

Abstract
Current treatment of solid tumors is limited by side effects that result from the nonspecific delivery of drugs to the tumor site. Alternative targeted therapeutic approaches for localized tumors would significantly reduce systemic toxicity. Peptide therapeutics are a promising new strategy for targeted cancer therapy because of the ease of peptide design and the specificity of peptides for their intracellular molecular targets. However, the utility of peptides is limited by their poor pharmacokinetic parameters and poor tissue and cellular membrane permeability in vivo. This review article summarizes the development of elastin-like polypeptide (ELP) as a potential carrier for thermally targeted delivery of therapeutic peptides (TP), and the use of cell penetrating peptides (CPP) to enhance the intracellular delivery of the ELP-fused TPs. CPP-fused ELPs have been used to deliver a peptide inhibitor of c-Myc function and a peptide mimetic of p21 in several cancer models in vitro, and both polypeptides are currently yielding promising results in in vivo models of breast and brain cancer.
Keywords: Elastin-like polypeptide, thermal targeting, therapeutic peptide, cell penetrating peptide, c-Myc, p21
1. Introduction
Cancer is a complex disease and, as it progresses, it can become aggressive, manifested by the invasion of cells from the primary tumor to the liver, lungs, brain and other organs. Tumor cell metastasis is a major cause of death amongst cancer patients, and in order to prevent this process, it is necessary to effectively treat primary solid tumors. However, current treatment for solid tumors is limited by the fact that only a small percentage of the administered dose of drug reaches the tumor site, while the rest of the drug is distributed throughout the body, which leads to increased toxicity in normal tissues.
Tumor tissues differ from normal tissues in anatomical and structural characteristics. They have a heterogeneous distribution of blood vessels and usually lack effective lymphatic drainage. These factors lead to an uneven and slowed blood flow and abnormal fluid dynamics in the tumor tissue. As a result, macromolecules and soluble polymeric carriers penetrate and accumulate preferentially in tumors relative to normal tissues. This phenomenon is called the enhanced permeability and retention (EPR) effect [1-3], and it is the key to the clinical success of anti-cancer macromolecular carrier systems. With the goals of increasing specificity and lowering systemic toxicity, many different systems such as macromolecular prodrugs, liposomes, and micro- and nano-particles have been developed (reviewed in [4-6]) to treat solid tumors. Several natural and synthetic water-soluble polymers, such as N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers, dextrans, poly(ethylene glycol) (PEG), and poly(l-glutamic acid) have been utilized successfully in clinical research or are in human clinical trials (reviewed in [3, 7, 8]). While clinically useful anti-tumor activity has been achieved by exploiting the EPR effect and using passive targeting by macromolecular drug delivery systems, further selectivity is possible by active targeting.
Bioconjugation of targeting moieties, such as peptide sequences or antibodies which have specific affinity to cancer cells, to the polymer backbone can further exploit differences between cancer and normal cells through selective receptor-mediated endocytosis. Another way of active targeting can be achieved using thermally responsive biopolymers, such as elastin-like polypeptide (ELP), that undergo an inverse phase transition [9-11]. When intravenously delivered, these thermally responsive polypeptides are likely to be cleared under physiological conditions (37 °C). However, they will aggregate and selectively accumulate in tumors where externally induced focused heat (40-42 °C) is applied. Work in the lab of Chilkoti using human tumors implanted in nude mice has clearly demonstrated that hyperthermia of the tumor results in increased accumulation of ELP polypeptides [12-14]. This method combines the established advantages of macromolecular carriers with the additional advantage of active thermal targeting, and it also introduces other synergistic effects of hyperthermia treatment. Hyperthermia preferentially increases the permeability of endothelial tumor vasculature to macromolecular drug carriers, which can further enhance the delivery of drugs to tumors [15-17]. Furthermore, hyperthermia enhances the cytotoxicity of some chemotherapeutic agents [18]. The clinical application of hyperthermia to increase tissue temperatures to 40 - 43 °C has been integrated in multimodal anti-cancer strategies [19-21], and due to substantial technical improvements, hyperthermia is becoming more accepted clinically. Selected increase of temperatures in superficial and deep-seated tumors is accomplished using microwave, radio-frequency, and high-intensity focused ultrasound. Therefore, the effect of hyperthermia combined with the therapeutic effect of a polymeric drug carrier might offer further synergistic advantages in treatment of localized tumors.
In order to deliver therapeutic cargo to intracellular molecular targets and demonstrate therapeutic efficacy, the ELP carrier must successfully overcome transport barriers to drug delivery that are posed by unique structural and physiological characteristics of tumors. Despite the beneficial EPR effect, which favors accumulation of macromolecular carriers in tumors, there are many factors opposing the delivery of drugs to tumors. A solid tumor does not simply exist as a mass of malignant cells, but contains tumor cells, normal cells, extracellular matrix, and tumor blood vessels. Leaky tumor blood vessels are generally some distance removed from target tumor cells, separated by stroma and other cell types. Tumor and stromal cells produce and assemble the extracellular matrix, which consists of a meshwork of collagens, proteoglycans, and other molecules that together represent significant barriers to penetration by therapeutics agents [22, 23]. Although the lack of lymphatic drainage reduces efflux of the drug away from the tumor, it also reduces redistribution and transport of the drug within the tumor by generating higher pressure in the interstitium than in the vasculature, which makes macromolecular uptake less efficient [24]. Furthermore, once macromolecular carriers reach the cancer cell, there is the additional obstacle of plasma membrane impermeability. The plasma membrane of eukaryotic cells is generally impermeable to therapeutic macromolecules such as oligonucleotides and proteins due to the large size and inherently poor penetration capabilities of these molecules. Cell penetrating peptides (CPP) can be used to overcome these transport barriers to drug delivery and permit noninvasive delivery of polypeptides to their appropriate intracellular molecular target.
CPPs are short peptides that can be cationic, amphipathic, or hydrophobic. These peptides have the ability to efficiently cross cellular plasma membranes and enter the cytoplasm. Furthermore, when CPPs are linked to oligonucleotides, proteins, or nanoparticles, they facilitate the transport of these entities across the cell membrane [25-27]. Thus, a number of investigators have assessed the use of CPPs for intracellular delivery of macromolecules (reviewed in [28, 29]). CPPs have shown efficacy in vivo for delivery of macromolecules to tumors and even across the blood brain barrier [30-34]. As discussed in this review, addition of CPPs to the ELP carrier may not only enhance its uptake into the tumor cells in vitro [35-38], but the CPPs also mediate escape of the polypeptide from the tumor vasculature and entry into the tumor cells.
