

Abstract
Purpose of review
This review summarizes recent research implicating Forkhead box (Fox)O1, a key transcription factor in glucose metabolism, in the regulation of hepatic lipid metabolism. Insulin dysregulation leading to hypertriglyceridemia is associated with increased hepatic VLDL secretion. FoxO1 is integrated in action with other regulatory factors in VLDL metabolism. The role of FoxO1 is defined in context of recent controversies.
Recent findings
FoxO1 regulates transcription of microsomal triglyceride transfer protein and apolipoprotein (apo)CIII involved in hepatic assembly and postsecretory catabolism of VLDL. Insulin activation of Akt leads to the phosphorylation of FoxO1 with nuclear exclusion and loss of transcriptional activity. Reduced insulin action increases FoxO1 activity and induces microsomal triglyceride transfer protein favoring VLDL assembly and induces apoCIII reducing peripheral triglyceride catabolism. This new mechanistic link between insulin resistance and VLDL overproduction and hypertriglyceridemia compounds effects of other known VLDL regulatory factors.
Summary
This review highlights recent advances in research of insulin regulation of hepatic VLDL metabolism. Formation of VLDL requires lipid, apoB structural protein, and microsomal triglyceride transfer protein. FoxO1 is a major factor in hepatic microsomal triglyceride ransfer protein regulation. A unifying hypothesis is presented linking regulation of the three necessary hepatic components for VLDL assembly with insulin action and insulin resistance.
Keywords: Akt, apoB, forkhead transcription factor, FoxO, lipoproteins, microsomal triglyceride transfer protein, very low-density lipoprotein
Introduction
Research relating Forkhead box (Fox)O1 to hepatic energy management has recently uncovered relationships between glucose and lipid metabolism regulated by insulin-dependent FoxO1 pathways. FoxO1 transcriptionally activates microsomal triglyceride transfer protein (MTP) and apolipoprotein (apo)CIII genes facilitating hepatic VLDL assembly and inhibiting postsecretory catabolism. Enhanced FoxO1 action parallels insulin resistance, which increases FoxO1 nuclear localization through reduced Akt phosphorylation of FoxO1. Considering similar actions in glucose metabolism, FoxO1 activity during insulin resistance favors characteristics of metabolic syndrome, which include both hypertriglyceridemia and hyperglycemia.
Overview
FoxO1 is a member of the FoxO family of proteins that are shown to play a role in adaptation to fasting by promoting the expression of genes involved in gluconeogenesis [1]. Insulin regulation of FoxO1 has gained importance with the understanding of the Akt pathway and the importance of insulin-dependent FoxO1 phosphorylation and subcellular distribution and its role in regulating hepatic glucose production. Resistance in the pathway favors FoxO1 nuclear localization and unbridled activation [2,3]. Recent interest in FoxO1 has focused on its role in hepatic lipid and lipoprotein pathways with the potential for FoxO1 to be the central link for regulation of and coordination of insulin action on carbohydrate and lipid metabolism. A parallel process of nuclear exclusion of FoxO1 eliminates activity on the MTP promoter reducing MTP protein and limiting VLDL assembly. In addition, lipogenesis is modulated by FoxO1 through sterol regulatory element binding protein 1c (SREBP-1c). FoxO1 metabolism thus affects two out of three major factors modulating VLDL assembly and hepatic lipid secretion. The third factor potentially rate-limiting in lipid export of hepatic VLDL is apoB. It is not known if there is a role for FoxO1 in regulating apoB production/availability. However, several publications suggest the potential for in-vivo transcriptional regulation of the apob gene [4], and it has been shown that both FoxO1 [5••] and FoxA2 [6] affect apob promoter activity in vitro. FoxO1 is a key regulator in the adaption of the liver to fasting and postprandial states with a net effect of balancing energy substrates between fasting and feeding. Teleologically, this avoids competition between intestinal and hepatic pathways in metabolic shifts created by feeding. A second type of balance occurs in shifts of these energy transitions modulated by resistance to insulin action. This resistance occurs with the metabolic syndrome and other insulin-resistance conditions affecting human diseases including diabetes and polycystic ovary disease. Modulation of insulin action could initially be considered adaptive in the sense of favoring energy export to peripheral tissues and storage of fatty acid (FA) as triglyceride by adipocytes with energy excess. At what point adaptation to energy excess becomes pathologic is not clear, and factors involved in developing diseases are under intense investigation.
Hypertriglyceridemia and hepatic overproduction of VLDL
Hypertriglyceridemia is a key feature of metabolic syndrome and is caused primarily by hepatic overproduction of VLDL by the liver with insulin resistance. Persistent hypertriglyceridemia is the proximal cause of disturbances in metabolism of plasma lipoproteins resulting in abnormal remodeling of lipoproteins leading to their increased atherogenicity. Hepatic VLDL assembly and secretion is a complex process involving many gene products including apoB, MTP, and lipid mobilizing and synthesizing proteins. Each of these components can become rate-limiting in VLDL synthesis. As hepatic apoB-100 (full-length apoB) derived from VLDL becomes the sole protein component of LDL, and because LDL and oxidized LDL are highly atherogenic, understanding how apoB and lipid are assembled into VLDL and enter the circulation, and how this process is regulated are important. FoxO1 has recently been shown to transcriptionally regulate the expression of MTP, one of the three essential components necessary in the VLDL assembly process. Increased MTP activity leads to hypersecretion of VLDL associated with insulin resistance [5••]. This surprising finding has provided a mechanistic link between insulin action/inaction, and development of hypertriglyceridemia.
FoxO1 is a nuclear transcription factor
FoxO1 is a nuclear transcription factor that is a family member of mammalian forkhead genes assigned to the O class. FoxO proteins are homologous to the transcription factor abnormal DAuer Formation-16 (DAF-16) in Caenorhabditis elgans that has been found to be a determinant in insulin signaling and extended life span [7]. Excellent reviews on FoxO1 have been presented [2,3,8•]. FoxO family members have a characteristic highly conserved DNA-binding motif termed ‘forkhead’ and include FoxO1 (FKHR), FoxO3a (FKHRL1), FoxO4 (AFX), and FoxO6 in mammals. The conserved forkhead domain is termed ‘forkhead box’ or ‘winged helix’ due to its butterfly appearance on X-ray crystallography. FoxO1 proteins bind DNA to the FoxO-recognized element T/C-G/A-A-A-A-C-A-A [9]. The precise conditions that lead to DNA binding are not fully understood. This element is a highly conserved insulin response element (IRE) that is found within the promoters of insulin-dependent genes including glucose 6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK), and insulin-like growth factor-binding protein 1 (IGFBP1). FoxO1 has two structural domains. The amino terminal forkhead domain is responsible for DNA binding, whereas the C-terminal transactivation domain is responsible for stimulating promoter activity. The C-terminal region controls the shuttling of FoxO1 into the cell nucleus [10]. FoxO1 as well as others in the family (FoxO3a, FoxO4, and FoxO6) are substrates of Akt/PKB and SGK kinases and play key roles in insulin action [3]. FoxO1 is inactivated by phosphorylation by Akt and its activity is regulated by acetylation/deacetylation and glucosylation [11–15,16•]. Phosphorylated FoxO1 is ubiquitinated and degraded by the proteasome in response to insulin-dependent Akt phosphorylation [8•].
