

Abstract
Material collected from a parkway in the city of Chicago afforded the isolation of a Nostoc species (UIC 10022A). The extract of this strain displayed significant inhibition of the 20S proteasome as well as antiproliferative activity against HT29, MCF7, NCI-H460, and SF268 cancer cell lines. A standardized dereplication protocol allowed for the rapid identification of three known (11-13) and nine new (1-9) chlorinated cylindrocyclophanes from less than 100 mg of organic extract. Scale-up isolation of 1-9 and 11-13 from a larger extract was guided by LC-UV-MS data. In addition, KBr enrichment of the culture media afforded the isolation of a brominated cylindrocyclophane (10). Biological evaluation of 1-5, 9, and 10-13 revealed a large range of activity against the 20S proteasome and allowed the determination of preliminary structure-activity relationships (SAR) of the cylindrocyclophane pharmacophore.
Cyanobacteria are considered a promising source for new pharmaceutical lead compounds and a large number of chemically diverse metabolites have been obtained from cyanobacteria.1-5 In particular, members of the genus Nostoc have been a valuable source of biologically active compounds. The most prominent example is the cryptophycins, which were first isolated from a lichen cyanobiont.6 These potent tubulin inhibitors eventually led to the development of a semi-synthetic clinical candidate.7-9 Other examples of compounds isolated from cyanobacteria of the genus Nostoc are the cytotoxic nostocyclopeptides,10 the antimicrobial nostocyclyne A,11 as well as protease inhibitors such as the insulapeptolides,12 nostopeptins,13, 14 and microviridins G and H.15 In all of these cases, the source organism was of freshwater or terrestrial origin.
As part of our ongoing effort to find novel bioactive natural products from cultured freshwater and terrestrial cyanobacteria from the U.S. Great Lakes region,16 we have evaluated cyanobacterial extracts for inhibition of the 20S proteasome. The 20S proteasome is the catalytic core of the proteasome complex and plays a pivotal role in the control of cell proliferation, apoptosis, and differentiation in a variety of normal and tumor cells.17-19 Currently, there is one proteasome inhibitor, bortezomib (Velcade) approved by the U.S. Food and Drug Administration.20 Bortezomib was first approved in May 2003 for the treatment of multiple myeloma, however it has been shown to be effective against other cancers, e.g. lymphoma, prostate, and lung cancers.17 Preliminary studies of the extract of Nostoc sp. (UIC 10022A), collected in Chicago, Illinois, revealed significant proteasome inhibitory activity as well as inhibition of growth in four cancer cell lines (HT-29, NCI-H460, SF268, and MCF7). HPLC MS/NMR dereplication allowed for identification of a series of cylindrocyclophanes as the active principles. Scale-up isolation using LC-UV-MS data to guide the fractionation yielded 9 new chlorinated cylindrocyclophanes. Herein we describe the isolation, structure determination and an in depth evaluation of the proteasome inhibitory activity of these new natural products.
Results and Discussion
The initial organic extract (83.5 mg) from a 4 L culture of Nostoc sp. (UIC 10022A) displayed significant, inhibitory activity against the 20S proteasome as well as antiproliferative activity against four cancer cell lines. This extract was subjected to a dereplication scheme, which included a Diaion VLC step with a subsequent semi-preparative HPLC fractionation and chemical analysis via ESI-TOF-MS and 1H NMR spectroscopy. These standardized steps revealed the presence of three previously reported compounds, cylindrocyclophanes A (11), C (12), and F (13)21 and 10 new analogues, cylindrocyclophanes A4-A1 (1-4), C4-C1 (5-8), and F4 (9). A second extract of strain UIC 10022A from a 20 L culture was subjected to Diaion and reversed-phase HPLC to yield 1-9 and 11-13 in larger quantities. The HRESIMS data acquired from the dereplication of the initial extract were used in combination with LC-UV-MS data acquired on the second extract to guide the isolation of 1-9 and 11-13.
