

Abstract
Four double-drug HIV NRTI / NNRTI inhibitors 15a-d of the type [d4U]-spacer-[HI-236] in which the spacer is varied as 1-butynyl (15a), propargyl-1-PEG (15b), propargyl-2-PEG (15c) and propargyl-4-PEG (15d) have been synthesized and biologically evaluated as RT inhibitors against HIV-1. The key step in their synthesis involved a Sonogashira coupling of 5-iodo d4U's benzoate with an alkynylated tethered HI-236 precursor followed by introduction of the HI-236 thiourea functionality. Biological evaluation in both cell-culture (MT-2 cells) as well as using an in vitro RT assay revealed 15a-c to be all more active than d4T. However, overall the results indicate the derivatives are acting as chain-extended NNRTIs in which for 15b-d the nucleoside component is likely situated outside of the pocket but with no evidence for any synergistic double binding between the NRTI and NNRTI sites. This is attributed, in part, to the lack of phosphorylation of the nucleoside component of the double drug as a result of kinase recognition failure, which is not improved upon with the phosphoramidate of 15d incorporating a 4-PEG spacer.
Keywords: HIV inhibitors, Reverse-Transcriptase, NRTI, NNRTI, Double-drug, HI-236, d4T, Antiviral
1. Introduction
HIV reverse transcriptase (RT)1 is a multifunctional enzyme responsible for the catalytic transformation of single-stranded HIV viral RNA into double-stranded DNA (dsDNA). It continues to be the principal drug-target for chemotherapy using combination therapy HAART2 (Highly Active Antiretroviral Therapy), in which RT inhibitors (RTIs) provide at least two of the three anti-HIV drugs3 used. Two types of RTIs have emerged as NRTIs3 (Nucleoside Inhibitors), which act as competitive inhibitors of DNA polymerization, and NNRTIs (Non-Nucleoside Inhibitors),4 which inhibit allosterically by binding into a hydrophobic pocket adjacent to the DNA substrate binding site.3 The close proximity (10-15 Å) of the two RTI sites inspired Arnold5 in 1993 and Steitz6 in 1994 to propose the double-drug concept for the first time. They postulated that a molecular entity comprising one of each type of inhibitor joined by a spacer might be able to interact simultaneously with the two sites on RT and in so doing provide a super-inhibitor via additive binding.7 Biochemical support to realize such a concept was subsequently provided by Anderson8a Goody8b and Wainberg8c who showed that binding of an NNRTI does not inhibit the binding processes of an NRTI. These research contributions effectively provided inspiration for a number of synthetic medicinal chemistry research efforts towards realizing such a concept, and in 1995 Camarasa9 published the first example of a bifunctional NRTI/NNRTI anti-HIV double-drug containing a non-cleavable spacer based on an [AZT]-spacer-[TSAO-T] or [AZT]-spacer-[HEPT] combination joined at N-3 of the base of each drug. TSAO-T10 derivatives interact with amino acids situated in both HIV-1 RT subunits (p51 and p66) at the dimer interface. A likely important interaction is with the carboxyl group of Glu 138, which lies at the entrance to the NNRTI pocket in the p51 sub-unit. Camarasa varied both the drug combinations as well as the tether length and did achieve some IC50 inhibition values in the 0.06-0.55 μM range against HIV-1.
However, it was concluded that the double-drugs inhibited as extended NNRTIs with no contribution apparent from the NRTI component. In a subsequent paper,11 the study was broadened to accommodate other NRTI's like d4T as well as different attachment points on the drugs, but still without demonstrating any activity at the NRTI site. In the following ten years or so, reports on both cleavable,12 in which the spacer is designed to be hydrolytically labile in order to release the individual drugs, as well as other non-cleavable double-drugs,13 also known as mixed site inhibitors,13b appeared in the literature but similarly without evidence of true synergy between the two inhibitors. For the cleavable type, systems have become extended to different drug targets14 other than RT, whereas for the non-cleavable type, RT remains to date as the only target15 studied for HIV in view of the uniqueness of having the NRTI and NNRTI sites in close proximity. Challenges regarding the successful realization of a truly synergistic double-drug RTI in the non-cleavable class have been primarily twofold as: i) where on the drugs to attach the linker – this has suffered from a lack of modeling data, and, ii) the lack of phosphorylation of the NRTI nucleoside prodrug contained within the double-drug in vitro. Recently, we have reported on our own efforts in the field of non-cleavable HIV double drugs using a d4U / HI-236 system. Our first prototype16 with a relatively short butynyl tether against HIV-1 (IIIB) in MT-2 cell culture using an MTT assay returned an EC50 of 250nM. This was only a fivefold reduction of the NNRTI (HI-236) EC50 and eightfold more potent than d4T (2 μM). Encouraged by this result we went on in a subsequent paper17 to explore the influence of tether type and length on the anti-HIV activity of C-2 aryl O-tethered HI-236 derivatives in order to establish the feasibility of projecting a tether out of the NNRTI pocket. A 2-PEG-propynyl substituent returned an EC50 of 390 nM suggesting feasibility of the concept. In this paper we extend these studies and report on results pertaining to a small family of [d4U]-spacer-[HI-236] double drugs as well as a phosphoramidate pro-drug of one of them.
2. Double-Drug Design Aspects
Regarding design aspects, d4U was chosen as the nucleoside in view of its synthetic versatility as well as its proven activity (as a d4T unit if not substituted at C-5 in the double-drug) in other double-drug systems.11 A water-friendly PEG (polyethylene glycol) spacer that could be varied in length and easily introduced via standard substitution methodology was chosen for the tether. The choice of HI-23618 as the NNRTI has been delineated earlier17 but essentially, apart from its potency, synthetic accessibility and ease of manipulation, its flexibility as a second-generation NNRTI was considered to be a crucial parameter for achieving communication between the two sites. Regarding the all-important issue of where to attach the tether, two important decisions clearly had to be made. C-5 on the pyrimidine base was chosen as the attachment point to the nucleoside drug as this was not only synthetically readily accessible but was also considered to offer minimal interference with both base-pairing as well as hindrance around the C-5′ site. These views were based on the work of Ruth and Chen,19 and the choice of the C-5 connecting group as alkynyl was considered to offer an attractive exit from the substrate binding site in view of its directionality away from the developing DNA strand. An alkynyl connection to the nucleoside also presented itself as an attractive synthetic option via the well-known and versatile Sonogashira Pd(0) cross-coupling protocol, a well-known methodology in the nucleoside field.11, 12d, 20 This left the all-important choice of NNRTI attachment. It was significant to us that Ladurée13a had failed to observe any appreciable activity with their trovirdine derivative-based double-drug, which is shown against our double-drug design together with HI-236 in Figure 1, both double drugs containing a non-cleavable linker.
Figure 1.

Structures of HI-236 and d4U-spacer-PETT double drugs.
Not withstanding the different (C-5 vs N-3) attachments to the individual nucleosides, the “para-like” configuration of the tethers attached to the piperazine nitrogens in Ladurée's case was felt to be extremely significant. In HI-236, the piperazine ring of the trovirdine derivative is replaced by a trisubstituted phenyl group in which the C-5 (see Figure 1 for numbering) methoxy group meta to the HI-236 thiourea tether at C-1 forges an important C-H / π interaction with the conserved Trp229 residue at the back of the NNRTI pocket. This allows the C-2 phenolic methoxyl to point its methyl group down towards the floor of the cavity where residues like V106 reside. Modeling reported in our 2008 paper17 illustrates such an arrangement as shown in Figure 2 (seen from either end of the pocket) for a derivative in which the C-2 methyl is replaced by a methoxycarbonylmethylene grouping. Importantly, it suggests that a C-2 ortho-substituent to the C-1 thiourea tether allows an exit possibility from the pocket and may well explain why Laduree's “para-like” arrangement on the piperazine in his d4U-spacer-trovirdine derivative double-drug failed to accommodate the tethered grouping to the nucleoside into the pocket. In this regard, one must bear in mind the importance of the directing role that the bromopyridyl group of the thiourea plays via tight hydrogen bonding towards the front of the pocket in Wing 1 with K101.17, 18a
Figure 2.
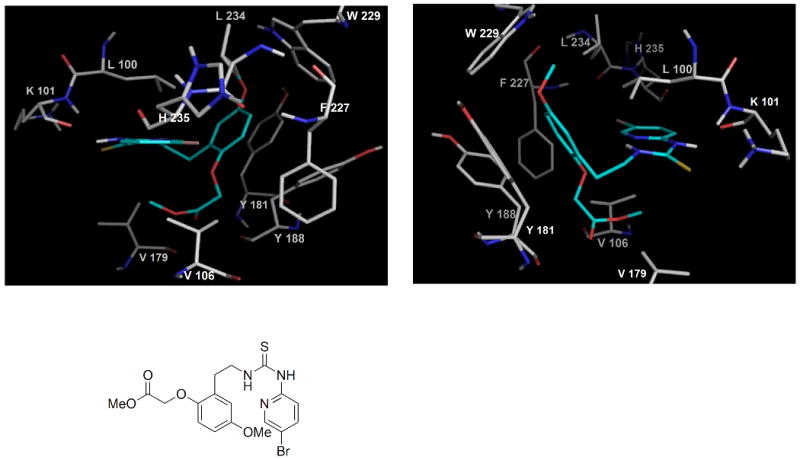
A C-2 O-alkylated HI-236 derivative modeled in the NNRTI pocket.17
Thus, based on these ideas and coupled with results from the earlier work mentioned previously,16, 17 we thought it feasible that a tethered [d4U]-spacer-[HI-236] might exit the pocket into the solvent channel close to Glu13821 (p51 sub-unit shown in Figure 3) and preferably closer to Tyr181 rather than Val179. On the assumption that the NNRTI would bind first, the NRTI of the double-drug would have to make its way to the substrate-binding site around the corner behind the hydrophobic back of the NNRTI pocket near to the conserved Trp229. Figure 3 depicts the NNRTI TMC12521 bound into the NNRTI pocket and helps to clarify this important issue.
Figure 3.
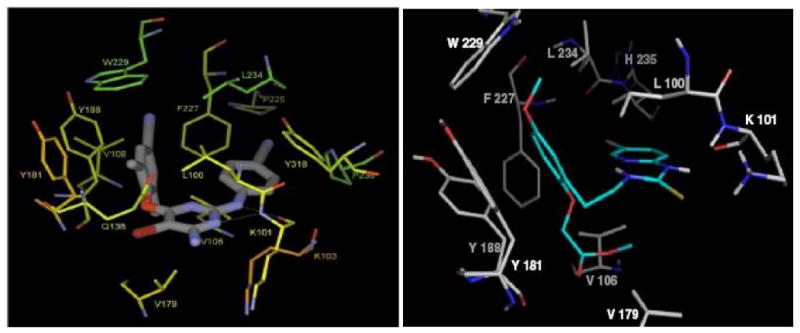
TMC125 in the NNRTI pocket21 showing Y181, Glu138 and Val179 compared to our modeling17 of an HI-236 derivative from Figure 2.
3. Chemistry
Typical of the art of total synthesis, the timing of key bond connections in the synthesis of the double-drugs proved to be crucial. A completely convergent synthesis via coupling of a tethered HI-236 alkyne to a protected derivative of 5-iodo-d4U using a Sonogashira Pd(0) coupling as the key and final step failed to give a significant yield of product, presumably due to interference from the nucleophilic thiourea sulfur. Thus, it was deemed necessary to bring the key coupling step forward in the sequence and introduce the HI-236 thiourea functionality late, and this approach gratifyingly turned out to be successful. Thus, the synthesis involved synthesis of two halves, a coupling step and an end-game as intimated in the retrosynthetic analysis shown in Figure 4.
Figure 4.

Retrosynthetic analysis of the d4U-spacer-HI-236 target.
For the right-hand tethered derivatives 7, the synthesis started with commercially available 2-hydroxy-5-methoxybenzaldehyde 1, which following a three-step sequence described by Glennon16, 22 involving phenolic hydroxyl protection with benzyl, a Henry aldol reaction and LAH-mediated reduction of both the double bond and the nitro group afforded amine 2 in gram quantities, Scheme 1. For practical reasons it was easier to isolate 2 as its N-Boc derivative 3 via a standard Boc protection step without using DMAP, which promoted di-Boc derivatisation. A 50% overall yield for the four-step sequence to afford 3 could be achieved on scale-up.
Scheme 1.

Reagents and conditions: (i) BnBr, K2CO3, EtOH, 80 °C; (ii) CH3NO2, NH4OAc, 70 °C; (iii) LiAlH4, THF, 70 °C; (iv) (Boc)2O, CH3CN, rt.
