

Abstract
Hepatitis C virus (HCV) affects an estimated 3% of the population and is a leading cause of chronic liver disease worldwide. Since HCV therapeutic and preventative options are limited, the development of new HCV antivirals has become a global health care concern. This has spurred the development of cell‐based infectious HCV high‐throughput screening assays to test the ability of compounds to inhibit HCV infection. This unit describes methods that may be used to assess the in vitro efficacy of HCV antivirals using a cell‐based high‐throughput fluorescence resonance energy transfer (FRET) HCV infection screening assay, which allows for the identification of inhibitors that target HCV at any step in the viral life cycle. Basic protocols are provided for compound screening during HCV infection and analysis of compound efficacy using an HCV FRET assay. Support protocols are provided for propagation of infectious HCV and measurement of viral infectivity. Curr. Protoc. Microbiol. 18:17.5.1‐17.5.27. © 2010 by John Wiley & Sons, Inc.
Keywords: hepatitis C virus, viral lifecycle, Huh7 cells, dimethylsulfoxide (DMSO), fluorescence resonance energy transfer (FRET), NS3 protease, antivirals, high‐throughput screening
Introduction
Hepatitis C virus (HCV) is a hepatotropic pathogen that causes liver disease, including fibrosis, cirrhosis, steatosis, insulin resistance, iron overload, and hepatocellular carcinoma. With an estimated 130 million people chronically infected, HCV is a major cause of liver disease worldwide and represents a global healthcare concern. Unfortunately, there is no preventative vaccine to protect against HCV infection, and the available IFN‐based standard of care therapy (pegylated IFN‐α plus ribavirin) is costly, poorly tolerated, associated with significant side effects, and only effective in a fraction of chronically infected individuals. Therefore, as with the other pathogens discussed within this chapter, the need to develop new‐generation HCV “antiinfectives” is a healthcare imperative. Although several small molecules that target HCV replication are in the clinical pipeline, the emergence of viral escape mutants indicates that a combination of anti‐HCV drugs targeting multiple aspects of infection will likely be required to successfully treat the majority of patients. Thus, this unit aims to facilitate the discovery of HCV antivirals by describing methods to test and evaluate promising antiviral compounds in vitro.
The ability to screen for HCV antivirals that inhibit any aspect of infection was made possible with the 2005 development of the first robust in vitro HCV infection models (Lindenbach et al., 2005; Wakita et al., 2005; Zhong et al., 2005), which recapitulate the entire viral life cycle and allow for HCV propagation in cell culture. This unit focuses on procedures for high‐throughput anti‐HCV compound screening. Basic Protocol 1 describes the screening of compounds for anti‐HCV activity in microtiter plate format using nondividing, synchronized hepatoma cells which allow for reproducible HCV infection in vitro using a low multiplicity of infection approach. Basic Protocol 2 describes a simple mix‐and‐read FRET‐based HCV NS3 protease assay which can be used to assess HCV infection and determine compound efficacy. Because stocks of infectious HCV are required for these basic screening protocols, support protocols detailing the preparation of the necessary HCV stocks are also provided. Support Protocol 1 explains how to generate infectious HCV from in vitro–transcribed HCV RNA, while Support Protocol 2 describes the further amplification of that original HCV stock, and Support Protocol 3 lists methods assessing the infectivity titer of the HCV stocks produced. Together, these protocols encompass several new approaches to compound screening and HCV antiviral drug discovery.
CAUTION: HCV is a Biosafety Level 2 (BSL‐2) pathogen. Follow all appropriate guidelines and regulations for the use and handling of pathogenic microorganisms. See unit mc01a01 and other pertinent resources (appendix mca01b) for more information.
NOTE: All solutions and equipment coming into contact with living cells must be sterile, and aseptic techniques should be used accordingly.
NOTE: All culture incubations should be performed in a humidified 37°C, 5% CO2 incubator unless otherwise specified.
Basic Protocol 1. Screening of Anti‐HCV Compounds in Hepatoma Cells
With the development of robust cell culture–based HCV infection systems (Lindenbach et al., 2005; Wakita et al., 2005; Zhong et al., 2005), it is now possible to screen for HCV inhibitors that act at any stage of the viral life cycle (e.g., entry, replication, assembly, egress, and spread). The following anti‐HCV compound screening protocol (Yu et al., 2009) accomplishes this with a low multiplicity of infection (MOI), 6‐day HCV infection approach, which consists of multiple rounds of infection and re‐infection. Adequate reproducibility, allowing for Z′ values greater than 0.6, is achieved by using nondividing, synchronized hepatoma cells.
Specifically, treatment of the human hepatoma cell line Huh7 with 1% DMSO induces cell growth arrest, allowing nondividing Huh7 cells to be maintained at a stable cell number for extended periods of time (Sainz and Chisari, 2006). Importantly, these nondividing cultures can be scaled down to a microtiter‐plate format for compound library screening, and support highly reproducible HCV infection from well to well, minimizing the sample‐to‐sample variability commonly associated with cell‐based viral assays (Yu et al., 2009). Once established, these nondividing Huh7 cell cultures are inoculated with cell culture–propagated HCV (HCVcc) at a low MOI, and the infection is allowed to progress over 6 days in the presence or absence of test compounds. Compounds are typically added three times throughout the course of infection: on days 0, 2, and 4 post infection. Treatment times and frequency can be adjusted (e.g., treatment at only day 2 and 4 post infection); however, inhibitors of HCV entry are more efficiently detected when a day‐0 treatment is included in the regimen. Following infection, cultures are lysed and HCV inhibition is assessed by NS3 protease FRET analysis (Basic Protocol 2).
Materials
Huh7 cells (Japan Health Science Research Resources Bank, cat. no. JCRB0403; http://www.jhsf.or.jp)
Huh7 cell maintenance medium (see recipe)
Huh7 cell maintenance medium with 1% (v/v) dimethyl sulfoxide (DMSO; tissue culture grade)
Compound library
HCVcc (stock titer ≥3.25 × 104; see Support Protocol 1 and 2)
FRET lysis buffer (see recipe), prechilled to 4°C
75‐cm2 or larger tissue culture flasks
96‐well clear flat‐bottom black microtiter BioCoat tissue culture plates (BD Biosciences)
96‐well U‐bottom microtiter plates
Plate seals
Additional reagents and equipment for growing mammalian cells (Phelan, 2007)
Prepare non‐growing Huh7 cell 96‐well screening plates cultures
-
1
Culture Huh7 cells in 75‐cm2 or larger tissue culture flasks in Huh7 cell maintenance medium until cells reach ∼90% confluence.
Huh7 cells double every 24 hr under these conditions.
Select an appropriate‐size tissue culture flask based on the number of 96‐well tissue culture screening plates to be seeded. A 150‐cm2 flask at 80% confluence contains ∼1×107 Huh7 cells, which is sufficient for ∼15 tissue culture plates.
-
2
Seed each well of a 96‐well clear flat‐bottom black microtiter BioCoat tissue culture plate with ∼7 × 103 Huh7 cells in 100 µl of Huh7 cell maintenance medium and incubate until 85% to 90% confluent.
A collagen type‐1 matrix is necessary for proper longevity of Huh7 cells cultured in the presence of 1% DMSO. Collagen‐coated 96‐well tissue culture plates, e.g., BioCoat from BD Biosciences, are commercially available, but coating plates with collagen in the laboratory is a viable alternative.
-
3
When the Huh7 cell monolayers are 85% to 90% confluent (in 1 to 2 days), decant the medium and replace it with 200 µl/well of Huh7 cell maintenance medium containing 1% DMSO. Continue incubation.
-
4
Continue culturing cells in Huh7 cell maintenance medium with 1% DMSO for 20 days. Decant the medium and replace with fresh Huh7 cell maintenance medium with 1% DMSO every 2 to 3 days.
In the presence of 1% DMSO, Huh7 cells will continue to divide until ∼6 day post treatment, at which time cells undergo cell‐cycle arrest (i.e., G0), with the number of cells/well reaching ∼6.5 × 104. During this time, Huh7 cell morphology changes, with the cells forming tightly packed monolayers of mono‐ and binucleated cells displaying the typical pavement‐like cytological features of primary hepatocytes; the cells are granulated and contain multiple nucleoli.
Prepare 96‐well test compound plates
-
5
Resuspend (or dilute) test and control compounds in DMSO or another appropriate diluent to 100× the desired final concentration.
DMSO is the recommend diluent for the 100× compounds, as once culture medium is added, the DMSO diluent will provide the final 1% DMSO required for the nongrowing Huh7 cell cultures.
-
6
For each set of 96 compounds (test and controls), transfer 2.2 µl of the 100× test or control compounds into one well of three replicate 96‐well U‐bottom microtiter plates (one for each treatment day, 0, 2, and 4) and store these compound aliquot plates according to the storage conditions specified for the compounds.
The layout of the 96‐well U‐bottom microtiter plate(s) will depend on the number of positive/negative controls and compounds to be tested. If using a compound library, manufacturers often provide one empty column for the addition of controls and blanks. At least one well must be left blank for a FRET buffer control. We also include, when possible, a blank well for a FRET water control.
The layout of the 96‐well U‐bottom microtiter plate(s) may also need to be adapted to avoid areas prone to “edge effect” (noise and variability of cell‐based assays corresponding to the edges of microtiter plates), if this proves problematic in the individual user's hands. Figure 1 illustrates an example of a standard plate layout in which two columns have been reserved for controls.
Figure 1.
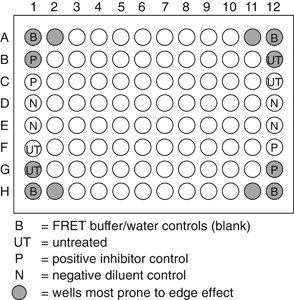
Sample plate layout for HTS anti‐HCV compound screening. This illustrates a standard plate layout in which two columns have been reserved for controls. However, compound libraries are often provided with only one empty column for the addition of controls. The layout chosen for individual screening campaigns will need to be adapted to accommodate these restrictions, and, if necessary, to avoid areas prone to “edge effect.”
Infect cells and screen compounds
-
7
On day 20 post DMSO treatment of the Huh7 cells, remove one set of the triplicate 96‐well test compound plates containing 2.2 µl/well of 100× test compound and controls (from step 6) and equilibrate to room temperature.
-
8
Add ∼108 µl of Huh7 cell maintenance medium (without DMSO) to each test compound well, such that the test compounds and controls will be diluted to 2× the desired final concentration (in 2% DMSO).
-
9
Immediately before use, dilute the virus inoculum in Huh7 cell maintenance medium (without DMSO) at an MOI of 0.05 ffu/cell.
Following 20 days in Huh7 cell maintenance medium with 1% DMSO, Huh7 cells will have reached a constant number of ∼65,000 cells/well. Thus, to achieve an MOI of 0.05 focus‐forming units (ffu)/cell, each well should be infected with ∼3250 ffu HCV diluted in 100 µl of in Huh7 cell maintenance medium (without DMSO). HCVcc should be thawed and diluted immediately prior to use, but can be kept on ice for an hour without loss of infectivity.
