

Abstract
The first evidence of a complex between glutathione and cobalamin, glutathionylcobalamin (GSCbl), was presented by Wagner and Bernhauer more than 40 years ago (Ann N Y Acad Sci, 1964, 112, 580). More recently, NMR and EXAFS solution studies by Brown et al (Biochemistry, 1993, 32, 8421) and Scheuring et al. (Biochemistry, 1994, 33, 6310), respectively, provided evidence that the glutathionyl moiety in GSCbl is bound to the cobalt center via a Co-S bond. Despite continued efforts, the structural analysis of glutathionylcobalamin in the solid state has remained elusive. Here we report the first atomic resolution crystal structure of GSCbl, refined to a crystallographic R-factor of 0.0683. The glutathione moiety is bound to the cobalt center through the sulfur atom as expected, with a Co-S bond distance of 2.295(1) A. This distance agrees with the distance obtained from the EXAFS analysis of GSCbl (2.280(5) Å). However, the bond to the axial α-5,6-dimethylbenzimidazole base (DMB), 2.074(3) Å, is significantly shorter than that determined from the EXAFS measurements (Co-N3B = 2.15(3) Å). The corrin fold angle is 24.7°, the highest ever reported for a cobalamin structure, and points in the direction of the β-face of the corrin, towards the glutathione (GS−). The GS− ligand has been modeled in two conformations, each featuring distinct hydrogen bonding interactions. In both conformations, the α-carboxylate group of the GS− ligand interacts with the generally rigid side chain a of the cobalamin molecule, resulting in two distinct conformations. A comparison with the structure of other thiolatocobalamins revealed high similarity in the positions of the atoms in the cysteinyl moiety, the fold of the corrin rings, and the Co-S bond distances.
Keywords: Glutathionylcobalamin, glutathione, thiolatocobalamin, Co-S bond, X-ray structure, synchrotron diffraction
Introduction
Cobalamins (Cbls) are cobalt complexes in which the metal center is coordinated by four equatorial donor nitrogens from a corrin ring macrocycle (Figure 1). In addition, the Co center is coordinated to a nitrogen atom from a 5,6-dimethylbenzimidazole (DMB) moiety at the lower (α) axial position. Displacement of the DMB moiety from Cbl generates the “base-off” conformation of the complex. The upper (β) axial position can be occupied by a large number of ligands including thiolate groups such as glutathionate (GS−), cysteinate (CysS−) and homocysteinate (HcyS−), giving rise to the thiolatocobalamin derivatives glutathionylcobalamin (GSCbl), cysteinylcobalamin (CysCbl), and homocysteinylCbl (HcyCbl), respectively.1-3 The Cbl complex presents a total of seven amide side chains, 3 acetamides and 4 propionamides, that project above and below the plane of the corrin ring. The conformation of the side chains is crucial for proper binding of the Cbl molecule to its transport proteins and Cbl-dependent enzymes.1,2,4,5
Figure 1.
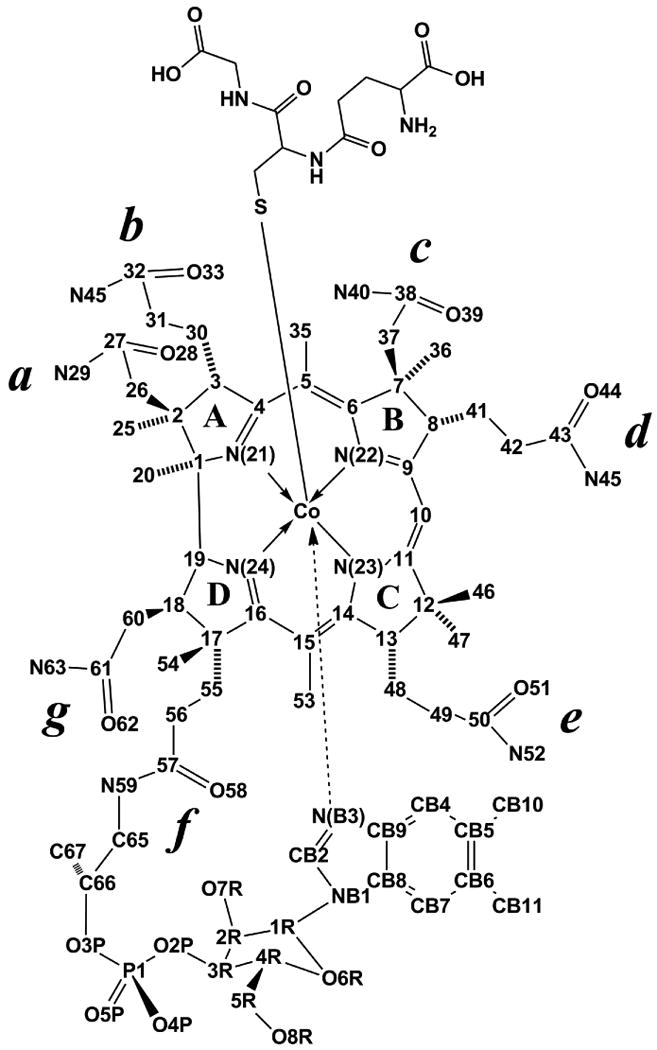
Chemical structure and atom numbering used for GSCbl.