In addition to physically targeting therapeutic agents to cancer cells, another method of specifically inhibiting cancer cell proliferation is to exploit their genetic abnormalities. In some cancer types, cancer cells have overexpressed and/or permanently active oncogenes, causing hyperactive growth and division and/or protection against apoptosis. Some cancer cells have inactivated or missing tumor suppressor genes, resulting in deregulation of the apoptotic pathway and loss of control over the cell cycle and DNA replication. Recent characterization of the genetic alterations that occur during carcinogenesis has identified many potential molecular targets for which to develop new therapeutics. One of the major advantages of therapeutic peptides is that they are much easier to design using a rational approach than small molecule drugs for stimulation or inhibition of a given protein/protein interaction. These peptides are derived from high-throughput screening or by using NMR or crystal structures of their molecular target and further optimized by a rational drug design approach. Such therapeutic peptides can be designed to bind almost any protein of interest with high affinity and specificity and can interfere with molecular pathways that are deregulated in cancer cells [39, 40]. The use of peptides to specifically inhibit aberrant oncogenic or tumor suppressor proteins should be more effective and have fewer side effects than current nonspecific cytotoxic drug treatments. However, the clinical efficacy of therapeutic peptides is limited by pharmacodynamic properties. When applied in vivo, therapeutic peptides are rapidly degraded in circulation, and their relatively large size and often charged nature makes them impermeable to cancer cell membranes [41, 42]. Therefore, in order to advance therapeutic peptides into the clinical setting, a suitable carrier system that can overcome these limitations and target the peptide to the tumor site and into the tumor cells is needed.
Attention is being focused on peptide delivery using macromolecular carriers. Micro- and nanospheres are being investigated for their ability to deliver bioactive peptides via the oral route, stabilizing and delivering them through absorption barriers in the gastrointestinal tract [43]. Liposome - peptide conjugates have been investigated, but the focus of this field is the conjugation of cell penetrating peptides to the surface of liposomes to enhance fusion with the cell membrane [44]. To overcome limitations of other macromolecular carriers and improve delivery of peptide therapeutics to solid tumors, our lab is working to develop an ELP-based thermally targeted peptide vector. Such a carrier would have all of the characteristics and advantages of existing soluble macromolecular carriers, but it would be also be capable of active targeting by application of local hyperthermia.
ELP has been used for thermally targeted delivery of small molecule drugs [37, 45-47], plasmid DNA [48], therapeutic peptides [35, 36, 38, 49, 50], and proteins [51] in various cancer models. This review focuses on the use of thermally responsive polypeptide carriers for hyperthermia targeted delivery of therapeutic peptides. In this article, we present a summary of results from our lab comparing cellular uptake mechanisms and efficiency of several different CPP-fused ELPs and discuss their potential therapeutic utility. We also describe use of ELP to deliver two therapeutic peptides, one that inhibits the function of the oncogenic transcription factor c-Myc [35, 36] and one that mimics the Cdk inhibitor p21 [38, 50].
2. The use of cell penetrating peptides for intracellular delivery of ELPs
The ELP drug carrier used for thermal targeting (ELP1) has a molecular weight of 59.1 kDa [52], and it enters eukaryotic cells at low levels by an endocytic mechanism. In an attempt to improve the efficiency of cellular internalization of ELP, we modified it at its N-terminus with several CPPs (Figure 1A) [38]. Originally, we used three CPPs: the penetratin peptide from the Drosophila transcription factor Antennapedia [53], The Tat peptide from the HIV-1 Tat protein [54], and the MTS (membrane translocating sequence) derived from Kaposi fibroblast growth factor (Figure 1B) [55]. In more recent studies, we have also used the Bac CPP derived from the bactenecin antimicrobial peptide [56].
Figure 1.

Schematic representation of the ELP-based peptide delivery vector. A. The thermally responsive ELP polypeptide is fused at its N-terminus to a cell penetrating peptide (CPP) to mediate uptake of the macromolecule across the plasma membrane and dictate intracellular localization. At the C-terminus, a therapeutic peptide is added. B. Table of CPPs used to date for intracellular delivery of ELP.
2.1. Comparing the efficiency of various CPPs for intracellular delivery of ELP
The ability of each CPP to enhance the cellular uptake of ELP was assessed using fluorescently labeled CPP-ELP polypeptides for flow cytometry and confocal microscopy. As shown in Figure 2A, each of the three CPPs produced brighter cell staining than the parent ELP polypeptide, and flow cytometry histograms of cell number versus fluorescence intensity were unimodal, indicating that all cells were bound equally by the CPP-ELPs. When the flow cytometry data was quantified, it was determined that, of the three CPPs tested, the penetratin peptide was by far the most efficient. At 30 M, the cellular association/uptake of the polypeptide was increased 1.7 fold for Tat-ELP, 2.6 fold for MTS-ELP, and 14.8 fold for Pen-ELP relative to the ELP polypeptide lacking a CPP. The flow cytometry assay used can not directly distinguish polypeptide that has been internalized by the cell from polypeptide bound to the cell surface. Therefore, we used the membrane impermeable dye trypan blue to quench the fluorescence of surface bound polypeptide, and calculated the fraction internalized by by dividing the quenched (intracellular) fluorescence by the unquenched (intracellular and extracellular) fluorescence. This calculation allows determination of the percentage of the total amount of polypeptide that is present inside the cell, but it does not give any indication of total polypeptide levels. Performing this assay at various time points after cellular exposure to the CPP-ELPs demonstrated that polypeptide internalization did occur. About 20% of all CPP-ELPs were internalized at the end of a 1 h treatment and, at 24 h after treatment, 60% – 80% of the polypeptides were present inside the cells (Figure 2C). All CPP-ELPs were internalized at a similar rate which did not differ from that of the ELP control, indicating that all polypeptides were internalized by a similar mechanism. Internalization and subcellular localization was further confirmed by confocal fluorescence microscopy, which revealed a punctate cytoplasmic distribution for all polypeptides 24 h after cellular exposure. Previous reports regarding the short CPP peptides have indicated that this internalization and subcellular distribution can be an artifact of cell fixation [57], but that is not the case with CPP-ELPs, as live cells showed identical internalization and localization results [50]. In summary, the data in Figure 2A and B demonstrate the cellular levels of polypeptide achieved with the various CPPs, and the data in Figure 2C represents the percentage of that total that is inside the cell at the indicated time. Taken together, these data demonstrate that all polypeptides were internalized at a similar rate, which suggests that they are all internalized by a similar mechanism, but much higher levels of the CPP-ELPs were delivered into the cell relative to the ELP control.
Figure 2.

A. Flow cytometry histograms showing relative fluorescence of HeLa cells after treatment with the indicated CPP-ELP-fluorescein (20μM) for 1 h at 37 °C. The results are representative of a typical experiment. B. Effect of CPP-ELP-fluorescein concentration on cellular uptake as expressed in relative fluorescence units (RFU) normalized to uptake of 5 μM ELP. Increasing concentrations of polypeptides were incubated with HeLa cells at 37 °C for 1 h. Results are represented as mean ± SEM of three independent experiments. C. Kinetics of internalization of CPP-ELPs. HeLa cells were incubated with 20 μM fluorescein labeled proteins for 1h. Cell fluorescence was measured by flow cytometry at 1, 2, 4 and 24 h after polypeptide exposure. The percentage internalized was calculated by dividing the trypan blue quenched (intracellular) fluorescence by the total unquenched fluorescence. The results represent the mean ± SEM of three independent experiments. Analysis of variance revealed that there is a difference in the initial internalization (1 and 2 hrs, p<0.05) between MTS-ELP and the other CPP-ELPs, but no CPP-ELPs are significantly different after 4 h and 24h incubation (p>0.17). D. Subcellular localization of CPP-ELPs. HeLa cells were treated with CPP-ELP-rhodamine (20 μM) for 1 h at 37 °C, and confocal images were taken 24 h later. Scale bar = 8 μm. Because of the considerable difference in fluorescence uptake between CPP-ELPs, the gain was adjusted individually during each image acquisition. Therefore, the fluorescence intensity of the images does not represent the relative amount of CPP-ELP in the cell.