-
10
Decant the medium from one Huh7 flat‐bottom black microtiter BioCoat Huh7 screening plate (in Huh7 cell maintenance medium with 1% DMSO; see step 4) and replace with 100 µl of 2× test or control compounds from the U‐bottom 96‐well compound plate (step 8).
To expedite transfer of compounds between microtiter plates, the use of a multichannel pipettor is advised. When using a multichannel pipettor to dispense compounds, care must be taken to ensure that equal volumes are taken up and dispensed.
-
11
Inoculate each well with 100 µl of the HCVcc inoculum from step 9, or mock inoculate with 100 µl of Huh7 cell maintenance medium alone.
The use of a multichannel pipettor and reagent reservoirs containing HCVcc inoculum and mock inoculum (e.g., medium alone) is advised. When using a multichannel pipettor to dispense virus, care must be taken to ensure equal‐volume dispensing. The use of aerosol filter pipet tips is recommended when handling virus.
The test and control compounds will now be at 1× concentration and the DMSO concentration will be 1%.
-
12
Incubate these HCV infected/compound treated screening plates for 48 hr.
We recommend that the plates not be stacked within the incubator, but rather laid out separately so that gas exchange is equal among all plates, as this reduces “edge effects” that are traditionally observed when using microtiter plates for cell‐based screening.
Replace test compound–containing medium
-
13
After 2 days, change the medium on the screening plates by removing another set of the triplicate 96‐well test compound plates (from step 6, containing 2.2 µl of 100× test compounds and controls) and equilibrating to room temperature.
-
14
Add ∼218 µl of Huh7 cell maintenance medium to each well to dilute the compounds to 1× treatment concentration.
-
15
Take the corresponding HCVcc‐infected 96‐well screening plates (step 11) from the incubator and set them in the hood.
-
16
One plate at a time, remove the used medium from each well of the HCVcc‐infected screening plates and replace with 200 µl of appropriate test or control compounds from the U‐bottom 96‐well plate containing the freshly diluted 1× compounds (step 14).
-
17
Incubate HCVcc‐infected screening plate(s) with the test compounds for 48 hr.
-
18
To replenish the drug containing–medium again on day 4, repeat steps 13 through 17.
Harvest medium and cells
-
19
Six days post infection, transfer medium from screening plates to 96‐well U‐bottom microtiter plates for future toxicity analysis.
-
20
Cover the 96‐well U‐bottom plate with a plate seal and store at −80°C.
The medium recovered on day 6 post infection (and/or at days prior to day 6) can be analyzed for drug‐induced toxicity using commercially available cellular cytotoxicity assays.
-
21
Place the 96‐well screening plate containing the cells on ice.
-
22
Add 50 µl of prechilled FRET lysis buffer to each well and incubate for 10 min on ice.
To ensure optimal reproducibility, monitor the time on ice closely, as all plates should incubate for exactly the same amount of time.
-
23
Cover the 96‐well clear flat‐bottom black microtiter BioCoat screening plate with a plate seal and store at −80°C immediately.
-
24
Repeat steps 19 through 23 for each screening plate.
Basic Protocol 2. Quantification of HCV Infection Using a Cell‐Based NS3 Protease Assay
Several groups have demonstrated the ability of the HCV NS3 protease to cleave synthetic peptides containing the enzymes natural viral cleavage sites (Bianchi et al., 1996; Kakiuchi et al., 1999). Optimal cleavage sequence and substrate length, and the ability to attach acceptor and donor fluorogenic moieties to the ends of these peptide substrates without loss of enzyme‐substrate interaction, have also been established. An improvement in the in vitro kinetic parameters of NS3 protease activity was made by Bianchi et al. (1996), who developed an NS4A/NS4B site–derived (DEMEECASHL) depsipeptide substrate, in which the standard amide bond at the cleavage site between the P1 and P1′ residues was replaced by an ester bond, resulting in a more readily cleaved peptide that can serve as an efficient NS3 FRET substrate for the continuous measurement of NS3 protease activity in a cell‐free assay using purified NS3 protease (Bianchi et al., 1996; Taliani et al., 1996). EDANS and DABCYL were selected as the donor/acceptor pair for this substrate because the high degree of spectral overlap between the fluorescent emission of EDANS and the absorption of DABCYL results in efficient proximity‐dependent quenching, and this HCV NS3 FRET substrate (commercially available from AnaSpec) has been used successfully by others to monitor HCV subgenomic replicon levels in cell lysates (O'Boyle et al., 2005). The following protocol, however, uses a new more sensitive 5‐FAM/QXL 520 NS3 FRET substrate (Fig. 2), which allows the enzymatic assay to be performed at higher wavelengths (excitation/emission = 490 nm/520 nm), avoiding cellular background fluorescence commonly detected when reading EDANS (AnaSpec). This improved substrate provides increased fluorescence quantum yield as well as longer emission wavelength and diminished autofluorescence, and is ten times more sensitive than the EDANS/DABCYL‐based assay, allowing for the detection of as little as 0.1 pmol of HCV NS3/4a protease.
Figure 2.
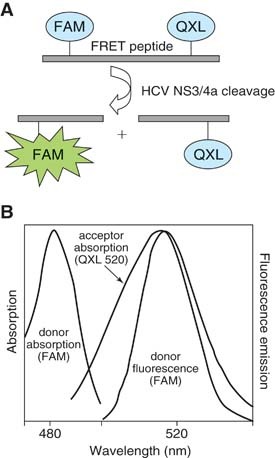
NS3 FRET peptide substrate. (A) The 5‐FAM/QXL 520 NS3 FRET substrate is an internally quenched peptide with a fluorescent donor (5‐FAM) and acceptor (QXL) on opposing sides of the NS3 protease cleavage site. (B) FRET protease assay. The donor absorbs energy at 490 nm and emits energy (i.e., fluorescence) at 520 nm. However, when in close contact on an intact peptide, the acceptor absorbs the 520‐nM energy emitted by the donor, preventing fluorescence. Cleavage of the peptide increases the distance between the fluorophores, resulting in proportional 5‐FAM fluorescence. Diagram and figure adapted from AnaSpec product information (http://www.anaspec.com/products/product.asp?id=30173&productid=13982).
Materials
Sample plate (Basic Protocol 1)
5‐FAM/QXL 520 NS3 FRET peptide substrate (AnaSpec; resuspend and distribute into single‐use aliquots upon receipt)
2× FRET assay buffer (AnaSpec)
1 M dithiothreitol (DTT; appendix mca02a)
Fluorescent microplate reader with 490‐nm excitation and 520‐nm emission filters
Shaking platform
Centrifuge with microtiter plate carrier
Set up assay
-
1
Program the fluorescent microplate reader with the necessary assay parameters and sample layout, taking into consideration any instrument‐specific settings.
The peptide substrate donor (5‐FAM) absorbs energy at 490 nm and emits energy at 520 nm. Thus, the excitation and emission parameters are 490 nm and 520 nm, respectively. Program the reader to measure fluorescence for at least thirty 150‐second cycles in kinetic mode at a constant temperature of 30°C. In addition, an injector to dispense the required volume of peptide substrate into each well should be utilized to ensure consistent results.
-
2
Remove one sample plate from −80°C.
-
3
Place the sample plate on ice and thaw on a slowly shaking platform shaker for 1 hr.
The method by which plates are thawed prior to analysis is critical for plate‐to‐plate reproducibility:
(i) Thaw on ice to avoid degradation of NS3 activity and minimize differential thaw patterns across the plate.
(ii) Shake plates to help minimize differential thaw patterns.
(iii) Every plate should be thawed for exactly the same period of time prior to assay.
-
4
At a time point 15 min before plate thawing is complete, take one aliquot of the FRET peptide substrate and the 1 M DTT from the freezer and take the 2× FRET assay buffer from refrigerator. Allow all to equilibrate to room temperature for 15 min.
A volume of 60 µl of the 5‐FAM/QXL 520 NS3 FRET substrate is sufficient for one entire 96‐well plate.
Perform assay
-
5
Remove the sample plate from ice (see step 3) and equilibrate to room temperature for 5 min.
-
6
While the sample plate is equilibrating to room temperature, prepare the diluted peptide substrate reagent by adding 300 µl of 1 M DTT and 60 µl of substrate to 5 ml of 2× FRET assay buffer in an appropriate‐sized tube. Mix well.
-
7
Centrifuge the sample plate 30 sec at 200 × g, room temperature.
-
8
Place the sample plate into the fluorescent microplate reader.
-
9
Prime the fluorescent microplate reader injector with the diluted peptide substrate reagent (step 6) as per instrument instructions.
If the fluorescent microplate reader to be used is not equipped with an injector, the diluted peptide substrate can be added to individual plates, immediately before running the program, with a multichannel pipettor.
-
10
Perform the FRET assay using a fluorescent microplate reader as programmed in step 1.
Typically, with a gain of 1390 and a required value of 20% relative fluorescence units (RFU), cycle 20 can be used as an endpoint analysis, as this cycle tends to reproducibly give the maximum signal‐to‐noise ratio; however, such parameters should be individually optimized, as 5‐FAM/QXL520 NS3 FRET substrate lots and fluorescent microplate readers can vary from laboratory to laboratory.
Analyze results
-
11
Once an appropriate cycle is determined for end point analysis, subtract the mean background (FRET lysis buffer alone) RFU values from all experimental RFU values. Set background‐corrected RFU values from nontreated HCV‐infected wells as 100% maximum activity, and express background‐corrected RFU signals from experimental wells as a percentage of the maximum.
Support Protocol 1. Generation of HCVcc from In Vitro–Transcribed RNA
Since its discovery in 1989, propagation of HCV in vitro was hampered because the infectious clones available did not replicate in cultured cells; however, in 2001 Wakita and colleagues cloned an HCV consensus genotype 2a genome (JFH‐1) from a Japanese Fulminant Hepatitis patient (Kato et al., 2001, 2003), which was subsequently found to produce infectious HCV in Huh7 cell culture (Lindenbach et al., 2005; Wakita et al., 2005; Zhong et al., 2005). Several infectious chimeric HCV clones have been produced since, and could also be used analogously to the JFH‐1 clone described herein.
The full length JFH‐1 viral consensus sequence has been cloned into the pUC DNA vector under the transcriptional control of the T7 promoter (Kato et al., 2001). pJFH‐1 is approximately 12.4 kb in length, expresses ampicillin resistance, and can be propagated in dam+ E. coli and selected for in the presence of 100 µg/ml ampicillin. Purified pJFH‐1 DNA is linearized at the end of HCV clone with the restriction enzyme XbaI to allow for in vitro transcription of the genomic JFH‐1 RNA with precise ends. This HCV JFH‐1 RNA is then electroporated into Huh7 cells to initiate HCV replication and infectious virus production. The virus secreted into the medium can be titered with infectious virus levels expressed as focus forming units (ffu) per ml or TCID50 (Support Protocol 3).