Regulation of FoxO1 transcriptional activation
The function of FoxO1 depends on the complement of transcription factors expressed in a particular cell [17•]. In response to insulin, activated Akt phosphorylates FoxO1 and results in cytoplasmic retention, which limits FoxO1 access to the nucleus and thereby inhibits FoxO1 transcriptional activity [18] (Fig. 1). For translocation out of the nucleus, FoxO1 as well as other FoxO proteins require phosphorylation of an N-terminal Akt site (T24) and a site present in the forkhead domain (S253) with phosphorylation influencing the function of the nuclear localization sequence [19] and association with 14-3-3 protein. Phosphorylation of the N-terminal site also is important for the disruption of the association of FoxO1 with coactivator p300. Without insulin stimulation, FoxO1 is dephosphorylated and translocates into the nucleus in which it is transcriptionally active.
Figure 1. Insulin signaling via PI3K to nuclear and lipoprotein assembly compartments in hepatocytes.
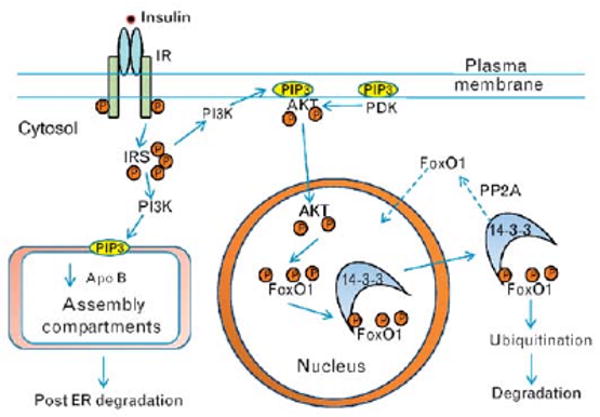
Glucose-stimulated insulin secretion results in high portal insulin that binds to hepatic insulin receptors and activates the tyrosine kinase of the β-subunit leading to autophosphorylation of the receptor and amplification of the tyrosine kinase activity. Activated insulin receptor results in multiple tyrosine phosphorylation of IRS which mediate the activation of PI3K in plasma membranes and in endoplasmic reticulum membranes, both sites containing substrates. PI3K activity results in the production of patches of phosphatidyinositide 3,4,5 tris phosphate (PIP3). In the endoplasmic reticulum, PIP3 interacts with the machinery involved in the assembly of TRLs leading to decreased apoB both by reducing apoB synthesis and by enhancing post-endoplasmic reticulum, presecretory proteolysis, and degradation within lysosomes. In the plasma membrane, PIP3 patches attract the serine/threonine kinase Akt (protein kinase B) through binding to its pleckstrin homology domain bringing Akt in proximity to phosphoinositide-dependent kinase (PDK) which lead to double phosphorylation of Akt (T308, S473) and Akt activation. Activated Akt in turn phosphorylates FoxO1 (Thr-24, Ser-256, Ser-319) with the net result of exclusion of FoxO1 from the nucleus and inactivation of FoxO1 transcriptional activity. Phosphorylated FoxO1 interacts with 14-3-3 protein which helps remove phosphorylated FoxO1 from the nucleus, and leads to FoxO1 ubiquitination and ultimately FoxO1 degradation through the 26S proteasome. IRS, insulin receptor substrates; PDK, phosphoinositide-dependent kinase; PI3K, phosphoinositide 3-kinase; TRL, triglyceride-rich lipoprotein.
Non-DNA binding functions of FoxO1
FoxO1 is involved in the regulation of a subset of genes through interactions with other transcription factors [17•]. Ramaswamy et al. [20] showed that when the DNA-binding function of FoxO is mutated, FoxO remained able to effectively regulate a certain subset of specific genes. There are a number of FoxO transcription factor binding partners including many involved in regulation of hepatic lipid metabolism including C/EBPs, peroxisome proliferators-activated receptor (PPAR)α, PPARγ, RAR, Smad, thyroid hormone receptor (TR), and PPAR gamma coactivator-1 (PGC1)α [17•,21–23]. PPARα inhibits FoxO1 transcriptional activation by decreasing DNA-binding capacity [24••]. Conversely, PPARα is inhibited by FoxO1 independently of ligand binding and inhibits the downstream target apoCIII, an important apolipoprotein regulating lipolysis [24••]. Recent studies of the IGFBP1 promoter support an additional function for FoxO1. FoxO1 binding to DNA opens up chromatin structure to potentiate and modulate active chromatin, which facilitates binding of additional transcription factors and preinitiation complexes [25].
Components necessary for VLDL secretion
There are numerous gene products that are involved in the synthesis and assembly of VLDL including those necessary to synthesize the phospholipid coating on all lipoproteins (Table 1). Three requisite components for VLDL assembly discussed here are apoB, MTP, and triglyceride.
Table 1. Elements involved in hepatic lipid metabolism.
| Pathway | Regulation | Postprandial | Insulin resistant |
|---|---|---|---|
| Hepatic VLDL metabolism | |||
| Insulin receptor (IR) | Insulin signal transmission | ↑ IR pY | ↓ IR pY |
| IR → IRS | Cytoplasmic signal transduction | ↑ IRS pY | ↓ IRS pY |
| IRS → PI3K | Activation of PI3K | ↑ PIP3 | ↓ PIP3 |
| PIP3 | ER signal | ↑apoB degraded | ↓ apoB degraded |
| PDKs → pAkt | Activation of Akt | ↑pT308/S473 | ↓T308/S473 |
| pAkt → pFoxO1 | Nuclear export and 14-3-3 binding | ↑pT32, pS253 | ↓phosphorylation Nuclear localization |
| IR → FoxO1-mediated pathway | |||
| ApoCIII | Regulator of lipolysis | ↓ (?) | ↑ |
| MTP | ApoB chaperone and lipid transfer | ↓ (?) | ↑ |
| ApoB | Structural VLDL protein | ↓ (?) | ↑ (?) |
| SREBP-1c | Master regulator of lipogenic genes | ↑ | ↑ |
| Other factors involved in VLDL metabolism | |||
| DGAT1 | Acylation of MG and DG | ? | ? |
| DGAT2 | Acylation of MG and DG | ? | ? |
| ATGL | TG lipolysis | ? | ? |
| ABHD5/CGI-58 | Cofactor for ATGL | ? | ? |
| Acylation of lysophosphatidic acid | |||
ABHD5/CGI-58, abhydrolase domain containing 5 also comparative genome identification 58; Akt, a retroviral oncogene encoding a serine-threonine kinase, 14-3-3 protein; apoCIII, apolipoprotein CIII; ATGL, adipose triglyceride lipase; DG, diacylglyceride; DGAT, diacylglyceride acyltransferase; ER, endoplasmic reticulum; IR, insulin receptor; IRS, insulin receptor substrate; MG, monoacylglyceride; MTP, microsomal triglyceride transfer protein; PDKs, phosphoinositide dependent kinases; PI3K, phosphatidylinositide 3-kinase; PIP3, phosphatidylinositide 3,4,5 tris-phosphate; pS, phosphoserine; pT, phosphothreonine; pY, tyrosine phosphorylation; SREBP-1C, sterol regulatory element binding protein 1C.
