

Abstract
The hallmark of many intracellular pore blockers such as tetra-alkylammonium compounds and local anesthetics is their ability to allosterically modify the movement of the voltage sensors in voltage-dependent ion channels. For instance, the voltage sensor of domain III is specifically stabilized in the activated state when sodium currents are blocked by local anesthetics. The molecular mechanism underlying this long-range interaction between the blocker-binding site in the pore and voltage sensors remains poorly understood. Here, using scanning mutagenesis in combination with voltage clamp fluorimetry, we systematically evaluate the role of the internal gating interface of domain III of the sodium channel. We find that several mutations in the S4–S5 linker and S5 and S6 helices dramatically reduce the stabilizing effect of lidocaine on the activation of domain III voltage sensor without significantly altering use-dependent block at saturating drug concentrations. In the wild-type skeletal muscle sodium channel, local anesthetic block is accompanied by a 21% reduction in the total gating charge. In contrast, point mutations in this critical intracellular region reduce this charge modification by local anesthetics. Our analysis of a simple model suggests that these mutations in the gating interface are likely to disrupt the various coupling interactions between the voltage sensor and the pore of the sodium channel. These findings provide a molecular framework for understanding the mechanisms underlying allosteric interactions between a drug-binding site and voltage sensors.
INTRODUCTION
The voltage-gated sodium channels determine the rising phase of an action potential and the length of the refractory period in nerves, skeletal muscle, and heart (Catterall, 2000; Hille, 2001). Local anesthetics along with antiarrhythmics and anticonvulsants comprise a group of compounds that are clinically one of the most important and widely used modifiers of sodium channel function (Fozzard et al., 2005). When used to treat peripheral pain, they simply block the pore of the sodium channels in resting state, thereby preventing further propagation of action potentials. On the other hand, the primary mode of action of anticonvulsants and antiarrhythmics is to block the sodium currents in a frequency-dependent manner (Strichartz, 1973). This phenomenon, termed as use-dependent block, makes the drug a more potent blocker when the sodium channels activate with a higher frequency. Thus, these “smart” compounds behave as low frequency filters in an electrical circuit, attenuating uncontrolled high frequency activity associated with disease states such as arrhythmias and epilepsies.
To account for these striking effects of local anesthetics on the sodium channel, it has been proposed that these drugs bind with a high affinity to the open/inactivated state relative to the closed state of the channel (Hille, 1977). Alternatively, the guarded receptor hypothesis postulates that the binding site for the local anesthetic is gated and becomes accessible only when the channel opens (Strichartz, 1973; Starmer et al., 1984). Although both of these models can rationalize the basic phenomenology, several lines of evidence suggest that the presence of a local anesthetic profoundly modifies the voltage-dependent gating behavior of the sodium channel. Gating current measurements show that the total charge associated with channel activation is reduced in the presence of local anesthetics (Keynes and Rojas, 1974; Peganov and Khodorov, 1977; Cahalan, 1978; Cahalan and Almers, 1979; Tanguy and Yeh, 1989; Hanck et al., 1994; Sheets and Hanck, 2003). Fluorescence–voltage (F-V) curves of probes attached to domain III voltage sensor were shifted to more hyperpolarized potentials upon the addition of lidocaine (Muroi and Chanda, 2009). Moreover, this modification of the voltage sensor is tightly linked to use-dependent block of sodium currents (Hanck et al., 2009; Muroi and Chanda, 2009). These findings suggest that the voltage sensor of domain III is stabilized in the activated state upon use-dependent block of sodium currents by local anesthetics. Structure–function studies implicate a cluster of residues in the S6 helices lining the central pore as the binding site primarily responsible for use-dependent block (Ragsdale et al., 1994; Wang and Wang, 1998; Wright et al., 1998; Wang et al., 2000; Yarov-Yarovoy et al., 2001, 2002). Because the voltage-sensing domain is likely to be a well-defined structural module separate from the pore domain (Long et al., 2005, 2007), these findings suggest that local anesthetic binding to the pore allosterically modulates the voltage sensors of the sodium channel. Similar studies in the inactivation-removed Shaker potassium channel show that the OFF gating current is reduced in the presence of tetra-ethylammonium ions (Bezanilla et al., 1991). Thus, allosteric modification of voltage gating by pore blockers may be a common feature of the voltage-dependent ion channel superfamily.
To determine molecular pathway–mediating interaction between the lidocaine-binding site and domain III voltage sensor of the sodium channel, we focused on the role of the intracellular gating interface. Studies in the Shaker potassium channel have identified two gating interfaces that are likely to be involved in electromechanical coupling of the voltage sensor and pore gates (Schoppa et al., 1992; Smith-Maxwell et al., 1998a,b; Ledwell and Aldrich, 1999; Hackos et al., 2002; Lu et al., 2002; Soler-Llavina et al., 2006; Lee et al., 2009). Mutagenesis and domain swap experiments have shown that residues in the S4–S5 linker and the intracellular end of S5 and S6 play a crucial role in coupling S6 segments to the voltage sensor movement. Lee et al. (2009) have suggested that the interface between S1 and the neighboring S5 segment is also involved in electromechanical coupling. In this study, we systematically mutated residues in the S4–S5 linker and S5 and S6 segments in domain III and examined the effect of lidocaine on the movement of this voltage sensor using voltage clamp fluorimetry and gating current measurements. The binding of lidocaine to the wild-type (WT) skeletal muscle sodium channel shifts the midpoints of the activation curves of domain III voltage sensor by >50 mV in the hyperpolarizing direction. We identify a set of residues that when mutated to a tryptophan dramatically reduce the effect of lidocaine on the steady-state properties of domain III voltage sensor. Measurements of gating currents show that lidocaine block of sodium currents in these mutants has a much smaller effect on the total gating charge modification compared with the WT sodium channels. Moreover, the maximal use-dependent block of the mutants is comparable to the WT channels. These findings suggest that the intracellular gating interface is a major pathway for allosteric modulation of voltage sensor movement by local anesthetics.
