

Abstract
Fascinating links are beginning to be discovered between mitochondrial function and cardiac physiology and disease in the context of diverse signaling mechanisms, energy production, and intersection with pathways producing reactive oxygen species. Proteins long known to drive mitochondrial fusion and fission are now reported to have emergent functions in intracellular calcium homeostasis, apoptosis, and vascular smooth muscle cell proliferation, all key issues in cardiac disease. Moreover, mitochondrial fusion has been demonstrated to be required for normal myofibril organization in skeletal muscle, and decreasing fission may confer protection against ischemic heart disease. These processes broaden the traditional role in energy production undertaken by mitochondria and provide new directions for potential therapeutic leads.
Keywords: Mitochondria, Fusion, Fission, ROS, Signaling
Introductory mini-review
Capture of proteobacteria by early eukaryotic cells resulted in a commensal and potentially symbiotic relationship. Evolution of the bacteria into modern day mitochondria provided a generating source of energy to the host eukaryotic cell, while the mitochondria might be viewed as having benefited through being able to transfer most of its genetic components to the more powerful and complex host genome. From the onset, it would have been vital to regulate mitochondrial biogenesis in relationship to the rate of cell division of the host cell—but in addition, as is now increasingly appreciated, mitochondria have become imbedded in fabric of eukaryotic cell biological processes, including signal transduction, vesicle trafficking, apoptosis, autophagy, lipid biosynthesis, and regulation of calcium levels [1].
Mitochondria were long viewed as relatively static organelles. This picture changed though, with the identification of a gene that regulated mitochondrial fusion in Drosophila and was critical for spermatogenesis [2]. The mammalian homologs, known as Mitofusin (Mfn) 1 and 2, were found to undertake similar roles in fusion and to be required, non-redundantly, for viability, as discussed in detail in the article in this special issue by R. Youle. Mutations in Mfn2 and in Opa1, another gene required for fusion, have been associated with neurological human diseases. Conversely, genes required for mitochondrial fission such as Drp1 have also been identified and similarly are required for viability [3, 4]. In the most fundamental sense, fission is required to ensure that the number of mitochondria is maintained subsequent to cell division; however, mitochondrial fusion and fission have become associated with many other functions (Fig. 1).
Fig. 1.
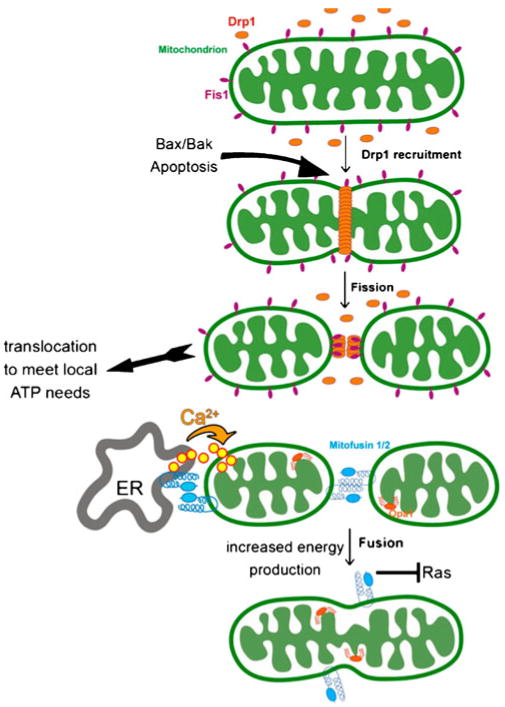
Mitochondrial fusion and fission connect to many other cell biological processes. Drp1, a key protein in mitochondrial fission, also recruits key components of the apoptotic pathway that function to trigger release of cytochrome C. Independent of apoptosis, fission is important to enable translocation of mitochondrial to subcellular locations at which ATP production is most needed. Mfn2, a key protein in fusion, also functions to tether the endoplasmic reticulum to mitochondria to facilitate calcium uptake and propagation during signaling events, and independently, to block Ras downstream pathways that lead to vascular smooth muscle cell hyperplasia and subsequent hypertension. Fusion itself is important for increasing the efficiency of energy production by mitochondria, for maintenance of mitochondrial DNA integrity, and proper functioning of neurons and skeletal muscle cells
Drp1, the dynamin-related protein that physically carries out the fission process, has been linked to the regulation of apoptosis, by interacting with components of the cell death pathway and ultimately triggering cytochrome c release from mitochondria [5, 6]; however, the regulation of fission is also important in normal cell physiology in the context of directing the production of ATP at the subcellular sites where it is most needed. Extended mitochondrial tubules are less mobile than small mitochondria, and the capacity to undergo fission is critical for movement of mitochondria to the trailing edge of migrating cells where myosin motors are assembled [7] or to synapses in response to nerve growth factor signaling [8].