The first evidence for the existence of a 1:1 molar complex of glutathione with cobalamin was presented by Wagner and Bernhauer in 1964.6 The authors noted that the reaction of aquacobalamin with glutathione produced a purple compound with UV-vis maxima at 287, 337, 374, 434 and 536 nm.6 It was also reported that in the presence of a molar excess of glutathione and iodomethane, GSCbl underwent alkylation to produce MeCbl. This finding led the authors to speculate that GSCbl could act as a precursor for the biosynthesis of MeCbl and AdoCbl in vivo.6 Intense research on the structure, reactivity, and potential biological roles of GSCbl followed thereafter in several laboratories.3,7-13
Prior to the biosynthesis of AdoCbl and MeCbl, the β-axial ligand of incoming dietary cobalamins (including exogenous MeCbl and AdoCbl) must undergo processing, that is, removal of the upper axial ligand to generate cob(II)alamin and/or cob(I)alamin, which serve as precursors for the de novo synthesis of AdoCbl and MeCbl.14 Early in vitro and ex vivo studies in our laboratory suggested a key role for glutathione in the processing and biosynthesis of AdoCbl and MeCbl.7,15-18
Recent studies have finally uncovered the mystery of how dietary cobalamins are processed in mammalian cells, prior to the biosynthesis of MeCbl and AdoCbl.19-22 Cobalamins are processed by the cblC gene product (also known as methylmalonic aciduria combined with homocystinuria type C, or MMACHC gene product), a thermolabile 32 kDa protein devoid of cofactors, which is highly conserved in eukaryotes.23-25 To attain this crucial function in vivo, MMACHC employs two mechanistically distinct strategies: a) removal of the cyanide group in CNCbl via reductive elimination, a reaction that requires reductases and NADPH to generate Cbl(II),19-22 and b) removal of the alkyl groups in MeCbl, AdoCbl and some non-natural alkylcobalamins analogues via nucleophilic attack of the Co-C bond by the thiolate form of glutathione, to produce Cbl(I) and the corresponding thioethers.20-21 The proposed involvement of GSH in the processing of dietary alkylcobalamins was therefore established, albeit GSCbl itself could not be detected as in intermediate in these in vitro reactions.21 We have previously shown that only small amounts of GSCbl can be isolated from cultured cells, suggesting that GSCbl likely exists as a transient species in the intracellular cobalamin pool.26
More than 50 cobalamin crystal structures have been reported to date since the original structural elucidation of AdoCbl by Lenhert and Hodgkin in 1961.27 There is a conspicuous lack of structural data for thiolatocobalamins. The first crystal structure of a thiolatocobalamin (γ-GluCysCbl) was reported only 9 years ago by Suto et al.11 The structure of γ-GluCysCbl revealed substantially long Co-S and Co-NB3 bonds (2.267(2) and 2.049(6), respectively) and a large corrin fold-angle (24.2°).11 The authors noted a highly disordered γ-GluCysS− ligand, which was resolved by modeling the structure to two conformations of roughly equal occupancy.11 Attempts to solve the crystal structure of GSCbl have been hampered by the substantial disorder of the glutathionyl moiety.2, 28 Herein, we describe the high-resolution X-ray crystal structure of the elusive GSCbl and provide a comparison with the structures of other thiolatocobalamins.
Experimental Section
General Procedures and Chemicals
Hydroxocobalamin hydrochloride (HOCbl•HCl) was purchased from Fluka. Stated purity by manufacturer is ≥96%. Reduced L-glutathione (γ-L-Glu-L-Cys-Gly, purity ≥98%), LiCl, NaCl, HEPES (4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid), TES (N-[Tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid) and MES (2-(N-morpholino)ethanesulfonic acid hemisodium salt, 4-morpholineethanesulfonic acid hemisodium salt) were purchased from Sigma. CsCl was purchased from Var Lac Oid Chemical Company, Inc. Highly purified water obtained from a MilliQ purification system (Millipore) was used throughout this study.
Crystallization of Glutathionylcobalamin
HOCbl•HCl (35 mg, 0.0253 mmol) was dissolved in 0.35 mL of water (final concentration, 72 mM). GSH (0.035 mL, 1.37 M) was added to the HOCbl•HCl solution, to yield a 1:1.9 molar mixture of HOCbl/GSH. Several crystallization mixtures were prepared in glass-vials, either in the presence or in the absence of 0.25, 0.5 and 1.0 mg of CsCl. The samples were placed in an ice-bath (4°C), protected from light. Single deep purple colored crystals suitable for X-ray analysis appeared after 72 h to 1 week of incubation. Addition of CsCl to the crystallization mixtures resulted in increased crystallization yield and crystal size. Attempts to obtain suitable crystals at room temperature were unsuccessful.
X-ray diffraction studies
Crystals of GSCbl were grown from their corresponding synthesis mixtures at 4°C. Crystals suitable for X-ray analysis appeared after 72 h to 1 week. The GSCbl crystals were transferred into paratone oil and any residual synthesis mixture was carefully removed by dragging the crystals through the oil. The crystals were then mounted in thin nylon loops on copper magnetic pins (Hampton Research) and flash-cooled in liquid nitrogen. X-ray diffraction experiments were carried out at beam line BL9-2 at the Stanford Synchrotron Radiation Lightsource (SSRL). Data from a single GSCbl crystal were collected on a MarMosaic 325 CCD detector using X-rays produced by a 16-pole wiggler insertion device through a flat Rh-coated collimating mirror, a liquid nitrogen-cooled double Si(111) crystal monochromator and a toroidal focusing mirror. The X-ray wavelength used for data collection was 0.79987 Å (15,500 eV). Two data sets were collected, both consisting of 90 images with an oscillation angle of 1.0 and with a crystal to detector distance of 95.0 mm. The first data set was recorded with an exposure time of 5 s and no beam attenuation, and the second data set had the same exposure time but the beam was attenuated by 75%. The two data sets were processed with the program XDS and scaled together with the program XSCALE.29 Symmetry-equivalent and Bijvoet pairs were not merged and no absorption correction was applied. A total of 39,643 reflections were measured, resulting in 19,768 unique reflections to a nominal resolution of 0.74 Å, with a merging R-factor of 0.057 for common reflections on all images.