MATERIALS AND METHODS
Molecular biology and oocyte expression
All mutants were generated in the background of the mutant (L1115C) rat skeletal muscle sodium channel (rNav 1.4), as described previously (Muroi et al., 2010). cDNAs of both α and β subunit of rNav 1.4 were linearized with NotI digestion (New England Biolabs, Inc.) and transcribed using the mMESSAGE mMACHINE T7 kit (Applied Biosystems). Equimolar ratios of α and β subunit mRNAs were coinjected into Xenopus laevis oocytes to a final volume of 50 nL. Oocytes were obtained from Nasco or surgically removed from Xenopus according to the guidelines of the Animal Care and Use Committee at the University of Wisconsin-Madison. After injection, the oocytes were kept at 18°C in a solution containing (in mM): 100 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, 0.1 DTT, and 0.2 EDTA, supplemented with 100 µg/ml gentamicin and 100 mg/ml bovine serum albumin. Measurements were performed within 1–7 d after injection.
Fluorescence and electrophysiology measurements
The oocytes were incubated for labeling with 10 µM tetramethyrhodamine maleimide (TMRM; Invitrogen) in a depolarizing solution (in mM: 110 KCl, 1.5 MgCl2, 0.8 CaCl2, and 10 HEPES, pH 7.5) on ice for 30 min. 10 mM TMRM stock solution was prepared in DMSO and stored at −20°C. Labeled oocytes were washed and stored at room temperature in 115 mM N-methyl glucamine (NMG) containing external solution until recording. All recordings were obtained at room temperature (22 ± 3°C). Fluorescence recordings were performed as described previously (Muroi and Chanda, 2009). In brief, the labeled oocytes were positioned in a modified cut-open oocyte setup (CA-1B; Dagan Corporation) and placed on a stage of an upright microscope (BX50WI; Olympus), and the current and fluorescence signals were simultaneously measured under voltage clamp conditions. The light from a xenon lamp source (Hamamatsu Photonics) was filtered with a 535/HQ50 bandpass filter and split using a Q565LP dichroic mirror (Chroma Technology Corp.). The emitted light was filtered with a 610/HQ75 bandpass filter and focused onto a PIN-020A photodiode (UDT Instruments) by a condenser lens. The photodiode was connected to the integrating headstage of a patch clamp amplifier (Axopatch 1B; Axon Instruments, Inc.). Gating current recordings were obtained using the cut-open oocyte technique. 10 µM tetrodotoxin (TTX) was first added to the external solution in the top and middle chambers, followed by lidocaine application. Measurements of use-dependent block were obtained using the two-electrode voltage clamp setup (OC-725C Oocyte Clamp; Warner Instruments).
Recording solutions
Fluorescence and gating current recordings obtained in the presence of external solution (105 mM NMG-Mes, 20 mM HEPES, and 2 mM Ca2+, pH 7.4) and internal solution (105 mM NMG-Mes, 20 mM HEPES, and 2 mM EGTA, pH 7.4) were used. Ionic current recordings were obtained in two-electrode voltage clamp setup, with an external solution composed of 21 mM NaMes, 84 mM NMG-Mes, 20 mM HEPES, and 2 mM Ca2+, adjusted to pH 7.4.
Data acquisition and analysis
Analogue signals were sampled at 250 kHz with a Digidata 1440 interface (MDS Analytical Technologies) and low-pass filtered at 10 kHz. For gating current measurements, linear leak and membrane capacitive current were subtracted online by a P/4 procedure with a subtraction holding potential of +50 mV. Gating currents were obtained by applying a 20-ms test pulse to voltages ranging from −130 to +40 mV, with a pre- and post-pulse of 50 ms at −130 mV. The holding potential was −80 mV.
Each fluorescence trace represents an average of 10 recordings obtained by a 20-ms test pulse from −120 mV after a 20-ms conditioning pulse at −10 mV, with a holding potential of −80 mV. The interval between the conditioning and test pulse was 100 ms. The conditioning pulse was used to ensure that the channel was fully blocked by lidocaine. Fluorescence intensity decay from photobleaching was corrected by fitting the trace to a single exponential or a straight line and subtracting it from the original trace. The data were subsequently low-pass filtered offline at 5 kHz.
F-V and gating charge–voltage (Q-V) curves were fitted to a single Boltzmann function:
where z is the apparent valence, e is the electronic charge, V1/2 is the half-maximal voltage, K is the Boltzmann constant, and T is the temperature.
To measure use-dependent block, 100 test pulses of 20 ms at −20 mV (10 Hz) from a holding potential of −80 mV were applied. The pulsing frequency was varied to measure frequency-dependent block. All data were acquired using Clampex (MDS Analytical Technologies) and analyzed with Clampfit, Excel (Microsoft), or Origin (GE Healthcare). Prism (GraphPad Software) was used for statistical analysis.
Homology modeling
The homology models of the sodium channel in the open state were developed using the crystal structure of the KV1.2/2.1 chimera (Protein Data Bank accession no. 2R9R) as a template. The sequences of the putative sodium channel domains were first aligned to the template sequences using ClustalW (Thompson et al., 1994), with default alignment parameters. The alignment was manually optimized to minimize the gaps within the putative transmembrane segments of the different sequences (Fig. S1). The homology modeling was performed using Modeller 9v4 (Sali and Blundell, 1993). 100 candidate models (S1 through S6) were generated for each of the four domains of the sodium channel. The final model for each domain was selected on the basis of the DOPE (discrete optimized potential energy) scores and the stereochemical quality of the candidate models, as assessed from the Ramachandran plots. The models of the four domains were organized symmetrically around the axis of K+ conduction by using an appropriate transformation of coordinates to generate the final tetrameric structure. To remove certain bad contacts/steric clashes, especially in the membrane reentrant loop (pore helix) between S5 and S6, it was necessary to move each of the four domains radially outward (by 1.5 Å). In vacuo energy minimization of this tetrameric structure was performed using NAMD 2.6 (Phillips et al., 2005), with the CHARMM 27 force field parameters and with all atomic charges switched off.
Modeling state occupancies
The expressions for the probability of the voltage sensor activation in the absence and presence of lidocaine, as derived from Scheme 2 (Eqs. 1 and 2), were fitted to the corresponding F-V data. The data fitting of both equations was performed simultaneously, so that one dataset served as a constraint for the other. Fitting was performed using the MATLAB curve-fitting toolbox (robust nonlinear least-squares method, Levenberg-Marquardt algorithm) to obtain the values of the different thermodynamic parameters. The parameters KV, KP, and KR are all voltage-dependent equilibrium constants, and their voltage dependence can be expressed as where i = V, P, or R. The voltage dependence of activation of the voltage sensor (with unbound lidocaine), zV, is related to that of the deactivation (or return) of the voltage sensor (with bound lidocaine), zR, as: zR = −zV. The binding affinity (or association constant) of lidocaine, KA (which is the product of the equilibrium binding constant and the concentration of lidocaine), and the coupling parameter θ are considered to be voltage independent.