2.2. The mechanism of CPP-ELP internalization
The mechanism of CPP internalization has been the subject of much debate. Mechanisms ranging from inverted micelles [58] to simple endocytosis [57, 59] have been proposed, and it is clear that the mechanism is dependent on the cargo attached to the CPP (reviewed in [29, 60]). Given the slow internalization rate observed for all the CPP-ELP polypeptides, we suspected that a simple endocytosis mechanism was at work to internalize these large polypeptides. To address this hypothesis, we employed several inhibitors of endocytosis in our flow cytometric cellular uptake assay. Incubation at 4 °C (Figure 3A) or ATP depletion (Figure 3B), both general inhibitors of endocytosis, significantly inhibited the cellular internalization of all the CPP-ELPs tested. In addition, using hyperosmolar sucrose to block the formation of clathrin-coated pits [61] also caused a significant inhibition of polypeptide internalization (Figure 3C). On the other hand, the use of methyl-β-cyclodextrin to deplete the membrane of cholesterol and block clathrin-independent endocytosis via caveolae [62] had very little effect on CPP-ELP internalization (Figure 3D). We concluded that ELP and CPP-ELP internalization occurs via a caveolae-independent endocytic mechanism.
Figure 3.

Effect of inhibitors of endocytosis on CPP-ELP uptake. A. Effect of low temperature incubation on polypeptide uptake. HeLa cells were incubated with CPP-ELP-fluorescein (20 μM) for 1 h at 37 °C and 4 °C. B. Effect of ATP depletion on polypeptide uptake. The intracellular ATP pool was depleted by pre-incubation with sodium azide and deoxyglucose, followed by treatment of cells with CPP-ELP-fluorescein (20 μM) for 1 h at 37 °C. C. Effect of hyperosmolar sucrose on polypeptide uptake. Cells were incubated with 0.45 M sucrose for 1 h along with polypeptide treatment (20 μM). D. Effect of methyl-β-cyclodextrin on polypeptide uptake. Cells were preincubated with methyl-β-cyclodextrin (5 mM) for 30 min before polypeptide treatment. In all cases, non-internalized protein was quenched using trypan blue prior to flow cytometric analysis, and the cellular uptake is shown as relative fluorescence normalized to uptake of ELP. The results represent the mean ± SEM of three independent experiments. A Student’s t-test was used to determine statistical significance (*, p<0.01; +, p<0.05).
2.3. The use of CPPs to deliver an ELP-fused TP to tumor cells in vivo
Previous studies have shown that ELP accumulation in tumor vasculature or interstitium can be increased with focused hyperthermia [12-14]. The next step is to determine if the use of CPPs allowed entry of the ELP carrier into the tumor cells, a property necessary for effective drug delivery. To test the ability of the Bac and Tat CPPs to enhance ELP uptake into tumor cells in vivo, rats bearing two subcutaneous C6 tumors were intravenously injected with Rhodamine-labeled Bac-ELP1-H1 or Tat-ELP1-H1 (CPP-ELPs with a c-Myc inhibitory peptide cargo, please section 3.3 see below). One tumor was heated above the polypeptide’s transition temperature for 60 min by illumination with infrared (IR) light, and the localization of the polypeptide in the tumor tissue was determined by fluorescence microscopy. As shown in Figure 4A, Bac-ELP1-H1 is present not only in the tumor blood vessels, but has also escaped circulation and entered the tumor cells. The polypeptide was also able to escape the vasculature and enter the tumor cells when the tumor was heated above the Tt. Similarly, Tat-ELP1-H1 was also able to escape the tumor vasculature and enter the tumor cells (Figure 4B). Current work is underway to determine the levels of each polypeptide in the tumor both with and without hyperthermia treatment in order to determine the optimal CPP for tumor delivery in vivo and to demonstrate the ability to thermally target the CPP-ELPs to the heated tumor.
Figure 4.

Intratumoral localization of the CPP-delivered ELP peptide carrier. Rhodamine-labeled Bac-ELP1-H1 (A.) or Tat-ELP1-H1 (B.) was injected IV, and one of two subcutaneous C6 tumors was heated with IR light for 60 min. 500 kDa FITC-dextran was injected 1 min prior to euthanasia in order to mark the perfused vessels, and tumors were frozen, sectioned, and stained with Hoechst 33342 to mark the cell nuclei. A representative section from multiple tumor sections from duplicate animals is shown. Scale bar = 30 μm.
3. ELP-based delivery of a c-Myc inhibitory peptide
c-Myc is a transcription factor that, when bound to its heterodimerization partner Max, controls the expression of a large number of genes. Overexpression of c-Myc can cause uncontrolled cell proliferation and cancer [63]. A peptide inhibitor of c-Myc function was first discovered by Draeger [64], who screened peptides derived from the helix-loop-helix and leucine zipper domains of c-Myc and Max for their ability to inhibit c-Myc binding to DNA. They found that a peptide from helix 1 (H1) of c-Myc, when two residues were mutated to alanine to increase helicity (H1-S6A, F8A), was able to inhibit the binding of purified c-Myc protein to DNA in vitro. Giorello et al. adapted this c-Myc peptide by fusing it to the penetratin peptide and tested its antiproliferative effects in breast cancer cells [65]. This cell penetrating version of the H1 peptide was capable of blocking the co-immunoprecipitation of c-Myc and Max and inhibiting proliferation and colony formation of MCF-7 breast cancer cells grown in culture. However, inhibition of cell proliferation required multiple treatments and was only observed after 11 days of peptide exposure. To address these potency issues, the authors attempted to generate a more stable retro-inverso peptide by synthesizing the peptide in reverse order out of D amino acids, and reported that this peptide was a more potent inhibitor of cell proliferation than the L peptide [66], but still required multiple treatments to achieve significant inhibition.
3.1. Pen-ELP-H1
In an effort to further improve the stability of the H1 peptide and to adapt it for thermal targeting, our lab fused the H1-S6A, F8A peptide to the C-terminus of the ELP carrier, and added the penetratin CPP to the N-terminus [35]. The Pen-ELP-H1 polypeptide was taken up by MCF-7 cells, and the cellular uptake was increased 13-fold when aggregation of the polypeptide was induced by hyperthermia treatment. This increase is due to the formation of polypeptide aggregates under hyperthermia conditions, which are capable of binding to the outer surface of the plasma membrane and being internalized by endocytosis. Pen-ELP-H1 localized to the cytoplasm, and a single 1 h exposure to the polypeptide resulted in significant inhibition of the cell proliferation rate. Furthermore, when the single exposure was combined with hyperthermia treatment, the antiproliferative effect of Pen-ELP1-H1 was enhanced 2-fold, while no antiproliferative effect was observed with control polypeptides lacking the Pen CPP or the H1 inhibitory peptide (Figure 5A). Pen-ELP2-H1, a control polypeptide that does not aggregate at 42 °C, showed similar inhibition to that seen with Pen-ELP1-H1 at 37 °C, demonstrating that the heat enhancement seen with Pen-ELP1-H1 was due to its aggregation and resultant enhanced uptake, not to non-specific effects of hyperthermia. The cytoplasmically localized Pen-ELP-H1 sequestered the endogenous c-Myc protein to the cytoplasm (Figure 5B), resulting in a reduction of c-Myc transcriptional activation as assessed by measuring the mRNA levels of c-Myc target genes (Figure 5C).