The GSCbl structure was solved by Patterson methods as implemented in the program SHELXS.30 The cobalt, phosphorus and some of the nitrogen atoms were readily located, with the remainder of the lighter atoms later identified by difference Fourier synthesis. The GSCbl model was constructed in three main stages. First, the entire Cbl moiety was built and refined by full-matrix least-squares methods using SHELXL30 with isotropic atomic displacement parameters (ADPs). Inspection of 2Fo-Fc and Fo-Fc electron density maps showed the location of the entire glutathione ligand, and these atoms were added in stage two and the model was refined with anisotropic ADPs. Additional difference Fourier synthesis identified solvent molecules that were subsequently added to the model in stage three. Fifteen water molecules were added to the GSCbl structure; four of these water molecules show some degree of disorder, which manifests as dumbbell-shaped electron density. Each of these four molecules were been modeled in two positions with partial site occupancy factors (SOFs), separated by between 1.1 and 1.24 Å. A correction for the anomalous scattering from cobalt at 15,500 eV was applied during refinement. All non-hydrogen atoms were refined with anisotropic ADPs and hydrogen atoms were added in idealized positions and refined in riding positions at the very end of the refinement. The final crystallographic R factor (R1) was 0.0683 for 18,744 reflections with Fo > 4σF. A summary of the refinement parameters is given in Table 1.
Table 1.
Refinement parameters for GSCbl.
| GSCbl | |
|---|---|
| Empirical formula | C72H104N16O20CoPS |
| H2O sites | |
| Formula weight | 1,850.02 |
| Crystal system | Orthorhombic |
| space group | P212121 |
| Unit cell dimensions: | |
| a [Å] | 16.23 |
| b [Å] | 21.08 |
| c [Å] | 25.78 |
| V [Å3] | 8,820.07 |
| Z | 4 |
| Dcalc [g cm-3] | 1.405 |
| μ [mm-1] | 0.32 |
| F(000) | 3852 |
| Crystal size [mm] | 0.4 × 0.2 × 0.1 |
| Temperature [K] | 100 |
| Wavelength [Å] | 0.79987 |
| # of unique reflections | 21,987 |
| # of reflections with I>4σI | 21,218 |
| R(int) | 0.0481 |
| Data/restraints/parameters | 21,218/1,084/1,375 |
| GOF on F2 | 1.106 |
| Final R indices: | |
| R1 (I>4σI / all data) | 0.0683 / 0.0709 |
| wR2 (all data) | 0.1824 |
| Largest diff. peak/hole [e/Å-3] | 0.96 and -1.11 |
The CCDC database (GSCbl: CCDC 779605) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Results and Discussion
Preparation of GSCbl Crystals
The production of high quality crystals was critical to our study due to the anticipated disorder in the glutathionyl moiety of GSCbl. Several attempts to prepare single crystals of GSCbl were made, both in the absence and in the presence of electrolytes, including LiCl, NaCl, and CsCl. The presence of electrolytes has been reported to improve crystal quality in some Cbls and related compounds.31,32 A number of buffer systems including HEPES, TES and MES were also utilized without success. Addition of 1, 2.5 and 5% v/v acetone to the crystallization mixture resulted in partial precipitation of GSCbl. Crystallization mixtures were incubated either at room temperature or at 4°C. Among these conditions, crystallization of a 1:1.9 mixture of HOCbl•HCl/GSH prepared in H2O, in the presence of various concentrations of CsCl at 4°C provided the best crystal yield and quality, suitable for X-ray analysis.
Crystals typically grew as long thin deep-red colored rods, approximately 0.5-1.0 mm in length. Several crystals were mounted, flash-cooled in liquid nitrogen and screened for diffraction quality. Crystals showing diffraction to greater than 0.8 Å and low mosaicity (typically less than 0.5°) were chosen for later diffraction data collection.
Analysis of the GSCbl Structure in the Solid State
Despite the increasing interest on the potential roles of GSCbl in biology,7,15-18,33-36 only a few Cbls that contain a Co-S axial bond have been crystallized,37 and only three that contain a Co-thiolate bond (γ-glutamylcysteinylCbl (γ-GluCysCbl),11 N-acetyl-L-cysteinylcobalamin (N-AcCysCbl),3 and the cis and trans isomers of Captopril-Cbl (CaptoCbl)38 have been structurally characterized. GSCbl crystallizes in the orthorhombic space group P212121 with cell dimensions a = 16.25 Å, b = 21.06 Å, c = 25.72 Å. The structure of the Cbl molecule, along with the lattice packing has been described exhaustively in the literature for this crystal form, and the structure of the Cbl moiety in GSCbl does not deviate markedly from the known Cbl structures. Briefly, the Cbl molecules in this crystal form are oriented such that the corrin ring plane is approximately parallel to the ab plane of the unit cell. Neighboring Cbl molecules are not perfectly parallel with each other, giving rise to layers of zig-zagged planes when viewed perpendicular to the bc plane. These planes are separated by layers of solvent molecules, and the GS− ligand and the axial DMB base extend into these solvent layers. In GSCbl, this solvent structure has been modeled as fifteen water molecules with either full or partial site occupancy. All of the solvent molecules are hydrogen bonded to either an oxygen or a nitrogen atom on the Cbl molecule or the GS− ligand.
The majority of Cbls which crystallize in P212121 have similar cell dimensions to GSCbl, and analysis of the crystal packing in this space group shows that they fall into one of four groups, cluster I, II, III and IV.2,31 This clustering is based upon the ratios of the c/a and b/a unit cell dimensions. For GSCbl, these ratios are c/a = 1.580 and b/a = 1.296, which shows that these crystals can be grouped in cluster I. Closer analysis of the Cbl complexes in cluster I shows that the four structures which contain a Co-thiolate bond (GSCbl, γ-GluCysCbl, N-AcCysCbl and CaptoCbl) have an average c/a ratio of 1.584, whereas all the other cluster I complexes have an average value around 1.51 - 1.52.2,31 It appears that the Co-thiolate Cbls may form their own sub-group of the typical cluster I packing.