Huh7 cells and Huh7‐derived clones [e.g., Huh7.5 (Lindenbach et al., 2005) and Huh7.5.1 cells (Zhong et al., 2005)] typically secrete 104 to 106 focus‐forming units (ffu) per ml following transfection with in vitro transcribed JFH‐1 RNA or infection with HCVcc (Support Protocol 2). Huh7 cells are hepatoma cells isolated from a Japanese male with a well differentiated hepatocellular carcinoma (Nakabayashi et al., 1982). While Huh7 cells have evolved during passaging in different laboratories, a recent screening of Huh7 cells from different laboratories suggests that the majority of Huh7 cells remain permissive for HCVcc infection (Sainz et al., 2009). The HCVcc propagation protocols described in this unit were optimized using a specific Huh7 cell line, but should be effective with most Huh7 cell lines after slight cell line–specific modifications.
Materials
pJFH‐1 (HCV JFH‐1 encoding plasmid; Kato et al., 2001)
XbaI restriction endonuclease and compatible 10× buffer
Buffered phenol, pH 7 to 8 (appendix mca02a)
Molecular‐biology‐grade nuclease‐free distilled H2O
25:24:1 phenol/chloroform/isoamyl alcohol (see appendix mca02a)
Chloroform
3 M sodium acetate, pH 7.0 (see appendix mca02a)
Isopropanol
75% (v/v) ethanol
100% (v/v) ethanol
-
High‐yield in vitro RNA transcription reagents (e.g., Ambion MEGAscript) including:
-
Gel electrophoresis apparatus and reagents including:
Molecular‐biology‐grade agarose
TE buffer (appendix mca02a)
1% (w/v) ethidium bromide (appendix mca02a)
10× sample loading buffer containing bromphenol blue (appendix mca02a)
DNA size ladder
Huh7 cells (Japan Health Science Research Resources Bank, cat. no. JCRB0403; http://www.jhsf.or.jp)
Huh7 cell maintenance medium (see recipe)
Dulbecco's phosphate buffered saline (DPBS) without calcium and magnesium (see recipe in appendix mca02a; prepare with tissue‐culture‐grade H2O and omit Ca2+ and Mg2+)
0.25% trypsin (tissue culture grade) with 2.21 mM EDTA in HBSS (see appendix mca02a for HBSS)
Serum‐free DMEM and/or Opti‐MEM (both available from Invitrogen), prechilled to 4°C
75‐cm2 (T‐75) or 150‐cm2 (T‐150) tissue culture flasks
50‐ and 15‐ml conical centrifuge tubes
Inverted light microscope
Refrigerated centrifuge, 4°C
0.4‐cm electroporation cuvettes
Electroporator
0.2‐µm cellulose acetate filters for sterilization
Additional reagents and equipment for determining DNA and RNA concentration (Gallagher and Desjardins, 2006), RNA purification by purification kit (Phenol‐Free Total RNA Purification Kit or Ribozol Plus RNA Purification Kit; AMRESCO Inc., http://www.amresco-inc.com/), lithium chloride precipitation (Diaz‐Ruiz and Kaper, 1978), spin‐column chromatography, or phenol:chloroform extraction and isopropanol precipitation (Chomczynski and Sacchi, 1987; Kingston et al., 1996), and determination of HCVcc infectivity titer (Support Protocol 3)
Linearize plasmid DNA
-
1
Digest pJFH‐1 with XbaI overnight at 37°C (according to enzyme manufacturer's protocols).
XbaI cuts the pJFH‐1 plasmid once and at the 3′ end of the HCV genome allowing for subsequent run‐off in vitro transcription. XbaI digestion results in a 4‐base‐pair 5′ overhang, which can be removed by treatment with mung bean nuclease as per the manufacturer's instructions; however, removal of the 5′ overhang is not necessary.
Digestion of 20 µg or more of pJFH‐1 DNA is recommended to facilitate efficient recovery after linearization and purification. Linearization of the ∼12.4‐kb plasmid template should be confirmed by agarose gel electrophoresis.
Purify linearized plasmid template
-
2
If the sample volume is less than 400 µl, adjust to at least 400 µl with nuclease‐free distilled water prior to purification.
-
3
Add 500 µl of buffered phenol (pH 7 to 8) and vortex sample vigorously.
CAUTION: Phenol and chloroform (see step 5) are hazardous and require use of protective gloves and eyewear. -
4
Microcentrifuge 10 min at maximum speed, room temperature.
-
5
Transfer upper aqueous phase containing linearized DNA to a new tube, add an equal volume of 25:24:1 phenol/chloroform/isoamyl alcohol, and vortex sample vigorously.
-
6
Microcentrifuge tubes 10 min at maximum speed, room temperature.
-
7
Transfer the upper aqueous phase containing linearized DNA to a new tube, add an equal volume of chloroform, and vortex sample vigorously.
-
8
Microcentrifuge 10 min at maximum speed, room temperature.
-
9
Transfer upper aqueous phase containing linearized DNA to a new tube.
-
10
Add 1/10th volume 3 M sodium acetate, pH 7.0, for a final concentration of 0.3 M sodium acetate (e.g., 44 µl 3 M sodium acetate, pH 7.0, into 400 µl sample) and vortex sample vigorously.
-
11
Add an equal volume of isopropanol and mix by vortexing and vigorously shaking.
Depending on sample volume and maximum tube capacity, one could also precipitate the DNA using 2 volumes of ethanol.
-
12
Precipitate plasmid DNA at −20°C.
At least 1 hr incubation is recommended, but overnight incubation may allow more complete precipitation if smaller amounts of DNA, e.g., less than 5 µg, were digested.
-
13
Pellet DNA by microcentrifugation for 30 min at maximum speed, 4°C.
-
14
Decant isopropanol and wash pellet with 500 µl 75% ethanol.
-
15
Centrifuge tubes 5 min at maximum speed, room temperature.
-
16
Decant 75% ethanol and wash pellet with 500 µl 100% ethanol.
-
17
Centrifuge tubes 5 min at maximum speed, room temperature.
-
18
Decant 100% ethanol. Aspirate any liquid that accumulates on the tube wall, being careful not to disturb the pellet, and let pellet dry at room temperature.
-
19
Resuspend the DNA pellet in nuclease‐free distilled water and determine DNA concentration by spectrophotometry.
The volume of nuclease‐free distilled water used is dependent on the amount of DNA that was originally digested; a concentration of 1 µg/µl is recommended. Samples are stable at this step and can be stored indefinitely at −20°C.
Transcribe JFH‐1 HCV RNA in vitro
-
20Use the linearized pJFH‐1 DNA as template to synthesize genomic HCV JFH‐1 RNA by preparing an in vitro RNA transcription reaction containing:
- 1× RNA transcription reaction buffer
- 7.5 mM each nucleoside triphosphate
- 20 U T7 RNA polymerase
- 0.1 to 1 µg linear DNA template (from step 19).
There are several commercially available high‐yield in vitro transcription kits intended for the synthesis of RNA transcripts of 0.5 kb and longer, which contain specific 10× buffers, polymerases, and other components necessary for RNA transcription. Each kit is different, requiring kit‐specific parameters and reagents; however, theoretically they all follow the same essential procedures.
-
21
Incubate at 37°C for 2 to 6 hr.
Relatively long incubations have proven to be effective for the synthesis of the large ∼9.6kb HCV RNA; however, the optimal incubation period will depend on the specific in vitro transcription reagent used, and needs to be empirically determined.
-
22
To remove the DNA template, add 2 U of DNase, mix well, and incubate at 37°C for 20 min.
-
23
Purify RNAs using one of the following methods:
- RNA purification kit (Phenol‐Free Total RNA Purification Kit or Ribozol Plus RNA Purification Kit; AMRESCO).
- Lithium chloride precipitation (Diaz‐Ruiz and Kaper, 1978).
- Spin‐column chromatography.
Purification methods include but are not limited to these methods. All methods have their particular benefits and all are convenient and effective ways to recover RNA. The authors routinely employ phenol:chloroform extraction and isopropanol precipitation.
-
24
Determine RNA concentration using a spectrophotometer (Gallagher and Desjardins, 2006).
RNA is unstable and is sensitive to both heat and RNases. When handling RNA, thaw and maintain all samples on ice, and wear gloves. All reagents should be of molecular‐biology grade and free of nucleases. Water should be nuclease‐free e.g., DEPC‐treated; see appendix mca02a).
Prepare cells
-
25
At a time point 2 days prior to transfection, seed 1.5 × 106 or 3 × 106 Huh7 cells into 75‐cm2 or 150‐cm2 tissue culture flask, respectively, in Huh7 cell maintenance medium, and incubate overnight.
The number of cells needed depends on the number of transfections to be performed; each transfection will require 4 × 106 cells. In a 75‐cm2 flask at 80% confluency, there are ∼5 × 106 Huh7 cells. In a 150‐cm2 flask at 80% confluency, there are ∼1 × 107 Huh7 cells.
-
26
On the day of transfection, trypsinize subconfluent Huh7 cells by first rinsing the monolayer (in a 75‐cm2 flask) with 10 ml room temperature DPBS (without Ca2+ or Mg2+), then removing the DPBS and adding 3 ml of 0.25% mM trypsin with 2.21 mM EDTA in HBSS (cells should become easily dislodged within 5 min). Watch cells closely to ensure proper trypsinization and stop digestion with Huh7 cell maintenance medium (which contains 10% FBS).
Huh7 cells will clump if over‐trypsinized and this can have a negative impact on transfection efficiency; therefore, watch cells closely to ensure proper trypsinization.
-
27
Transfer cells to a 50 ml centrifuge tube, bring volume up to 30 ml with prechilled (4°C) serum‐free DMEM or Opti‐MEM, and pellet cells for 5 min at 280 × g, 4°C, in a refrigerated centrifuge.
-
28
Aspirate medium off of the cell pellet, resuspend cells in 30 ml pre‐chilled serum‐free DMEM or Opti‐MEM, and pellet cells 5 min at 280 × g, 4°C, in a refrigerated centrifuge. Repeat an additional two times to remove all serum‐associated proteins.
-
29
After the third wash, remove supernatant and resuspend cell pellet in prechilled serum‐free DMEM or Opti‐MEM at a concentration of 1 × 107 cells/ml.
Transfect HCV RNA
-
30
For each transfection, prechill a 0.4 cm electroporation cuvette on ice.
-
31
Add ∼5 µg of RNA template (see steps 23 and 24) and 400 µl of resuspended cells (4 × 106 cells; see step 29) to the cuvette.
-
32
Electroporate the sample at 0.27 kV with a capacitance of 960 µF and a resistance of 100 Ω.
Electroporation conditions vary depending on the instrument and Huh7 cell line/clone used. Thus, electroporation conditions should be empirically determined prior to the start of these experiments. The use of a reporter plasmid construct, such as one encoding GFP, is recommended.
-
33
Remove the cuvette from the instrument and place it back on ice immediately. Gently transfer cells from cuvette to 12 ml of pre‐warmed Huh7 cell maintenance medium. Pipet up and down gently to break up cell clumps.