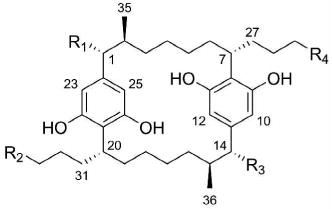 | ||||
|---|---|---|---|---|
| R1 | R2 | R3 | R4 | |
| 1 | OH | CHCl2 | OH | CHCl2 |
| 2 | OH | CH2Cl | OH | CHCl2 |
| 3 | OH | CH3 | OH | CHCl2 |
| 4 | OH | CH3 | OH | CH2Cl |
| 5 | OH | CHCl2 | H | CHCl2 |
| 6 | OH | CH2Cl | H | CHCl2 |
| 7 | OH | CH3 | H | CHCl2 |
| 8 | OH | CH3 | H | CH2Cl |
| 9 | H | CHCl2 | H | CHCl2 |
| 10 | OH | CHBr2 | OH | CHBr2 |
| 11 | OH | CH3 | OH | CH3 |
| 12 | OH | CH3 | H | CH3 |
| 13 | H | CH3 | H | CH3 |
Cylindrocyclophane A4 (1), the major metabolite, was obtained as a white, amorphous powder. The negative mode HRESI-TOF-MS data indicated a molecular formula of C36H52Cl4O6 ([M-H]-, m/z 719.2392) with an isotopic distribution pattern that was consistent with a tetrachlorinated molecule (70:100:53:13 M:M+2:M+4:M+6). The DEPTQ (Table 1) spectrum displayed only 18 carbon signals, indicating a symmetrical molecule. These results were consistent with a C2 axis of symmetry, which was also found in the previously described cylindrocyclophane A (11).21
Table 1.
NMR Data of Cylindrocyclophanes A4-A1 (1-4) in MeOH-d4
| position | cylindrocyclophane A4 (1) | cylindrocyclophane A3 (2) | cylindrocyclophane A2 (3) | cylindrocyclophane A1 (4) | ||||
|---|---|---|---|---|---|---|---|---|
| δCa, mult. | δHb (J in Hz) | δCc | δHb (J in Hz) | δCc | δHb (J in Hz) | δCc | δHb (J in Hz) | |
| 1/14 | 81.8, CH | 3.74, d (9.7) | 81.6 | 3.74, d (9.8) | 81.5 | 3.74, d(9.7) | 81.8 | 3.74, d (9.7) |
| 2/15 | 42.1, CH | 1.55, m | 41.9 | 1.55, m | 41.9 | 1.57, m | 41.7 | 1.56, m |
| 3/16a | 35.5, CH2 | 0.73, m | 35.0 | 0.74, m | 35.0 | 0.74, m | 35.0 | 0.75, m |
| 3/16b | 0.64, m | 0.63, m | 0.63, m | 0.63, m | ||||
| 4/17a | 29.9, CH2 | 1.42, m | 29.7 | 1.42, m | 29.7 | 1.42, m | 29.5 | 1.42, m |
| 4/17b | 0.83, m | 0.83, m | 0.82, m | 0.83, m | ||||
| 5/18a | 30.6, CH2 | 0.95, m | 30.3 | 0.96, m | 30.3 | 0.95, m | 30.5 | 0.95, m |
| 5/18b | 0.72, m | 0.72, m | 0.72, m | 0.72, m | ||||
| 6/19a | 35.2, CH2 | 2.07, m | 35.2 | 2.06, m | 35.3 | 2.05, m | 35.2 | 2.05, m |
| 6/19b | 1.29, m | 1.32, m | 1.32, m | 1.33, m | ||||
| 7/20 | 36.4, CH | 3.19, m | 36.3 | 3.19, m | 36.2 | 3.19, m | 36.5 | 3.17, m |
| 36.7 | 3.15, m | 36.6 | 3.15, m | |||||
| 8/21 | 116.7, C | - | 116.5 | - | 116.7 | - | 117.5 | - |
| 9/22 | 159.0, C | - | 158.8 | - | 158.7 | - | 159.5 | - |
| 10/23 | 105.0, CH | 6.25, d (1.2) | 104.8 | 6.25, br s | 104.7 | 6.25, br s | 104.8 | 6.24, br s |
| 11/24 | 144.2, C | - | 143.9 | - | 143.6 | - | 144.0 | - |
| 12/25 | 108.8, CH | 6.07, d (1.2) | 108.6 | 6.07, br s | 108.6 | 6.07, br s | 108.6 | 6.06, br s |
| 13/26 | 157.1, C | - | 156.9 | - | 156.6 | - | 157.5 | - |