The fusion process plays important roles in the signal transduction-mediated mitochondrial response to altering energy demands and substrate availability such as during insulin stimulation. Among other issues, larger mitochondria are more efficient producers of energy than smaller ones [9]; however, mitochondrial fusion is also important for maintaining the integrity of mitochondrial DNA and for facilitating mitochondria association with sarcomeres in muscle cells and, indirectly, normal organization of myofibril arrays [10]. Intriguingly, Mfn2, initially thought solely to mediate fusion through a mechanism involving trans-dimerization on the surface of closely apposed mitochondria, has acquired additional unexpected roles. Mfn2 can also be found on the endoplasmic reticulum (ER), where it again transdimerizes but this time to tether mitochondria in close approximation with the ER, enabling efficient capture of Ca2+ released by the ER during phospholipase C/inositol triphosphate (IP3)-generating signaling events and lipid transfer important for biosynthetic processes [11]; however, the interaction of mitochondria with the ER may be more complex than simply via Mfn2 dimerization, since other approaches have identified an additional protein complex called ER–mitochondria encounter structure that appears to perform a similar function [12, 13]. Finally, as described in the article by R.P. Xiao in this collection, Mfn2 was independently identified as a protein that suppresses Ras-mediated hyperproliferation of vascular smooth muscle cells in spontaneously hypertensive rats, via a mechanism, potentially involving the promotion of apoptosis that is distinct from its function in mediating mitochondrial fusion [14, 15].
This emerging more complicated view of mitochondrial fusion proteins is matched by the growing appreciation for how this originally distinct organelle has become integrated into the signaling and cell biological pathways that regulate other cellular organelles, for example, formation and trafficking of restricted cargo-laden small membrane vesicles is a hallmark of the secretory and membrane retrieval systems, but a similar process has now been described [16, 17] for trafficking between mitochondria and peroxisomes, organelles involved in the disposal of reactive oxygen species (ROS). Mitochondria have also been described to undergo “kiss and run” partial fusion events [18], similar to what is seen for secretory granules containing neurotransmitters at neuronal synapses. Finally, mitochondria have been found to undergo autophagy like other cellular components that need to be recycled, and the mechanism that regulates this involves a sensor on the mitochondrial surface that signals lack of mitochondrial fitness (mitochondrial potential). The sensor is destroyed via a cytosolic proteolysis event on healthy mitochondria, but remains intact and thus creates a signal that targets the mitochondria for destruction when they are impaired [19]. This process is reminiscent of the cytoplasmic proteolysis mechanisms that the ER uses to communicate ongoing unfolded protein stress events to the nucleus [20].
Although some of these events are coordinated in the mitochondrial matrix, a growing number involves signaling events on the mitochondrial surface as described above. Most of the signaling events involve recruitment of proteins, but pathways involving lipids are beginning to emerge as well, for example, cardiolipin functions as a signaling platform on the mitochondrial surface with vital functions at several steps during apoptosis [21], including recruitment of tBid [22]. Phosphatidic acid (PA), a lipid generated on the mitochondrial surface by the mitochondrial-specific phospholipase D MitoPLD, facilitates mitochondrial fusion [23], and diacylglycerol (DAG), which can be generated by dephosphorylation of PA or through phospholipase C-mediated hydrolysis of phosphatidylinositol-4, 5-bisphosphate (PI4,5P2), has been proposed to recruit protein kinase D to mitochondria in the setting of ROS generation to facilitate mitochondria to nucleus signaling [24].
How do these pathways connect to cardiac disease? There are many intriguing connections that have been identified, including the direct observation that loss of mitochondrial fusion results in abnormal myofibril organization and muscular atrophy in skeletal muscle [10]. Strikingly, the converse strategy, i.e., decreasing mitochondrial fission using a small molecule inhibitor of Drp1, is protective against ischemia/reperfusion-induced injury in the heart [25]. As noted above though, Drp1 mediates functions in addition to promotion of fission, and some of these may be critical in other contexts or in combination with fission, since a point mutation in Drp1 that alters its protein interactions causes dilated cardiomyopathy [4]. Thus, Drp1 activity and function may have to be manipulated and assessed carefully in the setting of potential therapeutic approaches. In the contributions to this special issue that follow, a number of topics are presented. JR Sowers discusses mitochondrial biogenesis and dysfunction in metabolic syndrome, and focuses on the only partially successful adaptive mitochondrial response in the setting of insulin resistance, when substrates for mitochondrial-driven energy production become imbalanced and consequences such as increased ROS occur. Intriguing links to this story may come to include ones such as Lipin, an enzyme that produces DAG from PA and has been linked to a Type II-like diabetes syndrome (fatty lipid dystrophy) [26] that involves abnormal mitochondrial functioning and could arise in part from altered lipid signaling on the mitochondrial surface. Alternately, Mfn2 is upregulated by insulin signaling pathways [27] and may act as a key player in insulin-dependent myogenesis by stimulating respiration, substrate oxidation, and OXPHOS subunit expression [28] or by inhibiting Ras-dependent mitogen-activated protein kinase pathways [14, 15].