Conversion of 3 to the tethered propargylated N-Boc derivatives 7 (Scheme 2) has been described previously17 and was found to be optimal by propargylating in the final step. Thus, hydrogenolytic debenzylation of 3 to phenol 4 followed by alkylation with a monobenzyl-protected PEG bromide using sodium hydride in THF or potassium carbonate as base in acetonitrile at reflux gave the protected PEG-alkylated phenols 5a-c. A second debenzylation followed by tosylation of the primary hydroxyl group gave tosylates 6a-c. Finally, nucleophilic displacement by an excess of propargyloxide ion returned the best results overall for obtaining the final propargylated products 7b-d, which were obtained as shown in Scheme 2, with the 4-PEG derivative considered to be well within striking distance of the substrate binding site. Each compound 7b-d was exhaustively characterized17 using 1H, 13C NMR spectroscopy as well as high resolution mass spectrometry (HRMS) and / or CHN combustion analysis. 13C NMR spectroscopy was particularly useful for identifying the carbons of the PEG units to confirm that the correct length of tether was in place in each case, while 1H NMR spectroscopy identified a characteristic fingerprint for the triad of aromatic signals with well-defined coupling relationships. Finally, the propargyl group could be easily discerned in both types of spectra. The exception in the library was the derivative without a PEG prepared by alkylation of phenol 4 with propargyl bromide, in which it was found that transformation into the final double-drug via Pd(0) coupling and thiourea introduction resulted in an unstable product following debenzoylation, presumably as a result of having the two drugs in close proximity as well as the reactive nature of the propargylic linker. Fortunately, the stability of the final double-drug could be improved by extending the propargyl tether by one carbon atom using 3-butynyl-1-tosylate in the alkylation to afford intermediate compound 7a as shown in Scheme 2.
Scheme 2.

Reagents and conditions: (i) H2, Pd/C, EtOH, rt; (ii) 3-butynyl-1-tosylate, K2CO3, CH3CN, 80 °C; (iii) Bn(PEG)nBr (n = 1, 2, 4), K2CO3, CH3CN, 80 °C or NaH, DME, 80 °C; (iv) TsCl, Et3N, DMAP, CH2Cl2, 0 °C-rt; (v) propargyl alcohol, NaH, THF, 70 °C.
For synthesis of the left-hand nucleoside partner, advantage was taken of the elegant Bristol-Myers Squibb process for converting 5-methyluridine into d4T23 based on seminal work by Fox involving uridine.24 This methodology could be carried out efficiently on a several-grm-scale with a single clean-up chromatography step at the end and was superior to other methodologies25 such as that using acetyl bromide.26 Thus, mesylation of the hydroxyl groups of uridine, followed by heating the product with sodium benzoate in acetamide furnished the 2′-anhydro nucleoside 8.24b Subsequent opening using HBr (from AcBr in MeOH) to bromomesylate 9 followed by zinc-mediated elimination furnished the 5′-benzoate of d4U 1026a as a crystalline solid. Interestingly, it was found that the zinc-elimination could be accelerated using 2% v/v conc HCl in the methanol solvent, presumably as a result of dissolution of surface oxide exposing metal surface, Scheme 3. Finally, iodination of 10 was accomplished using elemental iodine and ceric ammonium nitrate (CAN) in acetonitrile according to the procedure by Robins.27 Running the reaction at around 40 °C for about five hours secured a high yield of the 5-iodo derivative 11 after chromatography (82%) with minimal by-product formation involving iodination of the dideoxyribose double bond.
Scheme 3.

Reagents and conditions: (i) MsCl (3 eq), pyr, 0 °C; (ii) NaBz (3 eq), acetamide, 120 °C; (iii) CH3COBr (5 eq), EtOAc:MeOH (10:1), 70 °C; (iv) Zn, c HCl (2% by vol), MeOH, rt; (v) CAN (0.6 eq), I2 (0.6 eq), CH3CN, 35 °C.
With the two coupling partners 7 and 11 in hand, attention was turned towards the key Sonogashira coupling. Literature precedent for such a reaction is well established following early work by Robins28 and others,29, 30 and the topic for nucleosides has been recently reviewed.31 Generally, the most encountered conditions use either Pd(PPh3)4 and Pd(PPh3)2Cl2 as catalysts in varying mol % (around 20%) in deoxygenated DMF as solvent with triethylamine as base and with CuI in a 2:1 ratio to Pd catalyst. Excess alkyne ensures complete conversion of nucleoside in view of competing Glaser-type coupling of the alkyne, and one may form a furanopyrimidine cyclization by-product post Sonogashira coupling depending on the catalyst used.32
In the event, Pd(PPh3)4 was chosen as the Pd(0) catalyst. Generally, using Pd(PPh3)4 (10 mol %) with CuI (50%) at a higher ratio than normal with alkyne at 1.1 eq in a deoxygenated DMF / THF (1:2) solvent medium furnished coupled products 12a-d by tlc within 3-4 h at rt. Following the usual extractive work-up with aq. disodium EDTA to remove metal salts, chromatography furnished the requisite coupled products in around a 70 % isolated yield. Product integrity could be relatively easily discerned by observing representative resonances for each coupling partner in the 1H or 13C spectra. Importantly, the two NH signals could be discerned in the 1H NMR spectra (in CDCl3) at 8.50-9.00 for the pyrimidine signal and close to 5.00 ppm for the carbamate NH. Diagnostic peaks for key functional groups such as the triple bond could be identified in the IR spectrum, and HRMS using electrospray returned correct molecular ions in each case, Scheme 4.
Scheme 4.

Reagents and conditions: (i) Pd(PPh3)4 (10 mol %), CuI (50 mol %), Et3N (2 eq), DMF / THF (1:2), rt.
The end-game to the double-drug targets involved a three-step sequence involving Boc-deprotection, thiourea coupling and benzoate deprotection to the free nucleoside. Some concern was entertained regarding the two deprotection steps, particularly the first one involving trifluoroacetic acid as the standard reagent for Boc-deprotection. In the event, following some exhaustive optimization, it was established that exposing products 12a-d to TFA at 0 °C in DCM resulted in deprotection in about 2 hours to a more polar amine spot on TLC. In view of the anticipated water solubility of the amine, the final step was conducted without using an aqueous work-up. Thus, following addition of Hünig's base (EtN(i-Pr)2) and the complete removal of all volatiles, the residue was redissolved in THF and the thiocarbonyl reagent 13 added according to the original procedure33 described by the Eli Lilly group in their work on PETT NNRTIs. Reagent 13 could be readily prepared by reacting 2-amino-5-bromopyridine with 1,1′-thiocarbonyldiimidazole in acetonitrile at room temperature for 12 h to afford a precipitate that was used without purification. In our case, condensation between the amine and 13 could be realized in THF or DMF at room temperature overnight to afford the final double-drug targets 14 as their 5′-benzoate esters in about 60% over the two steps following chromatography. The lower temperature for condensation with 13 compared to the 100 °C (in DMF) used33 by the Eli-Lilly group made a significant improvement to the overall yield. Thereafter, methoxide-catalysed benzoate deprotection in MeOH at 0 °C furnished the final targets 15. Of crucial importance in this step in order to avoid degradation was the mode of isolation, for which the best procedure turned out to be quenching with a minimal amount of glacial acetic acid followed by direct rapid flash-chromatography. In such a way one could minimise cleavage of the nucleoside moiety. Once isolated, though, the double-drugs were stable enough for biological testing and evaluation purposes. All final double drug products were solids that could be exhaustively characterised by a full complement of spectroscopic techniques (see Experimental section). However, their recrystallization tended to promote some ribose nucleoside cleavage, so HRMS (ES) was used to successfully provide an accurate molecular ion in each case, Scheme 5.
Scheme 5.

Reagents and conditions: (i) TFA, CH2Cl2, 0 °C; (ii) EtN(i-Pr)2 followed by 13, THF, rt/overnight; (iii) NaOMe, MeOH, 0 °C.
The one derivative that eluded realization was the 4-PEG derivative 15d, as the product following TFA deprotection and condensation with 13 was found to lack the sugar ring by NMR spectroscopy. Since Sonogashira reactions are well known34 to proceed in the presence of unprotected hydroxyl or amino groups, it was decided to revise the order of events in the sequence and deprotect the Boc-protecting group of alkyne 7d first. Thereafter, following neutralization of TFA using K2CO3 in methanol and filtration of the salts the crude amine was subjected to the Sonogashira reaction and thiourea condensation steps to afford the 4-PEG double-drug 14d as its 5′-benzoate in 40% yield over the three steps after chromatography. Deprotection with methoxide as usual furnished the final free nucleoside double-drug 15d in a modest yield of 52%, Scheme 6.
Scheme 6.

Reagents and conditions: (i) TFA, CH2Cl2, 0 °C; (ii) 11, Pd(PPh3)4 (10 mol %), CuI (50 mol %), Et3N (2 eq), DMF / THF (1:2), rt; (iii) 13, THF, rt; (iv) NaOMe (cat), MeOH, 0 °C.
The 1H NMR and 13C NMR spectra illustrated in Figure 5 for 15d showing all of the required signals gave us great satisfaction.
Figure 5.
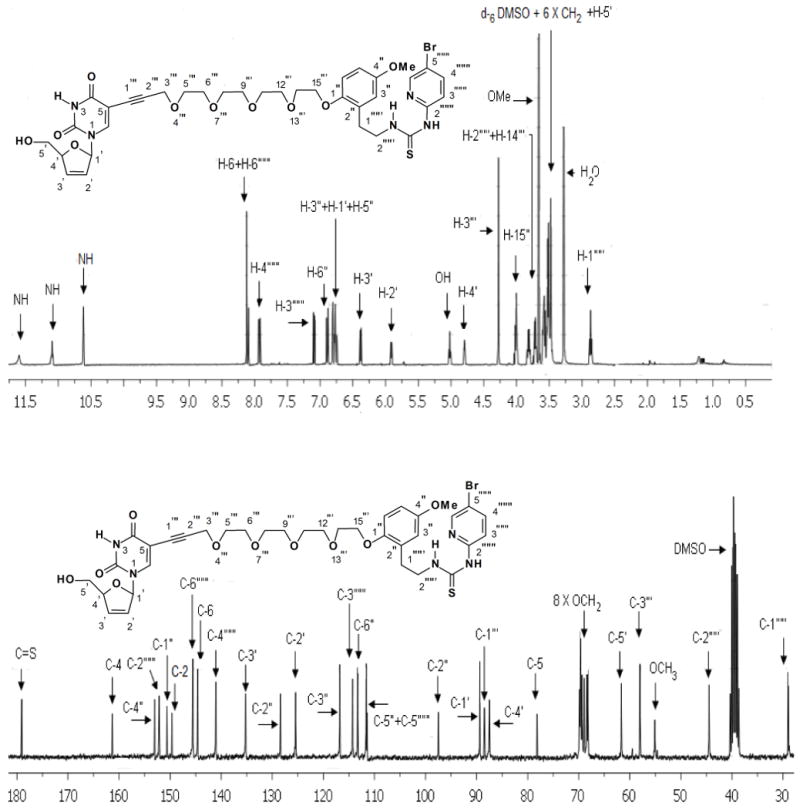
400 MHz 1H NMR and 75 MHz 13C NMR spectra of 15d in d6 DMSO
As a final piece of synthesis, it was decided to prepare a pronucleotide of 15d as the double-drug most likely to reach the DNA site in view of the 4-PEG spacer. The pronucleotide approach for enhancing nucleoside activity by bypassing the first rate-determining kinase-mediated phosphorylation step is well established with several variants on masked phosphate groupings. CycloSal35 and phosphoramidate36 functionalities provide two of the most frequently used options and we chose the latter in view of its proven ability to enhance the activity of d4T37 as well as the likelihood of accessing it from the nucleoside double-drug 15d in a single step. However, when 15d was reacted with p-tolyl methoxyalaninyl phosphorochloridate prepared38 according to a McGuigan published procedure with N-methylimidazole as a transfer base, no phosphoramidate could be isolated and only breakdown of the double-drug was observed by TLC. Given the relative robustness of the phosphoramidate grouping, it was thought that it could replace benzoate in the Sonogashira coupling. However, given that we doubted its stability towards TFA, we adopted the strategy developed for the 4-PEG double-drug 15d. Thus, Boc-deprotection of N-Boc alkyne 7d to the free amine as described before in Scheme 6 followed by Sonogashira coupling of the resultant amine with the phosphoramidate of 5-iodo-d4U 16 prepared from 5-iodo-d4U by a literature method,39 and finally coupling of the amino group of the coupled product with 13 gave the target thiourea double-drug phosphoramidate 17 as a nearly 50:50 mixture of diastereomers at phosphorus in an overall yield of 20% for the three steps, Scheme 7. Given our experience in deriving full assignments for the other double-drugs, 17 could be exhaustively characterized using a combination of both 1D and 2D 1H, 13C and 31P NMR spectroscopic techniques in spite of the diastereomeric mixture.