As noted above, the structure of the Cbl moiety in the GSCbl complex is comparable to structures reported previously. The four in-plane Co-N bonds are similar to those reported for other Co-S containing Cbls, and for most other Cbl complexes in general. These equatorial bonds appear to be rather insensitive to the nature of the axial ligand and the Co-ligand bond distance, as previously suggested.9 The GS− ligand is bound to the cobalt through the sulfur atom as expected, with a Co-S bond distance of 2.295(1) Å. This distance agrees remarkably well with the distance obtained from the EXAFS analysis of GSCbl (2.280(5) Å)9 (Table 2), and agrees well with comparable bond lengths in other Co-S and Co-thiolate Cbl structures, including N-AcCysCbl (Co-S = 2.250 Å), γ-GluCysCbl (2.267 Å) and CaptoCbl (2.282 Å) (Table 2). However the bond to the axial DMB base (Co-N = 2.074(3) Å) is significantly shorter than that determined from the EXAFS measurements (Co-N = 2.15(3) Å), and this is also consistent with the other Co-thiolate Cbl complexes (Table 2).
Table 2.
Comparison of the Co coordination sphere in GSCbl and other Co-S bonded Cbls.
| Cobalamin | Co-S (Å) | Co-NB3 (Å) | Fold angle (°) | Reference |
|---|---|---|---|---|
| X-ray crystallography | ||||
| GSCbl | 2.295(1) | 2.074(3) | 24.7(1) | This work |
| Na[γ-GluCysCbl] | 2.267(2) | 2.049(6) | 24.2 | 11 |
| Na[N-AcCysCbl] | 2.25 | 2.06 | 17.5 | 3 |
| CapSCbl-1 (trans isomer) | 2.282(3) | 2.106(5) | 14.9 | 38 |
| CapSCbl-2 (cis isomer) | 2.261(4) | 2.094(9) | 14.2 | 38 |
| NH4[SO3Cbl] | 2.231(1) | 2.134(4) | 16.3 | 39 |
| [(NH2)2CSCbl]Cl | 2.300(2) | 2.032(5) | 14.8 | 40 |
| NCSCbl | 2.250(4) | 1.994(4) | 14.9 | 31 |
| EXAFS | ||||
| GSCbl | 2.28±0.05 | 2.15±0.03 | ND | 9 |
| CysCbl | 2.34±0.03 | 2.13±0.04 | ND | 9 |
| SO3Cbl | 2.35±0.02 | 2.16±0.04 | ND | 9 |
The conformation of the corrin ring is generally determined by the fold angle around the Co-C10 axis, between the planes of the conjugated ring systems (plane 1: N21, C4, C5, C6, N22, C9, C10) and plane 2: N24, C16, C15, C14, N23, C11, C10). In GSCbl this corrin fold angle is 24.6° in the direction of the β face of the corrin (towards the glutathione). Figure 2 shows thermal ellipsoid plots of GSCbl drawn at 30 % probability. Analysis of other Cbl complexes shows that this fold angle is the highest of any reported, and comparable to the angles observed in the γ-GluCys-Cbl (24.2°) and N-AcCysCbl (22°) complexes1,3,11). Although it is tempting to try and correlate a larger corrin fold with a short axial DMB bond length (whereby the closer approach of the DMB pushes up on the corrin and increases the fold), there does not appear to be a correlation in the Co-thiolate Cbl complexes. This implies that other factors are more important in determining the extent to which the corrin folds away from the DMB base, and it may be related more to the interactions that the Cbl amide side chains make with the upper axial ligand. In γ-GluCysCbl for example, it was reported that the N40 atom on the c side chain (see Figure 1) is hydrogen bonded to the sulfur atom on the ligand,11 which could serve to pull that side of the corrin ring upwards to some extent. The same appears to be the case for the N-AcCysCbl complex (N40-S = 3.55 Å) despite the non-ideal hydrogen-bonding configuration at the sulfur atom.3 There is a similar potential hydrogen bonding contact in the GSCbl structure, where the N40-S distance is 3.31 Å, although as in the N-AcCysCbl structure, the C71–S70–N40 angle is only 93° and not ideal for efficient hydrogen bond formation.
Figure 2.
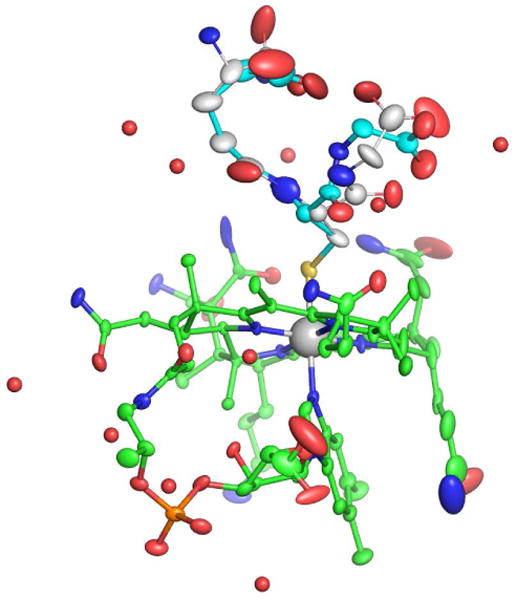
Thermal ellipsoid plot of GSCbl. The Cbl complex is colored green (C), red (O), blue (N), and cyan (P). The cobalt is shown as a gray sphere. The thermal ellipsoid was drawn at 30% probability for all the atoms except for the cobalt and the water molecules (shown as red spheres).