-
34
Culture cells in a 75‐cm2 culture flask at 37°C until the cells reach ∼80% confluence.
Harvest infectious HCVcc
-
35
Split cells split 1:3 when monolayers reach ∼80% confluence.
Conditioned culture medium can be harvested every time cells are split by collecting the conditioned medium into plastic centrifuge tubes. Virus‐containing medium can be stored at −80°C for future determination of viral titers (Support Protocol 3).
Virus production kinetics will vary following transfection, depending on the cell line/clone and HCV construct used, as well as the method of transfection. Some reports using chimeric virus constructs indicate that optimal viral titers are achieved by day 4 post transfection (Lindenbach et al., 2005), while others using the JFH‐1 viral clone indicate that maximal titers are achieved by day 18 post transfection (Zhong et al., 2005). As such, it is recommended that conditioned medium initially be collected over a large window of time to empirically determine, for each laboratory, the optimal harvest time based on the cell line/clone, viral construct, and transfection method utilized.
To ensure maximum viral titers, maintain cells at subconfluence. Each time cells are split 1:3 they can be expanded into additional flasks (i.e., the original T‐75 into three T‐75s) so that larger volumes of virus‐containing medium are available for collection.
HCV is not cytolytic, but can cause cytopathic effect in vitro when grown to high titers; hence, there will likely be a reduction in cell growth as well as some cell rounding in HCV‐electroporated cells as compared to control cultures.
-
36
After each cell split, add fresh Huh7 cell maintenance medium to flasks and return to the incubator.
If necessary, to help promote efficient viral spread, 1/3 of the used medium containing infectious virus can be returned to the flask after each cell passage.
-
37
Clarify conditioned medium containing HCVcc by centrifugation 10 min at 435 × g, 4°C, to remove cell debris, and filter supernatant through a 0.2‐µm cellulose acetate filter.
There may be excess Huh7 cells in the culture medium on days 1 to 2 post transfection, as efficient electroporation can often result in noticeable cell death.
-
38
Following filtration, dispense the culture fluid into aliquots and store indefinitely at −80°C.
Some researchers report storing HCVcc at 4°C without rapid loss of infectivity; however, the authors always store HCVcc stocks at −80°C. Storage in the presence of 10% fetal bovine serum (as contained in the Huh7maintenance medium in which it is collected) promotes viral stability with respect to repeat freeze‐thaw cycles. However, virus propagated in the absence of serum or purified away from serum proteins is less stable and will lose infectivity during repeated freeze‐thaw cycles.
-
39
Determine HCVcc infectivity titers (see Support Protocol 3).
Support Protocol 2. Generation of HCVcc from Infectious Virus
Huh7 cells can be infected with HCVcc and used to propagate infectious virus, in a manner that is similar to propagation methodologies utilized for other viruses. Depending on the Huh7 cell line and HCV viral clone used, maximal titers can be obtained 5 to 12 days following inoculation of Huh7 monolayers (or suspension cultures) with an MOI of 0.01 ffu/cell.
The advantages of this methodology over the generation of HCVcc from in vitro–transcribed RNA (Support Protocol 2) are (i) the ease of the procedure, (ii) the reduced time frame, and (iii) the ability to generate large volume stocks of HCVcc. Because RNA viruses accumulate mutations every time their genome is replicated, it is impossible to generate a clonal stock of HCVcc, and it is recommended that virus not be passaged multiple times after transfection, in order to limit the selection of specific adaptive mutations. The one‐passage protocol detailed in this unit was optimized to generate ∼54 ml of infectious JFH‐1 HCVcc per harvest day from Huh7 cells with titers ranging from 104 to106 ffu/ml; however, slight adjustments in cell confluence and harvest times may be necessary when growing different viral clones/mutants or when using Huh7 cell clones that exhibit more profound HCVcc‐induced CPE, such as Huh7.5 cells.
In all cases, for optimal virus yields, Huh7 cells should be maintained at subconfluence throughout the viral propagation process.
Materials
Huh7 cells (Japan Health Science Research Resources Bank, cat. no. JCRB0403; http://www.jhsf.or.jp)
Huh7 cell maintenance medium (see recipe)
Post‐transfection generated JFH‐1 HCVcc (Support Protocol 1)
Dulbecco's phosphate buffered saline (DPBS) without calcium and magnesium (see recipe in appendix mca02a; prepare with tissue‐culture‐grade H2O and omit Ca2+ and Mg2+)
0.25% trypsin (tissue culture grade) with 2.21 mM EDTA in HBSS (see appendix mca02a for HBSS)
75‐cm2 and 150‐cm2 tissue culture flasks
50‐ml conical centrifuge tubes (BD Falcon)
Refrigerated centrifuge
0.2‐µm cellulose acetate filter for sterilization
Prepare cells and initiating HCVcc infection
-
1
Seed 1.5 × 106 Huh7 cells in a 75‐cm2 starter flask in 12 ml Huh7 cell maintenance medium and incubate overnight (cells should be ∼60% confluent the next morning).
Several starter flasks can be seeded if larger volumes of HCVcc are necessary; scale quantities according to need.
-
2
Decant the culture medium and infect with post‐transfection generated HCVcc at an MOI of ∼0.01 ffu/cell in a volume of 10 ml of Huh7 cell maintenance medium.
Assuming ∼2 × 106 Huh7 cells, 20,000 ffu HCVcc is need to for inoculation.
-
3
When cells reach ∼90% confluence, on day 1 or 2 post‐infection (depending on how fast the cells are growing), remove medium from flask and place in 50 ml conical (∼12 ml), wash cells once with 10 ml DPBS (without Ca2+ or Mg2+), and trypsinize cells (in 75‐cm2 flask) at room temperature by adding 3 ml of 0.25% trypsin with 2.21 mM EDTA in HBSS (cells should become easily dislodged within 5 min). Watch cells closely to ensure proper trypsinization and stop digestion with Huh7 cell maintenance medium (which contains 10% FBS).
-
4
Resuspend trypsinized cells to a final volume of 10 ml with Huh7 cell maintenance medium and transfer to the conical tube containing the used medium. Bring the cell suspension volume up to 54 ml with Huh7 cell maintenance medium and transfer 18 ml of the resulting cell suspension to each of three 150‐cm2 tissue culture flasks (1:6 split). Incubate cells. Harvest infectious HCVcc.
-
5
Once some evidence of cytopathic effect is observed (for JFH‐1 in Huh7 cells, usually day 5 to 6 post infection), harvest culture medium daily by transferring the conditioned medium into plastic centrifuge tubes.
Maximum virus production kinetics varies depending on the cell line/clone and HCV construct, as well as the MOI. As such, it is recommended that conditioned medium initially be collected over a large window of time to empirically determine, for each laboratory, when peak titers are achieved. In the authors' laboratory, maximal JFH‐1 viral titers are achieved between days 7 to 12 post‐infection (p.i.) after an initial MOI of 0.01 ffu/cell.
To ensure maximum viral titers, maintain cells at subconfluence, splitting the cells 1:3 if the monolayers reach ∼90% confluence. Usually, splitting is required once at day 4 or 5 p.i.; however cells division should substantially slow such that subsequent splitting will no longer be necessary.
-
6
After each virus harvest, add 18 ml of fresh Huh7 cell maintenance medium to each flask and return cells to incubator.
If deemed necessary (i.e., lack of detectable CPE), 1/3 of the used medium containing infectious virus can be returned to the flask after each cell passage to help promote efficient viral spread.
-
7
Clarify conditioned medium containing HCVcc by centrifugation for 10 min at 435 ×8 g, 4°C, and filter through a 0.2‐µm cellulose acetate filter.
-
8
Store virus at −80°C.
Some researchers report storing HCVcc at 4°C without rapid loss of infectivity; however, the authors always store HCVcc stocks at −80°C. At this point, virus can be stored in 50‐ml plastic centrifuge tubes as well as several smaller 2‐ml plastic centrifuge tubes, which can be used for subsequent determination of infectivity titers. Once it is determined which medium harvests contain acceptable viral titers (Support Protocol 3), the medium from multiple harvests can be pooled, and appropriately sized aliquots can be made for future use. Again, several small aliquots of this pool should be made so that the infectivity titer of the pooled virus stock can be determined (Support Protocol 3).
-
9
Determine HCVcc infectivity titers (see Support Protocol 3).
Support Protocol 3. HCVcc Infectivity Titer Analysis
Viral infectivity can be assayed by several different techniques. For cytolytic viruses, like herpes viruses and coronaviruses, the most commonly used technique is plaque assay on cultured cell monolayers in the absence or presence of an inert overlay such as methylcellulose or agarose (Cooper, 1967). Viral infectivity titers are then expressed as plaque‐forming units (pfu)/ml, calculated based on the number of discrete plaques formed in the cell monolayers, the dilution of the inoculum, and the inoculum volume. HCVcc is noncytolytic and does not form plaques on Huh7 monolayers, but it does form small foci of HCV protein–positive cells, which can be visualized by immunostaining of fixed infected Huh7 monolayers and counted to calculate HCV infectivity titers expressed as focus‐forming units (ffu)/ml. To reduce satellite foci due to secondary spread during the assay period, this protocol includes the addition of a methylcellulose overlay. The methodology detailed in this unit describes a horseradish peroxidase–based staining procedure, which the authors have determined to be both user friendly and cost effective; however, the use of fluorescently‐labeled secondary antibodies has also been described (Zhong et al., 2005).
Materials
Huh7 cells (Japan Health Science Research Resources Bank, cat. no. JCRB0403)
Huh7 cell maintenance medium (see recipe)
HCVcc test samples (see protocols above)
Huh7 cell maintenance medium with 0.25% (w/v) methylcellulose (see recipe)
4% PFA fixation buffer (see recipe)
Dulbecco's phosphate buffered saline (DPBS) without calcium and magnesium (see recipe in appendix mca02a; prepare with tissue‐culture‐grade H2O and omit Ca2+ and Mg2+)
DPBS (without Ca2+ and Mg2+) containing 0.3% hydrogen peroxide (prepare immediately before use)
Blocking buffer (see recipe)
-
HCV‐specific primary antibody (Ab), e.g.:
Binding buffer (see recipe)
-
Appropriate HRP‐conjugated secondary antibody (Ab) such as:
DAKOCytomation EnVision+ System‐HRP labeled ready‐to‐use anti‐mouse antibody (for primary Ab raised in mouse)
Goat anti‐human HRP‐conjugated antibody (Pierce; for primary Ab raised in human)
AEC (3‐amino‐9‐ethylcarbazole) peroxidase substrate solution (BD Biosciences)
50% (v/v) glycerol in H2O
96‐well flat‐bottom tissue culture plates
96‐well U‐bottom microtiter plates
Inverted light microscope
Prepare cells
-
1
Seed ∼4 × 103 Huh7 cells into each required well of a 96‐well flat‐bottom tissue culture plate in 100 µl Huh7 cell maintenance medium, and incubate overnight.