For the first run of the fitting, the initial guesses of the different parameters were taken from our previous study (Muroi et al., 2010), and a set of optimized parameters was obtained. The robustness of the fit was checked by repeating the fitting procedure 1,000 times. For every iteration, six random numbers between 10−2 and 102 were generated. Each of the six parameters, obtained from the first run, was scaled separately by the random numbers. The rescaled parameter values were used as the initial estimates to repeat the fitting procedure. Thus, this random sampling algorithm allows us to efficiently search a six-dimensional parameter space spanning over four orders of magnitude and generate a probability distribution of the optimized value for each parameter. The mean and the percent standard deviation for the optimized parameters are reported in Table II. This fitting procedure was performed on the WT data and on one of the high impact mutants (A1145W). The simulated F-V traces are shown in Fig. 6.
Table II.
Parameters obtained from fitting Scheme 2 to the F-V data
| Construct | KV0 | ZV | KP0 | ZP | KA | KR |
| WT | 15.2 (4.3) | 1.5 (3.7) | 5.8 (1.7) | 0.36 (4.5) | 953.3 (0.9) | 0.00046 (6.9) |
| A1145W | 32.9 (1.8) | 1.18 (2.5) | 0.13 (5.6) | 0.22 (5.9) | 968.7 (1.4) | 0.018 (5.8) |
| K10 | Z1 | K20 | Z2 | KA | θ | |
| WT | 15.2 | 1.5 | 0.041 | 0.36 | 953.3 | 143 |
| A1145W | 32.9 | 1.18 | 0.076 | 0.22 | 968.7 | 1.7 |
The values of the parameters of Scheme 2, reported here, are the mean of the parameter values obtained from the 1,000 fitting procedures (refer to Materials and methods). The standard deviation, expressed as a percentage of the mean, of each parameter is reported in parentheses. From the fitted values of KV, KP, and KR, the intrinsic parameters K10, K20, and θ were derived using the relations provided in Eq. 3.
Figure 6.

Simulations of fluorescence activation curves of WT and a mutant before and after the addition of lidocaine. The equations describing the probability of activation of the voltage sensor were fitted to the WT data (A) and A1145W (B) to generate the activation curves shown above (refer to Materials and methods). Symbols represent the simulated data points, and the trend lines are fits of the simulated data with a single Boltzmann function.
Online supplemental material
Fig. S1 shows a sequence alignment of the four domains of the rNav 1.4 channel with Kv1.2/2.1 chimera and KcsA channel. Thermodynamic relationships between obligatory and nonobligatory coupling models are described in the supplemental text. Parameters obtained by fitting F-V curves to a Boltzmann function are shown in Table S1. The supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.201010438/DC1.
RESULTS
Mutations in the cytoplasmic gating interface alter the drug-induced shift in voltage sensor movement
The tryptophan-scanning mutagenesis was performed in the intracellular gating interface of domain III of the sodium channel (Fig. 1). This interface was formed in part by the S4–S5 linker region and the intracellular ends of the S5 and S6 transmembrane segments. The effect of these mutations on allosteric coupling was assessed by attaching a fluorophore, tetramethylrhodamine, near the extracellular end of the S4 segment of domain III and monitoring the changes in fluorescence intensity in response to lidocaine block of ionic currents. Previous studies have shown that the voltage-dependent fluorescence intensity changes from a probe on Leu1115 position in the muscle sodium channel correlate with its gating charge movement and are likely to be reporters of voltage-dependent conformational changes associated with the S4 of domain III movement (Chanda and Bezanilla, 2002).
Figure 1.

Sequence alignment of the putative transmembrane segments of domain III of the brain, nerve, and the muscle sodium channels (Nav1.2, Nav1.4, and Nav1.5) with a chimeric potassium channel (Kv1.2/2.1). The conserved positive charges of the S4 transmembrane segment are highlighted in green. The mutated regions in the S4–S5 linker, N terminus of S5, and C terminus of S6 are shown in red. The fluorophore was attached to an introduced cysteine at the L1115C (in orange) position.
55 mutants were generated in the background of the L1115C mutant sodium channel. This L1115C mutant is hereafter referred to as the WT channel. Most mutations were tryptophan substitutions, which because of their large side chain volume are likely to disrupt the tight packing at the interfaces. When, in some instances, tryptophan substitutions resulted in nonfunctional protein or low expression, alanine was substituted. The effect of these mutations on the conductance–voltage relationships and voltage-dependent fluorescence of domain III has been characterized in a recent study (Muroi et al., 2010). Fig. 2 A shows a comparison of representative voltage-dependent fluorescence traces before and after the addition of lidocaine in the WT and two mutants (A1145W and I1287W). Normalized F-V relationships were obtained by plotting the fluorescence intensities near the end of a 20-ms depolarizing pulse with respect to the membrane potential. 10 mM lidocaine in the bath ensures complete block of WT and mutant channels. To quantitatively assess the effect of these mutations, the data were fitted to a single Boltzmann function, and the values of the two parameters, the slope (z), and the voltage-eliciting half-maximal response (V1/2) were obtained. In the WT channel, the addition of lidocaine causes a dramatic shift in the F-V curve, such that the V1/2 is shifted to a more hyperpolarized potential by −56.1 mV (Fig. 2 B, left). Fig. 2 B (middle and right) shows two representative tryptophan mutants, A1145W and I1287W, where the drug-induced shifts of F-V curves by lidocaine were reduced. The effect of lidocaine on the F-V curves for all the mutants is summarized in Fig. 2 C and Table S1. Approximately half of the mutants show significantly reduced F-V shifts, and a majority of those are in the S4–S5 linker and S6 segment of domain III. The mutants that produce a <20-mV hyperpolarizing shift in V1/2 in the presence of lidocaine were designated as high impact mutants, whereas those that produce shifts >20 mV but <55 mV were designated as low impact mutants. Fig. 2 C shows that only seven (R1135W, V1142W, A1145W, S1276W, I1284W, I1287W, and K1295W) out of the 55 tested in this study show a <20-mV hyperpolarizing shift in the V1/2 in the presence of lidocaine. Mutations in 17 other sites were designated as low impact mutants.
Figure 2.