Apolipoprotein B
Apolipoprotein B is the integral protein component of VLDL and provides the scaffolding necessary to stabilize a triglyceride droplet within the secretory compartment and to allow the assembly of secretion-competent VLDL particles. Two forms of apoB are synthesized in mammals: apoB-100 and apoB-48. ApoB-100 represents 100% of the protein encoded by the gene, whereas apoB-48 is the N-terminal 48%. The two forms of apoB are synthesized from a 14.1 kb mRNA, and in humans the form synthesized is organ-specific. Human liver synthesizes apoB-100, the longer form, and human intestine synthesizes apoB-48, the shorter form. Differences in apoB-100 or apoB-48 synthesis are mainly based on the expression of a RNA-editing enzyme, apoB mRNA-editing catalytic component 1 (apobec-1) [26]. This enzyme, through localization mediated by apobec-1 complementation factor, introduces a nongenomic, in-frame, translational stop codon within the apoB mRNA sequence by converting a single C to U at nucleotide 6666. This stop codon results in the synthesis of a shortened protein that is roughly 48% of the N-terminal portion of apoB-100. Differences in cellular stability of apoB-100 vs. apoB-48 allow the intestine and rodent liver, which express apobec-1 and therefore synthesize apoB-48, to store preformed nascent apoB-48 for rapid formation of triglyceride-rich lipoproteins (TRL) [27•].
The secretion of apoB-lipoproteins by liver depends mainly on the level of cotranslational and posttranslational apoB degradation, which, in turn, depends upon the availability of lipids and insulin action. ApoB can undergo three forms of degradation including endoplasmic reticulum-associated degradation (ERAD) via the proteosome, post-endoplasmic reticulum presecretory proteolysis (PERPP) and postsecretory reuptake [28]. The role of apoB degradation in regulation of VLDL production has recently been reviewed [29•]. Under control situations, ERAD has a minimal role in the regulation of apoB secretion [27•]. However, under certain circumstances, increased delivery of oleic acid to hepatocytes can result in endoplasmic reticulum stress and decreased secretion of apoB [30] due to unfolded protein response [31•]. PERPP is a nonproteasomal, post-endoplasmic reticulum degradation pathway for apoB that occurs even when triglyceride is readily available. PERPP is responsible for the degradation of apoB mediated by n-3 fatty acids [32••], and is likely to be responsible for that mediated by insulin [33•]. PERPP is inhibited by wortmannin, a phosphatidylinositide 3-kinase (PI3K) inhibitor, suggesting the importance of activated PI3K in apoB degradation by this process [28]. PERPP of apoB occurs following aggregation of VLDL apoB leading to autophagosome formation and degradation via lysosomes and is responsible for the reduced secretion of apoB observed with n-3 fatty acid incubations [32••]. Loss of insulin signaling in livers of mice lacking hepatic insulin receptors results in increased secretion of VLDL apoB [34•], and similar increased lipoprotein secretion is observed in mice treated intravenously with wortmannin [35].
Microsomal triglyceride transfer protein
Microsomal triglyceride transfer protein is an 88 kDa protein found within the endoplasmic reticulum and Golgi compartment that is necessary for the proper assembly of VLDL and its importance in lipid metabolism has recently been reviewed [36•]. MTP has three main activities: lipid/phospholipid transfer activity, apoB chaperone activity, and membrane association. MTP activity requires protein disulfide isomerase (PDI), a subunit of the active enzyme. The requirement for MTP in VLDL assembly is exemplified in humans and animal models with genetic deficiency of MTP resulting in reduced ability to synthesize and secrete TRL leading to increased hepatic lipid deposition and associated disease [37,38].
Triglyceride
In addition to apoB and MTP, lipid availability is an important factor in efficiency of formation of apoB-lipoproteins. Reduced lipid availability results in targeting of apoB for degradation and decreased secretion [39]. Dietary FAs or FAs released from adipose tissue enter liver, and are re-esterifed by DGAT1 forming neutral lipid (triglyceride) droplets (Fig. 2). Most triglycerides are not incorporated en bloc into lipoproteins, but undergo a cycle of lipolysis and re-esterification [40] involving cellular phospholipid intermediates [41]. Entry of cytoplasmic triglyceride into the secretory pathway involves lipolysis of lipid droplets and re-esterification by DGAT2 near the sites of VLDL assembly creating nucleation sites that facilitate triglyceride transfer to apoB by MTP [36•]. In many regards, lipid droplets present in the cytosol of hepatocytes are similar structurally to plasma lipoproteins. Both are phospholipid monolayers with surface proteins that contain or attain appropriate cofactors necessary for their stability and catabolism. Approximately 2/3 of the triglyceride secreted as VLDL triglyceride can be derived from lipolysis/re-esterification.
Figure 2. Assembly of hepatic apoB-containing lipoproteins.