Figure 5.

A. Antiproliferative effect of Pen-ELP-H1. Proliferation of MCF-7 cells was determined 11 days after a single 1 h treatment with the indicated polypeptide (18 μM) at 37 °C or 42 °C. Cells were counted using the trypan blue dye exclusion assay. Results represent the mean ± SE of 3-5 experiments performed in duplicate. B. Effect of Pen-ELP1-H1 on c-Myc localization. The subcellular localization of c-Myc and Max was determined by confocal immunofluorescence microscopy in untreated cells (top row) and in cells treated with 18 μM Pen-ELP1 (middle row) or 18 μM Pen-ELP1-H1 (bottom row) for 1h. Images were taken 24 h after polypeptide treatment with a 100x oil immersion objective, scale bar = 8 μm. C. Effect of Pen-ELP1-H1 on transcriptional activation by c-Myc. The mRNA levels for the c-Myc responsive genes ODC (top panel) and LDH-A (middle panel) and a control gene GAPDH (bottom panel) were assayed by RT-PCR. MCF-7 cells were untreated (lane 1) or treated with 18 μM Pen-ELP1 (lane 2), ELP1-H1 (lane 3), or Pen-ELP1-H1 (lane 4) for 1h. RNA was purified 48 h after treatment. PCR products were analyzed by capillary electrophoresis using a Bioanalyzer Labchip with fluorescence detection. The fluorescence data was converted to a simulated gel using Agilent software. The experiment was repeated 2 times.
3.2. Pen-ELP-H1 enhances the potency of topoisomerase II inhibitors
In addition to its ability to inhibit cell proliferation directly, Pen-ELP-H1 also sensitized cells to the topoisomerase II inhibitors doxorubicin and etoposide [67]. The IC50 of both doxorubicin and etoposide were reduced 1.5 fold by pre-treating MCF-7 breast cancer cells with the Pen-ELP-H1 polypeptide. These results are promising because, if this effect is present in vivo, it could allow administration of the toxic drugs at lower doses, which would reduce side effects. The enhancement of potency was specific to the topoisomerase II inhibiting class of drugs, as no effect was observed on the potency of the topoisomerase I inhibitor camptothecin or the DNA alkylating agent cisplatin. It is likely that this effect is due to the ability of Pen-ELP-H1 to reduce expression levels of the enzyme ornithine decarboxylase (ODC) [35], which catalyzes the first step in polyamine biosynthesis. Lowering polyamine levels is hypothesized to influence topoisomerase II binding and cleavage of DNA by modulating chromatin structure [68-71]. The effect of Pen-ELP-H1 on topoisomerase II drug potency was not limited to MCF-7 cells, as similar effects were seen in both HeLa cervical carcinoma cells and MES-SA uterine sarcoma cells. These results demonstrate that, in addition to their potential as monotherapy agents, ELP-delivered TPs may also be useful in combination therapies with either classical chemotherapeutic drugs or with other TPs.
3.3. Optimizing the antiproliferative effect of the ELP-H1 polypeptide with alternative CPPs
As shown in the model in Figure 6, we concluded that the cytoplasmic Pen-ELP-H1 polypeptide bound to nascently translated c-Myc protein, thereby preventing its nuclear localization and interaction with Max. Though the penetratin-delivered polypeptide inhibited cell proliferation with a single treatment, a long incubation period (11 days) was needed to observe a significant reduction in cell number. Based on the model for c-Myc inhibition, we hypothesized that delivery of the H1 peptide directly into the nucleus could lead to a more efficient, and thus more potent, c-Myc inhibition. With this goal in mind, we synthesized other CPP-ELP-H1 constructs by utilizing the Tat and Bac CPPs [36]. As shown in Figure 7A, of the three CPP-ELP-H1 polypeptides tested, Pen-ELP-H1 was by far the most efficient for cellular association and uptake, which is consistent with our original results collected using the CPP-ELPs without the H1 peptide in HeLa cells. However, in spite of its lower cellular uptake, Bac-ELP-H1 was a far more potent inhibitor of MCF-7 cell proliferation than Pen-ELP-H1 or Tat-ELP-H1 (Figure 7B). When the subcellular localization of the CPP-ELP-H1 polypeptides was assessed by confocal fluorescence microscopy, we found that Bac-ELP-H1 was able to enter the nucleus of the cells (Figure 7C), a trait not seen for any other CPP-ELP-H1 constructs, and the percentage of the cells containing nuclear localized polypeptide increased with polypeptide concentration and with heat treatment [36]. As shown in Figure 6, we proposed that the ability of the Bac CPP to deliver a portion of the cellular polypeptide to the nucleus resulted in a more potent inhibition of cell proliferation even though the total intracellular polypeptide levels were lower compared to other CPP-ELP-H1 constructs.
Figure 6.

Proposed model for c-Myc inhibition by Pen-ELP-H1 and Bac-ELP-H1. Mitogen stimulation induces transcription of mRNA from the c-Myc gene. In the case of Pen-ELP-H1, newly translated c-Myc is bound by the polypeptide in the cytoplasm. Once bound, c-Myc can not be imported into the nucleus and interact with Max. In the case of Bac-ELP-H1, the polypeptide enters the nucleus and interferes directly with the c-Myc and Max interaction. Both situations result in the down-regulation of c-Myc-Max responsive genes and lead to inhibition of cell proliferation.
Figure 7.
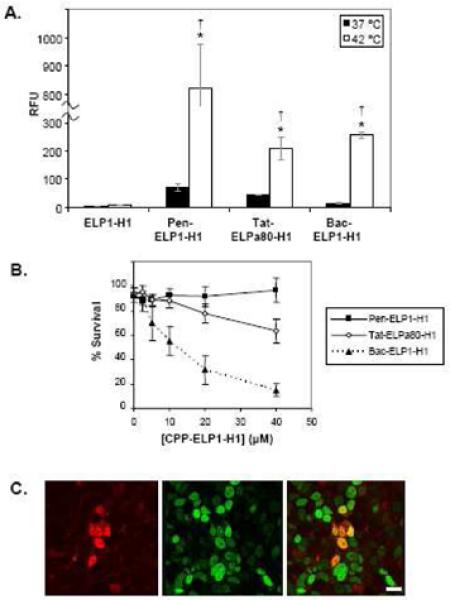
Optimization of ELP-H1 delivery with alternative CPPs. A. Cellular uptake of the CPP-ELP-H1 polypeptides. MCF-7 cells were treated for 1 h at 37 or 42 °C with fluorescein labeled polypeptides. Levels of each polypeptide were assessed using flow cytometry (n = 5,000 cells). Forward and side scatter gating were used to eliminate cell debris from the analysis, and fluorescence data was normalized to cellular autofluorescence and corrected for variations in labeling efficiency among the polypeptides. Data represent the average of 3 experiments; error bars, SEM.* Difference between 37 and 42 °C levels are statistically significant (ANOVA, p<0.01). † Difference is significant as compared to ELP at 42 °C. (ANOVA, p<0.01). B. MCF-7 proliferation after CPP-ELP-H1 treatment. MCF-7 cells were treated for 1 h at 37 or 42 °C with various concentrations of Pen-ELP1-H1, Tat-ELPa80-H1, or Bac-ELP1-H1, and the cell viability was determined after 7 days using the MTS assay. The data shown is an overlay of the 42 °C data for each CPP-ELP-H1. C. Subcellular localization of Bac-ELP1-H1. MCF-7 cells were treated for 1 h at 42 °C with rhodamine labeled Bac-ELP1-H1 (30 μM). 48 h after treatment, nuclei were stained with Sytox green and cells were imaged with a laser scanning confocal microscope. Scale bar = 20 μm.