Effect of lidocaine on the energetics of the domain III voltage sensor. (A) Time course of voltage-dependent fluorescence signals from domain III of WT and a couple of representative mutant sodium channels. Fluorescence traces were obtained before and after 10-mM lidocaine application by pulsing to a test potential (ranging from +50 to −210 mV) for 20 ms, with a prepulse to −120 mV for 20 ms. Each trace represents an average of 10 trials with an interval of 1 s between each pulse. The scale bars are provided as insets, and the percentage fluorescence changes (ΔF/F) are on the y axis. (B) Steady-state F-V curves before (filled symbols) and after (open symbols) 10-mM lidocaine application. The lines represent the best fits of the averaged data to a single Boltzmann function. The error bars represent the average ± SEM for each data point. (C) Summary of shifts in F-V curves of WT and various mutants upon the application of lidocaine. ΔV1/2 was obtained as: ΔV1/2 = V1/2(after lidocaine) − V1/2(before lidocaine). Each bar represents mean ± SEM of at least three independent oocyte measurements. The gray dashed line represents the shift in F-V curves of WT. To test whether these differences were statistically significant, a one-way ANOVA test was used, followed by a post-hoc Dunnett’s test. *, P < 0.001; #, P < 0.05.
Mutations in the gating interface alleviate gating charge modification by lidocaine
Gating charge studies show that local anesthetic block of sodium currents is accompanied by a 21% reduction in the total gating charge (Hanck et al., 1994; Sheets and Hanck, 2007; Muroi and Chanda, 2009) (Fig. 3 A). If a tryptophan mutation disrupts the interaction between local anesthetic-binding site and domain III voltage sensor, we would predict that the extent of charge modification by lidocaine will be diminished. This hypothesis was tested by measuring charge modification by lidocaine in six of the seven high impact mutants identified by fluorescence measurements. Use-dependent block in S1276W mutant was found to be highly impaired (data shown in the next section); therefore, gating currents for this mutant were not assessed. The Q-V relationships of six high impact mutants before and after the addition of 10 mM lidocaine are shown in Fig. 3 B. The effect of lidocaine on total gating charge in the mutant channels as compared with WT is summarized in Fig. 3 C, and the parameters for Boltzmann fits of the Q-V curves are in Table I. In the WT skeletal muscle sodium channel, lidocaine does not significantly alter the midpoints and the slope of the Q-V relationships, but reduces the maximum gating charge moved in response to large depolarization. The introduction of a tryptophan in the six high impact sites significantly reduces this gating charge modification by lidocaine. In the case of two of the mutants, A1145W and I1284W, the addition of lidocaine has almost no effect on gating charge movement. These findings indicate that mutations in key positions corresponding to the high impact sites in the cytoplasmic gating interface disrupt the interaction between the voltage sensor and the lidocaine-binding site.
Figure 3.

Effect of lidocaine on gating currents of WT and mutant sodium channels. (A; left) Representative ON gating currents of the WT channels and their integrals before and after 10-mM lidocaine application. (Right) Steady-state Q-V relationships of the WT channel before (filled symbols) and after (open symbols) lidocaine application. The gray dashed line represents the average reduction in the total gating charge after lidocaine application. The data represent mean ± SEM of at least three independent measurements. The line represents the best fit of the averaged data to a single Boltzmann function. (B) Steady-state Q-V relationships of mutant channels before (filled symbols) and after (open symbols) 10-mM lidocaine application. The gray dashed line represents the average reduction in the total gating charge after the application of lidocaine to the WT channel. The data represent mean ± SEM of at least three independent measurements, and the smooth lines represent the best fits of the averaged data to a single Boltzmann function. (C) Percentage reduction in the total gating charge upon the application of lidocaine in the WT and mutant channels. The total charge movement was measured by a pulse to +40 mV from −130 mV. The data represent mean ± SEM of at least three independent measurements.
Table I.
Effect of 10 mM lidocaine on the gating current properties of WT and mutant channels
| Channel | Control (TTX) | Lidocaine (+TTX) | Remaining gating charge | |||
| n | V1/2 | z | V1/2 | z | % | |
| WT | 6 | −63.8 ± 2.4 | 1.34 ± 0.02 | −63.3 ± 1.9 | 1.45 ± 0.03 | 79.0 ± 3.8 |
| R1135W | 4 | −65.9 ± 1.9 | 1.38 ± 0.06 | −63.8 ± 0.8 | 1.38 ± 0.06 | 91.4 ± 3.7 |
| V1142W | 4 | −50.4 ± 2.9 | 1.01 ± 0.06 | −57.6 ± 0.9 | 1.19 ± 0.03 | 90.0 ± 2.5 |
| A1145W | 3 | −56.0 ± 2.3 | 1.00 ± 0.03 | −54.3 ± 4.3 | 1.11 ± 0.03 | 96.2 ± 1.7 |
| I1284W | 3 | −57.3 ± 2.8 | 1.30 ± 0.11 | −54.2 ± 5.7 | 1.20 ± 0.00 | 100.0 ± 2.2 |
| I1287W | 3 | −55.0 ± 1.2 | 1.13 ± 0.03 | −59.2 ± 1.5 | 1.33 ± 0.01 | 92.0 ± 3.6 |
| K1295W | 4 | −53.7 ± 3.3 | 0.93 ± 0.01 | −59.2 ± 5.1 | 1.08 ± 0.02 | 91.2 ± 2.4 |
Boltzmann parameters from a first-order fit of the Q-V relationships. All measurements were done in the presence of TTX. Data represent the mean ± SEM of at least three independent experiments (n).
Effect of mutations on use-dependent block by lidocaine
Previous studies have shown that the ability of lidocaine to block sodium currents in a use-dependent manner was essential for modification of the voltage sensor movement (Hanck et al., 2009; Muroi and Chanda, 2009). Here, we test whether lidocaine at saturating concentrations can inhibit the mutant sodium currents in a use-dependent manner. Fig. 4 A shows a family of WT sodium current traces elicited by applying 100 depolarizing pulses to −20 mV from a holding potential of −80 mV (10-Hz frequency) in the presence of lidocaine. To obtain a dose–response curve for use-dependent block, the peak sodium current at the 100th pulse is plotted with respect to lidocaine concentration (Fig. 4 B). The estimated IC50 for the L1115C mutant skeletal muscle sodium channel is 20 µM, a value similar to the WT sodium channel (Wang et al., 2004). Use-dependent block for both high and low impact mutants was obtained in the presence of 1 mM lidocaine because the fluorescence and gating charge measurements were recorded under saturating conditions of lidocaine (Fig. 4 C). At this concentration of lidocaine, ∼70% of the WT sodium channels are blocked in a use-dependent manner. Most mutants exhibit similar use-dependent block, which suggests that the loss of voltage sensor modification is not because of their inability to bind to lidocaine. Only 4 out of the 25 show a significantly lower use-dependent block at these drug concentrations. The most dramatic effect was observed upon mutation of the Ser at position 1276, resulting in a significantly diminished use-dependent block of sodium currents. This mutant also exhibits a much smaller shift of F-V curve upon lidocaine addition (ΔV1/2 = 20 mV) relative to the WT channels. These findings are consistent with other studies showing that the Serine1276 in the skeletal muscle sodium channel is critical for use-dependent block (Wang et al., 2000).