| 27a | 33.8, CH2 | 2.07, m | 33.6 | 2.06, m | 33.6 | 2.07, m | 33.9 | 2.02, m |
| 27b | 1.48, m | 1.49, m | 1.50, m | 1.49, m | ||||
| 28 | 25.7, CH2 | 1.35, m | 25.4 | 1.36, m | 25.3, | 1.35, m | 26.3 | 1.26, m |
| 29a | 45.0, CH2 | 2.20, m | 44.8 | 2.20, m | 44.7 | 2.20, m | 33.6 | 1.76, m |
| 29b | 2.06, m | 2.06, m | 2.06, m | 1.65, m | ||||
| 30 | 75.4, CH | 5.82, t (6.2) | 75.1 | 5.82, t (6.1) | 75.0 | 5.82, t (6.1) | 45.7 | 3.43, t (7.4) |
| 31a | 33.8, CH2 | 2.07, m | 34.0 | 2.01, m | 34.5 | 1.94, m | 34.4 | 1.95, m |
| 31b | 1.48, m | 1.49, m | 1.47, m | 1.47, m | ||||
| 32a | 25.7, CH2 | 1.35, m | 26.3 | 1.25, m | 31.4 | 1.16, m | 31.4 | 1.15, m |
| 32b | 1.05, m | 1.05, m | ||||||
| 33a | 45.0, CH2 | 2.20, m | 33.8 | 1.75, m | 23.7 | 1.25, m | 23.8 | 1.20, m |
| 33b | 2.06, m | 1.64, m | ||||||
| 34 | 75.4, CH | 5.82, t (6.2) | 45.7 | 3.43, t (6.8) | 14.2 | 0.79, t (7.2) | 14.3 | 0.79, t (7.2) |
| 35/36 | 17.0, CH3 | 1.06, d (6.4) | 16.6 | 1.06, d (6.4) | 16.7 | 1.07, d (6.4) | 16.7 | 1.07, d (6.4) |
DEPTQ experiment recorded at 226 MHz.
Recorded at 600 MHz.
Determined indirectly using HSQC and HMBC.
The 1H NMR spectrum (Table 1) for 1 revealed a tetrasubstituted aromatic ring (δH 6.25 and 6.07), an oxygen-bearing benzylic methine (δH 3.74), a methyl doublet (δH 1.06), a methine triplet at δH 5.82, and 14 alkyl multiplets between δH 2.20 and 0.64. Analysis of the HSQC spectrum for 1 showed the downfield triplet (H-30/34, δH 5.82) was attached to a carbon (C-30/34) at δC 75.4. These chemical shifts were consistent with a dichlorinated methyl moiety.
Inspection of the 1H NMR signals for the aromatic protons (H-10/23, δH 6.25 and H-12/25, δH 6.07) of 1 revealed a small coupling constant of 1.2 Hz. This J value was consistent with a meta relationship. The HMBC spectrum revealed correlations from these protons (H-10/23 and H-12/25) to the hydroxyl-bearing phenolic carbons (C-9/22, δC 159.0 and C-13/26, δC 157.1) as well as the quaternary aromatic carbon (C-8/21, δC 116.7) meta to these protons. The HMBC spectrum for 1 also revealed correlations between the benzylic proton signal (H-7/20, δH 3.19) and the aromatic carbons C-8/21, C-9/22, and C-13/26, confirming that the carbons C-9/22 and C-13/26 were attached to C-8/21 and C-8/21 in turn was attached to C-7/20. The HMBC spectrum for 1 revealed a correlation between the oxygen bearing benzylic proton signal (H-1/14, δH 3.74) and the aromatic carbons C-11/24 (δC 144.2), C-10/23 (δC 105.0), and C-12/25 (δC 108.8). These data confirmed that the aromatic carbons (C-10/23 and C-12/25) were attached to C-11/24 and that C-11/24 was attached to C-1/14.