Infect cells
-
2
Quickly thaw virus sample to be titered in a 37°C water bath and transfer tube to ice.
-
3
Prepare a series of at least three 10‐fold dilutions of each virus sample in triplicate chilled Huh7 cell maintenance medium.
Dilutions can be done in 96‐well microtiter U‐bottom plates by serially transferring 20 µl of inoculum into wells containing 180 µl Huh7 cell maintenance medium using a multichannel pipettor (Fig. 3). It is important to mix well (by pipetting up and down) and to change pipet tips after each transfer.
Figure 3.
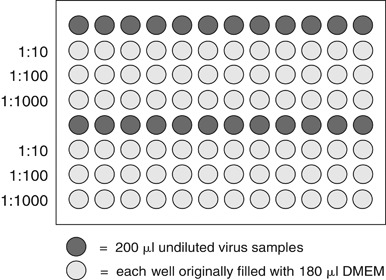
Sample plate layout for virus titration. 24 viral serial dilutions can be conveniently performed in each U‐bottom 96‐well microtiter plate. In this example, the wells in rows 1 and 5 are filled with the individual virus samples to be analyzed while the wells in rows 2 to 4 and 6 to 8 are each filled with 180 µl of Huh7 cell maintenance medium. Using a multichannel pipettor, three 1:10 dilutions can be made by transferring 20 µl between wells down each column, beginning with the undiluted virus row and moving on to the 1:10 row, the 1:100 row, and the 1:1000 row. It is important to thoroughly mix the virus dilution by pipetting up and down after each transfer, and to change pipet tips between each transfer.
-
4
When dilutions are complete, decant the medium from the Huh7 cell 96‐well tissue culture plate and transfer 50 µl of each viral dilution into triplicate cell‐containing wells.
Begin with the most dilute sample, in which case the same pipet tip can be used for increasing concentrations.
-
5
Incubate cells for 24 hr.
-
6
Overlay 150 µl of Huh7 cell maintenance medium containing 0.25% methylcellulose on each well.
Inoculum can be removed after 24 hr and monolayers overlaid with 200 µl of Huh7 cell maintenance medium containing 0.25 % methylcellulose; however, this additional step does not have a significant effect on final titer obtained.
-
7
Incubate the plates for an additional 48 hr.
Stain cells for virus
-
8
At a time point 72 hr post infection, remove medium from wells and add 300 µl of 4% PFA fixation buffer (pH 7) to each well.
-
9
Incubate for 20 min at room temperature in the HCV tissue culture hood.
Wells are filled completely in order to try and ensure inactivation of all virus on the sides of the well. Steps beyond this point can then be performed outside BSL‐2 containment.
-
10
Decant fixative and wash fixed cells three times with 200 µl of DPBS (without Mg2+ or Ca2+).
An extra permeabilization step can be added here if necessary (see Critical Parameters and Troubleshooting section, below).
-
11
To block endogenous peroxidase present in hepatocytes, add 100 µl of 0.3% hydrogen peroxide in DPBS to each well and incubate for 5 min at room temperature.
-
12
Decant peroxide and wash cells three times with 200 µl DPBS.
-
13
Add 50 µl blocking buffer to each well.
-
14
Incubate 1 hr at room temperature.
-
15
Decant blocking buffer.
-
16
Add 30 µl of primary Ab (diluted in binding buffer) to each well (see Table 1 for antibodies and dilutions).
Listed in Table 1 are academically available antibodies that target the HCV E2 glycoprotein (Law et al., 2008) or the viral NS5A protein (Lindenbach et al., 2005). In addition a third, commercially available antibody that targets the HCV Core protein is also listed. All three antibodies are equally suitable for the HRP‐based immunocytochemical procedure described within this unit.
Table 1.
HCVcc Infectivity Titer Assay: Antibodies and Dilutions
|
HCV antigen |
Primary antibody/dilution |
Secondary antibody/dilution |
|---|---|---|
|
E2 |
HRP‐conjugated goat anti‐human 1:1000 |
|
|
NS5A |
E910 (mouse anti‐HCV NS5A) 1:500 (Lindenbach et al., 2005) |
HRP‐conjugated goat anti‐mouse 1:1000 |
|
Core |
C750 (mouse anti‐HCV Core) 1:500 |
HRP‐conjugated goat anti‐mouse 1:1000 |
Also known as C1 (Zhong et al., 2005).
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
-
17
Incubate for 1 hr at room temperature.
-
18
Decant primary Ab/binding buffer and wash cells three times, each time with 200 µl/well DPBS.
-
19
Add 30 µl of secondary Ab (diluted in binding buffer) to each well (see Table 1 for antibodies and dilutions).
-
20
Incubate for 1 hr at room temperature.
-
21
Decant secondary Ab/binding buffer and wash cells three times with 200 µl/well DPBS.
-
22
At a time point 5 min prior to decanting the secondary Ab, prepare the AEC substrate according to the manufacturer's instructions.
-
23
Add 30 µl of the AEC substrate to each well.
-
24
Incubate for 20 to 40 min.
Incubation at 37°C accelerates the reaction time; however, incubation time can vary depending on the substrate used. As such, adhere to the manufacturer's instructions specific for the substrate used, or determine optimal incubation times empirically.
-
25
Decant AEC substrate and wash cells three times with 200 µl/well water.
-
26
Overlay each well with 100 µl of 50% (v/v) glycerol.
Wells can immediately be viewed with an inverted light microscope and foci quantified. Plates can be stored for ∼1 month at 4°C. For extended storage, the authors recommend using a plate seal to minimize evaporation.
Analyze titer results
-
27
To calculate a titer in focus‐forming units (ffu)/ml, under an inverted light microscope, select triplicate culture wells with a sufficient number of foci to give a reliable count, and count the foci.
For example, choose wells with ∼5 to 70 distinct foci.
-
28
Using the average number of foci in the triplicate wells, calculate the titer in ffu/ml using the following formula:
titer (ffu/ml) = (foci number) × (1/dilution factor of the virus preparation) (1/volume plated in ml).
For example, if an average of 20 foci were found at a 103 dilution after inoculation of 50 µl, then the ffu/ml = 20 ffu × (1/10−3) × (1/0.05 ml) = 4 × 105 ffu/ml.
-
29
Alternatively, to express infectivity titers as tissue culture infectious dose 50 (TCID50), select a culture well that expresses at least one HCV‐positive cell, and determine the TCID50 according to the method of Reed and Muench (Reed and Muench, 1938).
Reagents and Solutions
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see appendix mca02a; for suppliers, see mcaspl.
Binding buffer
400 ml Dulbecco's phosphate‐buffered saline (DPBS) without calcium and magnesium (see recipe in appendix mca02a; prepare with tissue‐culture‐grade H2O and omit Ca2+ and Mg2+)
15 g bovine serum albumin, Fraction V
2.5 ml 10% (v/v) Triton X‐100
Bring to 500 ml total volume with DPBS (without CaCl2 and MgCl2)
Filter sterilize using a 0.2‐µm cellulose acetate filter
Store up to 1 year at 4°C
Final composition: 1× DPBS/3% BSA/0.5% Triton X‐100.
Blocking buffer
400 ml Dulbecco's phosphate‐buffered saline (DPBS) without calcium and magnesium (see recipe in appendix mca02a; prepare with tissue‐culture‐grade H2O and omit Ca2+ and Mg2+)
15 g bovine serum albumin, Fraction V
2.5 ml 10% (v/v) Triton X‐100
50 ml fetal bovine serum (FBS; 10% final), heat‐inactivated (56°C for 30 min)
Bring to 500 ml total volume with DPBS (without CaCl2 and MgCl2)
Filter sterilize using a 0.2‐µm cellulose acetate filter
Store up to 1 year at 4°C
Final composition: 1× DPBS/3% BSA/0.5% Triton X‐100/10% FBS.
FRET lysis buffer
1.25% (v/v) Triton X‐100
50 mM Tris⋅Cl, pH 7.5 (appendix mca02a)
150 mM NaCl
2 mM EDTA
Store up to 1 year at 4°C
Huh7 cell maintenance medium
500 ml Dulbecco's high‐glucose minimal essential medium (DMEM)
5 ml 200 mM glutamine
5 ml nonessential amino acids
5 ml 1 M HEPES buffer
50 ml fetal bovine serum (FBS; 10% final), heat‐inactivated (56°C for 30 min)
5 ml pen/strep (10,000 U/ml penicillin G sodium and 10,000 µg/ml streptomycin sulfate)
Store up to 1 month at 4°C
Methylcellulose overlay, 0.25% in Huh7 cell maintenance medium
2% methylcellulose stock solution: Add 10 g of methylcellulose (Methocel MC, Fluka Bio‐Chemika, cat. no. 64625) to 500 ml water in a sterile 1‐liter bottle containing a stir bar. Stir at 4°C until methylcellulose partially goes into solution (∼4 hr). Do not heat; methylcellulose will only dissolve in cold solutions. Sterilize by autoclaving. Allow solution to return to room temperature (methylcellulose will solidify), shake vigorously to break up clumps, and transfer to 4°C overnight. It will not be possible to stir the methylcellulose, but it will gradually go into solution.
0.25% methylcellulose overlay: Add 1 part 2% methylcellulose stock solution to 7 parts Huh7 cell maintenance medium. Mix well and store at 4°C in the dark.
PFA fixation buffer, 4%
20 g paraformaldehyde (PFA)
400 ml Dulbecco's phosphate‐buffered saline (DPBS) without calcium and magnesium (see recipe in appendix mca02a; prepare with tissue‐culture‐grade H2O and omit Ca2+ and Mg2+)
Adjust pH to 7.4 with 10 N NaOH (PFA will not go into solution until pH is 7.4)
Aid dissolution by heating solution in fume hood (do not let solution rise above 65°C)
Bring to 500 ml total volume with DPBS (without CaCl2 and MgCl2)
Filter sterilize, wrap in aluminum foil, and store up to 1 year at 4°C
RNA transcription reaction buffer, 10×
20 mM Tris⋅Cl, pH 7.5 (appendix mca02a)
5 mM sodium chloride
9 mM magnesium chloride
Aliquot and store up to 1 year at −20°C
Commentary
Background Information
Hepatitis C virus (HCV) is a hepatotropic enveloped positive‐strand RNA virus and a primary causative agent of liver disease worldwide. Currently, HCV affects ∼170 million individuals globally, causing significant liver disease, including cirrhosis and hepatocellular carcinoma (HCC) in chronically infected individuals (Alter and Seeff, 2000). In fact, in the United States, HCV‐related HCC accounts for over 50% of HCC cases and over 30% of liver transplants performed annually.