Figure 4.

Use-dependent block of WT and mutant channels by lidocaine. (A) Representative ionic current traces of WT channel in the presence of different concentrations of lidocaine. Each trace was obtained by applying 100 pulses from −80 to −20 mV for 20 ms at 10 Hz from a holding potential of −80 mV. (B) Dose–response curve for use-dependent block of WT channel by lidocaine. The symbols represent mean ± SEM of at least three independent measurements, and the smooth line represents the best fit of the averaged data to the Hill equation. (C) Percent block of WT and mutant channels after 100 pulses in presence of 1 mM lidocaine. Gray dashed line represents the mean percent block of the WT channel (71.5 ± 2.2%; n = 5). The current protocol is the same as in A. In both B and C, the data represent mean ± SEM of at least three independent measurements.
Next, we examine the correlation between lidocaine-induced shifts of F-V curves and maximal use-dependent block (Fig. 5). For mutants A1149W and S1276W, use-dependent block at 1 mM lidocaine concentration is much smaller than that of the WT. Furthermore, fluorescence shift induced by lidocaine is also reduced compared to WT. A simple interpretation is that mutations at these positions alter use-dependent block, such that a smaller fraction of the channels compared to the WT is modified. The ionic currents of the other six high impact mutants, in contrast, were blocked in a use-dependent manner, like the WT channels as shown in Fig. 5 (filled circles). Thus, in these mutants the lidocaine block of the sodium currents appears to be largely decoupled from domain III voltage sensor modification. The majority of the low impact mutants show relatively intact use-dependent block but varying degree of lidocaine-induced shifts in F-V curves, suggesting that voltage sensor modification in these mutants is at least partially decoupled from lidocaine binding.
Figure 5.

Relationship between the ΔV1/2 of F-V curves and maximal use-dependent block by lidocaine. ΔV1/2 of F-V curves upon lidocaine addition and the maximal use-dependent block of ionic currents by lidocaine were obtained as described in the Results. The mutants whose ΔV1/2 shifts linearly correlate with use-dependent block are represented as open squares. The mutants that show the most reduction in ΔV1/2 without much change in use-dependent block are represented by filled circles, and the remaining mutants that show intermediate levels of reduction in the ΔV1/2 are represented by open symbols.
DISCUSSION
Many intracellular pore blockers allosterically modify the movement of the voltage-sensing charges in voltage-dependent ion channels. High resolution structural and functional studies of K+ and Na+ channels support the emerging view that S4 motions are transmitted through a gating interface encompassing the S4–S5 linker and the cytoplasmic ends of S5 and S6 helices (Lu et al., 2002; Long et al., 2005; Soler-Llavina et al., 2006; Lee et al., 2009; Muroi et al., 2010). The motivation for the present study is to investigate the role of this gating interface in coupling the conformation of the lidocaine-binding site to the movement of the voltage sensor in a sodium channel.
Although the notion that lidocaine binding to the sodium channel stabilizes the activated voltage sensor is relatively well established (Sheets and Hanck, 2003, 2007; Muroi and Chanda, 2009), its effect on the pore structure remains controversial. It has been suggested by several groups that lidocaine stabilizes the channel in the inactivated state (Scheuer, 1999). The affinity of the lidocaine to the inactivated cardiac sodium channel was estimated to be 10 µM, comparable to its therapeutic working concentration (Bean et al., 1983). The removal of inactivation in the squid sodium channels by pronase abolished use-dependent block by QX-314, suggesting that the local anesthetics bind mainly to the inactivated channels (Cahalan, 1978; Yeh, 1978). In contrast to these findings, Wang et al. (1987) reported that QX-314 retains its potency on inactivation-deficient sodium currents obtained by treatment with chloramine-T. Cysteine accessibility studies show that the use-dependent block does not involve accumulation of the fast inactivated channels (Vedantham and Cannon, 1999). Moreover, in another inactivation-deficient mutant sodium channel, the IC50 for lidocaine block was found to be comparable to those of the WT channels (Wang et al., 2004). Collectively, these findings suggest that the lidocaine may modulate the transitions that are associated with channel opening, rather than those leading to inactivated states.
Thermodynamic implications of mutations that disrupt allosteric modification
Next, we consider a simple qualitative model of an ion channel consisting of a voltage sensor and a pore, each of which can exist in two possible states (Scheme 1a). This system exists in four possible states: VRPC (pore closed and voltage sensor resting), VRPO (voltage sensor resting and pore open), VAPC (activated voltage sensor and closed pore), and VAPO (activated voltage sensor and open pore). This model can be described by three thermodynamic parameters: K1 is the intrinsic activation constant of the voltage sensor, K2 is the intrinsic activation constant of the pore, and θ represents the interaction between the activated voltage sensor and open pore. For the purposes of this analysis, we ignore the cross interaction terms as well as the interaction between the resting voltage sensor and closed pore.
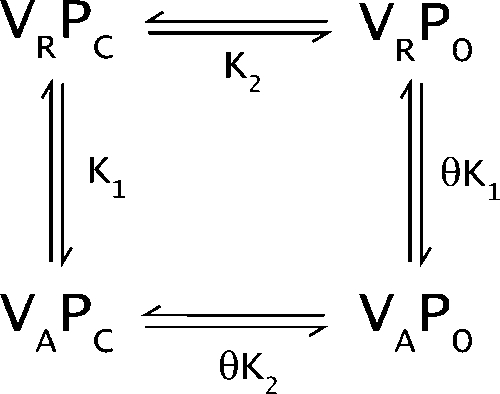 |
(SCHEME 1a) |
To consider the effects of local anesthetic on voltage sensor activation, we extend our initial scheme to include two new lidocaine-bound states: VAPOL and VRPOL. We assume that the lidocaine binds to the open pore (i.e., in channel states VRPO and VAPO), as suggested by studies on inactivation-deficient sodium channels (Wang et al., 2004) and quaternary ammonium (Armstrong, 1971). Studies in inactivation-deficient channels show that lidocaine-binding affinity is similar to the WT channels. For simplicity, we ignore the presence of inactivated states and lidocaine binding to the inactivated states. The addition of lidocaine modifies the above system, which is now described by Scheme 1b, where KA is the association constant of lidocaine with the open pore.