Analysis of the COSY spectrum for 1 revealed that H-1/14 correlated with H-2/15 (δH 1.55). H-2/15, in turn, correlated with the methyl moiety (H3-35/36, δH 1.06) as well as H-3b/16b (δH 0.64). The COSY data, in combination with HSQC data, also revealed correlations of H-3a/16a (δH 0.73) with H-4a/17a (δH 1.42), H-4b/17b (δH 0.83) with H-5b/18b (δH 0.72), H-5b/18b with H-6b/19b (δH 1.29), and H-6b/19b with H-7/10. The same process was used to assign the side chains (C-27/31 to C-30/34). Briefly, the COSY spectrum revealed the correlations from H-27b/31b (δH 1.48) to H2-28/32 (δH 1.35), H2-28/32 to H-29a/33a (δH 2.20), and H-29a/33a to H-30/34 (δH 5.82). Comparing these data with those previously reported by Moore et al.21 and Bui et al.22 we determined that 1 was 30,30,34,34-tetrachlorocylindrocyclophane A.
Cylindrocyclophanes A3-A1 (2-4) were also obtained as white, amorphous solids. The negative mode HRESI-TOF-MS data for 2-4 indicated molecular formulas of C36H53Cl3O6 ([M-H]-, m/z 685.2760), C36H54Cl2O6 ([M-H]-, m/z 651.3157) and C36H55ClO6 ([M-H]-, m/z 617.3666), respectively. For each compound, the observed isotopic distribution patterns were consistent with the predicted degree of chlorination. The 1H NMR spectra for 2-4 were similar to the spectrum of 1 indicating these compounds only differed by the degree of chlorination on C-30 and C-34. The 1H NMR spectrum for 2 revealed a similar triplet at δH 5.82 with half the relative intensity as the signal observed in cylindrocyclophane A4 and a new methylene triplet at δH 3.43 (H2-34). The HSQC spectrum for 2 displayed a correlation of H2-34 with δH 45.7 (C-34), which was consistent for 30,30,34-trichlorochlorocylindrocyclophane A. The 1H NMR spectrum for 4 revealed a methylene triplet at δH 3.43 (H2-30) and a methyl triplet at δH 0.79 (H3-34), which indicated a 30-chlorocylindrocyclophane A. The 1H NMR spectrum for 3 revealed a methine triplet at δH 5.82 (H-30) and a methyl triplet at δH 0.79 (H3-34). These data indicated that the correct structure of 3 was 30,30-dichlorocylindrocyclophane A. The 1H, HSQC, and HMBC NMR data for 2-4 were compared with the data acquired for 1 as well as data published by Moore et al.21 and Bui et al.22 to assign the chemical shifts for 2-4.
Cylindrocyclophanes C4-C (5-8, 12) were all obtained as white, amorphous solids. The negative mode HRESI-TOF-MS data for 5-8, 12 indicated molecular formulas of C36H52Cl4O5 ([M-H]-, m/z 703.2442), C36H53Cl3O5 ([M-H]-, m/z 669.2824), C36H54Cl2O5 ([M-H]-, m/z 635.3329), C36H55ClO5 ([M-H]-, m/z 601.3609) and C36H56O5 ([M-H]-, m/z 567.4019), respectively. Based upon the previously reported cylindrocyclophanes, it was proposed that 12 was cylindrocyclophane C as previously reported by Moore et al. and 5-8 were chlorinated analogs of 12.21
The 1H NMR spectra for 5-8 (Table 2) displayed similar signals as for 1-4 (Table 1), which indicated that these compounds followed the same chlorination pattern. However, a comparison of the molecular formulas revealed that the difference between these two series of compounds was that 5-8, 12 contained only five oxygen atoms while 1-4 contained six oxygen atoms. The 1H NMR spectrum for 7 revealed the same oxygen bearing benzylic proton (δH 3.75) as found in the 1H NMR spectra for 1-4, 11. However, the 1H NMR and HSQC spectra for 7 also revealed the presence of a benzylic methylene (δC 45.9, δH 2.61 and 1.83). From these data, it was deduced that C-1 was similar to C-1/14 found in 1-4, 11, however C-14 in 5-8, 12 lacked the hydroxy moiety. The lack of symmetry of C-1 and C-14 complicated the structure elucidation of 5-8, however the structures were solved using a combination of HMBC, COSY, and TOCSY data.