HCV is classified in the family Flaviviridae based on conservation of the viral RNA‐dependent RNA polymerase and genome organization (Lindenbach and Rice, 2005). The ∼9.6 kb RNA genome, flanked by highly structured 5′ and 3′ untranslated regions, encodes a single open reading frame, which is translated via a viral internal ribosome entry site into an ∼3010‐amino‐acid viral polyprotein. The viral polyprotein is co‐ and post‐translationally cleaved into structural and nonstructural (NS) proteins by both host and viral proteases. The NS viral proteins assemble on cellular membranes of the endoplasmic reticulum and mediate the formation of the viral RNA replication complex (also known as the membranous web) where negative strand RNA synthesis occurs (Gosert et al., 2003). The negative‐strand RNA then provides the template for ∼10‐fold amplification of positive‐strand genomic RNA, which is subsequently encapsidated by the viral nucleocapsid protein (i.e., Core), and directed into the cellular lipoprotein secretory pathway, where progeny virions further mature and become lipidated prior to exiting the cell (Lindenbach and Rice, 2005).
Since its discovery in 1989, the only effective treatment option for HCV has been interferon α (IFN‐α) treatment in combination with ribavirin (Glue et al., 2000). Unfortunately, this therapy has a wide spectrum of toxic side effects and is only effective in a subset of patients. With the number of HCV patients requiring treatment expected to significantly increase over the next decade (Williams, 2006), there is an obvious and immediate need for new and more effective HCV antivirals. As a result, high‐throughput‐screening (HTS) assays utilizing the recently developed infectious HCV cell culture system (Lindenbach et al., 2005; Wakita et al., 2005; Zhong et al., 2005) as a means to identify new anti‐HCV compounds that target all steps of the HCV life cycle are needed. This unit describes a unique HCV HTS cell‐based Fluorescence Resonance Energy Transfer (FRET) assay for antiviral compound screening (Yu et al., 2009), which combines the use of infectious HCVcc and non‐dividing hepatoma cell cultures (Sainz and Chisari, 2006; Choi et al., 2009) with a sensitive FRET‐based readout for the endogenous, virally‐encoded NS3 protease.
This assay offers several distinct advantages over conventional HCV antiviral compound screening assays. First, while traditional viral HTS assays typically aim to achieve a synchronized single‐cycle round of viral replication using a high multiplicity of infection (MOI) approach, this results in biased selection of inhibitors that target viral entry and replication over those compounds that inhibit later viral infection processes such as maturation, egress, and any subsequent cell‐to‐cell spread. To avoid this predisposition and to identify inhibitors of all aspects of the viral life cycle, the HCV HTS assay described herein is based on a low‐MOI 6‐day infection during which multiple rounds of HCV infection, and spreading of the virus throughout the culture, occur. As such, inhibitors that target any step of the viral life cycle can be successfully identified. Secondly, to accommodate this long, 6‐day infection, this HCV HTS assay makes use of nondividing, synchronized hepatoma cell cultures, which allow for highly reproducible, robust HCVcc infection over an extended period of time, virtually eliminating the well‐to‐well cell culture–related variability that often plagues cell‐based HTS strategies. Additionally, these ready‐for‐use cultures are easily maintained in and have an inherent tolerability to the common compound library diluent dimethylsulfoxide (DMSO). For the average researcher, it is also advantageous that the described HCV FRET assay can be performed with any standard infectious HCV clone (i.e., no special reporters need to be engineered into the viral genome) and makes use of a sensitive commercially available internally quenched NS3 peptide substrate.
Critical Parameters and Troubleshooting
Anti‐HCV compound screening protocol
Cell culture. As with all HCV‐related cell culture, the use of low‐passage Huh7 cells (or at least usage of cells within a defined HCV‐permissive cell passage window) is recommended. Additionally, it is imperative that cells be mycoplasma free and greater than 90% viable before use, as HCV infection can be affected by all these variables. Culturing of Huh7 cells in 1% DMSO to induce cell growth arrest involves some additional considerations. First, while this method has proven successful with numerous distinct Huh7 cell lines, it is recommended that each cell line be tested individually to ensure cells behave as expected (e.g., cell growth cessation and liver‐specific gene expression enhancement occurring at approximately day 6 post DMSO treatment). One potential problem with DMSO treatment is excess DMSO, which can result in Huh7 cell death and cytotoxicity. When preparing Huh7 cell maintenance medium with 1% (v/v) DMSO, accurately measure all volumes to ensure a final DMSO concentration of 1%. The use of tissue‐culture‐grade DMSO (Sigma) is recommended. Another potential problem is the overgrowth of Huh7 cells in the presence of 1% DMSO. This can happen when the Huh7 cell maintenance medium with 1% DMSO is initially added to cultures too late, when the cells are already confluent or over‐confluent rather than 85% to 90% confluent. The judgement regarding when to add the 1% DMSO–containing medium is subjective, and this should be determined empirically before beginning large‐scale experiments with DMSO‐treated cultures. Lastly, during the 20‐day incubation period, medium must be changed every 2 to 3 days, as Huh7 cells incubated in the presence of 1% DMSO are extremely metabolically active, and thus utilize nutrients at rates significantly faster than their actively dividing counterparts. As such, medium pH quickly becomes acidic, and nutrients are rapidly exhausted, particularly after 6 days of DMSO treatment; medium changes every 2 days are therefore necessary.
HCV infection. To ensure reproducibility throughout a single library screening effort, an HCVcc stock sufficient to complete the entire screen should be produced, and detailed 6‐day infection kinetics should be established prior to screening. Subsequent virus storage and handling techniques should also be standardized.
Compound screening. In this unit, we describe an approach that accommodates unstable compounds that must be kept frozen with minimum freeze/thaw cycles; however, handling of different compound libraries should be determined according to the stability and properties of the compounds being tested (e.g., it may be possible to use a deep‐well microtiter plate to predilute drugs in sufficient medium for all medium changes, and store this at 4°C during the 6‐day infection).
When performing the screen itself, standard positive and negative controls must be included on each individual screening plate as internal controls, so that results can be compared between plates. Positive controls could be known potent HCV inhibitors, such as IFN. Negative controls should include diluent only treatments.
FRET assay
The HCV FRET assay is a simple mix‐and‐read fluorescence‐based assay that measures HCV infectivity as a function of NS3 protease activity in a cell‐based system. Like all HTS assays, however, it is critical to carefully consider parameters such as lysis reagents, substrate activity, statistical validation of the assay, and data interpretation.
Lysis buffer. Complete lysis of cells is needed for proper quantification of NS3 protease activity. While standard growing Huh7 monolayers are efficiently lysed by traditional lysis buffers, including those provided by the manufacturer of the NS3 substrate (AnaSpec), lysis of DMSO‐Huh7 cultures requires more stringent conditions due to their tightly packed morphology. As such, individual lysis buffers were empirically tested, and it was determined that the FRET lysis buffer described in this unit (see Reagents and Solutions) allows for sufficient DMSO‐Huh7 cell culture lysis without reducing NS3 protease activity (Yu et al., 2009). In addition, it is recommended that each screening plate be frozen before FRET analysis, so that DMSO‐Huh7 cells undergo one freeze‐thaw cycle in the FRET lysis buffer.
Substrate. The 5‐FAM/QXL 520 NS3 FRET peptide substrate (AnaSpec) was chosen for its high sensitivity; however, any FRET peptide substrate containing the appropriate HCV NS3 cleavage site would be suitable as long as the final signal‐to‐noise ratio is acceptable, which is of particular importance when developing fluorescence‐based assays, as these typically have lower signal‐to‐background ratios than other commonly HTS assays (Zhang et al., 1999). Thus, if an alternate substrate is utilized, it is advisable to optimize signal‐to‐background ratios and to test multiple lysis buffers and read‐out parameters against an appropriate panel of positive and negative controls prior to performing a compound screening campaign.
Important for any fluorescently tagged peptide substrate is its proper storage and handling. Because fluorescently tagged peptide substrates are light and temperature sensitive, care should be taken to reduce exposure of the substrate to light and to avoid repeat freeze‐thaw cycles. To avoid these issues and control for lot‐to‐lot substrate variability, it is recommended, when feasible, to obtain bulk quantities of a particular peptide substrate lot and immediately aliquot it on ice into individual single‐use tubes, which can be kept at −80°C for long‐term storage. 60 µl of the 5‐FAM/QXL 520 NS3 FRET peptide substrate (AnaSpec) is sufficient to test an entire 96‐well microtiter sample plate, but aliquot volumes should be adjusted based on individual compound screening needs.
Statistical validation of HCV FRET assay. Critical to any HTS is reproducibility and consistent assay performance. The Z′ factor is a value that measures the quality of an assay by assessing its dynamic range as well as the variation within the assay by measuring the distance between the standard deviations for the positive (signal) and negative (noise) controls. This value also assesses well‐to‐well variability and the noise/error associated with the assay. For a HTS to be considered accurate, a Z′ value of greater than 0.5 is necessary. Following the procedures outlined in this unit, the HCV FRET described routinely exhibits a Z′ factor greater than 0.6 (Yu et al., 2009); however, this should be empirically determined for each user/laboratory.
Data interpretation. The success of any compound screening assay lies in the ability of the assay to distinguish hits from non‐hits. Thus, it is important to calculate an assay hit ratio to determine an appropriate hit window for identifying candidate anti‐HCV compounds, as described in Zhang et al. (1999). From a biological perspective, it is advisable to follow up with compounds that reproducibly inhibit HCV ≥5‐fold, relating to a 20% hit zone. If a 20% hit zone identifies an unmanageable number of hits, a more stringent hit window (e.g., 10%) can be established. Likewise, if too few compounds are identified using a 20% hit zone, the hit window can be increased (e.g., up to 30%) to accept more test compounds as potential inhibitors of HCVcc.
Propagation of HCVcc
In vitro–transcribed RNA quality. The ability to efficiently produce infectious HCVcc after transfection of in vitro–transcribed HCV RNA is dependent on the presence of sufficient quantities of full‐length HCV RNA genomes. The pJFH‐1 expression vector contains a T7 promoter, suitable for in vitro RNA transcription of the ∼9.6 kb RNA genome, but optimization may be necessary for optimal synthesis of the large HCV RNA transcript, even when using commercially available transcription kits according to the manufacturer's recommendations. It is highly recommended that in vitro–transcribed RNA be visualized by gel electrophoresis to assess RNA quality, as described (Brown et al., 2004). The presence of a predominant smear of RNAs of different sizes rather than a predominant single band (with slight smear of incomplete products) can indicate incomplete transcription or degradation, which can negatively impact intracellular HCV RNA replication following transfection. The addition of an RNase inhibitor and optimization of transcription reaction incubation period can help ensure that the RNA stock contains a high percent of intact HCV RNA genomes. If RNAs longer than genome length are observed, it may indicate that you have not completely linearized your template DNA and are thus getting continual RNA synthesis around an intact plasmid. This can be resolved by either redigesting the template or by incorporating a step in which the linearized template DNA is separated on an agarose gel before purification. Finally, because contaminants such as proteins or residual phenol, chloroform, or ethanol may reduce transfection efficiency, A 260/A 280 spectrophotometric analysis of the in vitro transcribed RNA is recommended to confirm RNA purity. Further purification including an optional proteinase K treatment step can be performed, if necessary, to obtain better RNA purity.
