar S, Reue K, Bergo MO, Young SG. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc Natl Acad Sci U S A. 2004;101:18111–18116. doi: 10.1073/pnas.0408558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gruber J, Lampe T, Osborn M, Weber K. RNAi of FACE1 protease results in growth inhibition of human cells expressing lamin A: implications for Hutchinson-Gilford progeria syndrome. J Cell Sci. 2005;118:689–696. doi: 10.1242/jcs.01652. [DOI] [PubMed] [Google Scholar]
- 65.Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci U S A. 2005;102:10291–10296. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, Miner JH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A. 2005;102:12873–12878. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:12879–12884. doi: 10.1073/pnas.0506001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mallampalli MP, Huyer G, Bendale P, Gelb MH, Michaelis S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:14416–14421. doi: 10.1073/pnas.0503712102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Glynn MW, Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet. 2005;14:2959–2969. doi: 10.1093/hmg/ddi326. [DOI] [PubMed] [Google Scholar]
- 71.Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, Young SG. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–1623. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- 72.Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest. 2006;116:2115–2121. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadinanos J, Lopez-Otin C, Tse HF, Hutchison C, Chen J, Cao Y, Cheah KS, Tryggvason K, Zhou Z. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11:780–785. doi: 10.1038/nm1266. [DOI] [PubMed] [Google Scholar]
- 74.Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J Cell Sci. 2006;119:4644–4649. doi: 10.1242/jcs.03263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Varela I, Cadinanos J, Pendas AM, Gutierrez-Fernandez A, Folgueras AR, Sanchez LM, Zhou Z, Rodriguez FJ, Stewart CL, Vega JA, Tryggvason K, Freije JM, Lopez-Otin C. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–568. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]
- 76.Delbarre E, Tramier M, Coppey-Moisan M, Gaillard C, Courvalin JC, Buendia B. The truncated prelamin A in Hutchinson-Gilford progeria syndrome alters segregation of A-type and B-type lamin homopolymers. Hum Mol Genet. 2006;15:1113–1122. doi: 10.1093/hmg/ddl026. [DOI] [PubMed] [Google Scholar]
- 77.Paradisi M, McClintock D, Boguslavsky RL, Pedicelli C, Worman HJ, Djabali K. Dermal fibroblasts in Hutchinson-Gilford progeria syndrome with the lamin A G608G mutation have dysmorphic nuclei and are hypersensitive to heat stress. BMC Cell Biol. 2005;6:27. doi: 10.1186/1471-2121-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006;103:10271–10276. doi: 10.1073/pnas.0601058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dhe-Paganon S, Werner ED, Chi YI, Shoelson SE. Structure of the globular tail of nuclear lamin. J Biol Chem. 2002;277:17381–17384. doi: 10.1074/jbc.C200038200. [DOI] [PubMed] [Google Scholar]
- 80.Krimm I, Östlund C, Gilquin B, Couprie J, Hossenlopp P, Mornon JP, Bonne G, Courvalin JC, Worman HJ, Zinn-Justin S. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies. Structure. 2002;10:811–823. doi: 10.1016/s0969-2126(02)00777-3. [DOI] [PubMed] [Google Scholar]
- 81.Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum Mol Genet. 2002;11:769–777. doi: 10.1093/hmg/11.7.769. [DOI] [PubMed] [Google Scholar]