Scheme 7.

Reagents and Conditions: (i) TFA, CH2Cl2, 0 °C; (ii) 16, Pd(PPh3)4 (10 mol %), CuI (50 mol %), Et3N (2 eq), DMF / THF (1:2), rt; (iii) 13, THF, rt.
4. Biological results and Discussion
The inhibitory activities of the bifunctional compounds 15a-d and phosphoramidate 17 together with HI-236 and d4T as controls were measured against HIV-1 (IIIB) replication in MT-2 cell culture using an MTT assay.40 The same compounds were also evaluated for their in vitro activity against RT directly in a steady-state RT inhibition assay using a D23/D36 primer/template in which the inhibition of incorporation of thymidine triphosphate (TTP) by each double-drug was measured as an IC50. The results are shown in Table 1 expressed in μM units. [d4U]-butyne-[HI-236] 15a showed a good inhibitory activity with an EC50 = 250 nM in the cell-culture assay as nine times more potent than d4T (EC50 = 2.3 μM) alone, and ca. six times less potent than HI-236 (EC50 = 0.042 μM) and thus closer in activity to the NNRTI component. The compound was ca. twofold less potent than HI-236 in the RT assay with IC50 values of HI-236 and 15a (38 and 61 nM respectively) both improving relative to the cell-culture EC50 results. This was not unexpected in view of aspects of cell permeability and the greater possibility for degradation in the cell-culture experiment. Lengthening the spacer resulted in a steady reduction in activity (0.25, 1.3, 1.9 and 3.1 μM for 15a-d respectively) in cell-culture. Notably, the 4-PEG derivative 15d still retained appreciable activity (3.1 μM), remarkably so for such a large molecule. As with 15a, the RT IC50 values for 15b-d were similarly lower than their EC50 cell-culture values, with nanomolar activities for 15b and 15c. Disappointingly, the 4-PEG phosphoramidate 17 showed no significant improvement (3.1 to 2.9 μM) in cell culture compared to its unphosphorylated counterpart 15d, effectively indicating that triphosphorylation of pro-drugs 15a-d and 17 in cell-culture does not occur. The superior activity of the 4-PEG double-drug nucleoside 15d in the RT assay compared with that of its phosphoramidate 17 (1.4 μM vs 2.3 μM) suggests binding of the 5′-hydroxyl group of 15d, which is considered to likely be taking place near to or at the substrate binding site.
Table 1.
A comparison of cell-culture versus in vitro HIV-1 RT inhibition (μM) for double-drugs 15a-d and 17.
| Compound | Cell-Culture EC501 (μM) | Cell-Culture CC502 (μM) | RT Assay3 (μM) |
|---|---|---|---|
| HI-236 | 0.042 | >1 | 0.038 ± 0.007 |
| d4T | 2.3 | >100 | 9.64 |
| 15a | 0.25 | 17 | 0.061±0.015 |
| 15b | 1.3 | 38 | 0.575 ± 0.14 |
| 15c | 1.9 | 43 | 0.850 ± 0.14 |
| 15d | 3.1 | 18 | 1.4 ± 0.5 |
| 17 | 2.9 | 11 | 2.3 ± 0.8 |
 |
12041 | N/A |
effective concentration that inhibits viral-mediated T-cell death by 50%, determined by averaging samples of each concentration in triplicate.
concentration that kills 50% of the T-cells, also determined by averaging triplicate samples.
concentration of inhibitor that inhibits by 50% the steady-state thymidine incorporation into a D23 / D36 primer-template as catalysed by RT.
Based on pre-steady-state kinetic analysis.
Our results indicate that the activity of double-drugs 15a-d and 17 is mainly due to the NNRTI component, with the size of the double-drugs suggesting that 15b-d and 17 have the nucleoside drug outside of the NNRTI pocket. Although the data doesn't allow a firm conclusion to be made regarding the possibility or nature of NRTI binding possibilities, the relatively potent cell-culture EC50 values of 1.3 μM and 1.9 μM for 1-PEG and 2-PEG derivatives 15b,c respectively (more active than d4T alone (2.3 μM)) strongly suggests the possibility of some cooperative binding outside of the pocket. If this is the case, the fact that Ladurée has shown the 5-alkynylated analogue 1841 (Table 1) to be completely inactive would support the view that the nucleoside part of the double-drugs 15b,c may well forge cooperative interactions away from the substrate binding site, although derivatives 15d and 17 containing the longer 4-PEG tether could well be interacting cooperatively near to the binding site based on anchoring from the NNRTI pocket. Such conclusions echo those of Monneret and co-workers based on the activities of their AZT-tether-HEPT bifunctionals in which activities also suggested out-of-pocket binding but without synergy between drug-sites, and to a lesser extent to those of Camarasa based on her nucleoside-spacer-TSAO double-drugs, since TSAO supposedly binds just outside of the conventional NNRTI pocket. The failure to achieve synergy in the double-drugs 15a-d as well as the other systems mentioned may be mainly ascribed to the inability of cellular kinases to recognize the nucleoside portion of the double drug.39a This conclusion unfortunately also applies to the monophosphate of 15d, assuming that phosphoramidate 17 is hydrolysed to such a mono-phosphate within the cell-culture as a by-pass of the first rate-limiting phosphorylation. On a positive note, our results suggest that it might be worthwhile to consider designing extended NNRTI inhibitors containing a second binding agent out of but near to the front of the pocket. Given the plethora of X-ray structures of NNRTIs available,4a it should be possible to model certain residues for this purpose, eg Glu138 on the outside of the pocket.10 The original double-drug suggestion by Arnold5 and Steitz6 didn't consider how the two drug-sites might communicate structurally in practice. Regarding the kinase recognition problem just mentioned, targeting protected triphosphates of double-drugs would present some serious challenges regarding their synthesis and stability. Moreover, communication between the two sites is made difficult if the intention is for the tether to exit the NNRTI binding pocket, since the front of the pocket points away from the substrate binding site. However, the possibility that the two drugs may communicate between the two sites by virtue of the tether exiting the NNRTI pocket from the back end close to Trp 229 provides a more direct connection. Results on this will be communicated in due course involving the diarylpyrimidine TMC-120 as the NNRTI component of such a double-drug system.
5. Experimental
5.1. General procedures for synthesis
Infrared (IR) absorptions were measured on a Perkin-Elmer Spectrum One FT-IR spectrometer. 1H NMR spectra were recorded on a Varian Mercury Spectrometer at 300 MHz or a Varian Unity Spectrometer at 400 MHz with Me4Si as internal standard. 13C NMR spectra were recorded at 75 MHz on a Varian Mercury Spectrometer or at 100 MHz on Varian Unity Spectrometer with Me4Si as internal standard. High resolution mass spectra were recorded on a VG70 SEQ micromass spectrometer. Melting points were determined using a Reichert-Jung Thermovar hot-stage microscope and are uncorrected. Analytical thin-layer chromatography (TLC) was performed on aluminium-backed silica-gel 60 F254 (70-230 mesh) plates. Column chromatography was performed with Merck silica-gel 60 (70-230 mesh). Compounds 2,22 5a,17 6a,17 6b,17 7b,17 7c,17 and 1333 have all been reported previously. 3-Butynyl 1-p-toluenesulfonate42 was prepared from the alcohol by a standard tosylation procedure. The monobenzyl PEG-Br alkylating agents for formation of 5b and 5c were prepared by standard mono-benzylation and bromination (using PPh3 and CBr4) methods from the corresponding glycol.
5.2. Synthesis of intermediates for the coupling partners 7 and 11
5.2.1. N-[2-(2-Benzyloxy-5-methoxyphenyl)ethyl]-tert-butylcarbamate 3
Di-tert-butyldicarbonate (5.22 g, 23.93 mmol) in acetonitrile (6 mL) was added to a solution of crude amine 2 (4.10 g, 15.95 mmol) in acetonitrile (50 mL) and the reaction mixture was stirred at rt for 20 h. Aqueous NH4Cl (100 mL) was added and the organic material extracted into EtOAc (3 × 100 mL). Following drying and evaporation of solvent the residue was subjected to column chromatography employing EtOAc / petroleum ether (15 / 85) to give carbamate 3 as colourless crystals (5.00 g, 88%); mp: 102-104 °C; IR (CHCl3): νmax 3449 (NH), 3007, 2935 (C-H), 1703 (C=O), 1501 (C=C), 1165 (C-N) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.45-7.34 (5H, m,), 6.85 (1H, d, J = 8.8 Hz,), 6.76 (1H, d, J = 2.8 Hz), 6.72 (1H, dd, J = 2.8, 8.8 Hz), 5.04 (2H, s,), 4.70 (1H, brs, NH), 3.78 (3H, s), 3.40 (2H, q, J = 6.4 Hz), 2.85 (2H, t, J = 6.4 Hz), 1.44 (9H, s); 13C NMR (75 MHz, CDCl3): δ 155.9 (C=O), 153.8, 150.9, 137.4, 129.2, 128.5, 127.8, 127.2, 116.8, 113.0, 112.0, 79.0, 70.8, 55.7, 40.8, 30.9, 28.4; HRMS (EI): m/z found 301.13383 [(M+ - t-butyl) + H]. C17H19NO4 requires 301.13409 [(M+ - t-butyl) + H]; Found C, 70.50; H, 7.62; N, 3.86. C21H27NO4 requires C, 70.56; H, 7.61; N, 3.92.
5.2.2. N-[2-(2-Hydroxy-5-methoxyphenyl)ethyl]-tert-butylcarbamate 4
The carbamate 3 (4.50 g, 12.61 mmol) was added to 10% palladium-on-carbon (0.22 g, 1.26 mmol) in ethanol. Hydrogen gas was introduced to the reaction at rt for 5 h. The palladium was filtered from the solution through Celite and the precipitate was washed with ethanol (3 × 100 mL). The solvent was removed under reduced pressure and the residue was subjected to column chromatography using EtOAc / petroleum ether (4 / 6) to give 4 as a colourless solid (2.50 g, 74%); mp: 115-117 °C; IR (CHCl3): νmax 3457, 3326 (NH, OH), 1686 (C=O), 1508 (C=C), 1210, 1166 (C-N) cm-1; 1H NMR (400 MHz, CDCl3): δ 6.99 (1H, brs, OH), 6.79 (1H, d, J = 8.4 Hz), 6.67 (1H, d, J = 2.8 Hz), 6.65 (1H, m), 5.00 (1H, brs, NH), 3.75 (3H, s), 3.33 (2H, q, J = 7.1 Hz), 2.81 (2H, t, J = 7.1 Hz), 1.46 (9H, s); 13C NMR (100 MHz, CDCl3): δ 157.0 (C=O), 153.2, 148.8, 126.0, 116.5, 116.2, 112.8, 80.0, 55.8, 41.0, 31.4, 28.4; HRMS (EI): m/z found 267.14329 (M+). C14H21NO4 (M+) requires 267.14706; Found C, 62.82; H, 7.92; N, 5.16. C14H21NO4 requires C, 62.94; H, 7.92; N, 5.24.
5.2.3. N-[2-(2-(5-Benzyloxy-3-oxapent-1-yloxy)-5-methoxyphenyl)ethyl]-tert-butylcarbamate 5b
2-(2-Benzyloxy)ethoxy-1-bromoethane (0.87 g, 3.36 mmol) in dry acetonitrile (10 mL) was added dropwise over 1h to a refluxing and stirring mixture of phenol 4 (0.45 g, 1.68 mmol) and anhydrous potassium carbonate (0.93 g, 6.70 mmol) in dry acetonitrile (20 mL). After 20h the mixture was filtered, the solvent evaporated and the residue subjected to silica-gel column chromatography using EtOAc / petroleum ether (1 / 9) to give 5b as a colourless oil (0.61 g, 81%.); IR (CHCl3): νmax 3442, 3377 (NH, OH), 3007, 2928 (C-H), 1697 (C=O), 1501 (C=O), 1227 (C-N) cm1; 1H NMR (400 MHz, CDCl3): δ 7.31 (4H, m), 7.21 (1H, m), 6.76 (1H, d, J = 8.7 Hz), 6.70 (1H, d, J = 2.8 Hz), 6.68 (1H, dd, J = 2.8, 8.7 Hz), 4.83 (1H, brs, NH), 4.55 (2H, s), 4.06 (2H, t, J = 4.8 Hz), 3.82 (2H, t, J = 4.8 Hz), 3.72 (3H, s), 3.70 (2H, m), 3.62 (2H, m), 3.31 (2H, q, J = 6.5 Hz), 2.76 (2H, t, J = 6.5 Hz), 1.38 (9H, s); 13C NMR (100 MHz, CDCl3): δ 156.0 (C=O), 153.8, 151.0, 138.2, 129.4, 128.4, 127.7, 127.6, 116.6, 113.1, 112.0, 78.8, 73.3, 70.8, 69.9, 69.5, 68.5, 55.6, 40.7, 31.0, 28.4; HRMS (EI): m/z found 445.24814 (M+). C25H35NO6 (M+) requires 445.24644.