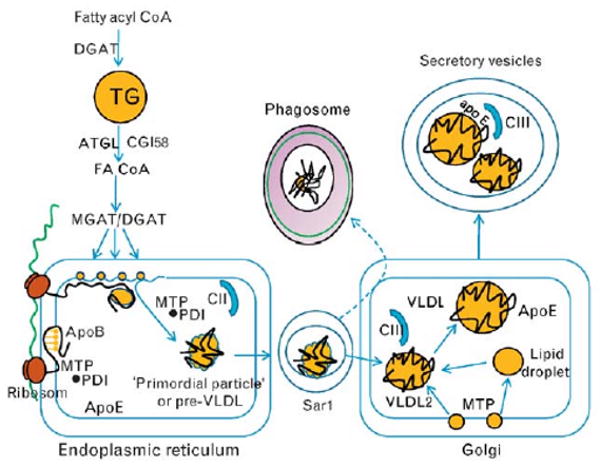
Triglycerides provide lipid precursors for the assembly of apoB-containing lipoproteins. FAs enter the liver and are first converted to fatty acyl CoA derivatives before entering triglyceride droplets for storage through the action of DGAT. Droplets are not assembled into lipoproteins en bloc, but undergo lipolysis and released FAs are re-esterified into triglyceride nucleation sites near the site of lipoprotein assembly in the endoplasmic reticulum. Lipolysis of lipid droplets occurs mainly through ATGL along with a coactivator, abhydrolase domain containing 5 (ABHD5, CGI58, comparative genome identification 58) which releases FAs that are re-esterified by monoacylglyceride and diacylglyceride acyl transferase (MGAT/DGAT) into nucleation sites within the endoplasmic reticulum membrane. The biogenesis of VLDL is initiated by the translation of apoB mRNA on ribosomes docked to the endoplasmic reticulum followed by translocation of the nascent apoB chain through the translocation channel. The first 20% of the N-terminal apoB peptide contains the lipoprotein-initiating domain which interacts with neutral lipid moieties within the inner leaflet. Thereafter, the orderly and sequential appearance of apoB peptide dictates subsequent interactions with MTP and its small MTP subunit, PDI. MTP acts as a chaperone for the N-terminal as lipoprotein formation proceeds, whereas shuttling neutral lipids (triglyceride and cholesteryl esters) and phospholipids from nucleation sites as the core expands into a primordial particle or pre-VLDL. Final phospholipid transfer to the particle by MTP releases through desorption the primordial particle as a secretion-competent nascent lipoprotein. Movement of lipoprotein particles from the endoplasmic reticulum into the secretory pathway involves Sar1-mediated liberation of COPII vesicles. The final maturation of VLDL takes place in the Golgi in which VLDL acquires additional triglyceride possibly through the action of MTP as well as lipoprotein surface components, apoE and apoCIII. ApoCIII blocks lipolytic action of lipoprotein lipase on VLDL triglyceride following VLDL secretion inhibiting the production of cholesteryl ester-rich LDL particles containing only apoB-100 as a core protein. ATGL, adipose triglyceride lipase; FAs, fatty acids; MTP, microsomal triglyceride transfer protein; PDI, protein disulfide isomerase.
Abhydrolase domain containing 5 (ABHD5), also known as CGI-58 for comparative genome identification 58, is a protein that facilitates lipolysis of triglyceride. Mutations in the ABHD5 gene cause Chanarin–Dorfman syndrome, which results in the accumulation of triglyceride in a number of tissues including liver. It has been suggested that ABHD5 is involved in the lipolysis of stored triglyceride destined for lipoprotein assembly. Although ABHD5 lacks triglyceride hydrolase activity [42], in-vitro studies indicate that it enhances the activity of adipose triglyceride lipase (ATGL), which is essential for mobilizing stored triglyceride into FA oxidation [43•]. Studies suggest that ABHD5 acts as a cofactor for ATGL and contributes to mobilization of triglyceride for lipoprotein assembly with apoB. ABHD5 deficiency in McArdle RH7777 cells results in reduced secretion of apoB, VLDL triglyceride, and cholesteryl esters supporting a role for ABHD5 in VLDL assembly [44•]. ABHD5 is a lysophosphatidic acid acyltransferase [45] suggesting its potential involvement in the generation of phospholipid intermediates during lipolysis/re-esterification and assembly into VLDL triglyceride.
Hepatic VLDL assembly and secretion
The liver exports triglyceride through the formation of an aqueous compatible macromolecular complex with apoB through a process that involves two steps (Fig. 2). The first step in the biogenesis of VLDL is initiated by the translation of apoB on ribosomes docked to the endoplasmic reticulum followed by translocation of the elongating protein into the endoplasmic reticulum lumen. Translation and subsequent translocation of the first 20% of the N-terminal ‘lipoprotein initiating domain region’ of apoB [46,47•] is followed by the sequential appearance and the orderly folding of the peptide while interacting with membranes and incorporating phospholipids and neutral lipids at triglyceride nucleation sites. The recruitment of phospholipid may be intrinsic to the N-terminus of apoB and may allow the protein to remodel the endoplasmic reticulum membrane into discrete protein lipid complexes. MTP acts as both a chaperone and as a lipid and phospholipid transfer protein directly interacting with the N-terminus of apoB [36•]. Continued apoB translation and translocation along with MTP activity produces a ‘primordial’ pre-VLDL particle containing a single molecule of apoB. The phospholipid transfer activity of MTP completes the surface coating and allows desorption from the membrane rendering the particle competent for secretion [36•]. The second step of assembly is necessary to form a mature ‘nascent’ VLDL particle and is not completely understood. This involves additional phospholipid remodeling, core expansion, and may involve fusion with preformed lipid droplets. The role of MTP in this second step is uncertain as is the site of final maturation. Approximately half of total VLDL triglyceride is added after pre-VLDL exits the endoplasmic reticulum and about 30–40% of VLDL phospholipid suggesting that lipid addition may occur in the Golgi possibly mediated by MTP [48]. However, there is evidence that selective inhibition of MTP activity does not affect apoB-100 secretion during the later stages of lipoprotein assembly [39]. This warrants further investigation. It is clear that MTP activity can be rate-limiting in VLDL formation as MTP overexpression increases and MTP deficiency decreases hepatic VLDL formation and secretion.
Insulin regulates the hepatic production of apoB-containing lipoproteins
Regulation of VLDL metabolism not only involves the availability of rate-limiting factors apoB and MTP and lipid, but also involves direct insulin-mediated regulation. Glucose-stimulated insulin secretion results in high portal insulin levels. Insulin binding activates the tyrosine kinase of hepatic insulin receptors that lead to autophosphorylation of the receptor and rapid activation of the tyrosine kinase followed by phosphorylation of insulin receptor substrates (IRS). Phosphorylated residues bind and activate class I phosphatidylinositide 3-kinase (PI3K). Activated PI3K phosphorylates phosphatidylinositide 4, 5 bisphosphate and generates the highly negatively charged phospholipid, phosphatidylinositide 3, 4, 5 trisphosphate (PIP3) within plasma membranes (Fig. 1). The generation of PIP3 patches attracts the threonine serine kinase, Akt, to membranes by interaction of PIP3 with the Akt pleckstrin homology domain. Akt localization leads to its phosphorylation by phosphoinositide-dependent kinases resulting in phosphorylation of Akt at threonine 308. A second phosphorylation of Akt at serine 473 takes place by way of mTOR complex 2 which leads to full activation of Akt [18]. The action of PI3K is antagonized by the PIP3 phosphatase, Phosphatase and tensin homologue deleted on chromosome 10 (PTEN). Our studies suggest that in hepatocytes, PI3K also translocates to intracellular membranes in which PIP3 is able to interact with the machinery involved in VLDL assembly [49]. The consequence is the inhibition of apoB synthesis and increased degradation of newly synthesized apoB in a postsecretory compartment along with the transient accumulation of intracellular triglyceride.
Intravenous injection of wortmannin, a PI3-kinase inhibitor, into mice expressing only apoB-100 due to the genetic deletion of apobec-1, results in marked increases in hepatic VLDL triglyceride and apoB-100 production [35]. Compared with insulin-suppressive effects following glucose injection, the enhancement of VLDL production by wortmannin is significantly greater than would be expected by simple loss of the inhibitory effect of insulin. The level of hepatic VLDL production with wortmannin is increased to that observed in insulin-resistant mice fed high fructose diets. A hypothesis to explain this observation is that the incremental augmentation by wortmannin (over-shoot) above that which would be expected by loss of insulin action alone is the result of the combination of loss of insulin inhibition and stimulation of MTP expression mediated by FoxO1.