- 82.Bonne G, Yaou RB, Beroud C, Boriani G, Brown S, de Visser M, Duboc D, Ellis J, Hausmanowa-Petrusewicz I, Lattanzi G, Merlini L, Morris G, Muntoni F, Opolski G, Pinto YM, Sangiuolo F, Toniolo D, Trembath R, van Berlo JH, van der Kooi AJ, Wehnert M. 108th ENMC International Workshop, 3rd Workshop of the MYO-CLUSTER project: EUROMEN, 7th International Emery-Dreifuss Muscular Dystrophy (EDMD) Workshop, 13–15 September 2002, Naarden, The Netherlands. Neuromuscul Disord. 2003;13:508–515. doi: 10.1016/s0960-8966(03)00063-4. [DOI] [PubMed] [Google Scholar]
- 83.Vytopil M, Benedetti S, Ricci E, Galluzzi G, Dello Russo A, Merlini L, Boriani G, Gallina M, Morandi L, Politano L, Moggio M, Chiveri L, Hausmanova-Petrusewicz I, Ricotti R, Vohanka S, Toman J, Toniolo D. Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J Med Genet. 2003;40:e132. doi: 10.1136/jmg.40.12.e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–920. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL, Martin D, Feneley MP, Fatkin D. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113:357–369. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacene E, Fromes Y, Toussaint M, Mura AM, Keller DI, Amthor H, Isnard R, Malissen M, Schwartz K, Bonne G. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet. 2005;14:155–169. doi: 10.1093/hmg/ddi017. [DOI] [PubMed] [Google Scholar]
- 87.Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet. 2005;14:2167–2180. doi: 10.1093/hmg/ddi221. [DOI] [PubMed] [Google Scholar]
- 88.Cutler DA, Sullivan T, Marcus-Samuels B, Stewart CL, Reitman ML. Characterization of adiposity and metabolism in Lmna-deficient mice. Biochem Biophys Res Commun. 2002;291:522–527. doi: 10.1006/bbrc.2002.6466. [DOI] [PubMed] [Google Scholar]
- 89.Östlund C, Bonne G, Schwartz K, Worman HJ. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J Cell Sci. 2001;114:4435–4445. doi: 10.1242/jcs.114.24.4435. [DOI] [PubMed] [Google Scholar]
- 90.Raharjo WH, Enarson P, Sullivan T, Stewart CL, Burke B. Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. J Cell Sci. 2001;114:4447–4457. doi: 10.1242/jcs.114.24.4447. [DOI] [PubMed] [Google Scholar]
- 91.Muchir A, Medioni J, Laluc M, Massart C, Arimura T, van der Kooi AJ, Desguerre I, Mayer M, Ferrer X, Briault S, Hirano M, Worman HJ, Mallet A, Wehnert M, Schwartz K, Bonne G. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve. 2004;30:444–450. doi: 10.1002/mus.20122. [DOI] [PubMed] [Google Scholar]
- 92.Wang Y, Herron AJ, Worman HJ. Pathology and nuclear abnormalities in hearts of transgenic mice expressing M371K lamin A encoded by an LMNA mutation causing Emery-Dreifuss muscular dystrophy. Hum Mol Genet. 2006;15:2479–2489. doi: 10.1093/hmg/ddl170. [DOI] [PubMed] [Google Scholar]
- 93.Vigouroux C, Auclair M, Dubosclard E, Pouchelet M, Capeau J, Courvalin JC, Buendia B. Nuclear envelope disorganization in fibroblasts from lipodystrophic patients with heterozygous R482Q/W mutations in the lamin A/C gene. J Cell Sci. 2001;114:4459–4468. doi: 10.1242/jcs.114.24.4459. [DOI] [PubMed] [Google Scholar]
- 94.Favreau C, Dubosclard E, Östlund C, Vigouroux C, Capeau J, Wehnert M, Higuet D, Worman HJ, Courvalin JC, Buendia B. Expression of lamin A mutated in the carboxyl-terminal tail generates an aberrant nuclear phenotype similar to that observed in cells from patients with Dunnigan-type partial lipodystrophy and Emery-Dreifuss muscular dystrophy. Exp Cell Res. 2003;282:14–23. doi: 10.1006/excr.2002.5669. [DOI] [PubMed] [Google Scholar]