Table 2.
NMR Data of Cylindrocyclophanes C4-C1 (5-8) in MeOH-d4
| position | cylindrocyclophane C4 (5) | cylindrocyclophane C3 (6) | cylindrocyclophane C2 (7) | cylindrocyclophane C1 (8) | ||||
|---|---|---|---|---|---|---|---|---|
| δCa | δHb (J in Hz) | δCa | δHb (J in Hz) | δCc, mutl. | δHb (J in Hz) | δCa | δHb (J in Hz) | |
| 1 | 80.9 | 3.75, d (9.8) | 81.7 | 3.75, d (9.6) | 81.8, CH | 3.75, d (9.6) | 81.4 | 3.75, d (10.4) |
| 2 | 41.2 | 1.57, m | 41.2 | 1.56, m | 42.0, CH | 1.56, m | 41.6 | 1.57, m |
| 3/16a | 35.6 | 0.75, m | 35.2 | 0.78, m | 35.2, CH2 | 0.75, m | 34.8 | 0.78, m |
| 3/16b | 0.64, m | 0.65, m | 0.62, m | 0.65, m | ||||
| 4/17a | 29.9 | 1.44, m | 30.0 | 1.43, m | 29.9, CH2 | 1.41, m | 29.7 | 1.43, m |
| 4/17b | 0.83, m | 0.81, m | 0.81, m | 0.81, m | ||||
| 5/18a | 30.6 | 0.96, m | 30.3 | 0.97, m | 30.7, CH2 | 0.96, m | 30.4 | 0.98, m |
| 5/18b | 0.75, m | 0.74, m | 0.74, m | 0.74, m | ||||
| 6/19a | 34.5 | 2.06, m | 35.3 | 2.04, m | 35.6, CH2 | 2.06, m | 35.1 | 2.02, m |
| 6/19b | 1.34, m | 1.33, m | 1.30, m | 1.32, m | ||||
| 7/20 | 35.6 | 3.19, m | 36.0 | 3.16, m | 36.3, CH2 | 3.18, m | 36.3 | 3.15, m |
| 8 | 114.6 | - | 115.0 | - | 115.0, C | - | 115.4 | - |
| 9 | 157.9 | - | 158.2 | - | 158.4, C | - | 158.7 | - |
| 10 | 107.4 | 6.03, s | 107.7 | 6.03, s | 107.9, CH | 6.02, s | 107.6 | 6.02, s |
| 11 | 140.9 | - | 141.2 | - | 141.3, C | - | 140.7 | - |
| 12 | 110.0 | 5.98, s | 109.7 | 5.98, s | 110.0, CH | 5.98, s | 109.7 | 5.97, s |
| 13 | 156.0 | - | 157.4 | - | 157.1, C | - | 157.2 | - |
| 14a | 45.8 | 2.61, dd (13.2, 3.6) | 45.5 | 2.61, dd (13.2, 3.6) | 45.9, CH2 | 2.61, dd (13.2, 3.6) | 45.5 | 2.61, dd (13.2, 3.6) |
| 14b | 1.82, dd (13.2, 11.7) | 1.81, dd (13.2, 11.7) | 1.83, dd (13.2, 11.7) | 1.81, dd (13.2, 11.7) | ||||
| 15 | 35.7 | 1.55, m | 36.5 | 1.59, m | 36.6, CH | 1.58, m | 36.5 | 1.58, m |
| 21 | 116.4 | - | 117.3 | - | 117.8, C | - | 117.3 | - |
| 22 | 158.7 | - | 157.2 | - | 158.9, C | - | 158.7 | - |
| 23 | 104.8 | 6.25, s | 104.8 | 6.25, s | 105.1, CH | 6.24, s | 104.7 | 6.24, s |