Analysis of the seven amide side chains incorporated in the corrin ring shows that several of them display a degree of structural disorder. Chains c and d (see Figure 1) have residual Fo-Fc electron density peaks associated with them, indicative of a higher degree of structural flexibility compared to the b, e, f and g side chains which all have very well defined electron density. Side chain a adopts two quite distinct conformations (Figure 3), clearly visible in 2Fo-Fc and Fo-Fc electron density maps, with a refined SOF ratio of 0.62:0.38. Although multiple conformations have been observed in the past for side chains b, c, e, f and g, chains a and d assume very similar conformations in all structures.32 Therefore, the observation of two clearly-defined conformations for side chain a is unprecedented, and is associated with the presence of the GS− moiety, and more importantly, with the conformational disorder observed in this ligand (discussed below). There is a partially-occupied water molecule (O21, refined SOF = 0.35) approximately 1.0 Å from the N29b atom of the minor side chain a conformation (Figure 3) which presumably could only occupy this site when side chain a adopts the major conformation.
Figure 3.
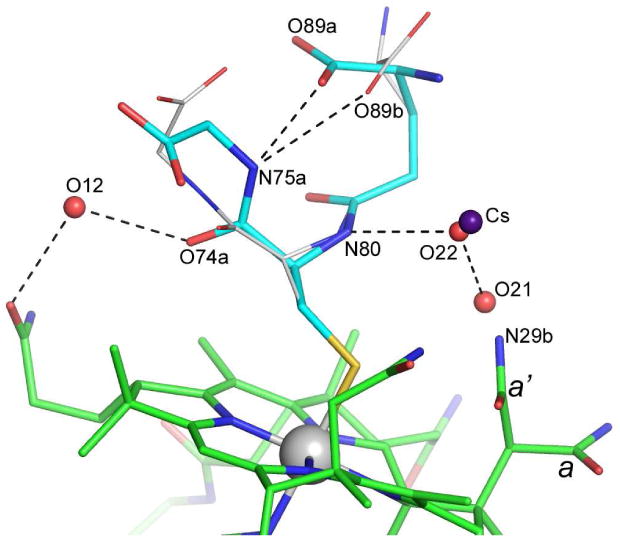
Alternate conformations of the GS− ligand in the GSCbl structure. Hydrogen bonding interactions are shown with dashed lines. The two alternate conformations of side chain a, are indicated as a and a′.
The GS− ligand has been modeled in two conformations. The atoms comprising the side chain of the cysteinyl residue (S70, C71), the amide bond between the cysteinyl residue and the γ-glutamyl moiety (N80, C81 and O82) and part of the γ-glutamyl side chain (C83 and C84) are well-defined and adopt a single rigid structure. The glutamyl residue shows disorder beyond the β-carbon (C84), such that the α-carbon (C85), α-amino nitrogen (N86) and the α-carboxylate group (C87, O88 and O89) adopt two distinct conformations. The two alternate conformations of the GS− ligand in the GSCbl structure are shown in Figure 3. During refinement, the atoms in the two conformations were treated independently with respect to their atomic coordinates and ADPs, however a single SOF was refined for each conformation such that the sum of the SOFs for equivalent atoms equaled unity. This approach assumed that there were only two major conformations of this part of the ligand, which upon analysis of the residual electron density appears to be valid. The final refined SOFs were 0.53 and 0.47 for the two components. The two conformations result from two bond rotations, one of approximately 30° about the C83-C84 bond, and the second of approximately 110° about the C84-C85 bond. This puts the two positions of the α-amino nitrogen N86 approximately 3.2 Å from each other, projecting out into the solvent channel. The corresponding positions of the α-carboxylate groups are significantly closer to each other such that one of the oxygen atoms (O88) is only 1 Å from its counterpart (Figure 3). Both α-carboxylate group conformations fold back over the top of the central cysteinyl residue and project towards the glycinyl end of the ligand. As noted above, side chain a of the Cbl moiety adopts two distinct conformations. The two conformational forms interact with the two α-carboxylate conformations in a symmetry-related molecule. In the major conformation of side chain a, the O28a atom makes a very close contact with the O89 atom of the α-carboxylate in one of its conformations, and it is very likely that the amide side chain only adopts this conformation when the α-carboxylate is in the second conformation. Since this conformational flexibility has never before been observed for the side chain a in any Cbl structures reported to date,32 it is conceivable that the conformational switching of the glutamyl residue of the GS− ligand subsequently drives the switching observed for this generally rigid side chain.
At the other end of the ligand the glycinyl residue also shows disorder which could be modeled as two distinct conformations, diverging at atom C72 which is equivalent to the α-carbon of the cysteinyl residue. The degree of disorder is somewhat less than that seen for the glutamyl group, with the group occupancies for the two conformations refining to a major component (SOF = 0.71) and a minor component (SOF = 0.29) (Figure 3). In the major component, the amide nitrogen of the glycinyl residue (N75a) is directed towards the α-carboxylate of the glutamyl group resulting in a strong hydrogen bond between N76a and O89b (2.49 Å) The O89b atom (the second position of α-carboxylate) also makes a hydrogen bond to N75a, albeit somewhat longer (3.09 Å). The fact that there are hydrogen bonding possibilities between the N75 atom and the disordered glutamyl α-carboxylate in both conformations (which are present in an almost 50:50 ratio based upon the refined group SOFs) may be the reason the glycinyl residue favors this conformation in a 70:30 ratio. In the minor component, the C72-C73 bond has rotated approximately 20°, the carbonyl oxygen (O74b) moves upwards away from the corrin ring, and the carboxylate group swings towards the glutamyl moiety. Although atom O78b appears to make a very close contact (2.45 Å) with O89a in this conformation, the likelihood is that the glycinyl residue only adopts this minor conformation when the glutamyl residue has adopted the second conformation, and the N75a-O88b hydrogen bonding interaction is longer and presumably weaker, enabling the C72-C73 bond to rotate.