- 95.Bechert K, Lagos-Quintana M, Harborth J, Weber K, Osborn M. Effects of expressing lamin A mutant protein causing Emery-Dreifuss muscular dystrophy and familial partial lipodystrophy in HeLa cells. Exp Cell Res. 2003;286:75–86. doi: 10.1016/s0014-4827(03)00104-6. [DOI] [PubMed] [Google Scholar]
- 96.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–378. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Broers JL, Peeters EA, Kuijpers HJ, Endert J, Bouten CV, Oomens CW, Baaijens FP, Ramaekers FC. Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum Mol Genet. 2004;13:2567–2580. doi: 10.1093/hmg/ddh295. [DOI] [PubMed] [Google Scholar]
- 99.Favreau C, Higuet D, Courvalin JC, Buendia B. Expression of a mutant lamin A that causes Emery-Dreifuss muscular dystrophy inhibits in vitro differentiation of C2C12 myoblasts. Mol Cell Biol. 2004;24:1481–1492. doi: 10.1128/MCB.24.4.1481-1492.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Markiewicz E, Ledran M, Hutchison CJ. Remodelling of the nuclear lamina and nucleoskeleton is required for skeletal muscle differentiation in vitro. J Cell Sci. 2005;118:409–420. doi: 10.1242/jcs.01630. [DOI] [PubMed] [Google Scholar]
- 101.Boguslavsky RL, Stewart CL, Worman HJ. Nuclear lamin A inhibits adipocyte differentiation: implications for Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2006;15:653–663. doi: 10.1093/hmg/ddi480. [DOI] [PubMed] [Google Scholar]
- 102.Capanni C, Mattioli E, Columbaro M, Lucarelli E, Parnaik VK, Novelli G, Wehnert M, Cenni V, Maraldi NM, Squarzoni S, Lattanzi G. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum Mol Genet. 2005;14:1489–1502. doi: 10.1093/hmg/ddi158. [DOI] [PubMed] [Google Scholar]
- 103.Schwankhaus JD, Katz DA, Eldridge R, Schlesinger S, McFarland H. Clinical and pathological features of an autosomal dominant, adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. Arch Neurol. 1994;51:757–766. doi: 10.1001/archneur.1994.00540200033013. [DOI] [PubMed] [Google Scholar]
- 104.Coffeen CM, McKenna CE, Koeppen AH, Plaster NM, Maragakis N, Mihalopoulos J, Schwankhaus JD, Flanigan KM, Gregg RG, Ptacek LJ, Fu YH. Genetic localization of an autosomal dominant leukodystrophy mimicking chronic progressive multiple sclerosis to chromosome 5q31. Hum Molec Genet. 2000;9:787–793. doi: 10.1093/hmg/9.5.787. [DOI] [PubMed] [Google Scholar]
- 105.Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38:1114–1123. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- 106.Barraquer-Ferre L. Lipodystrophie progressive: syndrome de Barraquer-Simons. Presse Med. 1935;86:1672–1674. [Google Scholar]
- 107.Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, Durrington PN. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet. 2006;79:383–389. doi: 10.1086/505885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K. Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci U S A. 2004;101:10428–10433. doi: 10.1073/pnas.0401424101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT. Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem. 2006;281:25768–25780. doi: 10.1074/jbc.M513511200. [DOI] [PubMed] [Google Scholar]