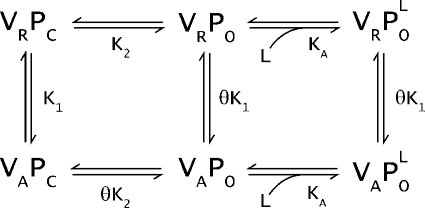 |
(SCHEME 1b) |
Studies on voltage-gated potassium channels suggest that the equilibrium constant for spontaneous opening of the pore is very low and, therefore, the state VRPO is sparsely populated (Hoshi et al., 1994). Therefore, Schemes 1a and 1b can be rewritten as a sequential model depicted in Schemes 2a and 2b.
 |
(SCHEME 2a) |
| (SCHEME 2b) |
The probabilities of activation of the voltage sensor, as derived from Scheme 2, with and without lidocaine are:
| (1) |
| (2) |
To determine the values of thermodynamic parameters described in Scheme 2, the equations for the probability of activation of the voltage sensor (Eqs. 1 and 2) were fitted to the WT F-V curve before and after the addition of lidocaine. The parameters that provide a reasonable fit to the WT data under both conditions are shown in Table II, and the simulated F-V curve of the WT is shown in Fig. 6 A.
Why does lidocaine binding cause a shift in the F-V curve? A comparison of Schemes 2a and 2b reveals that in the absence of lidocaine, the F-V curves reflect the transition between VRPC to VAPC. Saturating concentrations of lidocaine drive the equilibrium rightwards, and the system transits primarily between two possible states: VRPOL and VAPOL. Under these conditions, the F-V curves primarily track the transition between the states VRPOL and VAPOL. To understand the thermodynamic implication of this shift, we can see that the general model (Scheme 1) can be reduced to the sequential model when VRPO has negligible contribution (see supplemental text). Thus, the thermodynamic parameters in Scheme 2 are related to Scheme 1 as:
| (3a) |
| (3b) |
| (3c) |
| (3d) |
Examination of the above relationships shows that the equilibrium constant for VRPC to VAPC transition is K1, whereas that for the VRPOL to VAPOL transition is θK1. Because the voltage sensor and pore are positively coupled to each other (θ greater than 1), the activation of the voltage sensor is always favored in the presence of lidocaine. This analysis also shows that when θ tends to 1, the transitions VRPC to VAPC and VRPOL to VAPOL will become energetically identical. Under these conditions, the binding of lidocaine to the pore is going to have small or no effects on the movement of the voltage sensor.
The equations governing the probability of activation of the voltage sensor in the absence and presence of lidocaine (Eqs. 1 and 2) were fitted to the F-V traces of one of the high impact mutants, A1145W (Fig. 6 B). Examination of the fitted parameters for the mutant (Table II) shows that this mutation mostly affects the KR and KP terms. Expressing the KR and KP in terms of K1, K2, and θ (using Eq. 3) reveals that this interaction term is reduced by 80-fold in the A1145W mutant. This large change in coupling term accounts for most of the reduction in the lidocaine-induced shifts of F-V curves in mutant channels. We note that although the Monte Carlo–like-fitting algorithm efficiently samples a larger parameter space (varying over four orders of magnitude), we cannot eliminate the possibility that the equilibrium values reported in Table II are just one set of values that describe our data. Therefore, we cannot completely rule out other mechanisms to explain the lidocaine-induced fluorescence changes.
By examining the effect of these mutations on the probabilities of voltage sensor and pore opening in the absence of lidocaine, we can also make inferences regarding the thermodynamic effects of these mutations. The above analysis shows that the mutations where the interaction between activated voltage sensor and open pore is disrupted would exhibit small shifts in the F-V upon the addition of lidocaine. This would also cause rightward shifts in the F-V and G-V curves of this mutant in the absence of lidocaine, which was observed for the V1142W mutant (Muroi et al., 2010). A similar loss of coupling interactions between the resting-state voltage sensor and closed pore can account for small shifts in F-V curves upon lidocaine application (supplemental text). Three of the high impact mutants at I1284, I1287, and K1295 cause a leftward shift in the activation probability curves of the voltage sensor and pore opening, which suggests that these mutations disrupt the resting-state interactions between the voltage sensor and pore. The remaining two mutants, R1135W and A1145W, both cause opposite effects on the activation probabilities of the voltage sensor and pore. In these mutants, the closed state of the pore is stabilized while the voltage sensor is stabilized in the activated state. The effects of these mutants are consistent with the idea that these mutations disrupt resting-state and activated-state interactions (Muroi et al., 2010).
In sum, our analysis suggests that the mutations that disrupt coupling interactions between the voltage sensor and pore would reduce the lidocaine-induced shift in F-V curves. All sites identified by Muroi et al. (2010) except the S1134W cause reduced shifts in the F-V curves upon the addition of lidocaine. Moreover, only the residues that contribute both to the resting-state and activated-state interactions were identified in that study. By examining the effect of lidocaine on the mutations at the domain III intracellular gating interface, we are able to identify a larger set of residues that are important for voltage sensor and pore coupling. More specifically, this study also identifies the residues that mainly contribute to resting-state interactions and those that are involved in the activated-state interactions. Nonetheless, this interpretation should be considered tentative because it is based on the premise that lidocaine binding does not produce a channel pore with a very different structure, but primarily alters the energetics associated with pore opening and closing transition.
Mapping the mutations on a structural model of the sodium channel
The positions of the decoupling mutants were mapped onto a surface representation of a homology model of Nav1.4 (Fig. 7, A and B) and the isolated domain III (Fig. 7, C and D). The residues were color-coded based on the degree of shift in the domain III voltage sensor activation upon drug application. These mutants were mostly localized in the S4–S5 linker region and the lower end of the S6 segment. In the S4–S5 linker, they were exclusively limited to one face of the helix, indicating that this face plays a key role in coupling drug binding to voltage sensor activation. In contrast, such a clear pattern was not obvious in the S6 helix. Almost every residue in the stretch between L1282 to K1295 reduces the shifts in the domain III voltage sensor to a varying extent. One possibility is that the S6 helix, being in a tightly packed region near the bundle crossing, is likely to interact promiscuously with the neighboring S6 segments in addition to its own S5 helix and S4–S5 linker. The introduction of tryptophan into this region causes a leftward shift in the F-V and G-V curves (Muroi et al., 2010) in almost all the positions, consistent with the interpretation that these residues disrupt interactions between the voltage sensor and the pore in the resting state. Scanning mutagenesis studies in potassium channels show that most mutations in the S6 region near the bundle crossing stabilize the open pore, presumably by disrupting the packing in the closed state (Yifrach and MacKinnon, 2002; Wynia-Smith et al., 2008).