Transfection. The original reports describing generation of HCVcc from in vitro transcribed RNA utilized electroporation to transfect Huh7 cells (Lindenbach et al., 2005; Wakita et al., 2005; Zhong et al., 2005). While different Huh7 cell lines may respond optimally to slightly different electroporation conditions, the parameters described here should represent a reasonable starting point for optimization. Additionally, the choice of transfection method theoretically is not limited to electroporation, as other methodologies such as cationic lipid–mediated transfection using Lipofectamine 2000 (Invitrogen) have also been used with comparable efficacy (Zhong et al., 2005). Thus, any RNase‐free transfection protocol should be suitable as long as intact RNAs are delivered effectively to the target cells with efficiencies of at least 60%. In any case, it is advisable to first ensure (or at least determine in parallel) that your transfection protocol is effective by using plasmid vectors encoding a reporter gene, such as GFP.
HCV replication. Failure to produce detectable levels of infectious HCVcc may also be due to inadequate intracellular replication of transfected in vitro–transcribed HCV RNA. Although the majority of Huh7 cells are permissive for HCVcc infection and support robust HCV JFH‐1 replicon RNA replication (Sainz et al., 2009), specific laboratory cell lines may not support initiation of HCV RNA replication as efficiently. To assess intracellular HCV RNA replication following transfection, quantitation of HCV RNA by quantitative real‐time polymerase chain reaction (qRT‐PCR) can be performed as described (Sainz et al., 2009). Likewise, HCV protein levels (indicative of efficient RNA replication) can be determined by immunostaining, as described in Support Protocol 3.
Huh7 cells. Of major importance in the generation of high‐titer HCVcc stocks is the state of the cells during propagation. Under optimal cellular conditions, JFH‐1 HCVcc should grow to titers of 105 ffu/ml; however, over‐confluency, mycoplasma contamination, and other changes in the state of the host cell can negatively impact HCV RNA replication and de novo virus production (Pietschmann et al., 2001; Windisch et al., 2005; Nelson and Tang, 2006; Sainz and Chisari, 2006). Thus, it is critical to use Huh7 cells that have been maintained in a healthy growth state and that have not been allowed to become over‐confluent at the time of transfection, infection, or at any point thereafter. Moreover, during long‐term passaging, Huh7 cell permissiveness for HCVcc infection can fluctuate (Corcoran et al., unpub. observ.). Hence, particularly to ensure reproducibility throughout large‐scale HCV screening efforts, it is recommended that Huh7 cell passage be monitored such that cells are only cultured for a limited number of passages (i.e., ∼15 splits). At this time, cells should be discarded and an equivalent frozen aliquot thawed for continued use. Obviously, this requires that cells to be used be initially expanded and that large numbers of parallel aliquots be frozen for future use. Lastly, mycoplasma contamination should be avoided and cell lines treated if determined to be mycoplasma positive (see appendix mca03b).
Suboptimal viral titers. Although HCV is considered a noncytolytic virus, HCVcc infection in Huh7 cells does result in detectable cytopathic effect (CPE) characterized by a slowing of cell growth and the appearance of rounded, sometimes floating cells. The degree of CPE observed varies among different Huh7 cell lines (Sainz et al., 2009), but the onset of CPE usually coincides with maximal titer production. As such, harvesting virus during the initial onset of HCV‐induced CPE is recommended.
If titers achieved by the methods described in this unit are suboptimal for specific downstream purposes, higher titers can be obtained by concentrating viral stocks using centrifugal concentrators, such as Amicon Ultra‐4 Centrifugal Filter Units (Millipore). If harvested in serum‐free medium, virus can be concentrated at least 100‐fold without loss of infectivity.
Cell culture‐adapted mutations. As mentioned in Support Protocol 2, when propagating RNA viruses, mutations are incorporated into the viral genome every time it is replicated, due to intrinsic error rate and lack of proofreading function of the viral RNA–dependent RNA polymerase. Hence, during continuous passage in cell culture, selection for specific “cell‐culture adapted” mutants can occur (Zhong et al., 2006). While mutations that confer an advantage for viral growth in cell culture can randomly occur any time during infection, a significant shift in the virus population as a whole usually requires several weeks of continuous virus selection to occur. Thus, the HCVcc propagation methods described in Support Protocols 1 and 2 should not typically result in the emergence/selection of specific cell‐culture mutations. Bulk sequencing of the virus present in medium collected 18 days post transfection and 10 days post subsequent P1 infection has indicated no detectable selection of cell culture mutations in past virus stock preparations; however, only bulk sequencing can empirically confirm in a conclusive way whether an adaptive mutation has been selected within a specific virus stock.
Infectivity titer assay
The HCV infectivity titer assay described in Support Protocol 3 is based on standard titering protocols successfully utilized for numerous viruses (Condit, 2007); however, some HCV‐specific parameters should be noted.
Abnormal and/or difficult to count HCV foci. Although noncytolytic, HCV does form discrete foci on Huh7 monolayers. Ideally, by 72 hr post‐infection, when cells are fixed and stained, HCV protein positive foci consist of 5 to 10 grouped cells. However, sometimes individual foci are not readily distinguishable, due to the unexpected differences in the size or shape of the foci. Although foci size does vary depending on the specific Huh7 cell line used for titering (Sainz et al., 2009), it is more often the case that simple adjustments can be made to optimize foci formation. First, it can be very helpful to include a simple overlay to minimize secondary spread, which can give rise to satellite foci. Thus, the addition of 0.25% (w/v) methylcellulose is recommended to ensure the formation of countable discrete HCV foci. Additionally, if foci are too large at 72 hr post‐infection, cells can be fixed and stained earlier (e.g., at 48 hr post‐infection). In contrast, if foci are too small to reliably count, this may be the result of the cells being over‐confluent at the time of infection, suboptimal media conditions, use of high‐passage Huh7 cells, contamination with mycoplasma, or incubator CO2 levels exceeding 5%.
Inconsistent or suboptimal staining. As with all immunostaining methodologies, issues such as antibody specificity, cell permeability, and background may arise. The protocol described herein using the antibodies listed provided in Table 1 has been designed to ensure maximum staining and visualization of HCV foci. If inconsistent or suboptimal staining is encountered, all reagents should first be checked. Alternatively, standard changes in antibody dilutions, incubation periods, and incubation temperatures can be tested. Lastly, an additional permeabilization step consisting of a single 10‐min wash with ice‐cold 50% acetone/50% methanol (v/v) can be incorporated before Support Protocol 3, step 10.
Anticipated Results
The HCVcc FRET assay described herein is a useful and effective procedure for testing the efficacy of potential HCV antivirals against all aspects of the viral life cycle.
The use of non‐growing cell cultures provides a unique cell culture environment in which to test the efficacy of test compounds on HCV, and imparts the advantage of virtually eliminating the well‐to‐well variability routinely encountered in cell‐based compound screening assays that use actively dividing cell cultures. As early as 6 days post incubation in 1% DMSO, Huh7 cell should enter growth arrest, and, soon thereafter, form tightly packed monolayers of mono‐ and binucleated cells displaying the typical pavement‐like cytological features of primary hepatocytes. As previously described (Yu et al., 2009), these cells support robust HCVcc infection; therefore, following 6 days of infection, HCV NS3 protease activity as measured by FRET fluorescence should yield maximal RFU values if untreated, with a noise‐to‐background ratio of ∼6 to 8. Samples treated with compounds that inhibit HCV at any step in the viral life cycle (e.g., entry, uncoating, translation, replication, assembly, maturation, and egress) should yield RFU values significantly below those of the untreated HCV‐infected control wells. For example, interferon‐β, a potent inhibitor of HCV infection, reduces NS3 FRET RFU values by 90% to 95% as compared to untreated HCV‐infected control samples. It will not be uncommon to see variations in RFU values from experiment to experiment when working with different substrate lots (see Critical Parameters, “Substrate”); however, if inhibition is appropriately calculated relative to the proper controls, the percent reductions should be comparable from experiment to experiment.
Generation of HCVcc in Huh7 cells by either transfection of in vitro–transcribed HCV JFH‐1 RNA (Support Protocol 1) or by infection of Huh7 cells with HCVcc (Support Protocol 2) should yield titers between 104 and 106 ffu/ml. However, due to the variation inherent in HCVcc infection observed, due in part to the cell lines/clone utilized, the time post transfection or post infection that will yield maximal titers will vary. Nonetheless, following the protocols described within this unit, maximal JFH‐1 titers in Huh7 cells should be achieved by day 18 post transfection or day 12 post infection. Once HCVcc has been generated, the HCV infectivity titer assay (Support Protocol 3) can be used to determine viral titer expressed as ffu/ml. The assay should yield discrete HCV foci consisting of 5 to 10 HCV protein‐positive cells.
Time Considerations
For planning purposes, the durations of individual steps for each protocol are detailed in Table 2 and summarized below.
Table 2.
Time Requirements for Procedures Related to the Cell‐Based FRET Assay for HCV
|
Procedure |
Time required |
Protocol and step |
|---|---|---|
|
Plating microtiter plates |
1‐2 hr |
Basic Protocol 1, step 2 |
|
Incubation in the presence of 1% DMSO |
20 days |
Basic Protocol 1, steps 3‐4 |
|
Preparation of compounds |
1‐3 hr |
Basic Protocol 1, steps 5‐6 |
|
Treatments and HCVcc infection |
6 days |
Basic Protocol 1, steps 7‐18 |
|
Harvesting media and cells |
1 hr |
Basic Protocol 1, steps 19‐24 |
|
FRET assay preparation |
1 hr |
Basic Protocol 2, steps 1‐7 |
|
FRET assay |
2‐3 hr |
Basic Protocol 2, steps 8‐11 |
|
Linearization of plasmid DNA |
Overnight |
Support Protocol 1, step 1 |
|
Purification of linearized plasmid template |
3 hr‐overnight |
Support Protocol 1, steps 2‐19 |
|
In vitro transcription and purification |
Overnight |
Support Protocol 1, steps 20‐24 |
|
Plating cells for transfection |
1 hr |
Support Protocol 1, steps 25 |
|
Transfection |
2 hr |
Support Protocol 1, steps 26‐34 |
|
Splitting and reseeding transfected cells and harvesting HCVcc |
2‐18 days |
Support Protocol 1, steps 25‐39 |
|
Plating cells for HCVcc infection |
1 hr |
Support Protocol 2, step 1 |
|
Infection |
1 hr |
Support Protocol 2, step 2 |
|
Splitting and reseeding transfected cells and harvesting HCVcc |
2‐10 days |
Support Protocol 2, steps 3‐8 |
|
Plating cells for HCVcc titering |
1 hr |
Support Protocol 3, step 1 |
|
Preparing HCVcc test sample dilutions |
1 hr |
Support Protocol 3, step 2‐3 |
|
Infection |
1 hr |
Support Protocol 3, step 4 |
|
Incubation |
3 days |
Support Protocol 3, step 5‐7 |
|
Staining |
4 hr |
Support Protocol 3, steps 8‐26 |
|
Foci counting and titer analysis |
1 hr |
Support Protocol 3, steps 27‐28 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
FRET assay
The FRET assay described in this unit (Basic Protocols 1 and 2) can take several weeks to complete; however, the assay can be divided into manageable sub‐procedures, with stopping points along the way, which afford efficient execution of the entire assays from start to finish. As with any compound library screening endeavor, proper planning prior to beginning the assay is advised to ensure that needed reagents/compounds are available and appropriately diluted at the correct times, and that mandatory steps, such as preparation of cells and infections, occur at appropriate intervals throughout the assays. To this end, designing a strategic plan in advance of initiating the assay will prevent unanticipated errors and delays. The FRET assay has been divided into two basic protocols that require a total of ∼27 days (Basic Protocol 1) and 4 hr (Basic Protocol 2) to complete.