We are currently using Bac-ELP-H1 as a lead compound for in vivo investigation. In addition to its antiproliferative activity against breast cancer cells, this polypeptide is a potent inhibitor of ovarian cancer and glioma cell proliferation. Current efforts are testing biodistribution, tumor uptake, and tumor reduction using Bac-ELP-H1 in mouse breast cancer models and a rat glioma model. If successful, these efforts will demonstrate proof of principle for ELP-based thermally targeted delivery of a TP in vivo.
4. ELP-based delivery of a p21 mimetic peptide
p21 is a cyclin dependent kinase inhibitor (CKI) protein than functions to control progression through the cell cycle by modulating interactions between cyclins and cyclin dependent kinases (Cdk). Also, p21 can interact with the processivity factor of DNA polymerase δ, PCNA. p21 is a p53 controlled gene that is activated during the DNA damage response pathway, and loss of p21 activity (either due to p21 mutations or loss of p53 function) causes cells to lose the ability to arrest the cell cycle following DNA damage. Much effort has been placed on trying to find peptides which can mimic or restore p21 activity for use as anti-cancer agents, and the resulting peptides can be grouped into two classes: N-terminal peptides which inhibit Cdk2/cyclin E activation and C-terminal peptides which interact with PCNA and inhibit DNA replication (and also inhibit Cdk4/cyclin D1) (reviewed in [40]).
Of the C-terminal peptides, the region between amino acids 139 and 164 has received the most attention. Peptides from the 139-164 region were found to bind directly and specifically to PCNA [72-74] and were capable of inhibiting the repair of UV-damaged DNA in HeLa cell extracts. A peptide from the same region (141-160) has also been shown to bind to and inhibit Cdk4/cyclin D1 [75]. Peptides from the C-terminal region of p21 between amino acids 141 and 160 contain binding motifs for both PCNA (141-152) and Cdk4/cyclin D1 (155-160). Therefore, the use of the full length 141-160 peptide as an inhibitor may lead to two separate mechanisms of action. These peptides, when fused to the penetratin CPP, have shown antiproliferative activity against human keratinocyte-derived HaCaT cells [75], DLD1 colon cancer cells [76], and CA46 lymphoma cells [77]; and a GFP-fused p21 peptide inhibited proliferation of H1299 non-small cell lung carcinoma cells (p53 deletion), U2OS osteosarcoma cells (p53 wild type), and Saos2 osteocarcinoma cells (p53 deletion) [78].
4.1. ELP-fused p21 peptides
In order to facilitate targeted delivery, we fused the p21 139-164 peptide to the C-terminus of ELP. With the penetratin peptide at the C-terminus, the Pen-ELP-p21 polypeptide was taken up by HeLa cervical and SKOV-3 ovarian carcinoma cells and localized to the cytoplasm. We demonstrated that the Pen-ELP-p21 polypeptide, but not control polypeptides lacking the penetratin CPP or the p21 mimetic peptide, exhibited an antiproliferative effect in both HeLa and SKOV-3 cells (Figure 8A) [38]. Given that p21 is a nuclear protein and following the same logic applied to the c-Myc inhibitory peptide, we next synthesized Bac-ELP-p21 with the goal of enhancing the polypeptide’s nuclear delivery and potency. When exposed to SKOV-3 cells at 37 °C, Bac-ELP1-p21 had a modest inhibitory effect on cell proliferation. However, treatment of the cells with the polypeptide at 42 °C induced aggregation of the polypeptide and lead to binding of the aggregates to the plasma membrane followed by internalization by endocytosis. Under these treatment conditions, cell proliferation was abolished completely (Figure 8B, top panel). Bac-ELP1-p21 treatment combined with hyperthermia was also very effective for inhibition of MCF-7 breast cancer (Figure 8B, middle panel) and Panc-1 pancreatic cancer cell proliferation (Figure 8B, lower panel). As with the Bac-ELP-H1 construct, Bac-ELP-p21 localized to the nucleus in a large percentage of the target cells (Figure 8C), and nuclear localization was seen when treatment was carried out both above and below the polypeptides transition temperature. In order to confirm the mode of action of the ELP-delivered p21 peptide, we tested its ability to inhibit phosphorylation of the tumor suppressor protein Rb. When in the hypophosphorylated state, Rb inhibits progression through the cell cycle, thus functioning as a tumor suppressor. Phosphorylation causes Rb to release its inhibitory binding to the transcription factor E2F, and cell cycle progression occurs. As shown in Figure 8D, 24 h after treatment with Bac-ELP1-p21 for 1 h at 42°C, SKOV-3 cells showed a significant decrease in the pRb levels as compared to untreated or Bac-ELP1 treated cells. The total Rb level was unchanged, and β-tubulin blotting was used to confirm accurate gel loading. These results suggest that Bac-ELP1-p21 most likely blocks the cell cycle by inhibiting the phosphorylation of the Rb protein.
Figure 8.

CPP-ELP-p21 polypeptides. A. Inhibition of cell proliferation by Pen-ELP-p21. HeLa and SKOV-3 cells were exposed to the indicated concentration of Pen-ELP2-p21 at 37 °C for 1 h, and cell proliferation was determined 72 h later using the MTS assay. B. Inhibition of proliferation by Bac-ELP-p21. SKOV-3, MCF-7, and Panc-1 cells were exposed to the indicated concentration of Bac-ELP2-p21 at 37 °C or 42 °C for 1 h, and cell proliferation was determined 6 days later (SKOV-3) or 3 days later (MCF-7 and Panc-1) using the MTS assay. Data represent the mean ± SE of 3 independent experiments. C. Subcellular localization of rhodamine labeled Bac-ELP1-p21 in SKOV-3 cells as visualized by confocal microscopy. Cells were treated with 20 μM Bac-ELP1-p21 at 37°C or 42°C for 1 h. Confocal images were taken 24 h later. Tubulin was stained as a reference for cellular structure. Scale bar = 20 μm. The subcellular distribution of Bac-ELP1-p21 was also confirmed in live cells 24 h after a 1 h exposure at 37 or 42 °C (not shown). D. SDS-PAGE analysis of Rb protein in SKOV-3 cells following treatment with Bac-ELP1-p21. Cells were treated with the indicated polypeptide (30 μM) at 42°C for 1 h, harvested 24 h later, and lysed. Equal amounts of samples were loaded onto a 12% SDS gel and transferred to a blot which was probed with the indicated antibodies.
Bac-ELP-p21 is also being used as a lead molecule for in vivo testing. As shown in Figure 8, Bac-ELP-p21 is a potent inhibitor of SKOV-3, MCF-7, and Panc-1 cell proliferation. Current experiments are utilizing mouse and rat breast cancer models, a mouse ovarian cancer model, and mouse pancreatic cancer models to evaluate the delivery and therapeutic efficacy of Bac-ELP-p21.