| 24 | 143.7 | - | 144.1 | - | 143.8, C | - | 143.8 | - |
| 25 | 108.5 | 6.08, s | 108.7 | 6.08, s | 108.9, CH | 6.07,s | 108.5 | 6.07, s |
| 26 | 156.6 | - | 159.1 | - | 157.0, C | - | 157.2 | - |
| 27a | 35.0 | 2.06, m | 33.9 | 2.04, m | 35.5, CH2 | 2.05, m | 33.8 | 2.02, m |
| 27b | 1.54, m | 1.52, m | 1.51, m | 1.52, m | ||||
| 28 | 25.2 | 1.37, m | 25.4 | 1.39, m | 25.8, CH2 | 1.37, m | 26.3 | 1.28, m |
| 29a | 44.7 | 2.20, m | 45.0 | 2.20, m | 45.0, CH2 | 2.20, m | 33.6 | 1.75, m |
| 29b | 2.05, m | 2.06, m | 2.06, m | 1.66, m | ||||
| 30 | 75.1 | 5.82, t (6.2) | 75.2 | 5.83, t (6.2) | 75.4, CH | 5.82, t (6.2) | 45.5 | 3.44, t (7.0) |
| 31a | 35.0 | 2.06, m | 34.6 | 2.04, m | 34.9, CH2 | 1.93, m | 34.5 | 1.94, m |
| 31b | 1.54, m | 1.50, m | 1.45, m | 1.48, m | ||||
| 32a | 25.2 | 1.37, m | 26.8 | 1.25, m | 31.7, CH2 | 1.18, m | 31.5 | 1.16, m |
| 32b | 1.31, m | 1.02, m | 1.06, m | |||||
| 33a | 44.7 | 2.20, m | 33.8 | 1.75, m | 24.0, CH2 | 1.20, m | 23.5 | 1.29, m |
| 33b | 2.05, m | 1.65, m | 1.21, m | |||||
| 34 | 75.1 | 5.82,t (6.2) | 45.7 | 3.43, t (6.9) | 14.6, CH3 | 0.79, t (7.5) | 14.2 | 0.79, t (7.3) |
| 35 | 16.0 | 1.06,d (6.4) | 16.6 | 1.06, d (6.4) | 16.9, CH3 | 1.06, d (6.4) | 16.7 | 1.06, d (6.5) |
| 36 | 20.9 | 0.95,d (6.5) | 20.9 | 0.95, d (6.5) | 20.9, CH3 | 0.95, d (6.5) | 20.5 | 0.95, d (6.5) |
Determined indirectly using HSQC and HMBC.
Recorded at 600 MHz.
DEPTQ experiment recorded at 226 MHz.
The 1H NMR spectra for 5-8 displayed four different aromatic signals (δH ca. 6.24, 6.07, 6.02, and 5.98), as opposed to the two aromatic signals (δH 6.25, and 6.07) that were observed in the spectra of 1-4, 11. Correlations from the HMBC spectrum for 7 (Figure 1) revealed that protons H-23 (δH 6.24) and H-25 (δH 6.07) were part of the aromatic ring attached to C-1 (δC 81.8) and that protons H-10 (δH 6.02) and H-12 (δH 5.98) were part of the aromatic ring attached to C-14 (δC 45.9).
Figure 1.
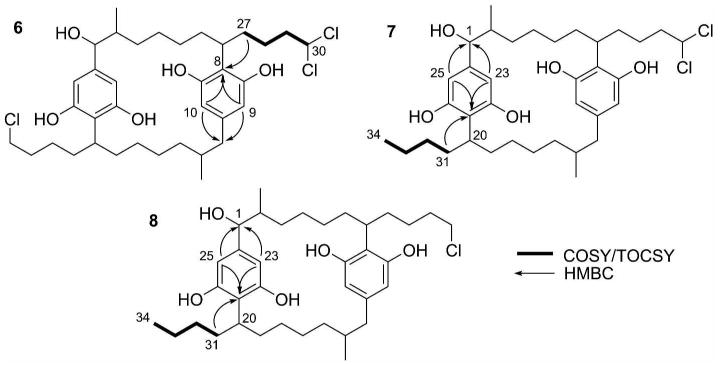
Key COSY, TOCSY, and HMBC correlations used for assignment of side-chain moieties.