The SOFs of some of the water molecules surrounding GS in GSCbl correlate well with the disorder seen in the GS− ligand. In the major conformation of the glycinyl residue, O74a accepts a hydrogen bond from water molecule O12, yet in the minor conformation, O12 would form a very close non-bonded contact with atom C76b; consequently, this water molecule is partially occupied with a refine SOF = 0.64. Another water molecule (O9) makes hydrogen bonds with the glycinyl carboxylate in both conformations and consequently has a higher refined SOF (0.77). At the other end of the GS− ligand, there are two partially-occupied water molecules which seem to respond to the disorder of the glutamyl moiety; O13 in its current location can only hydrogen bond to O88b but is interacts with two full occupancy water molecules, which could be why its refined SOF is 0.80. The location of water molecule O2, one of the disordered solvent molecules refined in two positions (with a SOF ratio of 0.60:0.40), is directly related to the disorder of GS in GSCbl. In one conformation of the glutamyl group, this water molecule forms hydrogen bonding interactions with an amide side chain of a symmetry-related corrin ring and two water molecules. However in the second glutamyl conformation, the N86 atom is much too close to this water position, so it must move 1.27 Å to its alternate position where it forms a hydrogen bond back to the N86 atom. Two water molecules make hydrogen bonding interactions with the amide bond between the cysteinyl residue and the γ-glutamyl moiety; O5 to N80 (3.20 Å) and O10 to O82 (2.67 Å) and both of these water molecule are fully occupied.
During the refinement of the GSCbl structure, the ADP of one water molecule refined to a low value compared to the surrounding water molecules and the atoms on the GS− and the Cbl moieties to which it was potentially hydrogen bonded. The distances to these potential hydrogen bonding partners were all relatively long (3.12, 3.23, 3.28 and 3.33 Å) except for one to the O88b atom of the GS− ligand in a symmetry-related molecule (2.71 Å). This suggested that if it were a water molecule it would not be very firmly anchored in the structure and therefore would most likely not have a small refined ADP. Furthermore, inspection of the difference electron density (calculated as Fo-Fc) showed considerable residual positive density at this water position. This electron density was initially fitted as two water molecules separated by 0.75 Å but upon subsequent refinement, the SOF ratio was 0.95:0.05 and there was still a significant amount of residual positive Fo-Fc electron density. Since CsCl was present in the crystallization medium, and the observed distances to neighboring atoms were consistent with Cs-O or Cs-N bond distances, a cesium atom was refined in the major position, retaining a water molecule (O22) in the second position since it made good hydrogen bonding contacts with the N80 atom of the GS− moiety (2.71 Å) and the water molecule O21 (2.35 Å) which occupies the position of the minor conformation of the corrin amide side chain a. Initially the ADP of the cesium was set to a value similar to the surrounding atoms and refined isotropically, along with the SOF. The ADP was then refined anisotropically and the resulting SOF refined to 0.15. The SOF of the water molecule refined to 0.69 and subsequent inspection of the Fo-Fc electron density showed no residual density. The cesium ion bridges between the N80 atom of the GS− ligand (3.23 Å), and the atom N29b of the a side chain (3.33 Å), and is in addition coordinated to a water molecule O13 (3.12 Å), O88b of a symmetry-related GS− ligand (2.71 Å) and N45 of a second symmetry-related Cbl molecule (3.24 Å).
Structural comparison with γ-GluCysCbl and N-AcCysCbl
The γ-GluCysCbl and N-AcCysCbl complexes are the closest structural neighbors to GSCbl. Superposition of these two structures onto GSCbl (based upon the 91 atoms in the Cbl molecule only) give a root-mean-square deviation (rmsd) in atomic positions of 0.112 Å and 0.096 Å for γ-GluCysCbl and N-AcCysCbl respectively. The similarity of the fold of the corrin rings in these three complexes is immediately obvious, particularly in GSCbl and γ-GluCysCbl which have almost identical fold angles. Not surprisingly, the positions of the atoms in the cysteinyl moiety overlay almost exactly in these three complexes (Figure 4), with the cysteinyl α-carboxylate groups in γ-GluCysCbl and N-AcCysCbl overlapping almost exactly with the major conformation of the Cys-Glu amide bond in glutathione. Calculating the rmsd based upon only the atoms which are equivalent between N-acetylcysteine (the smallest of the three ligands and the one with least disorder), γ-glutamyl-cysteine and glutathione, gives values between 0.093 and 0.147 Å. This is perhaps not surprisingly since this central piece of the three ligands is linked directly to the Cbl molecule via the Co-S bond, and is held more rigidly than the extremities of the ligands, which are clearly prone to disorder. The γ-glutamyl-cysteine and glutathione ligand deviate markedly in their respective conformations of the γ-glutamyl groups. As noted above, in GSCbl both α-carboxylate conformations fold back over the cysteinyl residue, whereas in γ-GluCysCbl both conformations of the α-carboxylate project into the solvent channels and make multiple hydrogen bonding interactions with water molecules.11
Figure 4.
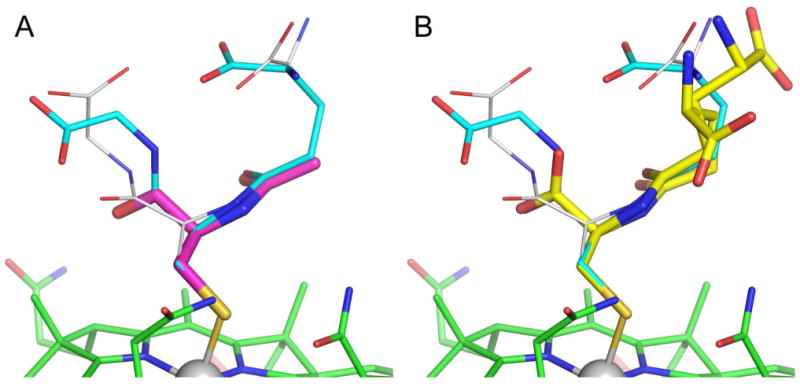
Superposition of the structures of GSCbl (cyan) with (A) N-AcCysCbl (magenta) and (B) γ-GluCysCbl (yellow).