5. Conclusion
The use of ELP for targeted peptide delivery has several unique features which makes it complementary and synergistic with existing targeting modalities. First, it has all advantages and characteristics of soluble macromolecules which accumulate in tumors due to a passive targeting EPR effect. Second, ELP based therapeutic peptide carriers are thermally responsive and therefore may be additionally actively targeted by application of local hyperthermia. Third, the addition of CPPs to the ELP carrier enhances uptake into the tumor cells in vitro [35-38], and CPPs also mediate the escape of the polypeptide from the tumor vasculature and the entry into the tumor cells. Furthermore, addition of a CPP not only increases cellular uptake of the ELP carrier, but the choice of CPP can also target ELP to the desired cellular compartment [36]. This allows the attached therapeutic peptide to reach its target protein efficiently, resulting in a potent inhibition of cancer cell proliferation. Finally, the use of therapeutic peptides designed to specifically interact with molecular targets which are aberrantly expressed or mutated only in cancer cells adds a third layer of targeting. As a result, the function of normal cells will be less affected by treatment, which will further reduce systemic side effects compared to conventional non-selective chemotherapeutics. In summary, ELP is an ideal drug carrier because it combines the advantages of active and passive targeting, is easy to procure in large, pure quantities, and has a modular design that is easy to modify for attachment of therapeutic agents.
6. Acknowledgements
This work presented here was supported by NIH grant R21 CA113813-01A2, R43 CA135799-01A2, and a Wendy Will Case Cancer Foundation grant to DR; and Department of Defense (DOD) Breast Cancer Research Program (BCRP) Era of Hope Postdoctoral Award W81XWH-08-1-0647 to GLB, III. We would also like to thank Emily Thomas for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target. 2007;15(7-8):457–464. doi: 10.1080/10611860701539584. [DOI] [PubMed] [Google Scholar]
- [2].Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11(17-18):812–818. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- [3].Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- [4].Talelli M, Rijcken CJ, van Nostrum CF, Storm G, Hennink WE. Micelles based on HPMA copolymers. Adv Drug Deliv Rev. 2009 doi: 10.1016/j.addr.2009.11.029. [DOI] [PubMed] [Google Scholar]
- [5].Matsumura Y. Poly (amino acid) micelle nanocarriers in preclinical and clinical studies. Adv Drug Deliv Rev. 2008;60(8):899–914. doi: 10.1016/j.addr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- [6].Vicent MJ, Duncan R. Polymer conjugates: nanosized medicines for treating cancer. Trends Biotechnol. 2006;24(1):39–47. doi: 10.1016/j.tibtech.2005.11.006. [DOI] [PubMed] [Google Scholar]
- [7].Hu X, Jing X. Biodegradable amphiphilic polymer-drug conjugate micelles. Expert Opin Drug Deliv. 2009;6(10):1079–1090. doi: 10.1517/17425240903158917. [DOI] [PubMed] [Google Scholar]
- [8].Khare P, Jain A, Gulbake A, Soni V, Jain NK, Jain SK. Bioconjugates: harnessing potential for effective therapeutics. Crit Rev Ther Drug Carrier Syst. 2009;26(2):119–155. doi: 10.1615/critrevtherdrugcarriersyst.v26.i2.10. [DOI] [PubMed] [Google Scholar]
- [9].Urry DW, Luan C-H, Parker TM, Gowda DC, Prasad KU, Reid MC, Safavy A. Temperature of Polypeptide Inverse Temperature Transition Depends on Mean Residue Hydrophobicity. J. Am. Chem. Soc. 1991;113:4346–4348. [Google Scholar]
- [10].Urry DW, Trapane TL, Prasad KU. Phase-structure transitions of the elastin polypentapeptide-water system within the framework of composition-temperature studies. Biopolymers. 1985;24(12):2345–2356. doi: 10.1002/bip.360241212. [DOI] [PubMed] [Google Scholar]
- [11].Urry DW. Free energy transduction in polypeptides and proteins based on inverse temperature transitions. Prog Biophys Mol Biol. 1992;57(1):23–57. doi: 10.1016/0079-6107(92)90003-o. [DOI] [PubMed] [Google Scholar]
- [12].Meyer DE, Kong GA, Dewhirst MW, Zalutsky MR, Chilkoti A. Targeting a Genetically Engineered Elastin-like Polypeptide to Solid Tumors by Local Hyperthermia. Cancer Res. 2001;61(4):1548–1554. [PubMed] [Google Scholar]
- [13].Dreher MR, Liu W, Michelich CR, Dewhirst MW, Chilkoti A. Thermal cycling enhances the accumulation of a temperature-sensitive biopolymer in solid tumors. Cancer Res. 2007;67(9):4418–4424. doi: 10.1158/0008-5472.CAN-06-4444. [DOI] [PubMed] [Google Scholar]
- [14].Liu W, Dreher MR, Furgeson DY, Peixoto KV, Yuan H, Zalutsky MR, Chilkoti A. Tumor accumulation, degradation and pharmacokinetics of elastin-like polypeptides in nude mice. J Control Release. 2006;116(2):170–178. doi: 10.1016/j.jconrel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- [15].Issels RD. Regional hyperthermia combined with systemic chemotherapy of locally advanced sarcomas: preclinical aspects and clinical results. Recent Results Cancer Res. 1995;138:81–90. doi: 10.1007/978-3-642-78768-3_10. [DOI] [PubMed] [Google Scholar]
- [16].Feyerabend T, Steeves R, Wiedemann GJ, Richter E, Robins HI. Rationale and clinical status of local hyperthermia, radiation, and chemotherapy in locally advanced malignancies. Anticancer Res. 1997;17(4B):2895–2897. [PubMed] [Google Scholar]
- [17].van Vulpen M, Raaymakers BW, de Leeuw AA, van de Kamer JB, van Moorselaar RJ, Hobbelink MG, Battermann JJ, Lagendijk JJ. Prostate perfusion in patients with locally advanced prostate carcinoma treated with different hyperthermia techniques. J Urol. 2002;168(4 Pt 1):1597–1602. doi: 10.1016/S0022-5347(05)64527-2. [DOI] [PubMed] [Google Scholar]
- [18].Issels RD. Regional hyperthermia in high-risk soft tissue sarcomas. Curr Opin Oncol. 2008;20(4):438–443. doi: 10.1097/CCO.0b013e3283025e50. [DOI] [PubMed] [Google Scholar]
- [19].Falk MH, Issels RD. Hyperthermia in oncology. Int J Hyperthermia. 2001;17(1):1–18. doi: 10.1080/02656730150201552. [DOI] [PubMed] [Google Scholar]
- [20].Dewhirst MW, Prosnitz L, Thrall D, Prescott D, Clegg S, Charles C, MacFall J, Rosner G, Samulski T, Gillette E, LaRue S. Hyperthermic treatment of malignant diseases: current status and a view toward the future. Semin. Oncol. 1997;24(6):616–625. [PubMed] [Google Scholar]
- [21].Takahashi I, Emi Y, Hasuda S, Kakeji Y, Maehara Y, Sugimachi K. Clinical application of hyperthermia combined with anticancer drugs for the treatment of solid tumors. Surgery. 2002;131(1 Suppl):S78–84. doi: 10.1067/msy.2002.119308. [DOI] [PubMed] [Google Scholar]