Figure 7.

Mapping the mutations that perturb allosteric modification by lidocaine. (A) Surface representation of the Nav1.4 sodium channel model with mutations that disrupt the communication between the lidocaine-binding site and voltage sensor, viewed from the intracellular side. A color gradient from yellow to red was used to represent ΔV1/2 changes induced upon the addition of lidocaine. (B) Same as in A after the structure was rotated by 90°. The domains I and IV are hidden from view by the other two domains. (C) Surface representation of domain III of the Nav1.4 channel. The other three domains were removed for clarity. The residues were color-coded as in A. (D) A close-up view of the S4–S5 linker, N terminus of S5, and C terminus of S6, with transparent surfaces and side chains colored as in A. All of these structures were drawn using PyMol (DeLano Scientific LLC).
Preliminary model of allosteric modulation
Finally, we consider a working model to rationalize the modification of voltage-dependent gating by intracellular pore blockers (Fig. 8). The main assumption is that lidocaine-like quaternary ammonium compounds upon binding to the sodium channel prevent the pore from fully closing (Armstrong, 1971). Furthermore, we have shown recently that lidocaine has the largest effect on the S6 segment of domain III (Muroi and Chanda, 2009). This imposes an energetic cost on the return of the domain III S6 segment to a closed position when the membrane is repolarized. This destabilization of the closed pore by lidocaine binding would propagate to the domain III voltage sensor because they are coupled. Therefore, lidocaine binding would shift the voltage-dependent activation of the domain III voltage sensor to more hyperpolarized potentials. Our finding that a strong hyperpolarization can drive the voltage sensor to a resting-state conformation suggests that either the pore gates are able to close even when the lidocaine is bound to the channel, or that an intermediate state corresponding to a resting domain III voltage sensor and S6 domain III in an open conformation is populated. At present, we are unable to clearly distinguish between these two possibilities because of a lack of a conformational probe for tracking the dynamics of individual pore segments. Despite these limitations, our study provides strong evidence that the intracellular gating interface is a major pathway for allosteric coupling between the drug-binding site and the voltage-sensing domains.
Figure 8.

Plausible model for voltage sensor modification by local anesthetics. Upon depolarization, the voltage sensors move outward and the pore of the sodium channel opens. For clarity, only domains I and III are shown. Lidocaine binds to the open pore with high affinity (open lidocaine bound). When the membrane is repolarized, the domain III pore helices are unable to close because of lidocaine occupying the binding site and destabilizing the resting state of the domain III voltage sensor relative to the open state. Strong hyperpolarization drives the domain III voltage sensor to a resting-like conformation, even though the lidocaine-binding site is occupied.
Acknowledgments
We thank Kristina Schuldt for excellent technical assistance. We also thank the members of the Chanda laboratory for their comments and discussions.
This project was supported by funds from the National Institutes of Health (grant RO1-GM084140) and Shaw Scientist award (to B. Chanda).
Christopher Miller served as editor.
Footnotes
Abbreviations used in this paper:
- F-V
- fluorescence–voltage
- Q-V
- charge–voltage
- TTX
- tetrodotoxin
- WT
- wild type
References
- Armstrong C.M. 1971. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 58:413–437 10.1085/jgp.58.4.413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean B.P., Cohen C.J., Tsien R.W. 1983. Lidocaine block of cardiac sodium channels. J. Gen. Physiol. 81:613–642 10.1085/jgp.81.5.613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F., Perozo E., Papazian D.M., Stefani E. 1991. Molecular basis of gating charge immobilization in Shaker potassium channels. Science. 254:679–683 10.1126/science.1948047 [DOI] [PubMed] [Google Scholar]
- Cahalan M.D. 1978. Local anesthetic block of sodium channels in normal and pronase-treated squid giant axons. Biophys. J. 23:285–311 10.1016/S0006-3495(78)85449-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan M.D., Almers W. 1979. Interactions between quaternary lidocaine, the sodium channel gates, and tetrodotoxin. Biophys. J. 27:39–55 10.1016/S0006-3495(79)85201-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W.A. 2000. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 26:13–25 10.1016/S0896-6273(00)81133-2 [DOI] [PubMed] [Google Scholar]
- Chanda B., Bezanilla F. 2002. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J. Gen. Physiol. 120:629–645 10.1085/jgp.20028679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fozzard H.A., Lee P.J., Lipkind G.M. 2005. Mechanism of local anesthetic drug action on voltage-gated sodium channels. Curr. Pharm. Des. 11:2671–2686 10.2174/1381612054546833 [DOI] [PubMed] [Google Scholar]
- Hackos D.H., Chang T.H., Swartz K.J. 2002. Scanning the intracellular S6 activation gate in the shaker K+ channel. J. Gen. Physiol. 119:521–532 10.1085/jgp.20028569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanck D.A., Makielski J.C., Sheets M.F. 1994. Kinetic effects of quaternary lidocaine block of cardiac sodium channels: a gating current study. J. Gen. Physiol. 103:19–43 10.1085/jgp.103.1.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanck D.A., Nikitina E., McNulty M.M., Fozzard H.A., Lipkind G.M., Sheets M.F. 2009. Using lidocaine and benzocaine to link sodium channel molecular conformations to state-dependent antiarrhythmic drug affinity. Circ. Res. 105:492–499 10.1161/CIRCRESAHA.109.198572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. 1977. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69:497–515 10.1085/jgp.69.4.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. 2001. Ion channels of excitable membranes. Third edition Sinauer Associates, Sunderland, MA: 814 pp [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. 1994. Shaker potassium channel gating. I: transitions near the open state. J. Gen. Physiol. 103:249–278 10.1085/jgp.103.2.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keynes R.D., Rojas E. 1974. Kinetics and steady-state properties of the charged system controlling sodium conductance in the squid giant axon. J. Physiol. 239:393–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledwell J.L., Aldrich R.W. 1999. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 113:389–414 10.1085/jgp.113.3.389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.Y., Banerjee A., MacKinnon R. 2009. Two separate interfaces between the voltage sensor and pore are required for the function of voltage-dependent K(+) channels. PLoS Biol. 7:e47 10.1371/journal.pbio.1000047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S.B., Campbell E.B., Mackinnon R. 2005. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 309:903–908 10.1126/science.1116270 [DOI] [PubMed] [Google Scholar]