5.2.4. N-[2-(2-(11-Benzyloxy-3,6.9-trioxaundec-1-yloxy)-5-methoxyphenyl)ethyl]-tert-butylcarbamate 5c
11-Benzyloxy-1-bromo-3,6.9-trioxaundecane (0.91 g, 2.62 mmol) in dry acetonitrile (10 mL) was added dropwise over 1h to a refluxing and stirring mixture of phenol 4 (0.35 g, 1.31 mmol) and anhydrous potassium carbonate (0.72 g, 5.20 mmol) in dry acetonitrile (20 mL). After 20h the mixture was filtered, the solvent evaporated and the residue subjected to silica-gel column chromatography using EtOAc / petroleum ether (1 / 9) to give 5c as a colourless oil (0.56 g, 80%.); IR (CHCl3): νmax 3681, 3449 (N-H), 3021, 2913 (C-H), 1700 (C=O), 1501 (C=C) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.34 (4H, m), 7.21 (1H, m), 6.78 (1H, d, J = 8.8 Hz), 6.72 (1H, d, J = 3.0 Hz), 6.70 (1H, dd, J = 3.0 Hz, 8.8 Hz), 4.90 (1H, brs, NH), 4.57 (2H, s), 4.08 (2H, t, J = 4.9 Hz), 3.83 (2H, t, J = 4.9 Hz), 3.76 (3H, s), 3.72−3.68 (10H, m), 3.64 (2H, m), 3.35 (2H, q, J = 6.7 Hz), 2.80 (2H, t, J = 6.7 Hz), 1.43 (9H, s); 13C NMR (100 MHz, CDCl3): δ 155.9 (C=O), 153.7, 151.0, 138.3, 129.3, 128.3, 127.6, 127.5, 116.6, 113.0, 111.9, 78.7, 73.1, 78.7, 70.6, 70.6, 69.8, 69.4, 68.4, 55.6, 40.6, 31.0, 28.4; HRMS (ES): m/z found 534.3036 (M+ + H). C29H44NO8 requires (M+ + H) 534.3067.
5.2.5. N-[2-(2-(11-p-Toluenesulphonyloxy-3,6.9-trioxaundec-1-yloxy)-5-methoxyphenyl)ethyl]-tert-butylcarbamate 6c
Triethylamine (0.18 mL, 1.36 mmol) was added to a stirring solution in dry CH2Cl2 (5 mL) at 0 °C of the alcohol (0.50 g, 1.13 mmol) obtained from hydrogenolysis of 5c. p-Toluenesulfonyl chloride (0.26 g, 1.36 mmol) was added together with a catalytic amount of DMAP (20 mg). The reaction mixture was stirred at rt for 16 h, then diluted with CH2Cl2 (80 mL) and the organic layer washed with aq NH4Cl (25 mL), water (30 mL), dried over MgSO4 and the solvent removed under reduced pressure. Purification by column chromatography EtOAc in petroleum ether (8 / 2) gave 6c as a colorless oil (0.58 g, 84%); IR (CHCl3): νmax 3377 (NH), 2928 (C-H), 1707 (C=O), 1501 (C=C), 1223 (C-N), 1176 (O-SO2-) cm-1; 1H NMR (300 MHz, CDCl3): δ 7.77 (2H, d, J = 8.4 Hz), 7.31 (2H, d, J = 8.4 Hz), 6.76 (1H, d, J = 9.2 Hz), 6.68 (2H, m), 4.85 (1H, brs, NH), 4.14 (2H, t, J = 4.8 Hz), 4.06 (2H, t, J = 4.8 Hz), 3.81 (2H, t, J = 4.8 Hz), 3.73 (3H, s), 3.69−3.57 (10H, m), 3.32 (2H, q, J = 6.5 Hz), 2.77 (2H, t, J = 6.5 Hz), 2.42 (3H, s), 1.40 (9H, s); 13C NMR (75 MHz, CDCl3): δ 155.9 (C=O), 153.8, 151.0, 144.7, 133.0, 129.7, 129.3, 127.9, 116.6, 113.0, 111.9, 78.8, 70.7 (3 × CH2O), 70.5 (CH2O), 69.8 (CH2O), 69.2 (CH2O), 68.6 (CH2O), 68.4 (CH2O), 55.6, 40.6, 31.0, 28.4, 21.5; HRMS (ES): m/z found 598.2673 (M+ + H), C29H44NO10S requires (M+ + H) 598.2686.
5.2.6. N-[2-(2-(3-butynyl-1-oxy)-5-methoxyphenyl)ethyl]-tert-butylcarbamate 7a16
3-Butynyl-1-p-toluenesulfonate (1.34 g, 6.00 mmol), phenol 4 (0.80 g, 3.00 mmol) and anhydrous potassium carbonate (1.70 g, 12.0 mmol) were refluxed in dry acetonitrile (30 mL). After 20h the mixture was filtered, the solvent evaporated and the residue subjected to silica-gel column chromatography using EtOAc / petroleum ether (1 / 9) to give to give 7a as colourless needles (0.57 g, 61%); mp: 76-77 °C; IR (CHCl3): νmax 3455 (NH), 3309 (≡CH), 2413 (C≡C), 1707 (C=O), 1602 (C=C) cm-1; 1H NMR (300 MHz, CDCl3): δ 6.75 (3H, m), 4.70 (1H, brs, NH), 4.05 (2H, t, J = 6.8 Hz), 3.75 (3H, s), 3.36 (2H, q, J = 6.6 Hz), 2.79 (2H, t, J = 6.6 Hz), 2.66 (2H, dt, J = 2.7, 6.8 Hz), 2.03 (1H, t, J = 2.7 Hz), 1.42 (9H, s); 13C NMR (75 MHz, CDCl3): δ 155.9 (C=O), 153.9, 150.6, 129.3, 116.8, 112.8, 112.0, 80.7, 78.9, 69.8, 66.8, 55.6, 40.7, 30.9, 28.4, 19.7; HRMS (EI): m/z found 319.17756 (M+). C18H25NO4 (M+) requires 319.17836; Found: C, 67.10; H, 7.66; N, 3.67. C18H25NO4 requires C, 67.89; H, 7.89; N, 4.39.
5.2.7 N-[2-(2-(11-propargyloxy-3,6.9-trioxaundec-1-yloxy)-5-methoxyphenyl)ethyl]-tert-butylcarbamate 7d
To a stirred suspension of NaH (0.12 g, 60% in mineral oil, 3.00 mmol) in THF (10 mL) at 0 °C was added propargyl alcohol (252 mg, 4.50 mmol) dissolved in THF (2 mL) dropwise. The mixture was refluxed for 30 min and a solution of 6c (435 mg, 0.73 mmol) in THF (10 mL) then added dropwise. After refluxing for 20 h, a solution of aq. NH4Cl (20 mL) was added and the organic material extracted into EtOAc (3 × 30 mL). The combined organic extracts were washed with water (2 × 20 mL), dried (MgSO4) and the solvent removed to afford a residue, which was purified by column chromatography (15% EtOAc in petroleum ether) to afford 7d (0.290 g, 83%) as a colourless oil; IR (CHCl3): νmax 3449 (NH), 3304 (≡CH), 3007, 2920 (C-H), 2116 (C≡C), 1703 (C=O), 1501 (C=C), 1227 (C-N) cm-1; 1H NMR (300 MHz, CDCl3): δ 6.77 (1H, d, J = 8.8 Hz), 6.68 (2H, m), 4.88 (1H, brs, NH), 4.18 (2H, d, J = 2.1 Hz), 4.07 (2H, t, J = 4.8 Hz), 3.82 (2H, t, J = 4.8 Hz), 3.74 (3H, s), 3.72–3.64 (12H, m, 6 × CH2O), 3.34 (2H, q, J = 6.5 Hz), 2.78 (2H, t, J = 6.5 Hz), 2.40 (1H, t, J = 2.1 Hz), 1.41 (9H, s); 13C NMR (100 MHz, CDCl3): δ 156.0 (C=O), 153.8, 151.0, 129.3, 116.6, 113.0, 112.0, 79.7, 78.8, 74.4, 70.8 (CH2O), 70.7 (CH2O), 70.6 (2 × CH2O), 70.4 (CH2O), 69.9 (CH2O), 69.1 (CH2O), 68.5 (CH2O), 58.3, 55.6, 40.6, 31.0, 28.4; HRMS (ES): m/z found 482.2732 (M+ + H). C25H40NO8 requires (M+ + H) 482.2754.
5.2.8. 5′-Benzoyl-5-iodo-d4U 11
To a solution of 5′-O-benzoyl-d4U 10 (0.88 g, 2.8 mmol)26a in CH3CN (30 mL), were added cerium ammonium (IV) nitrate (0.92 g, 1.68 mmol) and iodine (0.43 g, 1.68 mmol). The mixture was stirred at 35 °C for 4 h before being diluted with a solution of sodium bisulfite (50 mL) and extracted with EtOAc (3 × 50 mL). The organic layer was dried over MgSO4 and the solvent reduced under vacuum. Recrystallization from CH2Cl2 / pet ether gave 11 as colourless needles (1.05 g, 85%): mp 168-169 °C (EtOAc, MeOH); 1H NMR (300 MHz, CDCl3): δ 11.78 (1H, brs, NH), 7.96 (2H, d, J = 7.9 Hz), 7.73 (1H, s), 7.67 (1H, m), 7.55 (2H, t, J = 7.9 Hz), 6.76 (1H, m), 6.54 (1H, dt, J = 1.7, 6.0 Hz), 6.12 (1H, dq, J = 1.5, 6.0 Hz), 5.18 (1H, m), 4.52 (2H, m); 13C NMR (75 MHz, CDCl3): δ 165.5, 160.2, 150.1, 144.0, 133.5, 133.3, 129.2, 129.0, 128.7, 126.5, 90.0, 84.0, 69.6, 65.3; HRMS (ES): m/z found 440.9930 (M+ + H). C16H14IN2O5 requires (M+ + H) 440.9942; Found C, 43.66; H, 2.98; N, 6.36. C16H13IN2O5 requires C, 43.12; H, 2.97; N, 6.04.
5.3. General procedure for Sonogashira coupling for compounds 7a-c to afford 12a-c
A solution of 5′-benzoyl-5-iodo-d4U 11 (0.5 mmol, 1 eq) in dry DMF (2.5 mL) was added to a stirred solution of triethylamine (1.0 mmol, 2 eq) and alkyne 7a-d (0.6 mmol, 1.2 eq) in THF (4 mL). The mixture was thoroughly degassed with nitrogen for 1 h. CuI (0.25 mmol, 0.5 eq) and Pd(PPh3)4 (0.05 mmol, 0.1 eq) were added to the degassed solution under a nitrogen atmosphere. The mixture was left stirring at rt for 4 h before adding 5% aq disodium EDTA (30 mL) and extracting with CHCl3 (3 × 30 mL). Washing with water (15 mL), drying over MgSO4, filtration and solvent evaporation under reduced pressure gave a residue that was purified by column chromatography employing EtOAc in petroleum ether mixtures to give the product as a yellow oil.