- [22].Jain RK. Transport of molecules, particles, and cells in solid tumors. Annu Rev Biomed Eng. 1999;1:241–263. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- [23].Jain RK. Delivery of novel therapeutic agents in tumors: physiological barriers and strategies. J Natl Cancer Inst. 1989;81(8):570–576. doi: 10.1093/jnci/81.8.570. [DOI] [PubMed] [Google Scholar]
- [24].Jain RK. Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 1987;6(4):559–593. doi: 10.1007/BF00047468. [DOI] [PubMed] [Google Scholar]
- [25].Nori A, Kopecek J. Intracellular targeting of polymer-bound drugs for cancer chemotherapy. Adv Drug Deliv Rev. 2005;57(4):609–636. doi: 10.1016/j.addr.2004.10.006. [DOI] [PubMed] [Google Scholar]
- [26].Snyder EL, Dowdy SF. Cell penetrating peptides in drug delivery. Pharm Res. 2004;21(3):389–393. doi: 10.1023/B:PHAM.0000019289.61978.f5. [DOI] [PubMed] [Google Scholar]
- [27].Temsamani J, Vidal P. The use of cell-penetrating peptides for drug delivery. Drug Discov Today. 2004;9(23):1012–1019. doi: 10.1016/S1359-6446(04)03279-9. [DOI] [PubMed] [Google Scholar]
- [28].Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev. 2005;57(4):637–651. doi: 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- [29].Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol. 2009;157(2):195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285(5433):1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- [31].Rousselle C, Clair P, Lefauconnier JM, Kaczorek M, Scherrmann JM, Temsamani J. New advances in the transport of doxorubicin through the blood-brain barrier by a peptide vector-mediated strategy. Mol Pharmacol. 2000;57(4):679–686. doi: 10.1124/mol.57.4.679. [DOI] [PubMed] [Google Scholar]
- [32].Drin G, Rousselle C, Scherrmann JM, Rees AR, Temsamani J. Peptide delivery to the brain via adsorptive-mediated endocytosis: advances with SynB vectors. AAPS PharmSci. 2002;4(4):E26. doi: 10.1208/ps040426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Adenot M, Merida P, Lahana R. Applications of a blood-brain barrier technology platform to predict CNS penetration of various chemotherapeutic agents. 2. Cationic peptide vectors for brain delivery. Chemotherapy. 2007;53(1):73–76. doi: 10.1159/000098422. [DOI] [PubMed] [Google Scholar]
- [34].Vives E, Schmidt J, Pelegrin A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim Biophys Acta. 2008;1786(2):126–138. doi: 10.1016/j.bbcan.2008.03.001. [DOI] [PubMed] [Google Scholar]
- [35].Bidwell GL, 3rd, Raucher D. Application of thermally responsive polypeptides directed against c-Myc transcriptional function for cancer therapy. Mol Cancer Ther. 2005;4(7):1076–1085. doi: 10.1158/1535-7163.MCT-04-0253. [DOI] [PubMed] [Google Scholar]
- [36].Bidwell GL, 3rd, Davis AN, Raucher D. Targeting a c-Myc inhibitory polypeptide to specific intracellular compartments using cell penetrating peptides. J Control Release. 2009;135(1):2–10. doi: 10.1016/j.jconrel.2008.11.015. [DOI] [PubMed] [Google Scholar]
- [37].Bidwell GL, 3rd, Fokt I, Priebe W, Raucher D. Development of elastin-like polypeptide for thermally targeted delivery of doxorubicin. Biochem Pharmacol. 2007;73(5):620–631. doi: 10.1016/j.bcp.2006.10.028. [DOI] [PubMed] [Google Scholar]
- [38].Massodi I, Bidwell GL, 3rd, Raucher D. Evaluation of cell penetrating peptides fused to elastin-like polypeptide for drug delivery. J Control Release. 2005;108(2-3):396–408. doi: 10.1016/j.jconrel.2005.08.007. [DOI] [PubMed] [Google Scholar]
- [39].Bidwell GL, 3rd, Raucher D. Therapeutic peptides for cancer therapy. Part I - peptide inhibitors of signal transduction cascades. Expert Opin Drug Deliv. 2009;6(10):1033–1047. doi: 10.1517/17425240903143745. [DOI] [PubMed] [Google Scholar]
- [40].Raucher D, Moktan S, Massodi I, Bidwell GL., 3rd Therapeutic peptides for cancer therapy. Part II - cell cycle inhibitory peptides and apoptosis-inducing peptides. Expert Opin Drug Deliv. 2009;6(10):1049–1064. doi: 10.1517/17425240903158909. [DOI] [PubMed] [Google Scholar]
- [41].Lipka E, Crison J, Amidon GL. Transmembrane transport of peptide type compounds: prospects for oral delivery. J Control Release. 1996;39(2-3):121–129. doi: 10.1016/0168-3659(95)00145-x. [DOI] [PubMed] [Google Scholar]
- [42].Talmadge JE. Pharmacodynamic aspects of peptide administration biological response modifiers. Adv Drug Deliv Rev. 1998;33(3):241–252. doi: 10.1016/s0169-409x(98)00032-5. [DOI] [PubMed] [Google Scholar]
- [43].Jung T, Kamm W, Breitenbach A, Kaiserling E, Xiao JX, Kissel T. Biodegradable nanoparticles for oral delivery of peptides: is there a role for polymers to affect mucosal uptake? Eur J Pharm Biopharm. 2000;50(1):147–160. doi: 10.1016/s0939-6411(00)00084-9. [DOI] [PubMed] [Google Scholar]
- [44].Torchilin VP, Levchenko TS. TAT-liposomes: a novel intracellular drug carrier. Curr Protein Pept Sci. 2003;4(2):133–140. doi: 10.2174/1389203033487298. [DOI] [PubMed] [Google Scholar]
- [45].MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat Mater. 2009;8(12):993–999. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dreher MR, Raucher D, Balu N, Michael Colvin O, Ludeman SM, Chilkoti A. Evaluation of an elastin-like polypeptide-doxorubicin conjugate for cancer therapy. J Control Release. 2003;91(1-2):31–43. doi: 10.1016/s0168-3659(03)00216-5. [DOI] [PubMed] [Google Scholar]
- [47].Bidwell GL, 3rd, Davis AN, Fokt I, Priebe W, Raucher D. A thermally targeted elastin-like polypeptide-doxorubicin conjugate overcomes drug resistance. Invest New Drugs. 2007;25(4):313–326. doi: 10.1007/s10637-007-9053-8. [DOI] [PubMed] [Google Scholar]
- [48].Chen TH, Bae Y, Furgeson DY. Intelligent biosynthetic nanobiomaterials (IBNs) for hyperthermic gene delivery. Pharm Res. 2008;25(3):683–691. doi: 10.1007/s11095-007-9382-5. [DOI] [PubMed] [Google Scholar]
- [49].Massodi I, Thomas E, Raucher D. Application of thermally responsive elastin-like polypeptide fused to a lactoferrin-derived peptide for treatment of pancreatic cancer. Molecules. 2009;14(6):1999–2015. doi: 10.3390/molecules14061999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Massodi I, Moktan S, Rawat A, Bidwell GL, 3rd, Raucher D. Inhibition of ovarian cancer cell proliferation by a cell cycle inhibitory peptide fused to a thermally responsive polypeptide carrier. Int J Cancer. 2010;126(2):533–544. doi: 10.1002/ijc.24725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shamji MF, Chen J, Friedman AH, Richardson WJ, Chilkoti A, Setton LA. Synthesis and characterization of a thermally-responsive tumor necrosis factor antagonist. J Control Release. 2008;129(3):179–186. doi: 10.1016/j.jconrel.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Meyer DE, Chilkoti A. Genetically encoded synthesis of protein-based polymers with precisely specified molecular weight and sequence by recursive directional ligation: examples from the elastin-like polypeptide system. Biomacromolecules. 2002;3(2):357–367. doi: 10.1021/bm015630n. [DOI] [PubMed] [Google Scholar]