In GSCbl there are two indirect water-mediated interactions between the glutathione and Cbl moiety which do not exist in γ-GluCysCbl. Firstly, water molecule O22 (which shares partial occupancy of a site with the cesium ion) is hydrogen bonded to the N80 atom of the glutathione (as noted above), and also makes a hydrogen bond to the either N29b atom of the a side chain of the corrin in its minor conformation, or to the water molecule which replaces the N29b atom when this amide group adopts the major conformation. Secondly, the partially occupied water molecule O12 interacts with O74a of glutathione and with the e side chain of the corrin, donating a hydrogen bond to O51.
Solution studies have suggested that in GSCbl, the glutamate α-amino group may be hydrogen bonded to the f side chain carbonyl oxygen (O58).8 However, no intramolecular hydrogen-bonding interactions were seen in the γ-GluCys-Cbl structure.11 In the GSCbl structure there is also no possibility of an interaction between the α-amino nitrogen and the f side chain since these atoms are between 11 and 11.5 Å apart. However this could be simply an artifact of the crystallization. If the γ-glutamyl moiety were able to extend down the side of the corrin ring rather than away from it as seen in the γ-GluCysCbl and GSCbl crystal structures, it is conceivable that an interaction could occur. This would require an approximately 20° rotation of the entire glutathione ligand about the axis the through the Co-S bond and concerted changes in the C72-N80, C81-C83 and C83-C84 and C84-C85 torsion angles but it is possible to bring the α-amino nitrogen and O58 to within approximately 3.5 Å of each other. The rotation about the Co-S bond is troublesome, in that the GS− ligand currently sits in the most energetically favored position on the β face of the corrin and presumably this orientation would be equally as favored in solution.
In conclusion, the X-ray crystal structure of the elusive GSCbl was obtained at atomic resolution and refined to a low crystallographic R-factor (0.0683). The glutathione moiety is bound to the cobalt center through the sulfur atom as expected, with Co-S and Co-N3B bond distances of 2.295(1) Å and 2.074(3) Å, respectively. The corrin fold angle is 24.7°, the highest ever reported for a Cbl structure, and points in the direction of the β-face of the corrin, towards the GS moiety. The GS− ligand was modeled in two alternate conformations, which resulted from rotation of approximately 30° and 110° about the C83-C84 and C84-C85 bonds, respectively. In both conformations, the α-carboxylate group of GS− ligand interacts with the generally rigid side chain a of the Cbl molecule in a symmetry-related molecule. A partially-occupied cesium ion, derived from the crystallization medium, bridges between the GS and the minor conformation of the a side chain. Despite substantial differences found in the pattern and number of hydrogen bonding interactions, GSCbl displayed high structural similarity with other thiolatocobalamins, primarily in the coordination sphere of the cobalt, the cysteinyl moiety and the large fold of the corrin ring.
Supplementary Material
Acknowledgments
This research was funded by the National Heart, Lung and Blood Institute of the National Institutes of Health (HL52234 and HL71907 to DWJ). SSRL is a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy (BES, BER) and by the National Institutes of Health (NCRR, BTP, NIGMS).
Footnotes
There was an error in the corrin fold angle of NaCysCbl (17.4°) reported in Ref 3; the correct value is 22 °.
References
- 1.Brown KL. Chem Rev. 2005;105:2075–2149. doi: 10.1021/cr030720z. [DOI] [PubMed] [Google Scholar]
- 2.Randaccio L, Geremia S, Nardin G, Wuerges J. Coord Chem Rev. 2006;250:1332–1350. [Google Scholar]
- 3.Suarez-Moreira E, Hannibal L, Smith CA, Chavez RA, Jacobsen DW, Brasch NE. Dalton Trans. 2006:5269–5277. doi: 10.1039/b610158e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garau G, Fedosov SN, Petersen TE, Geremia S, Randaccio L. Acta Crystallogr D Biol Crystallogr. 2001;57:1890–1892. doi: 10.1107/s0907444901015360. [DOI] [PubMed] [Google Scholar]
- 5.Ludwig ML, Evans PR. Chemistry and Biochemistry of B12. Wiley-IEEE; New York: 1999. X-ray crystallography of B12 enzymes: methylmalonyl-CoA mutase and methionine synthase; p. 921. [Google Scholar]
- 6.Wagner F, Bernhauer K. Ann N Y Acad Sci. 1964;112:580–589. doi: 10.1111/j.1749-6632.1964.tb45034.x. [DOI] [PubMed] [Google Scholar]
- 7.Pezacka E, Green R, Jacobsen DW. Biochem Biophys Res Commun. 1990;169:443–450. doi: 10.1016/0006-291x(90)90351-m. [DOI] [PubMed] [Google Scholar]
- 8.Brown KL, Zou X, Savon SR, Jacobsen DW. Biochemistry. 1993;32:8421–8428. doi: 10.1021/bi00084a006. [DOI] [PubMed] [Google Scholar]