5.3.1. 5-{4-[2-(2-tert-Butoxycarbonylaminoethyl)-4-methoxyphenoxy]but-1-ynyl}-5′-O-benzoyl-2′,3′-didehydro-2′,3′-dideoxyuridine 12a
Using 11 (70 mg, 0.20 mmol) with alkyne 7a (0.10 g, 0.30 mmol) gave 12a (95 mg, 94%): [α]D -14.4 (c 1.60, CHCl3); IR (CHCl3): νmax 3692, 3606, 3451 (NH), 3011, 2934 (C-H), 2243 (C≡C), 1707 (C=O), 1502 (C=C) cm-1; 1H NMR (300 MHz, CDCl3): δ 8.79 (1H, brs), 8.00 (2H, d, J = 7.7 Hz), 7.61 (1H, s), 7.53 (1H, t, J = 7.7 Hz), 7.42 (2H, t, J = 7.7 Hz), 6.91 (1H, m), 6.70 (3H, m), 6.39 (1H, dt, J = 1.7, 5.8 Hz), 5.99 (1H, dq, J = 1.4, 5.8 Hz), 5.20 (1H, m), 4.83 (1H, brs), 4.64 (1H, dd, J = 4.3, 12.5 Hz), 4.50 (1H, dd, J = 3.0, 12.5 Hz), 3.94 (2H, t, J = 6.8 Hz), 3.74 (3H, s), 3.32 (2H, m), 2.77 (2H, t, J = 6.9 Hz), 2.68 (2H, t, J = 6.8 Hz), 1.40 (9H, s); 13C NMR (75 MHz, CDCl3): δ 166.2, 161.4, 153.9, 153.9, 150.5, 149.4, 141.6, 133.5, 133.3, 132.0, 129.7, 129.2, 128.6, 127.0, 116.7, 112.9, 112.0, 100.7, 91.3, 90.7, 85.0, 78.8, 72.4, 66.7, 65.1, 55.6, 40.6, 31.0, 28.4, 20.8; FAB HRMS: m/z found 654.24300 [M+Na]+. C34H37N3O9Na [M+Na]+ requires 654.24274.
5.3.2. 5-{6-[2-(2-tert-Butoxycarbonylaminoethyl)-4-methoxyphenoxy]hexa-4-oxa-1-ynyl}-5′-O-benzoyl-2′, 3′-didehydro-2′, 3′-dideoxyuridine 12b
Using 11 (0.30 g, 0.68 mmol) with alkyne 7b (0.26 g, 0.75 mmol) gave 12b (0.34 g, 76%): [α]D +16.2 (c 1.0, CHCl3); IR (CHCl3): νmax 3449, 3384 (NH), 3007, 2928 (C-H), 2246 (C≡C), 1711 (C=O), 1501 (C=C), 1169 (C-N) cm-1; 1H NMR (300 MHz, CDCl3): δ 8.42 (1H, brs), 8.01 (2H, d, J = 7.5 Hz), 7.68 (1H, s), 7.56 (1H, m), 7.45 (2H, t, J = 7.5 Hz), 6.92 (1H, m), 6.76 (1H, d, J = 8.7 Hz), 6.68 (2H, m), 6.40 (1H, dt, J = 1.7, 6.0 Hz), 6.00 (1H, dq, J = 1.3, 6.0 Hz), 5.21 (1H, m), 4.94 (1H, brs), 4.69 (1H, dd, J = 4.2, 12.5 Hz), 4.50 (1H, dd, J = 3.0, 12.5 Hz), 4.27 (2H, s), 4.07 (2H, t, J = 4.5 Hz), 3.85 (2H, m), 3.74 (3H, s), 3.33 (2H, m), 2.78 (2H, t, J = 6.9 Hz), 1.42 (9H, s); 13C NMR (75 MHz, CDCl3): δ 166.2, 161.3, 156.0, 153.8, 151.0, 149.4, 142.2, 133.6, 133.5, 129.7, 129.3, 129.2, 128.6, 127.0, 116.5, 113.0, 111.9, 99.9, 90.8, 90.0, 85.1, 78.8, 77.2, 68.4, 68.2, 65.0, 59.0, 55.6, 40.6, 30.8, 28.4; HRMS (ES): m/z found 662.2698 (M+ + H), C35H40N3O10 requires (M+ + H) 662.2714.
5.3.3. 5-{9-[2-(2-tert-Butoxycarbonylaminoethyl)-4-methoxyphenoxy]nona-4,7-dioxa-1-ynyl}-5′-O-benzoyl-2′, 3′-didehydro-2′, 3′-dideoxyuridine 12c
Using 11 (0.20 g, 0.46 mmol) with alkyne 7c (0.20 g, 0.51 mmol) gave 12c (0.23 g, 72%): [α]D +32.1 (c 1.0, CHCl3); IR (CHCl3): νmax 3674, 3377 (NH), 3014 (C-H), 2225 (C≡C), 1711 (C=O), 1501 (C=C), 1212 (C-N) cm-1; 1H NMR (400 MHz, CDCl3): δ 8.82 (1H, brs), 8.02 (2H, d, J = 7.6 Hz), 7.68 (1H, s), 7.56 (1H, m), 7.46 (2H, t, J = 7.6 Hz), 6.92 (1H, m), 6.77 (1H, d, J = 8.8 Hz), 6.69 (2H, m), 6.39 (1H, dt, J = 1.5, 6.0 Hz), 5.99 (1H, dq, J = 1.3, 6.0 Hz), 5.21 (1H, m), 4.97 (1H, brs), 4.70 (1H, dd, J = 4.2, 12.4 Hz), 4.51 (1H, dd, J = 2.9, 12.4 Hz), 4.25 (2H, s), 4.08 (2H, t, J = 4.9 Hz), 3.84 (2H, t, J = 4.9 Hz), 3.75 (3H, m), 3.73 (4H, m), 3.35 (2H, m), 2.80 (2H, t, J = 6.9 Hz), 1.43 (9H, s); 13C NMR (75 MHz, CDCl3): δ 166.2, 161.1, 156.0, 153.7, 151.0, 149.4, 142.2, 133.5, 133.4, 129.7, 129.3, 129.2, 128.6, 127.0, 116.5, 113.0, 112.0, 99.9, 90.7, 90.1, 85.1, 78.9, 77.2, 70.5, 69.8, 69.1, 68.4, 65.0, 58.9, 55.6, 40.6, 31.0, 28.4; HRMS (ES): m/z found 706.2958 (M+ + H). C37H44N3O11 requires (M+ + H) 706.2976.
5.4 General Procedure for the Synthesis of Thioureas 14a-d
Trifluoroacetic acid (0.20 mL) was added to a solution of compounds 12a-c (0.2 mmol) in CH2Cl2 (3 mL) at 0 °C, and the solution stirred for 2 h. Diisopropylethylamine (0.30 mL) was added, the solvent evaporated in vacuo and the crude amine dried under vacuum for 1 h, after which it was dissolved in THF (5 mL), thiourea 13 (0.24 mmol, 1.2 eq) added and the reaction mixture stirred at room temperature for 20 h. Following evaporation of the solvent, the residue was subjected directly to column chromatography using (EtOAc in pet ether = 7:3) to give the desired products 14a-c as pale-yellow solids.
5.4.1 5-{4-[2-(2-(5-Bromo-2-pyridinyl)aminothiocarbonylamino)ethyl)-4-methoxyphenoxy]but-1-ynyl}-5′-O-benzoyl-2′,3′-didehydro-2′,3′-dideoxyuridine 14a
Using 12a (100 mg, 0.16 mmol) with 13 (54 mg, 0.19 mmol) gave 14a (72 mg, 60%); mp 95-97 °C; [α]D - 9.9 (c 0.8, CHCl3); IR (CHCl3): νmax 3934, 3681 (NH), 2297 (C≡C), 1714 (C=O), 1602 (C=C), 1418 (C=S), 1212 (C-N) cm-1; 1H NMR (400 MHz, CDCl3): δ 10.92 (1H, t, J = 2.7 Hz), 9.75 (1H, brs), 9.13 (1H, s), 8.03 (2H, m), 7.91 (1H, d, J = 2.3 Hz), 7.61 (1H, s), 7.56 (1H, m), 7.45 (2H, m), 7.28 (1H, dd, J = 2.3, 8.8 Hz), 6.96 (1H, m), 6.80 (1H, m), 6.76 (2H, m), 6.62 (1H, d, J = 8.8 Hz), 6.37 (1H, dt, J = 1.6, 6.0 Hz), 6.09 (1H, m), 5.19 (1H, m), 4.65 (1H, dd, J = 4.2, 12.3 Hz), 4.53 (1H, dd, J = 3.8, 12.3 Hz), 3.97 (4H, m), 3.79 (3H, s), 3.03 (1H, m), 2.88 (1H, m), 2.71 (2H, t, J = 6.3 Hz); 13C NMR (100 MHz, CDCl3): δ 179.1 (C=S), 164.7, 162.2, 153.6, 151.6, 150.9, 149.3, 146.3, 141.8, 140.6, 133.3, 133.1, 129.7, 129.3, 128.8, 128.6, 127.7, 118.3, 113.3, 112.0, 111.6, 111.2, 100.6, 91.3, 90.7, 84.9, 72.6, 66.5, 65.3, 55.5, 45.4, 30.3, 20.9; HRMS (ES): m/z found 746.1262 (M+ + H). C35H33N5O7BrS requires (M+ + H) 746.1284.
5.4.2. 5-{6-[2-(2-(5-Bromo-2-pyridinylaminothiocarbonylamino)ethyl)-4-methoxyphenoxy]hexa-4-oxa-1-ynyl}-5′-O-benzoyl-2′,3′-didehydro-2′,3′-dideoxyuridine 14b
Using 12b (0.20 g, 0.30 mmol) with 13 (0.11 g, 0.39 mmol) gave 14b (0.13 g, 60%); mp: 73-76 °C; [α]D − 20.2 (c 1.0, CHCl3); IR (CHCl3): νmax 3601, 3391 (NH), 2957 (C-H), 2254 (-C≡C-), 1718 (C=O), 1501 (C=C), 1462 (C=S) cm-1; 1H NMR (300 MHz, CDCl3): δ 10.82 (1H, m), 9.00 (1H, brs), 8.84 (1H, brs), 8.03 (1H, d, J = 2.4 Hz), 8.00 (2H, m), 7.70 (1H, s), 7.65 (1H, dd, J = 2.4, 8.8 Hz), 7.56 (1H, , m), 7.45 (2H, m), 6.93 (1H, m), 6.87 (1H, d, J = 8.8 Hz), 6.79 (1H, d, J = 2.8 Hz), 6.77 (1H, d, J = 8.8 Hz), 6.73 (1H, dd, J = 2.8, 8.8 Hz), 6.40 (1H, dt, J = 1.7, 6.0 Hz), 6.02 (1H, dq, J = 1.4, 6.0 Hz), 5.21 (1H, m), 4.68 (1H, dd, J = 4.3, 12.5 Hz), 4.54 (1H, dd, J = 2.9, 12.5 Hz), 4.25 (2H, s), 4.00 (4H, m), 3.83 (2H, m), 3.75 (3H, s), 2.98 (2H, m); 13C NMR (100 MHz, CDCl3): δ 179.2 (C=S), 166.3, 161.6, 153.7, 151.8, 151.2, 149.4, 146.6, 142.6, 141.0, 133.6, 133.4, 29.7, 129.2, 128.9, 128.7, 127.0, 117.5, 113.6, 112.8, 112.5, 111.6, 100.0, 90.9, 90.0, 85.2, 77.2, 68.5, 68.3, 65.1, 59.2, 55.6, 45.7, 29.9; HRMS (ES): m/z found 776.1379 (M+ + H). C36H35N5O8SBr requires (M+ + H) 776.1390.
5.4.3. 5-{9-[2-(2-(5-Bromo-2-pyridinylaminothiocarbonylamino)ethyl)-4-methoxyphenoxy]nona-4,7-dioxa-1-ynyl}-5′-O-benzoyl-2′,3′-didehydro-2′,3′-dideoxyuridine 14c
Using 12c (0.15 g, 0.21 mmol) with 13 (76 mg, 0.27 mmol) gave 14c (0.10 g, 63%); mp 88-91 °C; [α]D - 18.5 (c 1.0, CHCl3); IR (CHCl3): νmax 3406 (NH), 3029, 2949 (C-H), 2152 (-C≡C-), 1718, 1671 (C=O), 1501 (C=C), 1469 (C=S), 1126 (C-N) cm-1; 1H NMR (300 MHz, CDCl3): δ 11.01 (1H, t, J = 4.8 Hz), 9.29 (1H, brs), 8.94 (1H, brs), 8.04 (1H, d, J = 2.4 Hz), 8.00 (2H, d, J = 7.4 Hz), 7.67 (1H, s), 7.65 (1H, dd, J = 2.4, 8.8 Hz), 7.56 (1H, m), 7.43 (2H, t, J = 7.4 Hz), 6.91 (1H, m), 6.80 (1H, d, J = 9.2 Hz), 6.79 (1H, d, J = 2.9 Hz), 6.77 (1H, d, J = 8.8 Hz), 6.73 (1H, dd, J = 2.9, 9.2 Hz), 6.38 (1H, dt, J = 1.6, 6.0 Hz), 5.98 (1H, m), 5.19 (1H, m), 4.67 (1H, dd, J = 4.3, 12.5 Hz), 4.50 (1H, dd, J = 3.0, 12.5 Hz), 4.21 (2H, s), 4.03 (2H, t, J = 4.7 Hz), 3.98 (2H, q, J = 6.6 Hz), 3.82 (2H, t, J = 4.7 Hz), 3.74 (3H, s), 3.71-3.64 (4H, m), 2.97 (2H, t, J = 6.6 Hz); 13C (75 MHz, CDCl3): δ 179.2 (C=S), 166.2, 161.4, 153.6, 151.7, 151.2, 149.5, 146.6, 142.5, 141.0, 133.5, 133.4, 129.7, 129.2, 128.8, 128.6, 127.0, 117.5, 113.5, 112.7, 112.5, 111.5, 100.0, 90.8, 90.1, 85.1, 77.0, 70.6, 69.8, 69.2, 68.4, 65.1, 59.0, 55.5, 45.6, 29.8; HRMS (ES): m/z found 820.1638 (M+ + H). C38H39N5O9SBr requires (M+ + H) 820.1652.