- [53].Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. Journal of Biological Chemistry. 1994;269(14):10444–10450. [PubMed] [Google Scholar]
- [54].Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272(25):16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- [55].Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem. 1995;270(24):14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- [56].Sadler K, Eom KD, Yang JL, Dimitrova Y, Tam JP. Translocating proline-rich peptides from the antimicrobial peptide bactenecin 7. Biochemistry. 2002;41(48):14150–14157. doi: 10.1021/bi026661l. [DOI] [PubMed] [Google Scholar]
- [57].Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278(1):585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- [58].Derossi D, Chassaing G, Prochiantz A. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 1998;8(2):84–87. [PubMed] [Google Scholar]
- [59].Vives E, Richard JP, Rispal C, Lebleu B. TAT peptide internalization: seeking the mechanism of entry. Curr Protein Pept Sci. 2003;4(2):125–132. doi: 10.2174/1389203033487306. [DOI] [PubMed] [Google Scholar]
- [60].Edenhofer F. Protein transduction revisited: novel insights into the mechanism underlying intracellular delivery of proteins. Curr Pharm Des. 2008;14(34):3628–3636. doi: 10.2174/138161208786898833. [DOI] [PubMed] [Google Scholar]
- [61].Heuser JE, Anderson RG. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108(2):389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Thyberg J. Caveolae and cholesterol distribution in vascular smooth muscle cells of different phenotypes. J Histochem Cytochem. 2002;50(2):185–195. doi: 10.1177/002215540205000206. [DOI] [PubMed] [Google Scholar]
- [63].Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2(10):764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- [64].Draeger LJ, Mullen GP. Interaction of the bHLH-zip domain of c-Myc with H1-type peptides. Characterization of helicity in the H1 peptides by NMR. J Biol Chem. 1994;269(3):1785–1793. [PubMed] [Google Scholar]
- [65].Giorello L, Clerico L, Pescarolo MP, Vikhanskaya F, Salmona M, Colella G, Bruno S, Mancuso T, Bagnasco L, Russo P, Parodi S. Inhibition of cancer cell growth and c-Myc transcriptional activity by a c-Myc helix 1-type peptide fused to an internalization sequence. Cancer Res. 1998;58(16):3654–3659. [PubMed] [Google Scholar]
- [66].Pescarolo MP, Bagnasco L, Malacarne D, Melchiori A, Valente P, Millo E, Bruno S, Basso S, Parodi S. A retro-inverso peptide homologous to helix 1 of c-Myc is a potent and specific inhibitor of proliferation in different cellular systems. Faseb J. 2001;15(1):31–33. doi: 10.1096/fj.00-0422fje. [DOI] [PubMed] [Google Scholar]
- [67].Bidwell GL, 3rd, Raucher D. Enhancing the antiproliferative effect of topoisomerase II inhibitors using a polypeptide inhibitor of c-Myc. Biochem Pharmacol. 2006;71(3):248–256. doi: 10.1016/j.bcp.2005.10.041. [DOI] [PubMed] [Google Scholar]
- [68].Bakic M, Chan D, Freireich EJ, Marton LJ, Zwelling LA. Effect of polyamine depletion by alpha-difluoromethylornithine or (2R,5R)-6-heptyne-2,5-diamine on drug-induced topoisomerase II-mediated DNA cleavage and cytotoxicity in human and murine leukemia cells. Cancer Res. 1987;47(24 Pt 1):6437–6443. [PubMed] [Google Scholar]
- [69].Desiderio MA, Bergamaschi D, Mascellani E, De Feudis P, Erba E, D’Incalci M. Treatment with inhibitors of polyamine biosynthesis, which selectively lower intracellular spermine, does not affect the activity of alkylating agents but antagonizes the cytotoxicity of DNA topoisomerase II inhibitors. Br J Cancer. 1997;75(7):1028–1034. doi: 10.1038/bjc.1997.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Feuerstein BG, Pattabiraman N, Marton LJ. Spermine-DNA interactions: a theoretical study. Proc Natl Acad Sci U S A. 1986;83(16):5948–5952. doi: 10.1073/pnas.83.16.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hung DT, Marton LJ, Deen DF, Shafer RH. Depletion of intracellular polyamines may alter DNA conformation in 9L rat brain tumor cells. Science. 1983;221(4608):368–370. doi: 10.1126/science.6408733. [DOI] [PubMed] [Google Scholar]
- [72].Warbrick E, Lane DP, Glover DM, Cox LS. A small peptide inhibitor of DNA replication defines the site of interaction between the cyclin-dependent kinase inhibitor p21WAF1 and proliferating cell nuclear antigen. Curr Biol. 1995;5(3):275–282. doi: 10.1016/s0960-9822(95)00058-3. [DOI] [PubMed] [Google Scholar]
- [73].Chen IT, Akamatsu M, Smith ML, Lung FD, Duba D, Roller PP, Fornace AJ, Jr., O’Connor PM. Characterization of p21Cip1/Waf1 peptide domains required for cyclin E/Cdk2 and PCNA interaction. Oncogene. 1996;12(3):595–607. [PubMed] [Google Scholar]
- [74].Pan ZQ, Reardon JT, Li L, Flores-Rozas H, Legerski R, Sancar A, Hurwitz J. Inhibition of nucleotide excision repair by the cyclin-dependent kinase inhibitor p21. J Biol Chem. 1995;270(37):22008–22016. doi: 10.1074/jbc.270.37.22008. [DOI] [PubMed] [Google Scholar]
- [75].Ball KL, Lain S, Fahraeus R, Smythe C, Lane DP. Cell-cycle arrest and inhibition of Cdk4 activity by small peptides based on the carboxy-terminal domain of p21WAF1. Curr Biol. 1997;7(1):71–80. doi: 10.1016/s0960-9822(06)00029-7. [DOI] [PubMed] [Google Scholar]
- [76].Cayrol C, Knibiehler M, Ducommun B. p21 binding to PCNA causes G1 and G2 cell cycle arrest in p53-deficient cells. Oncogene. 1998;16(3):311–320. doi: 10.1038/sj.onc.1201543. [DOI] [PubMed] [Google Scholar]
- [77].Mutoh M, Lung FD, Long YQ, Roller PP, Sikorski RS, O’Connor PM. A p21(Waf1/Cip1)carboxyl-terminal peptide exhibited cyclin-dependent kinase-inhibitory activity and cytotoxicity when introduced into human cells. Cancer Res. 1999;59(14):3480–3488. [PubMed] [Google Scholar]
- [78].Mattock H, Lane DP, Warbrick E. Inhibition of cell proliferation by the PCNA-binding region of p21 expressed as a GFP miniprotein. Exp Cell Res. 2001;265(2):234–241. doi: 10.1006/excr.2001.5160. [DOI] [PubMed] [Google Scholar]