- 9.Scheuring EM, Sagi I, Chance MR. Biochemistry. 1994;33:6310–6315. doi: 10.1021/bi00186a034. [DOI] [PubMed] [Google Scholar]
- 10.Brasch NE, Hsu TL, Doll KM, Finke RG. J Inorg Biochem. 1999;76:197–209. doi: 10.1016/s0162-0134(99)00128-2. [DOI] [PubMed] [Google Scholar]
- 11.Suto RK, Brasch NE, Anderson OP, Finke RG. Inorg Chem. 2001;40:2686–2692. doi: 10.1021/ic001365n. [DOI] [PubMed] [Google Scholar]
- 12.Zheng D, Birke RL. J Am Chem Soc. 2002;124:9066–9067. doi: 10.1021/ja017684a. [DOI] [PubMed] [Google Scholar]
- 13.Xia L, Cregan AG, Berben LA, Brasch NE. Inorg Chem. 2004;43:6848–6857. doi: 10.1021/ic040022c. [DOI] [PubMed] [Google Scholar]
- 14.Chu RC, Begley JA, Colligan PD, Hall CA. Metabolism. 1993;42:315–319. doi: 10.1016/0026-0495(93)90080-8. [DOI] [PubMed] [Google Scholar]
- 15.Pezacka EH, Denison CJ, Green R, Jacobsen DW. J Cell Biol. 1988;107:860a. [Google Scholar]
- 16.Pezacka EH, Green R, Jacobsen DW. FASEB J. 1990;4:A2126. [Google Scholar]
- 17.Jacobsen DW, Pezacka EH, Brown KL. J Inorg Biochem. 1993;50:47–63. doi: 10.1016/0162-0134(93)80013-y. [DOI] [PubMed] [Google Scholar]
- 18.Pezacka EH. Biochim Biophys Acta. 1993;1157:167–177. doi: 10.1016/0304-4165(93)90061-c. [DOI] [PubMed] [Google Scholar]
- 19.Kim J, Gherasim C, Banerjee R. Proc Natl Acad Sci U S A. 2008;105:14551–14554. doi: 10.1073/pnas.0805989105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hannibal L, Kim J, Brasch NE, Wang S, Rosenblatt DS, Banerjee R, Jacobsen DW. Mol Genet Metab. 2009;97:260–266. doi: 10.1016/j.ymgme.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J, Hannibal L, Gherasim C, Jacobsen DW, Banerjee R. J Biol Chem. 2009;284:33418–33424. doi: 10.1074/jbc.M109.057877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Froese DS, Zhang J, Healy S, Gravel RA. Mol Genet Metab. 2009;98:338–343. doi: 10.1016/j.ymgme.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 23.Lerner-Ellis JP, Tirone JC, Pawelek PD, Dore C, Atkinson JL, Watkins D, Morel CF, Fujiwara TM, Moras E, Hosack AR, Dunbar GV, Antonicka H, Forgetta V, Dobson CM, Leclerc D, Gravel RA, Shoubridge EA, Coulton JW, Lepage P, Rommens JM, Morgan K, Rosenblatt DS. Nat Genet. 2006;38:93–100. doi: 10.1038/ng1683. [DOI] [PubMed] [Google Scholar]
- 24.Morel CF, Lerner-Ellis JP, Rosenblatt DS. Mol Genet Metab. 2006;88:315–321. doi: 10.1016/j.ymgme.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Lerner-Ellis JP, Anastasio N, Liu J, Coelho D, Suormala T, Stucki M, Loewy AD, Gurd S, Grundberg E, Morel CF, Watkins D, Baumgartner MR, Pastinen T, Rosenblatt DS, Fowler B. Hum Mutat. 2009;30:1072–1081. doi: 10.1002/humu.21001. [DOI] [PubMed] [Google Scholar]
- 26.Hannibal L, Axhemi A, Glushchenko AV, Moreira ES, Brasch NE, Jacobsen DW. Clin Chem Lab Med. 2008;46:1739–1746. doi: 10.1515/CCLM.2008.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lenhert PG, Hodgkin DC. Nature. 1961;192:937–938. doi: 10.1038/192937a0. [DOI] [PubMed] [Google Scholar]
- 28.Hsu TLC, Brasch NE, Finke RG. Inorg Chem. 1998;37:5109–5116. [Google Scholar]
- 29.Kabsch W. J Appl Cryst. 1993;2:795–800. [Google Scholar]
- 30.Sheldrick GM, Schneider TR. Method Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 31.Garau G, Geremia S, Marzilli LG, Nardin G, Randaccio L, Tauzher G. Acta Crystallogr B. 2003;59:51–59. doi: 10.1107/s0108768102019353. [DOI] [PubMed] [Google Scholar]
- 32.Randaccio L, Furlan M, Geremia S, Slouf M, Srnova I, Toffoli D. Inorg Chem. 2000;39:3403–3413. doi: 10.1021/ic0001199. [DOI] [PubMed] [Google Scholar]
- 33.Pezacka EH, Rosenblatt DS. Intracellular metabolism of cobalamin. Altered activities of β-axial-ligand transferase and microsomal cob(III)alamin reductase in cbl C and cbl D fibroblasts. In: Bhatt, James, Besser, Bottazzo, Keen, editors. Advances in Thomas Addison's Diseases. J Endocrinol Ltd.; Bristol: 1994. pp. 315–323. [Google Scholar]
- 34.Watson WP, Munter T, Golding BT. Chem Res Toxicol. 2004;17:1562–1567. doi: 10.1021/tx0497898. [DOI] [PubMed] [Google Scholar]
- 35.McCaddon A, Regland B, Hudson P, Davies G. Neurology. 2002;58:1395–1399. doi: 10.1212/wnl.58.9.1395. [DOI] [PubMed] [Google Scholar]
- 36.Birch CS, Brasch NE, McCaddon A, Williams JH. Free Radic Biol Med. 2009;47:184–188. doi: 10.1016/j.freeradbiomed.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 37.Randaccio L, Geremia S, Stener M, Toffoli D, Zangrando E. Eur J Inorg Chem. 2002:93–103. [Google Scholar]
- 38.Mukherjee R, McCaddon A, Smith CA, Brasch NE. Inorg Chem. 2009;48:9526–9534. doi: 10.1021/ic900891y. [DOI] [PubMed] [Google Scholar]
- 39.Randaccio L, Geremia S, Nardin G, Shlouf MS, Srnova i. Inorg Chem. 1999;38:4087–4092. [Google Scholar]
- 40.Zou X, Brown KL. Inorg Chim Acta. 1998;267:305–308. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.