5.4.4. 5-{15-[2-(2-(5-Bromo-2-pyridinylaminothiocarbonylamino)ethyl)-4-methoxyphenoxy]pentadeca-4,7,10,13-tetraoxa-1-ynyl}-5′-O-benzoyl-2′,3′-didehydro-2′,3′-dideoxyuridine 14d
Trifluoroacetic acid (0.20 mL) was added to a solution of alkyne 7d (0.12 g, 0.25 mmol) in CH2Cl2 (3 mL) at 0 °C, and the solution stirred for 2 h. Anhydrous K2CO3 (0.10 g, 0.75 mmol) was added, the mixture stirred for a further 15 min then filtered through Celite. Solvent evaporation in vacuo and drying of the residue under vacuum for 1 h gave the crude amine, which was dissolved in dry THF (4 mL) with DMF (2 mL) and 11 (0.10 g, 0.23 mmol) and triethylamine (0.06 mL, 0.46 mmol) were added. The mixture was thoroughly degassed with nitrogen for 1h. CuI (23 mg, 0.12 mmol) and Pd(PPh3)4 (27 mg, 0.02 mmol) were added to the degassed solution under a nitrogen atmosphere and the mixture was left stirring at rt for 4h. The reaction mixture was then dissolved in MeOH: CHCl3 (1: 4) (30 mL), which was washed with portions (15 mL) of 5% aq disodium EDTA, water (10 mL) and dried over MgSO4. Filtration, solvent removal under reduced pressure and column chromatography of the residue employing EtOAc / MeOH / Et3N (5 /4 / 1) gave the coupled amine product, which was reacted with thiourea 13 (85 mg, 0.30 mmol) in dry THF (5 mL) at room temperature for 20 h. Following evaporation of solvent, the residue was subjected directly to column chromatography using EtOAc / pet ether (9 / 1) to afford the double-drug 14d as a solid (76 mg, 40% over the 3 steps); mp 112-115 °C; [α]D - 24.1 (c 1.0, CHCl3); IR (CHCl3): νmax 3377, 3217 (NH), 3007, 2920 (C-H), 2239 (-C≡C-), 1722, 1675 (C=O), 1501 (C=C), 1465 (C=S), 1227 (C-N) cm-1; 1H NMR (400 MHz, CDCl3): δ 11.09 (1H, t, J = 5.1 Hz), 8.88 (1H, brs), 8.76 (1H, brs), 8.05 (1H, d, J = 2.4 Hz), 8.02 (2H, d, J = 7.6 Hz), 7.68 (1H, s), 7.68 (1H, dd, J = 2.4, 8.8 Hz), 7.58 (1H, m), 7.46 (2H, t, J = 7.6 Hz), 6.93 (1H, m) 6.81 (1H, d, J = 2.8 Hz), 6.78 (1H, d, J = 8.8 Hz), 6.75 (1H, dd, J = 2.8, 8.8 Hz), 6.74 (1H, d, J = 8.8 Hz), 6.41 (1H, dt, J = 1.7, 6.0 Hz), 6.00 (1H, dq, J = 1.2, 6.0 Hz), 5.22 (1H, m,), 4.70 (1H, dd, J = 4.2, 12.5 Hz), 4.52 (1H, dd, J = 3.0, 12.5 Hz), 4.21 (2H, s), 4.02 (4H, m), 3.83 (2H, t, J = 5.8 Hz), 3.77 (3H, s), 3.71-3.64 (12H, m), 2.99 (2H, t, J = 6.6 Hz); 13C NMR (75 MHz, CDCl3): δ 179.1 (C=S), 166.2, 161.3, 153.5, 151.8, 151.2, 149.5, 146.4, 142.3, 141.0, 133.5, 133.4, 129.6, 129.1, 128.8, 128.6, 126.9, 117.5, 113.6, 112.7, 112.4, 111.5, 100.0, 90.7, 90.1, 85.1, 76.9, 70.8, 70.6, 70.5, 70.5, 70.3, 69.8, 69.1, 68.4, 65.0, 58.9, 55.5, 45.7, 29.8; HRMS (ES): m/z found 908.2178 (M+ + H). C42H47N5O11SBr requires (M+ + H) 908.2176.
5.5. General Procedure for benzoate deprotection of 14a-d to afford 15a-d
A solution of NaOMe in methanol (0.03 mL, 2M, 0.06 mmol, 0.6 eq) was added to a solution of the nucleoside (0.1 mmol) in MeOH (2 mL) at 0 °C. The mixture was stirred for 30 min, acetic acid (0.05 mL) was added, and the crude mixture was diluted with CH2Cl2 (1 mL) and subjected directly to column chromatography employing MeOH/ CH2Cl2 (2 / 8) to give the desired product as a pale-yellow solid.
5.5.1. 5-{4-[2-(2-(5-Bromo-2-pyridinylaminothiocarbonylamino)ethyl)-4-methoxyphenoxy]but-1-ynyl}-2′,3′-didehydro-2′,3′-dideoxyuridine 15a16
14a (83 mg, 0.11 mmol) gave 15a (43 mg, 60%); mp 121-122 °C; [α]D + 22.4 (c 1.10, CHCl3); IR (CHCl3): νmax 3934, 3688 (NH), 2304 (-C≡C), 1696 (C=O), 1606 (C=C), 1425 (C=S) cm-1; 1H NMR (400 MHz, CDCl3): δ 11.05 (1H, t, J = 4.2 Hz), 8.70 (1H, brs), 7.99 (1H, d, J = 2.6 Hz), 7.97 (1H, s), 7.56 (1H, dd, J = 2.6, 8.8 Hz), 6.97 (1H, m), 6.80 (1H, m), 6.77 (2H, m), 6.66 (1H, d, J = 8.8 Hz), 6.37 (1H, dt, J = 1.5, 5.8 Hz), 5.86 (1H, m), 4.93 (1H, m), 4.05 (2H, t, J = 5.9 Hz), 3.95 (2H, m), 3.80 (2H, m), 3.78 (3H, s), 2.98 (2H, m), 2.84 (2H, t, J = 5.8 Hz); 13C NMR (100 MHz, CDCl3): δ 178.9, 162.2, 153.4, 151.5, 150.9, 149.1, 146.6, 143.6, 140.8, 135.6, 129.3, 128.9, 118.0, 113.4, 112.8, 111.4, 110.8, 100.3, 91.3, 90.4, 87.7, 72.5, 66.8, 62.9, 55.6, 45.7, 30.2, 21.0; HRMS (ES): m/z found 642.1008 (M+ + H). C28H29N5O6BrS requires (M+ + H) 642.1022.
5.5.2. 5-{6-[2-(2-(5-Bromo-2-pyridinylaminothiocarbonylamino)ethyl)-4-methoxyphenoxy]hexa-4-oxa-1-ynyl}-2′,3′-didehydro-2′,3′-dideoxyuridine 15b
14b (100 mg, 0.14 mmol) gave 15b (59 mg, 69%); mp 76-79 °C; [α]D + 49.1 (c 1.0, CHCl3); IR (CHCl3): νmax 3681, 3594, 3391 (NH, OH), 3022, 2928 (C-H), 2239 (-C≡C-), 1715, 1697 (C=O), 1501 (C=C), 1465 (C=S), 1216 (C-N) cm-1; 1H NMR (300 MHz, CDCl3): δ 11.09 (1H, t, J = 5.1 Hz), 9.44 (1H, brs), 9.09 (1H, brs), 8.14 (1H, s), 8.00 (1H, d, J = 2.4 Hz), 7.65 (1H, dd, J = 2.4, 8.7 Hz), 6.97 (1H, m), 6.83 (1H, d, J = 8.7 Hz), 6.75 (3H, m), 6.35 (1H, dt, J = 1.7, 6.0 Hz), 5.85 (1H, m), 4.93 (1H, m), 4.38 (2H, s), 4.06−3.78 (8H, m), 3.75 (3H, s), 3.36 (1H, brs), 2.97 (2H, t, J = 6.6 Hz); 13C NMR (100 MHz, CDCl3): δ 178.8 (C=S), 161.8, 153.7, 151.8, 151.1, 149.9, 146.4, 144.7, 141.0, 135.1, 128.9, 126.0, 117.5, 113.8, 112.9, 112.6, 111.6, 99.3, 90.3, 89.4, 87.7, 77.5, 68.4, 68.4, 62.8, 59.3, 55.6, 45.8, 30.0; HRMS (ES): m/z found 672.1139 (M+ + H). C29H31N5O7SBr requires (M+ + H) 672.1128.
5.5.3 5-{9-[2-(2-(5-Bromo-2-pyridinylaminothiocarbonylamino)ethyl)-4-methoxyphenoxy]nona-4,7-dioxa-1-ynyl}-2′,3′-didehydro-2′,3′-dideoxyuridine 15c
14c (80 mg, 0.11 mmol) gave 15c (37 mg, 54%); mp 130-134 °C; [α]D + 41.1 (c 1.0, DMSO); IR (DMSO): νmax 3442, 3260 (NH, OH), 2928 (C-H), 2246, 2123 (-C≡C-), 1700 (C=O), 1462 (C=S), 1223 (C-N) cm-1; 1H NMR (300 MHz, DMSO-d6): δ 11.50 (1H, brs),1.05 (1H, t, J = 5.0 Hz),10.52 (1H, brs), 8.11 (1H, d, J = 2.6 Hz), 8.10 (1H, s), 7.93 (1H, dd, J = 2.6, 8.7 Hz), 7.12 (1H, d, J = 8.7 Hz), 6.92 (1H, d, J = 8.7 Hz), 6.82 (1H, d, J = 3.0 Hz), 6.80 (1H, m), 6.77 (1H, dd, J = 3.0, 8.7 Hz), 6.40 (1H, dt, J = 1.7, 6.0 Hz), 5.93 (1H, dq, J = 1.4 Hz, 6.0 Hz), 4.94 (1H, t, J = 5.0 Hz), 4.81 (1H, m), 4.31 (2H, s), 4.03 (2H, t, J = 4.9 Hz), 3.84 (2H, q, J = 6.6 Hz), 3.74 (2H, t, J = 4.9 Hz), 3.68 (3H, s), 3.66-3.58 (6H, m, H-5′), 2.90 (2H, t, J = 6.6 Hz); 13C NMR (75 MHz, DMSO-d6): δ 179.1 (C=S), 161.3, 153.1, 152.1, 150.6, 149.5, 145.6, 144.5, 141.0, 135.2, 128.4, 127.4, 116.8, 114.3, 113.4, 111.6, 111.4, 97.5, 89.4, 88.5, 87.5, 78.1, 69.5, 68.9, 68.5, 68.2, 61.7, 58.0, 55.1, 44.5, 28.9; HRMS (ES): m/z found 716.1389 (M+ + H), C31H35N5O8SBr requires (M+ + H) 716.1390.