The precise mechanism by which insulin leads to the turnover of apoB-containing lipoproteins in a post-endoplasmic reticulum compartment has yet to be discovered [50,51•]. The product phospholipid of PI3K action, PIP3, exhibits unusual physical properties on the basis of its highly charged structure accounting for its activity in promoting localization and activation of factors involved in signaling. This same physical property might have unique functions in mechanisms involving lipoproteins and apoB in terms of alteration of protein structure and lipoprotein surface properties which could have significant impact on the VLDL assembly process in particular, lipid mobilization, lipid membrane transfer, and triglyceride and phospholipid transfers mediated by MTP.
In liver, insulin acts on FAs similarly to glucose, that is promoting storage of glucose as glycogen, and FAs as triglyceride during feeding. The diversion of energy substrates towards storage results in decreased hepatic VLDL secretion and decreased hepatic glucose release. Decreased VLDL during feeding also limits elevation of plasma triglyceride during the prandial period, when intestinal fat is absorbed and chylomicrons are produced. This pathway favors delivery of chylomicron FAs to adipose tissue. Insulin action allows excess glucose, after maximization of glycogen storage, to be converted into FA and triglyceride by activation of lipogenesis through induction of SREBP-1c.
FoxO1 and hepatic lipid metabolism
FoxO1 is an important transcription factor that plays a key role in regulating hepatic glucose production during fasting. Recent studies suggest that FoxO1 may coordinately regulate lipid metabolism through effects on VLDL.
FoxO1 and hepatic VLDL production
Recent studies using multiple model systems have linked increased hepatic nuclear FoxO1 activity with increased MTP expression and enhanced secretion of VLDL triglyceride and apoB [5••] and reviewed in [52••] (Fig. 3). As insulin acts to reduce nuclear FoxO1 activity following activation of Akt, these results support that in insulin-resistance states, the increased production of VLDL apoB-containing lipoproteins is related to unrestrained FoxO1-dependent MTP transcription. Five lines of evidence support this concept. First, MTP expression at both mRNA and protein levels are induced by FoxO1 and inhibited by insulin. Second, induction of MTP is associated with FoxO1 occupancy of the mttp promoter. Third, mutation of the FoxO1-binding site leads to loss of insulin-dependent regulation of MTP expression. Fourth, mice expressing a constitutively active FoxO1 transgene in liver have increased MTP expression, augmented hepatic VLDL production, and hypertriglyceridemia. Fifth, knock-down of hepatic FoxO1 using RNA interference suppresses hepatic MTP expression and reduces hepatic VLDL production. Liver-specific expression of active FoxO1 has not always produced consistent results, and findings of FoxO1 gain and loss of function models on hepatic lipid metabolism have recently been summarized [53•]. In one model, FoxO1 induced lipid transport genes and decreased expression of genes important for glycolysis and lipid/sterol synthesis resulting in decreased serum triglyceride levels during the postprandial period compared with control mice [54]. Differences are likely to be due to a number of factors including mouse strain differences (C57BL6 vs. FVB/N), level of overexpression or insufficiency of FoxO1, as well as dietary differences including experimental conditions (fed vs. fasting).
Figure 3. FoxO1 effects on hepatic lipid-related genes.
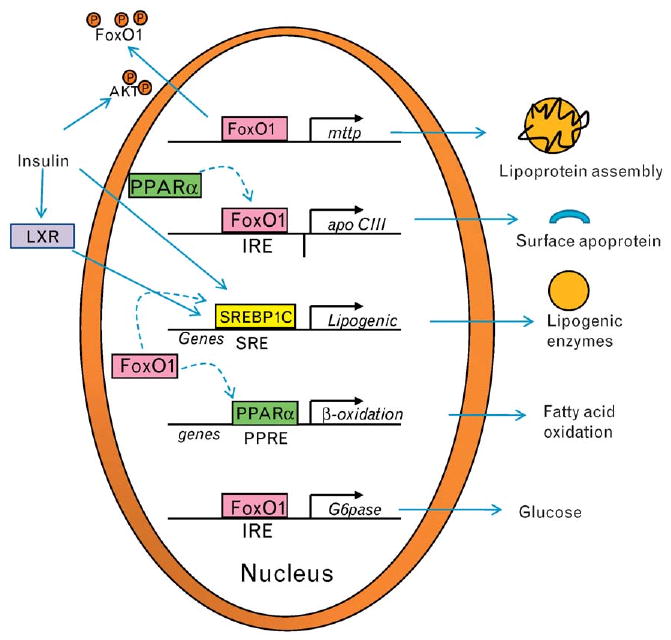
FoxO1 binds to promoter regions of the mttp and apoCIII genes leading to increased mRNA levels and protein expression. MTP activity enhances the assembly of apoB-containing lipoproteins. ApoCIII acts as an inhibitor of lipolysis once lipoproteins have reached the plasma compartment by preventing interaction of lipoprotein lipase with the neutral lipid core. FoxO1 also acts through transrepression (dotted arrows) blocking transcription mediated by a number of transcription factors including PPARα, PPARγ, and SREBP-1c. MTP, microsomal triglyceride transfer protein; PPAR, peroxisome proliferator-activated receptor; SREBP-1c, sterol regulatory element binding protein 1c.
Evidence has been presented that MTP is a direct transcriptional target of another forkhead transcription factor FoxA2, and with PGC1β may regulate apoB and VLDL section in hepatocytes in a similar fashion as FoxO1, as FoxA2 is also a target of insulin-activated Akt leading to phosphorylation and nuclear exclusion [6]. Whether the FoxA2 pathway is important in insulin-mediated inhibition of VLDL, apoB secretion is not known, however, as FoxA2 is predominantly localized in the cytoplasm under hyperinsulinemic conditions, the role of FoxA2 in the transcriptional upregulation of MTP in insulin resistant states is unclear. Net changes in MTP mRNA and protein have to be understood in the context of the relatively long half-life of the MTP protein of 4 d [55•]. Despite its long half-life, MTP mRNA and protein demonstrate diurnal variation, while food and light are able to regulate MTP expression and plasma lipid levels [55•]. These discrepancies warrant further investigation.
Insulin and apob gene expression
The apob gene is believed to be constitutively expressed as hepatic mRNA levels in vivo tend to be stable in most animal systems. However, older studies suggest that apoB mRNA abundance can be influenced by insulin in vivo. Hepatic apoB mRNA levels in livers derived from streptozotocin-induced hypoinsulinemic diabetes in rats are similar to control. However, following intensive insulin therapy to normalize blood glucose, hepatic apoB mRNA levels double [56]. These data suggest that in the context of insulin resistance produced by insulin injection, apoB mRNA levels are upregulated possibly by increased apob gene transcription. Recently, an increase in apob gene transcription has been shown definitively using nuclear run-on assays in vivo in livers derived from rats following induction of betaine homocysteine methyl transferase (BHMT) [4]. The increase in apoB mRNA levels observed with induction of BHMT was confirmed in vitro in HepG2 cells with BHMT overexpression [57].