- 110.Boriani G, Gallina M, Merlini L, Bonne G, Toniolo D, Amati S, Biffi M, Martignani C, Frabetti L, Bonvicini M, Rapezzi C, Branzi A. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: a long-term longitudinal study. Stroke. 2003;34:901–908. doi: 10.1161/01.STR.0000064322.47667.49. [DOI] [PubMed] [Google Scholar]
- 111.Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM, Duboc D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006;354:209–210. doi: 10.1056/NEJMc052632. [DOI] [PubMed] [Google Scholar]
- 112.Clements L, Manilal S, Love DR, Morris GE. Direct interaction between emerin and lamin A. Biochem Biophys Res Commun. 2000;267:709–714. doi: 10.1006/bbrc.1999.2023. [DOI] [PubMed] [Google Scholar]
- 113.Sakaki M, Koike H, Takahashi N, Sasagawa N, Tomioka S, Arahata K, Ishiura S. Interaction between emerin and nuclear lamins. J Biochem. 2001;129:321–327. doi: 10.1093/oxfordjournals.jbchem.a002860. [DOI] [PubMed] [Google Scholar]
- 114.Fairley EA, Riddell A, Ellis JA, Kendrick-Jones J. The cell cycle dependent mislocalisation of emerin may contribute to the Emery-Dreifuss muscular dystrophy phenotype. J Cell Sci. 2002;115:341–354. doi: 10.1242/jcs.115.2.341. [DOI] [PubMed] [Google Scholar]
- 115.Muchir A, van Engelen BG, Lammens M, Mislow JM, McNally E, Schwartz K, Bonne G. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp Cell Res. 2003;291:352–362. doi: 10.1016/j.yexcr.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 116.Muchir A, Massart C, van Engelen BG, Lammens M, Bonne G, Worman HJ. Proteasome-mediated degradation of integral inner nuclear membrane protein emerin in fibroblasts lacking A-type lamins. Biochem Biophys Res Commun. 2006;351:1011–1017. doi: 10.1016/j.bbrc.2006.10.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Holaska JM, Rais-Bahrami S, Wilson KL. Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum Mol Genet. 2006;15:3459–3472. doi: 10.1093/hmg/ddl423. [DOI] [PubMed] [Google Scholar]
- 118.Muntoni F, Bonne G, Goldfarb LG, Mercuri E, Piercy RJ, Burke M, Yaou RB, Richard P, Recan D, Shatunov A, Sewry CA, Brown SC. Disease severity in dominant Emery Dreifuss is increased by mutations in both emerin and desmin proteins. Brain. 2006;129:1260–1268. doi: 10.1093/brain/awl062. [DOI] [PubMed] [Google Scholar]
- 119.Melcon G, Kozlov S, Cutler DA, Sullivan T, Hernandez L, Zhao P, Mitchell S, Nader G, Bakay M, Rottman JN, Hoffman EP, Stewart CL. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum Mol Genet. 2006;15:637–651. doi: 10.1093/hmg/ddi479. [DOI] [PubMed] [Google Scholar]
- 120.Ozawa R, Hayashi YK, Ogawa M, Kurokawa R, Matsumoto H, Noguchi S, Nonaka I, Nishino I. Emerin-lacking mice show minimal motor and cardiac dysfunctions with nuclear-associated vacuoles. Am J Pathol. 2006;168:907–917. doi: 10.2353/ajpath.2006.050564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol. 2005;170:781–791. doi: 10.1083/jcb.200502148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bakay M, Wang Z, Melcon G, Schiltz L, Xuan J, Zhao P, Sartorelli V, Seo J, Pegoraro E, Angelini C, Shneiderman B, Escolar D, Chen YW, Winokur ST, Pachman LM, Fan C, Mandler R, Nevo Y, Gordon E, Zhu Y, Dong Y, Wang Y, Hoffman EP. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain. 2006;129:996–1013. doi: 10.1093/brain/awl023. [DOI] [PubMed] [Google Scholar]
- 123.Worman HJ, Evans CD, Blobel G. The lamin B receptor of the nuclear envelope inner membrane: a polytopic protein with eight potential transmembrane domains. J Cell Biol. 1990;111:1535–1542. doi: 10.1083/jcb.111.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ye Q, Worman HJ. Primary structure analysis and lamin B and DNA binding of human LBR, an integral protein of the nuclear envelope inner membrane. J Biol Chem. 1994;269:11306–113011. [PubMed] [Google Scholar]