5.5.4. 5-{15-[2-(2-(5-Bromo-2-pyridinylamino thiocarbonylamino)ethyl)-4-methoxyphenoxy]pentadeca-4,7,10,13-tetraoxa-1-ynyl}-2′,3′-didehydro-2′,3′-dideoxyuridine 15d
14d (70 mg, 0.08 mmol) gave 15d (32 mg, 52%); mp 63-65 °C; [α]D + 53.2 (c 1.0, DMSO); IR (DMSO): νmax 3442, 3283 (NH, OH), 2246, 2123 (C≡C), 1660 (C=O), 1469 (C=S), 1226 (C-N) cm-1; 1H NMR (400 MHz, DMSO-d6): δ 11.60 (1H, brs) 11.09 (1H, t, J = 4.8 Hz), 10.62 (1H, brs), 8.12 (1H, s), 8.09 (1H, d, J = 2.4 Hz), 7.93 (1H, dd, J = 2.4, 8.8 Hz), 7.09 (1H, d, J = 8.8 Hz), 6.89 (1H, d, J = 8.8 Hz), 6.81 (1H, d, J = 3.2 Hz), 6.78 (1H, m), 6.75 (1H, dd, J = 3.2, 8.8 Hz), 6.38 (1H, dt, J = 1.7, 6.0 Hz), 5.91 (1H, dq, J = 1.4, 6.0 Hz), 5.02 (1H, t, J = 5.2 Hz), 4.79 (1H, m), 4.28 (2H, s), 4.00 (2H, t, J = 4.9 Hz), 3.81 (2H, q, J = 6.6 Hz), 3.71 (2H, t, J = 4.9 Hz), 3.66 (3H, s), 3.60-3.47 (14H, m), 2.87 (2H, t, J = 6.6 Hz); 13C NMR (75 MHz, DMSO-d6): δ 179.1 (C=S), 161.4, 153.0, 152.2, 150.6, 149.6, 145.6, 144.6, 141.1, 135.2, 128.4, 125.5, 116.8, 114.3, 113.3, 111.6, 111.5, 97.5, 89.4, 88.5, 87.5, 78.2, 69.8, 69.7, 69.6, 69.6, 69.4, 68.9, 68.4, 68.2, 61.7, 58.0, 55.1, 44.5, 29.0; HRMS (ES): m/z found 804.1932 (M+ + H). C35H43N5O10SBr requires (M+ + H) 804.1914.
5.6 2′,3′-Didehydro-2′,3′-dideoxy-5-iodouridine (5-iodo-d4U)
To a solution of 11 (0.80 g, 1.82 mmol) in dry MeOH (15 mL), was added a solution of NaOMe in methanol (0.90 mL, 2M, 1.80 mmol) at 0 °C. The mixture was stirred at rt for 2 h before being diluted with aq NH4Cl (25 mL) and then extracted using CHCl3: MeOH (4:1) (3 × 40 mL). Drying (MgSO4), evaporation of solvent and purification of the residue on silica-gel chromatography using EtOAc as eluent gave 2′,3′-didehydro-2′,3′-dideoxy-5-iodouridine as a colourless solid (0.45 g, 74%); mp 176-177 °C; 1H NMR (300 MHz, DMSO-d6): δ 11.58 (1H, brs), 8.23 (1H, s), 6.78 (1H, m), 6.40 (1H, dt, J = 1.7, 6.0 Hz), 5.94 (1H, dq, J = 1.4, 6.0 Hz), 5.01 (1H, t, J = 4.9 Hz), 4.83 (1H, m), 3.62 (2H, m); 13C NMR (75 MHz, DMSO-d6): δ 160.3, 150.3, 145.7, 135.2, 125.7, 89.1, 87.4, 68.6, 61.5.
5.7. 2′,3′-Didehydro-2′,3′-dideoxy-5-iodouridine-5′-[p-tolylmethoxyalaninyl phosphate] 16
p-Tolyl methoxylalaninyl phosphorochloridate38 (1.77 g, 6.42 mmol) and 2′,3′-didehydro-2′,3′-dideoxy-5-iodouridine (0.72 g, 2.14 mmol) were dissolved in THF (25 mL) and N-methylimidazole (1.02 mL, 12.84 mmol) was added with vigorous stirring. After 24 h at rt the solvent was removed under vacuum. The residue was dissolved in CHCl3 (100 mL) and washed with hydrochloric acid solution (1M, 2 × 30 mL), aq NaHCO3 (2 × 30 mL), and then water (3 × 30 mL). The organic layer was dried over MgSO4 and the solvent evaporated under vacuum. Purification of the residue by chromatography on silica-gel eluting with 3% MeOH in CHCl3 gave 16 (0.45 g, 35 %) as a 1:1 mixture of diastereoisomers and as a colourless solid; mp 40-44 °C; IR (CHCl3): νmax 3384 (NH), 3007 (C-H), 1743, 1704 (C=O), 1505 (C=C), 1245 (P=O) cm-1; 1H NMR (400 MHz, CDCl3): δ (some peaks are split due to diastereoisomers at P) 8.91 (1H, brs), 7.92, 7.90 (1H, s), 7.11 (4H, m), 6.94, 6.92 (1H, m), 6.38, 6.32 (1H, dt, J = 1.7, 6.0 Hz), 5.94, 5.86 (1H, dq, J = 1.3, 6.0 Hz), 5.05 (1H, m), 4.46-3.82 (4H, m), 3.72, 3.71 (3H, s), 2.31 (3H, s), 1.41, 1.36 (3H, d, J = 7.6 Hz); 13C NMR (75 MHz, CDCl3): δ (values bearing an asterisk are given as an average of peaks, split due to diastereoisomers and/or C-P coupling) 173.9*, 159.9*, 150.3, 148.2*, 144.6*, 134.6, 133.7*, 133.0*, 126.9*, 120.0*, 90.2*, 85.3*, 69.0*, 66.6*, 52.4*, 50.0*, 21.0*, 20.8*; 31P NMR (CDCl3): δ 3.7, 3.5 (1:1); HRMS (ES): m/z found 592.0356 (M+ + H). C20H24N3O8PI requires (M+ + H) 592.0346.
5.8. 5-{15-[2-(2-(5-Bromo-2-pyridinylaminothiocarbonylamino)ethyl)-4-methoxyphenoxy]pentadeca-4,7,10,13-tetraoxa-1-ynyl}-2′,3-didehydro-2′,3′-dideoxy-5-iodouridine-5′-5′-(p-methylphenyl methoxyalaninyl phosphate)-2′,3′-didehydro-2′,3′-dideoxyuridine 17
Trifluoroacetic acid (0.20 mL) was added to a solution of alkyne 7d (92 mg, 0.19 mmol) in CH2Cl2 (2 mL) at 0 °C, and the solution stirred for 2 h. Anhydrous K2CO3 (80 mg, 0.57 mmol) was added, the mixture was stirred for a further 15 min and then filtered through Celite, The solvent was then evaporated in vacuo, the crude amine dried under vacuum for 1 h, dissolved in dry THF (3 mL) with DMF (2 mL), and 5-iodouridine-5′-[p-methylphenyl methoxyalaninyl phosphate] 16 (0.10 g, 0.17 mmol), followed by triethylamine (0.05 mL, 0.34 mmol) were added, The mixture was thoroughly degassed with nitrogen for 1h. CuI (17 mg, 0.09 mmol) and Pd(PPh3)4 (20 mg, 0.02 mmol) were then added to the degassed solution under a nitrogen atmosphere. The mixture was left stirring at rt for 2h, after which a mixture of MeOH: CHCl3 (1: 4, 30 mL) was added and washed with portions (2 × 10 mL) of 5% aq disodium EDTA, water (10 mL) and then dried over MgSO4. Filtration and solvent evaporation under reduced pressure gave a crude product, which was flashed through a silica-gel column employing EtOAc/ MeOH/ Et3N (5/ 4/ 1) as eluent. The coupled amine product was dissolved in dry THF (3 mL), thiourea 13 (65 mg, 0.23 mmol) was added and the mixture stirred at room temperature for 20 h. Following evaporation of solvent, the residue was subjected directly to column chromatography using EtOAc / MeOH (9 / 1) to give 17 as a pale-yellow solid (36 mg, 20% over the 3 steps); mp 51-54 °C; [α]D +4.4 (c 1.0, CHCl3); IR (CHCl3): νmax 3391 (NH), 3007, 2928 (C-H), 1707 (C=O), 1505 (C=C), 1462 (C=S), 1259 (P=O), 1223 (C-N) cm-1; 1H NMR (300 MHz, CDCl3): δ (some peaks are split due to diastereoisomers at P) 11.11 (1H, t, J = 4.9 Hz, NH), 9.11 (1H, brs, NH), 9.05, 9.00 (1H, s, NH), 8.03 (1H, d, J = 2.4 Hz), 7.79, 7.78 (1H, s), 7.66, 7.63 (1H, dd, J = 2.4, 8.8 Hz), 7.08 (4H, s), 6.96, 6.94 (1H, m), 6.80 (1H, d, J = 3.0 Hz), 6.78 (1H, d, J = 8.8 Hz), 6.74 (1H, d, J = 8.8 Hz), 6.72 (1H, dd, J = 3.0, 8.8 Hz), 6.37, 6.27 (1H, dt, J = 1.7, 6.0 Hz), 5.91, 5.81 (1H, dq, J = 1.4, 6.0 Hz), 5.02 (1H, m), 4.41-3.97 (10H, m), 3.81 (2H, t, J = 4.9 Hz), 3.75 (3H, s), 3.68, 3.67 (3H, s), 3.70-3.57 (12H, m), 2.97 (2H, t, J = 6.6 Hz), 2.28 (3H, s), 1.37, 1.33 (3H, d, J = 6.3 Hz); 13C NMR (75 MHz, CDCl3): δ (values bearing an asterisk are given as an average of peaks, split due to diastereoisomers and/or C-P coupling) 179.2 (C=S), 173.9*, 161.2, 153.6, 151.9, 151.3, 149.6, 148.3*, 146.5, 143.1*, 141.0, 134.7, 133.8, 130.1*, 128.8, 126.7*, 120.0*, 117.5, 113.6, 112.7, 112.4, 111.5, 99.9, 91.3*, 90.2, 85.4*, 77.2, 70.8, 70.6, 70.5 (2 × OCH2), 70.2*, 69.9, 69.2*, 68.4, 66.7 (d, Jcp = 4.5 Hz, C-5′), 58.9, 55.6, 52.5, 50.3*, 45.8, 29.8, 20.9*, 20.7; 31P NMR (CDCl3): δ 3.63, 3.56 (1:1); HRMS (ES): m/z found 1059.2548 (M+ + H). C46H57N6O14SBrP requires (M+ + H) 1059.2574.
5.9. Anti-HIV Evaluation
The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: MT-2 cells and HTLV-IIIB/H9 from Dr. Robert Gallo.
Antiviral activity and cellular toxicity were determined using the MTT colorimetric method40 in the following way. MT-2 cells43 at a concentration of 1 × 105 cells per milliliter were infected with wild type HIV-IIIB44 at a multiplicity of infection (MOI) of 0.1. Infected and mock-infected cells were incubated in growth medium (RPMI 1640, 10% dFBS, kanamycin) for 5 days with varying concentrations of each compound being tested in triplicate in a 96-well plate. MTT, a cell-permeable tetrazolium dye was then added to each well. After 5 hours, acidified isopropanol was added to lyse the cells and stop the reaction. The plates were gently shaken overnight, and the absorbance measured at 595 nm on a plate reader. The average of these triplicate samples were then plotted versus inhibitor concentration to generate dose-response curves. The 50% effective concentration (EC50) and 50% cytotoxic concentration (CC50) of the compounds were defined as the concentrations required to inhibit viral replication and to reduce the number of viable cells by 50%, respectively.
4.9.1 Steady-state IC50 determination
6 nM RT (active sites based on pre-steady-state active site determination) was pre-incubated for at least 15 minutes with 1 μM 5′-radiolabeled primer/template prior to mixing with appropriate concentrations of inhibitor and allowed to incubate for a minimum of 15 additional minutes on ice. DMSO concentrations were kept constant at less than 2%. DMSO alone was added as a no inhibitor control for each set of experiments. Reactions were initiated by the addition of 5 μM dTTP and 10 mM MgCl2 and were quenched after 15 minutes at 37° C with 0.3 M EDTA. All concentrations represent final concentrations after mixing. Reaction products were subjected to 20% denaturing polyacrylamide gel-electrophoresis and quantitated on a Bio-Rad Molecular Imager FX. Product formation was plotted as a function of inhibitor concentration and fitted to a hyperbola to generate IC50 curves. IC50 values are defined as the concentration of inhibitor that inhibits steady-state single nucleotide incorporation by 50%.