As discussed above, hepatic apoB production is regulated mostly at the posttranslational level by lipid availability, which is counteracted by insulin. This model accounts for the acute inhibitory effect of insulin on hepatic VLDL-triglyceride secretion to limit postprandial plasma lipid excursion. Recently, it was demonstrated that hepatic apoB mRNA is stimulated by FoxO1 and inhibited by insulin in HepG2 cells [5••]. Furthermore, hepatic FoxO1 activity is augmented under fasting conditions and suppressed in response to refeeding [24••]. Likewise, hepatic apoB mRNA expression is induced in the liver with elevated FoxO1 production [5••]. These data suggest an additional mechanism by which the liver evolves to control hepatic apoB production by regulating the apoB mRNA pool at the transcriptional level. Consistent with this idea is the presence of a putative IRE motif within the 5′ proximal promoter region of apob genes in mice, rats, and humans (Fig. 4). Further studies are clearly warranted to validate that this conserved IRE motif is responsible for mediating FoxO1-dependent regulation of apoB mRNA expression.
Figure 4. Conservation of the putative IRE motif in the apob promoter.
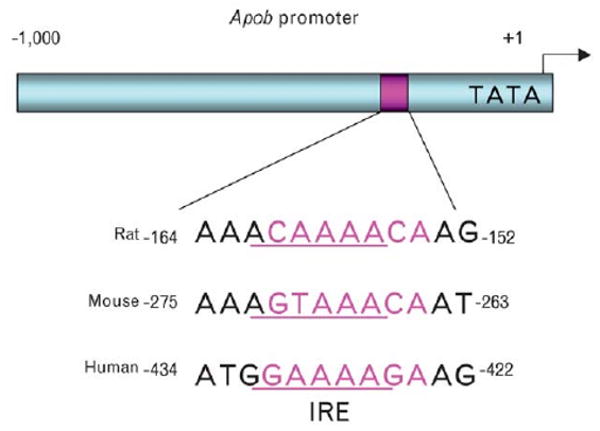
The promoter sequences of rat, mouse, and human apob genes were aligned for the comparison of IRE, the DNA motif that is responsible for binding FoxO1 and mediating the inhibitory effect of insulin on target gene expression. The putative IRE motif along with its nucleotide positions is indicated. IRE, insulin-responsive element.
Conclusion and future directions
VLDL are assembled in hepatocytes and are secreted from the liver in a pathway that is tightly regulated by insulin. Hepatic VLDL production is inhibited in response to increased insulin action. This effect acts to limit postprandial plasma triglyceride excursion. Conversely, with reduced insulin action, VLDL production is enhanced and increased circulating VLDL serves to transport lipids to peripheral tissues for energy. These reciprocal mechanisms are crucial for maintaining lipid homeostasis. Recent findings of FoxO1 as a key regulator of hepatic MTP provide an important insight into the mechanism of insulin-dependent regulation of VLDL secretion. In the absence of insulin action, FoxO1 resides in the nucleus and binds as a trans-activator to IRE, enhancing promoter activity. In response to insulin, FoxO1 is phosphorylated through the PI3K pathway, resulting in its nuclear exclusion and inhibition of target gene expression. This model helps explain why insulin resistance is so strongly associated with hepatic VLDL overproduction and hypertriglyceridemia. It follows that impaired ability of FoxO1 to undergo insulin-dependent protein trafficking can result in sustained nuclear localization and continued expression of MTP in hepatic insulin resistance contributing to the development of hypertriglyceridemia in patients with metabolic syndrome. Although FoxO1 is shown to stimulate hepatic apoB mRNA expression in vitro correlating with increased VLDL apoB secretion in cultured HepG2 cells, it remains to be confirmed whether the apob gene is a FoxO1 target. Likewise, a putative IRE motif is detected within the promoters of mouse, rat, and human apoB genes (Fig. 4). It remains uncertain whether this IRE motif is able to bind FoxO1 and mediate insulin-dependent regulation of apoB-lipoprotein production. Further studies are needed to investigate the mechanism of insulin-dependent degradation of apoB-lipoproteins.
Acknowledgments
We would like to thank Charles Sparks for critical reading of this manuscript. This work was supported in part by grants from the American Diabetes Association (J.D.S. and H.H.D.) and NIH grants DK078131 (J.D.S.) and DK066301 (H.H.D.).
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (pp. 247 – 248).
- 1.Matsumoto M, Pocai A, Rossetti L, et al. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 n liver. Cell Metab. 2007;6:208–216. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 3.Barthel A, Schmoll D, Unterman TG. FoxO proteins in insulin action and metabolism. Trends Endocrinol Metab. 2005;16:183–189. doi: 10.1016/j.tem.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 4.Sparks JD, Collins HL, Chirieac DV, et al. Hepatic very-low-density lipoprotein and apolipoprotein B production are increased following in vivo induction of betaine-homocysteine S-methyltransferase. Biochem J. 2006;395:363–371. doi: 10.1042/BJ20051966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5••.Kamagate A, Qu S, Perdomo G, et al. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J Clin Invest. 2008;118:2347–2364. doi: 10.1172/JCI32914. [DOI] [PMC free article] [PubMed] [Google Scholar]; Authors provide strong evidence that nuclear FoxO1-stimulated transcription of MTP results in enhanced VLDL production under conditions of insulin resistance.
- 6.Wolfrum C, Stoffel M. Coactivation of Foxa2 through Pgc-1beta promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metab. 2006;3:99–110. doi: 10.1016/j.cmet.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 7.van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8:440–450. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 8•.Maiese K, Chong ZZ, Shang YC. OutFOXOing disease and disability: the therapeutic potential of targeting FoxO proteins. Trends Mol Med. 2008;14:219–227. doi: 10.1016/j.molmed.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article discusses the role of FoxO proteins in disease and suggests the potential for new therapies targeting FoxO proteins.
- 9.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 10.Van Der Heide LP, Hoekman MF, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J. 2004;380:297–309. doi: 10.1042/BJ20040167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perrot V, Rechler MM. The coactivator p300 directly acetylates the forkhead transcription factor Foxo1 and stimulates Foxo1-induced transcription. Mol Endocrinol. 2005;19:2283–2298. doi: 10.1210/me.2004-0292. [DOI] [PubMed] [Google Scholar]
- 12.Jing E, Gesta S, Kahn CR. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007;6:105–114. doi: 10.1016/j.cmet.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- 14.Daitoku H, Hatta M, Matsuzaki H, et al. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004;101:10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Heide LP, Smidt MP. Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem Sci. 2005;30:81–86. doi: 10.1016/j.tibs.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 16•.Kuo M, Zilberfarb V, Gangneux N. O-glycosylation of FoxO1 increases its transcriptional activity towards the glucose 6-phosphatase gene. FEBS Lett. 2008;582:829–834. doi: 10.1016/j.febslet.2008.02.010. [DOI] [PubMed] [Google Scholar]; This article shows that glucosamine induces O-glycosylation of FoxO1 which enhances the transcription of G6Pase, a key enzyme in hepatic glucose production, suggesting a mechanism for glucotoxicity in chronic hyperglycemia in diabetes.