Therefore, all procedures can be completed either in tandem or can be alternated. For example, step 24 at the end of Basic Protocol 1 represents a stopping point where sample plates can be stored indefinitely. Thus, the authors advise that Basic Protocol 2 be performed once a sufficient number of sample plates for FRET analysis have been accumulated. This will ensure more consistent FRET results between assay plates, as all fluorescence‐related procedures will be performed together and over the course of a few days rather than staggered across several weeks.
In addition, it is advised that preparation of cell culture plates be staggered such that a manageable number of plates are handled at one time. Since cells must incubate for 3 weeks (i.e., 20 days) in the presence of 1% DMSO, batches of plates can be seeded weekly, thus reducing the number of plates to be treated and infected at one time. The aforementioned suggestions, however, are dependent on the number of compounds to be tested and liquid handling equipment available.
Propagation and titering of HCVcc
In addition to the FRET assay described in Basic Protocols 1 and 2, Support Protocols 1 through 3 detail procedures for generating HCVcc and determining infectivity titer. Generation of HCVcc can be accomplished by transfection of Huh7 cells with in vitro–transcribed HCV JFH‐1 RNA (Support Protocol 1), or by infection of cells with HCVcc (Support Protocol 2), requiring 7 to 21 days or 10 days, respectively. Once HCVcc has been generated, infectivity titer analysis can be performed (Support Protocol 3) within 4 days, with foci‐counting and titer‐analysis time varying depending on the method of detection (HRP versus immunofluorescence) and the number of wells to be counted.
Literature Cited
- Alter, H.J. and Seeff, L.B. 2000. Recovery, persistence, and sequelae in hepatitis C virus infection: A perspective on long‐term outcome. Semin. Liver Dis. 20:17‐35. [DOI] [PubMed] [Google Scholar]
- Bianchi, E. , Steinkuhler, C. , Taliani, M. , Urbani, A. , Francesco, R.D. , and Pessi, A. 1996. Synthetic depsipeptide substrates for the assay of human hepatitis C virus protease. Anal. Biochem. 237:239‐244. [DOI] [PubMed] [Google Scholar]
- Brown, T. , Mackey, K. , and Du, T. 2004. Analysis of RNA by northern and slot blot hybridization. Curr. Protoc. Mol. Biol. 67: 4.9.1‐4.9.19. [DOI] [PubMed] [Google Scholar]
- Choi, S. , Jr. Sainz, B., , Corcoran, P. , Uprichard, S. , and Jeong, H. 2009. Characterization of increased drug metabolism activity in dimethyl sulfoxide (DMSO)‐treated Huh7 hepatoma cells. Xenobiotica 39:205‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski, P. and Sacchi, N. 1987. Single‐step method of RNA isolation by acid guanidinium thiocyanate‐phenol‐chloroform extraction. AnalBiochem 162:156‐159. [DOI] [PubMed] [Google Scholar]
- Condit, R. 2007. Principles of virology In Field's Virology (Fields B., Knipe D., Howley P., and Griffin D., eds.) pp. 25‐55. Lippincott Williams & Wilkins, Philadelphia. [Google Scholar]
- Cooper, P.D. 1967. The plaque assay of animal viruses In Methods in Virology (K. Maramorosch, K. and H. Koprowski, eds.) pp. 243‐311. Academic Press, New York; London. [Google Scholar]
- Diaz‐Ruiz, J. R. and Kaper, J. M. 1978. Isolation of viral double‐stranded RNAs using a LiCl fractionation procedure. Prep. Biochem. 8:1‐17. [DOI] [PubMed] [Google Scholar]
- Gallagher, S.R. and Desjardins, P.R. 2006. Quantitation of DNA and RNA by absorption and fluorescence spectroscopy. Curr. Protoc. Mol. Biol. 76:A.3D.1‐A.3D.21. [DOI] [PubMed] [Google Scholar]
- Glue, P. , Fang, J.W. , Rouzier‐Panis, R. , Raffanel, C. , Sabo, R. , Gupta, S.K. , Salfi, M. , and Jacobs, S. 2000. Pegylated interferon‐alpha2b: Pharmacokinetics, pharmacodynamics, safety, and preliminary efficacy data. Hepatitis C Intervention Therapy Group. Clin. Pharmacol. Ther. 68:556‐567. [DOI] [PubMed] [Google Scholar]
- Gosert, R. , Egger, D. , Lohmann, V. , Bartenschlager, R. , Blum, H.E. , Bienz, K. , and Moradpour, D. 2003. Identification of the hepatitis C virus RNA replication complex in Huh‐7 cells harboring subgenomic replicons. J. Virol. 77:5487‐5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakiuchi, N. , Nishikawa, S. , Hattori, M. , and Shimotohno, K. 1999. A high throughput assay of the hepatitis C virus nonstructural protein 3 serine proteinase. J. Virol. Methods 80:77‐84. [DOI] [PubMed] [Google Scholar]
- Kato, T. , Furusaka, A. , Miyamoto, M. , Date, T. , Yasui, K. , Hiramoto, J. , Nagayama, K. , Tanaka, T. , and Wakita, T. 2001. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J. Med. Virol. 64:334‐339. [DOI] [PubMed] [Google Scholar]
- Kato, T. , Date, T. , Miyamoto, M. , Furusaka, A. , Tokushige, K. , Mizokami, M. , and Wakita, T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808‐1817. [DOI] [PubMed] [Google Scholar]
- Kingston, R. , Chomczynski, P. , and Sacchi, N. 1996. Guanidine methods for total RNA preparation. Curr. Protoc. Mol. Biol. 36:4.2.1‐4.2.9. [DOI] [PubMed] [Google Scholar]
- Law, M. , Maruyama, T. , Lewis, J. , Giang, E. , Tarr, A.W. , Stamataki, Z. , Gastaminza, P. , Chisari, F.V. , Jones, I.M. , Fox, R.I. , Ball, J.K. , McKeating, J.A. , Kneteman, N.M. , and Burton, D.R. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25‐27. [DOI] [PubMed] [Google Scholar]
- Lindenbach, B.D. and Rice, C.M. 2005. Unravelling hepatitis C virus replication from genome to function. Nature 436:933‐938. [DOI] [PubMed] [Google Scholar]
- Lindenbach, B.D. , Evans, M.J. , Syder, A.J. , Wölk, B. , Tellinghuisen, T.L. , Liu, C.C. , Maruyama, T. , Hynes, R.O. , Burton, D.R. , McKeating, J.A. , and Rice, C.M. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623‐626. [DOI] [PubMed] [Google Scholar]
- Nakabayashi, H. , Taketa, K. , Miyano, K. , Yamane, T. , and Sato, J. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858‐3863. [PubMed] [Google Scholar]
- Nelson, H. B. and Tang, H. 2006. Effect of cell growth on hepatitis C virus (HCV) replication and a mechanism of cell confluence‐based inhibition of HCV RNA and protein expression. J. Virol. 80:1181‐1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2nd O'Boyle, D.R. , Nower, P.T. , Lemm, J.A. , Valeras, L. , Sun, J.H. , Rigat, K. , Colonno, R. , and Gao, M. 2005. Development of a cell‐based high‐throughput specificity screen using a hepatitis C virus‐bovine viral diarrhea virus dual replicon assay. Antimicrob. Agents Chemother. 49:1346‐1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan, M. 2007. Basic techniques in mammalian cell culture. Curr. Protoc. Cell Biol. 36:1.1.1‐1.1.18. [DOI] [PubMed] [Google Scholar]
- Pietschmann, T. , Lohmann, V. , Rutter, G. , Kurpanek, K. , and Bartenschlager, R. 2001. Characterization of cell lines carrying self‐replicating hepatitis C virus RNAs. J. Virol. 75:1252‐1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, L.J. and Muench, H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27:493. [Google Scholar]
- Jr. Sainz, B. and Chisari, F.V. 2006. Production of infectious hepatitis C virus by well‐differentiated, growth‐arrested human hepatoma‐derived cells. J. Virol. 80:10253‐10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jr. Sainz, B. , Barretto, N. , and Uprichard, S.L. 2009. Hepatitis C virus infection in phenotypically distinct Huh7 cell lines. PLoS ONE 4:e6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliani, M. , Bianchi, E. , Narjes, F. , Fossatelli, M. , Urbani, A. , Steinkühler, C. , De Francesco, R. , and Pessi, A. 1996. A continuous assay of hepatitis C virus protease based on resonance energy transfer depsipeptide substrates. Anal. Biochem. 240:60‐67. [DOI] [PubMed] [Google Scholar]
- Wakita, T. , Pietschmann, T. , Kato, T. , Date, T. , Miyamoto, M. , Zhao, Z. , Murthy, K. , Habermann, A. , Kräusslich, H.G. , Mizokami, M. , Bartenschlager, R. , and Liang, T.J. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, R. 2006. Global challenges in liver disease. Hepatology 44:521‐526. [DOI] [PubMed] [Google Scholar]
- Windisch, M.P. , Frese, M. , Kaul, A. , Trippler, M. , Lohmann, V. , and Bartenschlager, R. 2005. Dissecting the interferon‐induced inhibition of hepatitis C virus replication by using a novel host cell line. J. Virol. 79:13778‐13793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, X. , Jr. Sainz, B. and Uprichard, S.L. 2009. Development of a cell‐based hepatitis C virus infection fluorescent resonance energy transfer assay for high‐throughput antiviral compound screening. Antimicrob. Agents Chemother. 53:4311‐4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J.H. , Chung, T.D. , and Oldenburg, K.R. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67‐73. [DOI] [PubMed] [Google Scholar]
- Zhong, J. , Gastaminza, P. , Cheng, G. , Kapadia, S. , Kato, T. , Burton, D.R. , Wieland, S.F. , Uprichard, S.L. , Wakita, T. , and Chisari, F.V. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U.S.A. 102:9294‐9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, J. , Gastaminza, P. , Chung, J. , Stamataki, Z. , Isogawa, M. , Cheng, G. , McKeating, J.A. , and Chisari, F.V. 2006. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J. Virol. 80:11082‐11093. [DOI] [PMC free article] [PubMed] [Google Scholar]