Further analysis of the HMBC spectrum for 7 revealed that the carbon at δC 117.8 ppm (C-21) was part of the aromatic ring containing H-23 (δH 6.24) and H-25 (δH 6.07). The HMBC spectrum for 7 also revealed a correlation between C-21 (δC 117.8) and H-31a (δH 1.93). Analysis of the TOCSY spectrum for 7 revealed that H-31a (δH 1.93) correlated to H3-34 (δH 0.79), thus the side chain of C-31 to C-34 terminated in methyl moiety (Figure 1). Therefore, by a process of elimination, the side chain of C-27 to C-30 terminated in the dichloromethyl moiety.
The HMBC spectrum acquired for 8 revealed similar correlations between C-21 (δC 117.3) and H-23 (δH 6.07), H-25 (δH 6.24), and H-31b (δH 1.48). This, combined with the H31-H34 fragment determined by COSY, allowed for the proper assignment of the methyl at C-34, thus the chloromethyl moiety was assigned to position C-30. For compound 6, the HMBC correlation between C-8 (δC 115.0) and H-27b (δH 1.52) was used in conjunction with the H27-H30 COSY fragment to properly assign the dichloromethyl moiety to position C-30. The structure elucidation of 5 was simplified by the fact that C-30 and C-34 were both dichloromethyl moieties, thus both side chains (C-27 to C-30 and C-31 to C-34) were, in essence, identical. The NMR data for 5-8 were compared with the data reported by Moore et al.21 and Bui et al.22 to properly assign all the spectroscopic data and to confirm the structures of cylindrocyclophane C4 (5), cylindrocyclophane C3 (6), cylindrocyclophane C2 (7), and cylindrocyclophane C1 (8).
Cylindrocyclophane F (13) was identified as a compound previously described by Moore et al. using 1H NMR and MS data ([M-H]-, m/z 551.4057).21 Cylindrocyclophane F4 (9) was obtained as a white, amorphous solid. The negative mode HRESI-TOF-MS data for 9 indicated the molecular formula of C36H52O4Cl4 ([M-H]-, m/z 687.2510), respectively. The 1H NMR spectrum for 9 (Table 3) displayed signals similar to those for 1 and 5; most notable was the presence of a methine triplet at δH 5.83 which correlated with δC 75.1 in the HSQC spectrum. As shown in 1 and 5, these signals were consistent with a dichlorinated methyl moiety. The 1H NMR spectrum for 9 lacked the signal of the oxygen attached benzylic methine found in 1 (H-1/14, δH 3.74) and 5 (H-1, δH 3.75). Instead, the 1H NMR data for 9 revealed two signals (δH 2.58 and 1.83) and associated HSQC signal (δC 45.7) that were consistent with a benzylic methylene as found in 5 (14, δC 45.8, δH 2.61 and 1.82). The NMR spectra of 9 displayed only half of the expected NMR signals, revealing a similar C2 axis of symmetry as found in 1. This simplified the structure elucidation of 9. The NMR data for 9 were compared with the data acquired for 1-8 to determine that 9 was 30,30,34,34-tetrachlorocylindrocyclophane F. Due to the low yield of 9 (approximately 0.1 mg), it was not possible to gain sufficient signal to noise in the HMBC spectrum to be able to assign the carbons (C-9/22 and C-13/26).
Table 3.