- Long S.B., Tao X., Campbell E.B., MacKinnon R. 2007. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 450:376–382 10.1038/nature06265 [DOI] [PubMed] [Google Scholar]
- Lu Z., Klem A.M., Ramu Y. 2002. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 120:663–676 10.1085/jgp.20028696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muroi Y., Chanda B. 2009. Local anesthetics disrupt energetic coupling between the voltage-sensing segments of a sodium channel. J. Gen. Physiol. 133:1–15 10.1085/jgp.200810103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muroi Y., Arcisio-Miranda M., Chowdhury S., Chanda B. 2010. Molecular determinants of coupling between the domain III voltage sensor and pore of a sodium channel. Nat. Struct. Mol. Biol. 17:230–237 10.1038/nsmb.1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peganov E.M., Khodorov B.I. 1977. Gating current in the membrane of nodes of Ranvier under conditions of linear alteration of the membrane potential. Biull. Eksp. Biol. Med. 84:515–518 [PubMed] [Google Scholar]
- Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale D.S., McPhee J.C., Scheuer T., Catterall W.A. 1994. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 265:1724–1728 10.1126/science.8085162 [DOI] [PubMed] [Google Scholar]
- Sali A., Blundell T.L. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- Scheuer T. 1999. Commentary: a revised view of local anesthetic action: what channel state is really stabilized? J. Gen. Physiol. 113:3–6 10.1085/jgp.113.1.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa N.E., McCormack K., Tanouye M.A., Sigworth F.J. 1992. The size of gating charge in wild-type and mutant Shaker potassium channels. Science. 255:1712–1715 10.1126/science.1553560 [DOI] [PubMed] [Google Scholar]
- Sheets M.F., Hanck D.A. 2003. Molecular action of lidocaine on the voltage sensors of sodium channels. J. Gen. Physiol. 121:163–175 10.1085/jgp.20028651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets M.F., Hanck D.A. 2007. Outward stabilization of the S4 segments in domains III and IV enhances lidocaine block of sodium channels. J. Physiol. 582:317–334 10.1113/jphysiol.2007.134262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Maxwell C.J., Ledwell J.L., Aldrich R.W. 1998a. Role of the S4 in cooperativity of voltage-dependent potassium channel activation. J. Gen. Physiol. 111:399–420 10.1085/jgp.111.3.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Maxwell C.J., Ledwell J.L., Aldrich R.W. 1998b. Uncharged S4 residues and cooperativity in voltage-dependent potassium channel activation. J. Gen. Physiol. 111:421–439 10.1085/jgp.111.3.421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Llavina G.J., Chang T.H., Swartz K.J. 2006. Functional interactions at the interface between voltage-sensing and pore domains in the Shaker K(v) channel. Neuron. 52:623–634 10.1016/j.neuron.2006.10.005 [DOI] [PubMed] [Google Scholar]
- Starmer C.F., Grant A.O., Strauss H.C. 1984. Mechanisms of use-dependent block of sodium channels in excitable membranes by local anesthetics. Biophys. J. 46:15–27 10.1016/S0006-3495(84)83994-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strichartz G.R. 1973. The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. J. Gen. Physiol. 62:37–57 10.1085/jgp.62.1.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanguy J., Yeh J.Z. 1989. QX-314 restores gating charge immobilization abolished by chloramine-T treatment in squid giant axons. Biophys. J. 56:421–427 10.1016/S0006-3495(89)82688-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedantham V., Cannon S.C. 1999. The position of the fast-inactivation gate during lidocaine block of voltage-gated Na+ channels. J. Gen. Physiol. 113:7–16 10.1085/jgp.113.1.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.K., Brodwick M.S., Eaton D.C., Strichartz G.R. 1987. Inhibition of sodium currents by local anesthetics in chloramine T-treated squid axons. The role of channel activation. J. Gen. Physiol. 89:645–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.Y., Wang G.K. 1998. Point mutations in segment I-S6 render voltage-gated Na+ channels resistant to batrachotoxin. Proc. Natl. Acad. Sci. USA. 95:2653–2658 10.1073/pnas.95.5.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.Y., Nau C., Wang G.K. 2000. Residues in Na(+) channel D3-S6 segment modulate both batrachotoxin and local anesthetic affinities. Biophys. J. 79:1379–1387 10.1016/S0006-3495(00)76390-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.Y., Mitchell J., Moczydlowski E., Wang G.K. 2004. Block of inactivation-deficient Na+ channels by local anesthetics in stably transfected mammalian cells: evidence for drug binding along the activation pathway. J. Gen. Physiol. 124:691–701 10.1085/jgp.200409128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S.N., Wang S.Y., Wang G.K. 1998. Lysine point mutations in Na+ channel D4-S6 reduce inactivated channel block by local anesthetics. Mol. Pharmacol. 54:733–739 [PubMed] [Google Scholar]
- Wynia-Smith S.L., Gillian-Daniel A.L., Satyshur K.A., Robertson G.A. 2008. hERG gating microdomains defined by S6 mutagenesis and molecular modeling. J. Gen. Physiol. 132:507–520 10.1085/jgp.200810083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V., Brown J., Sharp E.M., Clare J.J., Scheuer T., Catterall W.A. 2001. Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na(+) channel alpha subunit. J. Biol. Chem. 276:20–27 10.1074/jbc.M006992200 [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V., McPhee J.C., Idsvoog D., Pate C., Scheuer T., Catterall W.A. 2002. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J. Biol. Chem. 277:35393–35401 10.1074/jbc.M206126200 [DOI] [PubMed] [Google Scholar]
- Yeh J.Z. 1978. Sodium inactivation mechanism modulates QX-314 block of sodium channels in squid axons. Biophys. J. 24:569–574 10.1016/S0006-3495(78)85403-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yifrach O., MacKinnon R. 2002. Energetics of pore opening in a voltage-gated K(+) channel. Cell. 111:231–239 10.1016/S0092-8674(02)01013-9 [DOI] [PubMed] [Google Scholar]