- 17•.van der Vos KE, Coffer PJ. FOXO-binding partners: it takes two to tango. Oncogene. 2008;27:2289–2299. doi: 10.1038/onc.2008.22. [DOI] [PubMed] [Google Scholar]; This excellent article reviews FoxO1-binding partners and summarizes the role of interactions of FoxO1 with other transcription factors in regulation of glucose and fatty acid metabolism.
- 18.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brownawell AM, Kops GJ, Macara IG, Burgering BM. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol Cell Biol. 2001;21:3534–3546. doi: 10.1128/MCB.21.10.3534-3546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramaswamy S, Nakamura N, Sansal I, et al. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2:81–91. doi: 10.1016/s1535-6108(02)00086-7. [DOI] [PubMed] [Google Scholar]
- 21.Puigserver P, Rhee J, Donovan J, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 22.Dowell P, Otto TC, Adi S, Lane MD. Convergence of peroxisome proliferator-activated receptor gamma and Foxo1 signaling pathways. J Biol Chem. 2003;278:45485–45491. doi: 10.1074/jbc.M309069200. [DOI] [PubMed] [Google Scholar]
- 23.Armoni M, Harel C, Karni S, et al. FOXO1 represses peroxisome proliferator-activated receptor-gamma1 and -gamma2 gene promoters in primary adipocytes. A novel paradigm to increase insulin sensitivity. J Biol Chem. 2006;281:19881–19891. doi: 10.1074/jbc.M600320200. [DOI] [PubMed] [Google Scholar]
- 24••.Qu S, Su D, Altomonte J. PPARα mediates the hypolipidemic action of fibrates by antagonizing FoxO1. Am J Physiol Endocrinol Metab. 2007;292:E421–E434. doi: 10.1152/ajpendo.00157.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that nuclear FoxO1 is favored in fructose-induced insulin resistance in hamsters, which correlates with augmented hepatic apoCIII production. Treatment with fenofibrate reversed hypertriglyceridemia by functionally antagonizing FoxO1 through direct physical interaction.
- 25.Hatta M, Cirillo LA. Chromatin opening and stable perturbation of core histone:DNA contacts by FoxO1. J Biol Chem. 2007;282:35583–35593. doi: 10.1074/jbc.M704735200. [DOI] [PubMed] [Google Scholar]
- 26.Davidson NO, Shelness GS. Apolipoprotein B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annu Rev Nutr. 2000;20:169–193. doi: 10.1146/annurev.nutr.20.1.169. [DOI] [PubMed] [Google Scholar]
- 27•.Williams KJ. Molecular processes that handle – and mishandle – dietary lipids. J Clin Invest. 2008;118:3247–3259. doi: 10.1172/JCI35206. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a comprehensive and beautifully illustrated review article that summarizes current knowledge of dietary lipid and intestinal lipid metabolism and details unresolved questions.
- 28.Fisher EA, Pan M, Chen X, et al. The triple threat to nascent apolipoprotein B. Evidence for multiple, distinct degradative pathways. J Biol Chem. 2001;276:27855–27863. doi: 10.1074/jbc.M008885200. [DOI] [PubMed] [Google Scholar]
- 29•.Ginsberg HN, Fisher EA. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J Lipid Res. 2009;50 doi: 10.1194/jlr.R800090-JLR200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is an up-to-date review of the role of apoB degradation in regulating VLDL particle secretion that summarizes recent studies involving FA-induced endoplasmic reticulum stress mediated ERAD and n-3 induced PERPP degradation pathways.
- 30.Sparks JD, Collins HL, Sabio I, et al. Effects of fatty acids on apolipoprotein B secretion by McArdle RH-7777 rat hepatoma cells. Biochim Biophys Acta. 1997;1347:51–61. doi: 10.1016/s0005-2760(97)00050-7. [DOI] [PubMed] [Google Scholar]
- 31•.Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B-100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest. 2008;118:316–332. doi: 10.1172/JCI32752. [DOI] [PMC free article] [PubMed] [Google Scholar]; Evidence is provided that oleic acid can induce endoplasmic reticulum stress in vivo and can impact levels of secretion of VLDL apoB-100 suggesting the potential for fatty acids to limit triglyceride export and contribute to hepatic steatosis development.
- 32••.Pan M, Maitin V, Parathath S. Presecretory oxidation, aggregation, and autophagic destruction of apoprotein-B: a pathway for late-stage quality control. Proc Natl Acad Sci U S A. 2008;105:5862–5867. doi: 10.1073/pnas.0707460104. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article provides strong evidence for regulation of apoB by PERPP stimulated by polyunsaturated fatty acids. Data demonstrate that after exit from the endoplasmic reticulum, apoB undergoes PERPP following oxidant-dependent late-stage aggregation resulting in autophagosome formation and degradation of apoB in lysosomes.
- 33•.Sparks JD, Sparks CE. Overindulgence and metabolic syndrome: is FoxO1 a missing link? J Clin Invest. 2008;118:2012–2015. doi: 10.1172/JCI35693. [DOI] [PMC free article] [PubMed] [Google Scholar]; A commentary on the potential role of FoxO1 in promoting hepatic overproduction of VLDL based on increased expression of MTP. The possible role of FoxO1 in short-term insulin-suppressive effects on hepatic VLDL is discussed.
- 34•.Biddinger SB, Hernandez-Ono A, Rask-Madsen C. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7:125–134. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; Loss of hepatic insulin receptors in mice is used as a model of pure hepatic insulin resistance. In these mice, there is increased hepatic VLDL production, dyslipidemia, and increased risk of atherosclerosis associated with metabolic syndrome. Results support that lack of insulin action results in augmentation of VLDL output by liver.
- 35.Chirieac DV, Davidson NO, Sparks CE, Sparks JD. PI3-kinase activity modulates apoB available for hepatic VLDL production in apobec-1 −/− mice. Am J Physiol Gastrointest Liver Physiol. 2006;291:G382–G388. doi: 10.1152/ajpgi.00472.2005. [DOI] [PubMed] [Google Scholar]
- 36•.Hussain MM, Rava P, Pan X. Microsomal triglyceride transfer protein in plasma and cellular lipid metabolism. Curr Opin Lipidol. 2008;19:277–284. doi: 10.1097/MOL.0b013e3282feea85. [DOI] [PubMed] [Google Scholar]; This study summarizes recent advances in the regulation of MTP and its role in apoB-containing lipoprotein assembly.