Supplementary Material
Acknowledgments
We thank the National Research Foundation of South Africa, the Third World Organization for Women in Science and the University of Cape Town for financial support. We also thank the National Institutes of Health (GM49551) for support to K.S.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Souza TML, Rodrigues DQ, Ferreira VF, Marques IP, Santos F, da Costa C, Cunha AC, Vieira de Souza MCB, Frugulhetti IC de Palmer Paixao, Bou-Habib DC, Fontes CFL. Current HIV Research. 2009;7:327. doi: 10.2174/157016209788347958. [DOI] [PubMed] [Google Scholar]
- 2.Marsden MD, Zack JA. J Antimicrob Chemother. 2009;63:7. doi: 10.1093/jac/dkn455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehellou Y, De Clercq E. J Med Chem. 2010;53:521. doi: 10.1021/jm900492g. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zhan P, Liu X, Li Z, Pannecouque C, De Clercq E. Curr Med Chem. 2009;16:3903. doi: 10.2174/092986709789178019. [DOI] [PubMed] [Google Scholar]; (b) Basavapathruni A, Anderson KS. Curr Pharm Des. 2006;12:1857. doi: 10.2174/138161206776873617. [DOI] [PubMed] [Google Scholar]
- 5.Nanni RG, Ding J, Jacobo-Molina A, Hughes SH, Arnold E. Perspect Drug Discovery Des. 1993;1:129. [Google Scholar]
- 6.Smerdon SJ, Jäger J, Wang J, Kohlstaedt LA, Chirino AJ, Friedman JM, Rice PA, Steitz TA. Proc Natl Acad Sci USA. 1994;91:3911. doi: 10.1073/pnas.91.9.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jencks WP. Proc Natl Acad Sci USA. 1981;78:4046. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Spence R, Kati W, Anderson KS, Johnson KA. Science. 1995;267:988. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rittinger K, Divita G, Goody RS. Proc Natl Acad Sci USA. 1995;92:8046. doi: 10.1073/pnas.92.17.8046. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gu Z, Quan Y, Li Z, Arts EJ, Wainberg MA. J Biol Chem. 1995;270:31046. doi: 10.1074/jbc.270.52.31046. [DOI] [PubMed] [Google Scholar]
- 9.Velázquez S, Alvarez R, San-Félix A, Jimeno ML, De Clercq E, Balzarini J, Camarasa MJ. J Med Chem. 1995;38:1641–1649. doi: 10.1021/jm00010a008. [DOI] [PubMed] [Google Scholar]
- 10.Tomassi C, Van Nhien AN, Marco-Contelles J, Balzarini J, Pannecouque C, De Clercq E, Soriano E, Postel D. Bioorg Med Chem Lett. 2008;18:2277–2281. doi: 10.1016/j.bmcl.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 11.Velázquez S, Tuñón V, Jimeno ML, Chamorro C, De Clercq E, Balzarini J, Camarasa MJ. J Med Chem. 1999;42:5188. doi: 10.1021/jm991092+. [DOI] [PubMed] [Google Scholar]
- 12.(a) Fossey C, Vu AH, Vidu A, Zarafu I, Laduree D, Schmidt S, Laumond G, Aubertin AM. J Enz Inhib Med Chem. 2007;22:591. doi: 10.1080/14756360701425386. [DOI] [PubMed] [Google Scholar]; (b) Pedersen L, Jørgensen PT, Nielsen S, Hansen TH, Nielsen J, Pedersen EB. J Med Chem. 2005;48:1211. doi: 10.1021/jm040845b. [DOI] [PubMed] [Google Scholar]; (c) Velázquez S, Lobatón E, De Clercq E, Koontz DL, Mellors JW, Balzarini J, Camarasa MJ. J Med Chem. 2004;47:3418. doi: 10.1021/jm031045o. [DOI] [PubMed] [Google Scholar]; (d) Sugeac E, Fossey C, Ladurée D, Schmidt S, Laumond G, Aubertin AM. J Enz Inhib Med Chem. 2003;18:175. doi: 10.1080/1475636032000069846. [DOI] [PubMed] [Google Scholar]
- 13.(a) Gavriliu D, Fossey C, Ciurea A, Delbederi Z, Sugeac E, Ladurée D, Schmidt S, Laumond G, Aubertin AM. Nucleos Nucleot Nucleic Acids. 2002;21:505. doi: 10.1081/NCN-120015066. [DOI] [PubMed] [Google Scholar]; (b) Pontikis R, Dollé V, Guillaumel J, Dechaux E, Note R, Nguyen CH, Legraverend M, Bisagni E, Aubertin AM, Grierson DS, Monneret C. J Med Chem. 2000;43:1927. doi: 10.1021/jm991125l. [DOI] [PubMed] [Google Scholar]; (c) Renoud-Grappin M, Fossey C, Fontaine G, Ladurée D, Aubertin AM, Kirn A. Antiviral Chem Chemother. 1998;9:205. doi: 10.1177/095632029800900302. [DOI] [PubMed] [Google Scholar]
- 14.Muhanji CI, Hunter R. Curr Med Chem. 2007;14:127. doi: 10.2174/092986707780597952. [DOI] [PubMed] [Google Scholar]
- 15.An interesting variation based on targeting both IN and RT in the form of a non-cleavable portmanteau inhibitor. Wang Z, Bennett EM, Wilson DJ, Salomon C, Vince R. J Med Chem. 2007;50:3416–3419. doi: 10.1021/jm070512p. [DOI] [PubMed] [Google Scholar]
- 16.Hunter R, Muhanji CI, Hale I, Bailey CM, Basavapathruni A, Anderson KS. Bioorg Med Chem Lett. 2007;17:2614. doi: 10.1016/j.bmcl.2007.01.107. [DOI] [PubMed] [Google Scholar]
- 17.Hunter R, Younis Y, Muhanji CI, Curtin TL, Naidoo KJ, Petersen M, Bailey CM, Basavapathruni A, Anderson KS. Bioorg Med Chem. 2008;16:10270. doi: 10.1016/j.bmc.2008.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Mao C, Sudbeck EA, Venkatachalam TK, Uckun FM. Biochem Pharmacol. 2000;60:1251. doi: 10.1016/s0006-2952(00)00408-1. [DOI] [PubMed] [Google Scholar]; (b) Mao C, Sudbeck EA, Venkatachalam TK, Uckun FM. Bioorg Med Chem Lett. 1999;9:1593. doi: 10.1016/s0960-894x(99)00235-8. [DOI] [PubMed] [Google Scholar]; (c) Vig R, Mao C, Venkatachalam TK, Tuel-Ahlgren L, Sudbeck EA, Uckun FM. Bioorg Med Chem Lett. 1998;8:1461. doi: 10.1016/s0960-894x(98)00250-9. [DOI] [PubMed] [Google Scholar]
- 19.(a) Ruth JL, Cheng YC. J Biol Chem. 1982:10261. [PubMed] [Google Scholar]; (b) Ruth JL, Cheng YC. Mol Pharmacol. 1981;20:415. [PubMed] [Google Scholar]
- 20.Chinchilla R, Najera C. Chem Rev. 2007;107:874. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 21.Pauwels R. Current Opinion in Pharmacology. 2004;4:437. doi: 10.1016/j.coph.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 22.Glennon RA, Liebowitz SM, Leming-Doot D, Rosecrans JA. J Med Chem. 1980;23:990. doi: 10.1021/jm00183a006. [DOI] [PubMed] [Google Scholar]
- 23.(a) Chen BC, Quinlan SL, Ried JG, Spector RH. Tetrahedron Lett. 1998;39:729. [Google Scholar]; (b) Chen BC, Quinlan SL, Stark DR, Reid G, Audia VH, George JG, Eisenreich E, Brundidge SP, Racha S, Spector RH. Tetrahedron Lett. 1995;36:7957. [Google Scholar]
- 24.(a) Codington JF, Doerr IL, Fox JJ. J Org Chem. 1964;29:558. [Google Scholar]; (b) Codington JF, Fecher R, Fox JJ. J Amer Chem Soc. 1960;82:2794. [Google Scholar]
- 25.Sheng J, Hassan AEA, Huang Z. J Org Chem. 2008;73:3725. doi: 10.1021/jo7025806. [DOI] [PubMed] [Google Scholar]
- 26.(a) Starrett JE, Jr, Tortolani DR, Baker DC, Omar MT, Hebbler AK, Wos JA, Martin JC, Mansuri MM. Nucleosides & Nucleotides. 1990;9:885. [Google Scholar]; (b) Mansuri MM, Starrett JE, Jr, Wos JA, Tortolani DR, Brodfuehrer PR, Howell HG, Martin JC. J Org Chem. 1989;54:4780. [Google Scholar]
- 27.Asakura J, Robins MJ. J Org Chem. 1990;55:4928–4933. [Google Scholar]
- 28.Robins MJ, Barr PJ. J Org Chem. 1983;48:1854. [Google Scholar]
- 29.Hobbs FW., Jr J Org Chem. 1989;54:3420. [Google Scholar]
- 30.Crisp G, Flynn BL. J Org Chem. 1993;58:6614. [Google Scholar]
- 31.Agrofolio LA, Gillaizeau I, Saito Y. Chem Rev. 2003;103:1875. doi: 10.1021/cr010374q. [DOI] [PubMed] [Google Scholar]
- 32.Kelleher MR, McGuigan C, Bidet O, Carangio A, Weldon H, Andrei G, Snoeck R, De Clercq E, Balzarini J. Nucleosides, Nucleotides & Nucleic Acids. 2005;24:643. doi: 10.1081/ncn-200060122. [DOI] [PubMed] [Google Scholar]
- 33.Bell FW, Cantrell AS, Högberg M, Jaskunas SR, Johansson NG, Jordan CL, Kinnick MD, Lind P, Morin JM, Jr, Noréen R, Öberg B, Palkowitz JA, Parrish CA, Pranc P, Sahlberg C, Ternansky RJ, Vasileff RT, Vrang L, West SJ, Zhang H, Zhou XX. J Med Chem. 1995;38:4929. doi: 10.1021/jm00025a010. [DOI] [PubMed] [Google Scholar]
- 34.(a) Robins MJ, Vinayak RS, Wood SG. Tetrahedron Lett. 1990;31:3731. [Google Scholar]; (b) Olivi N, Spruyt P, Peyrat JF, Alami M, Brion JD. Tetrahedron Lett. 2004;45:2607. [Google Scholar]
- 35.Meier C. Eur J Org Chem. 2006:1081. [Google Scholar]
- 36.Balzarini J, Karlsson A, Aquaro S, Perno CF, Cahard D, Naesens L, De Clercq E, McGuigan C. Proc Natl Acad Sci USA. 1996;93:7295. doi: 10.1073/pnas.93.14.7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siddiqui AQ, Ballatore C, McGuigan C, De Clercq E, Balzarini J. J Med Chem. 1999;42:393. doi: 10.1021/jm9803931. [DOI] [PubMed] [Google Scholar]
- 38.McGuigan C, Pathirana R, Balzarini J, De Clercq E. J Med Chem. 1993;36:1048. doi: 10.1021/jm00060a013. [DOI] [PubMed] [Google Scholar]
- 39.(a) Mehellou Y, Balzarini J, McGuigan C. Org Biomol Chem. 2009;7:2548. doi: 10.1039/b904276h. [DOI] [PubMed] [Google Scholar]; (b) Mehellou Y, McGuigan C, Brancale A, Balzarini J. Bioorg Med Chem Lett. 2007;17:3666. doi: 10.1016/j.bmcl.2007.04.043. [DOI] [PubMed] [Google Scholar]; (c) McGuigan C, Tsang HW, Cahard D, Turner K, Velazquez S, Salgado A, Bidois L, Naesens L, De Clercq E, Balzarini J. Antiviral Res. 1997;35:195. doi: 10.1016/s0166-3542(97)00029-6. [DOI] [PubMed] [Google Scholar]
- 40.Pannecouque C, Daelemans D, De Clercq E. Nature Protocols. 2008;3:427. doi: 10.1038/nprot.2007.517. [DOI] [PubMed] [Google Scholar]
- 41.Ciurea A, Fossey C, Gavriliu D, Delbederi Z, Sugeac E, Ladurée D, Schmidt S, Laumond G, Aubertin AM. J Enzyme Inhib. 2004;19:511. doi: 10.1080/14756360412331280527. [DOI] [PubMed] [Google Scholar]
- 42.Deng BL, Hartman TL, Buckheit RW, Jr, Pannecouque C, De Clercq E, Fanwick PE, Cushman M. J Med Chem. 2005;48:6140. doi: 10.1021/jm050452s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.(a) Haertle T, Carrera CJ, Wasson DB, Sowers LC, Richmann DD, Carson DA. J Biol Chem. 1988;263:5870. [PubMed] [Google Scholar]; (b) Harada S, Koyanagi Y, Yamamoto N. Science. 1985;229:563. doi: 10.1126/science.2992081. [DOI] [PubMed] [Google Scholar]
- 44.(a) Popovic M, Read-Connole E, Gallo R. Lancet. 1984;ii:1472. doi: 10.1016/s0140-6736(84)91666-0. [DOI] [PubMed] [Google Scholar]; (b) Popovic M, Sarngadharan MG, Read E, Gallo RC. Science. 1984;224:497. doi: 10.1126/science.6200935. [DOI] [PubMed] [Google Scholar]; (c) Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K, Ivanoff L, Petteway SRJ, Pearson ML, Lautenberger JA, Papas TS, Ghrayab J, Chang NT, Gallo RC, Wong-Stall F. Nature. 1985;313:277. doi: 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.