Potentially related to these phenomena, as discussed by Ungvari, increases in mitochondrial oxidative stress cause vascular disease in aging. Archer, Michelakis, and colleagues in other articles in this series note that decreases in superoxide dismutase 2 result in a reduced redox environment that promotes pulmonary artery hyperplasia [29, 30]. Moreover, impaired mitochondrial oxidative function triggers a glycolytic shift in both the smooth muscle cells in pulmonary arterial hypertension and the right ventricular myocytes in the associated right ventricular hypertrophy. The glycolytic shift can be reversed by inhibitors of pyruvate dehydrogenase kinase such as dichloroacetate, leading to increased glucose oxidation and an associated decrease in pulmonary artery smooth muscle cell proliferation that leads to regression of the pulmonary artery hyperplasia [31] and an improvement in RV myocyte function. In the context of the pathways described above, the redox alterations could decrease Mfn2, causing mitochondrial fragmentation and decreased function, and loss of suppression of vascular or pulmonary artery smooth muscle cell proliferation, or perhaps even more complexities in these intersecting pathways remain to be discovered.
Acknowledgments
This study is supported by the NIH awards GM071520 and GM084251.
Footnotes
Conflict of interests The author declares no conflict of interests related to this study.
References
- 1.Soubannier V, McBride HM. Positioning mitochondrial plasticity within cellular signaling cascades. Biochim Biophys Acta. 2009;1793:154–170. doi: 10.1016/j.bbamcr.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Hales KG, Fuller MT. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997;90:121–129. doi: 10.1016/s0092-8674(00)80319-0. [DOI] [PubMed] [Google Scholar]
- 3.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashrafian H, Docherty L, Leo V, Towlson C, Neilan M, Steeples V, Lygate CA, Hough T, Townsend S, Williams D, Wells S, Norris D, Glyn-Jones S, Land J, Barbaric I, Lalanne Z, Denny P, Szumska D, Bhattacharya S, Griffin JL, Hargreaves I, Fernandez-Fuentes N, Cheeseman M, Watkins H, Dear TN. A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet. 2010;6:e1001000. doi: 10.1371/journal.pgen.1001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka A, Youle RJ. A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Mol Cell. 2008;29:409–410. doi: 10.1016/j.molcel.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Campello S, Lacalle RA, Bettella M, Manes S, Scorrano L, Viola A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med. 2006;203:2879–2886. doi: 10.1084/jem.20061877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amiri M, Hollenbeck PJ. Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev Neurobiol. 2008;68:1348–1361. doi: 10.1002/dneu.20668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 12.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kornmann B, Walter P. ERMES-mediated ER-mitochondria contacts: molecular hubs for the regulation of mitochondrial biology. J Cell Sci. 2010;123:1389–1393. doi: 10.1242/jcs.058636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, Li P, Qiu X, Wen S, Xiao RP, Tang J. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol. 2004;6:872–883. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- 15.Guo X, Chen KH, Guo Y, Liao H, Tang J, Xiao RP. Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ Res. 2007;101:1113–1122. doi: 10.1161/CIRCRESAHA.107.157644. [DOI] [PubMed] [Google Scholar]
- 16.Andrade-Navarro MA, Sanchez-Pulido L, McBride HM. Mitochondrial vesicles: an ancient process providing new links to peroxisomes. Curr Opin Cell Biol. 2009;21:560–567. doi: 10.1016/j.ceb.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol. 2008;18:102–108. doi: 10.1016/j.cub.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Weaver D, Shirihai O, Hajnoczky G. Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion–fission dynamics. EMBO J. 2009;28:3074–3089. doi: 10.1038/emboj.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 21.Schug ZT, Gottlieb E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta. 2009;1788:2022–2031. doi: 10.1016/j.bbamem.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 22.Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;2:754–761. doi: 10.1038/35036395. [DOI] [PubMed] [Google Scholar]
- 23.Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. 2006;8:1255–1262. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- 24.Cowell CF, Doppler H, Yan IK, Hausser A, Umezawa Y, Storz P. Mitochondrial diacylglycerol initiates protein-kinase D1-mediated ROS signaling. J Cell Sci. 2009;122:919–928. doi: 10.1242/jcs.041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 26.Reue K, Dwyer JR. Lipin proteins and metabolic homeostasis. J Lipid Res. 2009;50(Suppl):S109–S114. doi: 10.1194/jlr.R800052-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soriano FX, Liesa M, Bach D, Chan DC, Palacin M, Zorzano A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator–activated receptor–gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes. 2006;55:1783–1791. doi: 10.2337/db05-0509. [DOI] [PubMed] [Google Scholar]
- 28.Pawlikowska P, Gajkowska B, Orzechowski A. Mitofusin 2 (Mfn2): a key player in insulin-dependent myogenesis in vitro. Cell Tissue Res. 2007;327:571–581. doi: 10.1007/s00441-006-0320-3. [DOI] [PubMed] [Google Scholar]
- 29.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 30.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thebaud B, Husain AN, Cipriani N, Rehman J. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension. A basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121(24):2661–2671. doi: 10.1161/CIRCULATIONAHA.109.916098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105:244–250. doi: 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]