- 37.Wetterau JR, Aggerbeck LP, Bouma ME, et al. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999–1001. doi: 10.1126/science.1439810. [DOI] [PubMed] [Google Scholar]
- 38.Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma M, Wetterau JR. The role of the microsomal triglygeride transfer protein in abetalipoproteinemia. Annu Rev Nutr. 2000;20:663–697. doi: 10.1146/annurev.nutr.20.1.663. [DOI] [PubMed] [Google Scholar]
- 39.Pan M, Liang JS, Fisher EA, Ginsberg HN. The late addition of core lipids to nascent apolipoprotein B100, resulting in the assembly and secretion of triglyceride-rich lipoproteins, is independent of both microsomal triglyceride transfer protein activity and new triglyceride synthesis. J Biol Chem. 2002;277:4413–4421. doi: 10.1074/jbc.M107460200. [DOI] [PubMed] [Google Scholar]
- 40.Wiggins D, Gibbons GF. The lipolysis/esterification cycle of hepatic triacylglycerol. Its role in the secretion of very-low-density lipoprotein and its response to hormones and sulphonylureas. Biochem J. 1992;284(Pt 2):457–462. doi: 10.1042/bj2840457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiggins D, Gibbons GF. Origin of hepatic very-low-density lipoprotein triacylglycerol: the contribution of cellular phospholipid. Biochem J. 1996;320(Pt 2):673–679. doi: 10.1042/bj3200673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lass A, Zimmermann R, Haemmerle G, et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin–Dorfman syndrome. Cell Metab. 2006;3:309–319. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 43•.Reid BN, Ables GP, Otlivanchik OA. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J Biol Chem. 2008;283:13087–13099. doi: 10.1074/jbc.M800533200. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hormone-sensitive lipase and adipose triglyceride lipase are important in lipolysis of triglyceride lipid droplets, mobilization of fatty acids, and channeling them to oxidation pathways without increasing hepatic VLDL secretion.
- 44•.Caviglia JM, Sparks JD, Toraskar N. ABHD5/CGI58 facilitates the assembly and secretion of apolipoprotein B lipoproteins by McA RH7777 rat hepatoma cells. Biochim Biophys Acta. 2009;1791:198–205. doi: 10.1016/j.bbalip.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; Evidence is provided for the role of ABHD5/CGI58 as a cofactor for ATGL in the lipolysis component of the lipolysis/re-esterification cycle and may be involved in channeling fatty acids for VLDL triglyceride assembly and secretion.
- 45.Ghosh AK, Ramakrishnan G, Chandramohan C, Rajasekharan R. CGI-58, the causative gene for Chanarin–Dorfman syndrome, mediates acylation of lysophosphatidic acid. J Biol Chem. 2008;283:24525–24533. doi: 10.1074/jbc.M801783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ledford AS, Weinberg RB, Cook VR, et al. Self-association and lipid binding properties of the lipoprotein initiating domain of apolipoprotein B. J Biol Chem. 2006;281:8871–8876. doi: 10.1074/jbc.M507657200. [DOI] [PubMed] [Google Scholar]
- 47•.Manchekar M, Richardson PE, Sun Z, et al. Charged amino acid residues 997-1000 of human apolipoprotein B100 are critical for the initiation of lipoprotein assembly and the formation of a stable lipidated primordial particle in McA-RH7777 cells. J Biol Chem. 2008;283:29251–29265. doi: 10.1074/jbc.M804912200. [DOI] [PMC free article] [PubMed] [Google Scholar]; Authors provide evidence for the importance of residues 717–720 and 997–1000 in the formation of the lipoprotein initiating domain of apoB possibly by a hair-pin-bridge mechanism that completes initial stages of primordial particle formation.
- 48.Swift LL, Zhu MY, Kakkad B, et al. Subcellular localization of microsomal triglyceride transfer protein. J Lipid Res. 2003;44:1841–1849. doi: 10.1194/jlr.M300276-JLR200. [DOI] [PubMed] [Google Scholar]
- 49.Phung TL, Roncone A, Jensen KL, et al. Phosphoinositide 3-kinase activity is necessary for insulin-dependent inhibition of apolipoprotein B secretion by rat hepatocytes and localizes to the endoplasmic reticulum. J Biol Chem. 1997;272:30693–30702. doi: 10.1074/jbc.272.49.30693. [DOI] [PubMed] [Google Scholar]
- 50.Fisher EA, Ginsberg HN. Complexity in the secretory pathway: the assembly and secretion of apolipoprotein B-containing lipoproteins. J Biol Chem. 2002;277:17377–17380. doi: 10.1074/jbc.R100068200. [DOI] [PubMed] [Google Scholar]
- 51•.Brodsky JL, Fisher EA. The many intersecting pathways underlying apolipoprotein B secretion and degradation. Trends Endocrinol Metab. 2008;19:254–259. doi: 10.1016/j.tem.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is an excellent review article on how VLDL secretion by liver is regulated by apoB degradation pathways with a focus on endoplasmic reticulum-associated degradation and autophagy.
- 52••.Kamagate A, Dong HH. FoxO1 integrates insulin signaling to VLDL production. Cell Cycle. 2008;7:3162–3170. doi: 10.4161/cc.7.20.6882. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is an excellent review of FoxO1 in hepatic VLDL metabolism supporting the idea that FoxO1 is involved in insulin regulation of hepatic production and VLDL metabolism in parallel mechanisms with hepatic glucose production with both involving transcriptional regulation.
- 53•.Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27:2320–2336. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]; This review of FoxO1 contains a summary of FoxO1 gain and loss of function studies in hepatic lipid metabolism.
- 54.Zhang W, Patil S, Chauhan B, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem. 2006;281:10105–10117. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 55•.Pan X, Hussain MM. Diurnal regulation of microsomal triglyceride transfer protein and plasma lipid levels. J Biol Chem. 2007;282:24707–24719. doi: 10.1074/jbc.M701305200. [DOI] [PubMed] [Google Scholar]; This article presents surprising evidence demonstrating that hepatic and intestinal MTP activity, MTP protein levels and plasma lipid levels undergo diurnal regulation and are affected by environmental factors such as food and light. MTP changes are associated with changes in mttp gene transcription.
- 56.Sparks JD, Sparks CE, Bolognino M, et al. Effects of nonketotic streptozotocin diabetes on apolipoprotein B synthesis and secretion by primary cultures of rat hepatocytes. J Clin Invest. 1988;82:37–43. doi: 10.1172/JCI113597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ji C, Shinohara M, Kuhlenkamp J, et al. Mechanisms of protection by the betaine-homocysteine methyltransferase/betaine system in HepG2 cells and primary mouse hepatocytes. Hepatology. 2007;46:1586–1596. doi: 10.1002/hep.21854. [DOI] [PMC free article] [PubMed] [Google Scholar]