- 125.Worman HJ, Yuan J, Blobel G, Georgatos SD. A lamin B receptor in the nuclear envelope. Proc Natl Acad Sci U S A. 1988;85:8531–8534. doi: 10.1073/pnas.85.22.8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ye Q, Worman HJ. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J Biol Chem. 1996;271:14653–14656. doi: 10.1074/jbc.271.25.14653. [DOI] [PubMed] [Google Scholar]
- 127.Holmer L, Pezhman A, Worman HJ. The human lamin B receptor/sterol reductase multigene family. Genomics. 1998;54:469–476. doi: 10.1006/geno.1998.5615. [DOI] [PubMed] [Google Scholar]
- 128.Hoffmann K, Dreger CK, Olins AL, Olins DE, Shultz LD, Lucke B, Karl H, Kaps R, Muller D, Vaya A, Aznar J, Ware RE, Sotelo-Cruz N, Lindner TH, Herrmann H, Reis A, Sperling K. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huet anomaly) Nat Genet. 2002;31:410–414. doi: 10.1038/ng925. [DOI] [PubMed] [Google Scholar]
- 129.Waterham HR, Koster J, Mooyer P, Noort GG, Kelley RI, Wilcox WR, Wanders RJ, Hennekam RC, Oosterwijk JC. Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3 beta-hydroxysterol delta 14-reductase deficiency due to mutations in the lamin B receptor gene. Am J Hum Genet. 2003;72:1013–1017. doi: 10.1086/373938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shultz LD, Lyons BL, Burzenski LM, Gott B, Samuels R, Schweitzer PA, Dreger C, Herrmann H, Kalscheuer V, Olins AL, Olins DE, Sperling K, Hoffmann K. Mutations at the mouse ichthyosis locus are within the lamin B receptor gene: a single gene model for human Pelger-Huet anomaly. Hum Mol Genet. 2003;12:61–69. doi: 10.1093/hmg/ddg003. [DOI] [PubMed] [Google Scholar]
- 131.Lin F, Blake DL, Callebaut I, Skerjanc IS, Holmer L, McBurney MW, Paulin-Levasseur M, Worman HJ. MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and emerin. J Biol Chem. 2000;275:4840–4847. doi: 10.1074/jbc.275.7.4840. [DOI] [PubMed] [Google Scholar]
- 132.Mansharamani M, Wilson KL. Direct binding of nuclear membrane protein MAN1 to emerin in vitro and two modes of binding to barrier-to-autointegration factor. J Biol Chem. 2005;280:13863–13870. doi: 10.1074/jbc.M413020200. [DOI] [PubMed] [Google Scholar]
- 133.Osada S, Ohmori SY, Taira M. XMAN1, an inner nuclear membrane protein, antagonizes BMP signaling by interacting with Smad1 in Xenopus embryos. Development. 2003;130:1783–1794. doi: 10.1242/dev.00401. [DOI] [PubMed] [Google Scholar]
- 134.Raju GP, Dimova N, Klein PS, Huang HC. SANE, a novel LEM domain protein, regulates bone morphogenetic protein signaling through interaction with Smad1. J Biol Chem. 2003;278:428–437. doi: 10.1074/jbc.M210505200. [DOI] [PubMed] [Google Scholar]
- 135.Hellemans J, Preobrazhenska O, Willaert A, Debeer P, Verdonk PC, Costa T, Janssens K, Menten B, Van Roy N, Vermeulen SJ, Savarirayan R, Van Hul W, Vanhoenacker F, Huylebroeck D, De Paepe A, Naeyaert JM, Vandesompele J, Speleman F, Verschueren K, Coucke PJ, Mortier GR. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet. 2004;36:1213–1218. doi: 10.1038/ng1453. [DOI] [PubMed] [Google Scholar]
- 136.Lin F, Morrison JM, Wu W, Worman HJ. MAN1, an integral protein of the inner nuclear membrane, binds Smad2 and Smad3 and antagonizes transforming growth factor-beta signaling. Hum Mol Genet. 2005;14:437–445. doi: 10.1093/hmg/ddi040. [DOI] [PubMed] [Google Scholar]
- 137.Pan D, Estevez-Salmeron LD, Stroschein SL, Zhu X, He J, Zhou S, Luo K. The integral inner nuclear membrane protein MAN1 physically interacts with the R-Smad proteins to repress signaling by the transforming growth factor-{beta} superfamily of cytokines. J Biol Chem. 2005;280:15992–16001. doi: 10.1074/jbc.M411234200. [DOI] [PubMed] [Google Scholar]
- 138.Hellemans J, Debeer P, Wright M, Janecke A, Kjaer KW, Verdonk PC, Savarirayan R, Basel L, Moss C, Roth J, David A, De Paepe A, Coucke P. Germline LEMD3 mutations are rare in sporadic patients with isolated melorheostosis. Hum Mutat. 2006;27:290. doi: 10.1002/humu.9403. [DOI] [PubMed] [Google Scholar]
- 139.Mumm S, Wenkert D, Zhang X, McAlister WH, Mier RJ, Whyte MP. Deactivating germline mutations in LEMD3 cause osteopoikilosis and Buschke-Ollendorff syndrome, but not sporadic melorheostosis. J Bone Miner Res. 2007;22:243–250. doi: 10.1359/jbmr.061102. [DOI] [PubMed] [Google Scholar]
- 140.Caputo S, Couprie J, Duband-Goulet I, Konde E, Lin F, Braud S, Gondry M, Gilquin B, Worman HJ, Zinn-Justin S. The carboxyl-terminal nucleoplasmic region of MAN1 exhibits a DNA binding winged helix domain. J Biol Chem. 2006;281:18208–18215. doi: 10.1074/jbc.M601980200. [DOI] [PubMed] [Google Scholar]
- 141.Ishimura A, Ng JK, Taira M, Young SG, Osada S. Man1, an inner nuclear membrane protein, regulates vascular remodeling by modulating transforming growth factor beta signaling. Development. 2006;133:3919–3928. doi: 10.1242/dev.02538. [DOI] [PubMed] [Google Scholar]
- 142.Taylor MR, Slavov D, Gajewski A, Vlcek S, Ku L, Fain PR, Carniel E, Di Lenarda A, Sinagra G, Boucek MM, Cavanaugh J, Graw SL, Ruegg P, Feiger J, Zhu X, Ferguson DA, Bristow MR, Gotzmann J, Foisner R, Mestroni L. Familial Cardiomyopathy Registry Research Group, Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat. 2005;26:566–574. doi: 10.1002/humu.20250. [DOI] [PubMed] [Google Scholar]
- 143.Gros-Louis F, Dupre N, Dion P, Fox MA, Laurent S, Verreault S, Sanes JR, Bouchard JP, Rouleau GA. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. 2007;39:80–85. doi: 10.1038/ng1927. [DOI] [PubMed] [Google Scholar]
- 144.Worman HJ, Gundersen GG. Here come the SUNs: a nucleocytoskeletal missing link. Trends Cell Biol. 2006;16:67–69. doi: 10.1016/j.tcb.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 145.Cronshaw JM, Matunis MJ. The nuclear pore complex protein ALADIN is mislocalized in triple A syndrome. Proc Natl Acad Sci U S A. 2003;100:5823–5827. doi: 10.1073/pnas.1031047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 147.Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci U S A. 2004;101:847–852. doi: 10.1073/pnas.0304375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Naismith TV, Heuser JE, Breakefield XO, Hanson PI. TorsinA in the nuclear envelope. Proc Natl Acad Sci U S A. 2004;101:7612–7617. doi: 10.1073/pnas.0308760101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gonzalez-Alegre P, Paulson HL. Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J Neurosci. 2004;24:2593–2601. doi: 10.1523/JNEUROSCI.4461-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Goodchild RE, Dauer WT. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J Cell Biol. 2005;168:855–862. doi: 10.1083/jcb.200411026. [DOI] [PMC free article] [PubMed] [Google Scholar]