NMR Data of Cylindrocyclophanes F4 (9) and AB4 (10) in MeOH-d4
| position | cylindrocyclophane F4 (9) | cylindrocyclophane AB4 (10) | ||
|---|---|---|---|---|
| δCa | δHb (J in Hz) | δCc | δHb (J in Hz) | |
| 1/14a | 45.7 | 2.58, dd (13.6, 3.9) | 81.8, CH | 3.74, d (9.6) |
| 1/14b | 1.83, dd (13.6, 11.0) | |||
| 2/15 | 36.5 | 1.57, m | 42.1, CH | 1.55, m |
| 3/16a | 36.5 | 1.02, m | 35.2, CH2 | 0.75, m |
| 3/16b | 0.65, m | 0.64, m | ||
| 4/17a | 29.8 | 1.44, m | 29.7, CH2 | 1.43, m |
| 4/17b | 0.80, m | 0.83, m | ||
| 5/18a | 30.2 | 0.99, m | 30.6, CH2 | 0.95, m |
| 5/18b | 0.78, m | 0.73, m | ||
| 6/19a | 35.3 | 2.01, m | 35.2, CH2 | 2.07, m |
| 6/19b | 1.32, m | 1.33, m | ||
| 7/20 | 36.0 | 3.16, m | 36.4, CH | 3.20, m |
| 8/21 | 114.2 | - | 116.7, C | - |
| 9/22 | n.d.d | - | 159.0, C | - |
| 10/23 | 107.7 | 6.02, s | 105.0, CH | 6.25, s |
| 11/24 | 141.4 | - | 144.2, C | - |
| 12/25 | 109.7 | 5.99, s | 108.8, CH | 6.07, s |
| 13/26 | n.d. d | - | 157.1, C | - |
| 27/31a | 33.5 | 2.06, m | 33.5, CH2 | 2.07, m |
| 27/31b | 1.50, m | 1.51, m | ||
| 28/32 | 25.6 | 1.37, m | 27.8, CH2 | 1.36, m |
| 29/33a | 44.8 | 2.21, m | 46.9, CH2 | 2.41, m |
| 29/33b | 2.06, m | 2.26, m | ||
| 30/34 | 75.1 | 5.83, t (6.2) | 48.0, CH | 5.80, t (6.8) |
| 35/36 | 20.6 | 0.94, d (6.6) | 17.0, CH3 | 1.07, d (6.4) |
Determined indirectly using HSQC and HMBC.
Recorded at 600 MHz.
DEPTQ experiment recorded at 226 MHz.
These atoms could not be assigned due to insufficient signal to noise in the HMBC data.
Configurational Analysis
The report by Moore et al. determined the absolute configuration of 11 using data acquired via Mosher's method and comparing the results with the X-ray crystallography data of nostocyclophane D.21, 23 The same report also determined the absolute configurations of 12 and 13, by a comparison of circular dichroism data. In the end, all three compounds were found to have the same absolute configuration at C-1, C-2, C-7, C-14, C-15, and C-20.21 A similar relationship was found for 1-9. The 3JH-1/14,H-2/15 values for 1-4 ranged from 9.8-9.7 Hz. These values were similar to the value reported (10.3 Hz) by Moore et al. for 11.23 This large coupling constant (> 7 Hz) indicates that H-1/14 and H-2/15 are in an anti- conformation. Additional experiments are needed to fully elucidate the relative conformation and configuration of these atoms, however the [α]D values of 1-3 ranged from -26 to -27. The optical rotation of 4 could not be measured due to the low yield of the compound. The [α]D values for 1-3 were similar to the value (-20) reported for 11.23 In addition, the CD spectrum of 1 was similar to the CD spectrum reported for cylindrocyclophane A.23 Based on these similarities, we submit that 1-4 have the same absolute configuration at C-1/14, C-2/15, and C-7/20 as 11.
The 3JH-1,H-2 values for 5-8 showed similar values (9.6-10.4 Hz), which is also consistent with an anti-conformation of H-1 and H-2. The [α]D values for cylindrocyclophanes C1-C4 (-37 to -44) were also similar to the value reported for 12 (-40).23 Based on these similarities, we submit that 5-8 also have the same absolute configuration at C-1/14, C-2/15, and C-7/20 as 12.
The yield of 9 was not sufficient to determine the [α]D value for this compound. However, given the stereochemical similarities found for 1-8 and 11-13, we submit that 9 should have the same absolute configuration at C-1/14, C-2/15, and C-7/20 as the previously reported 13, because these compounds likely share a common biosynthesis.
Biosynthetic Incorporation of Bromine
Incorporation of halogens, especially chlorine and bromine, is not uncommon in the secondary metabolism of cyanobacteria.24-26 However, it is important to note that these molecules are often isolated from marine cyanobacteria whereas, the chlorinated cylindrocyclophanes were isolated from a terrestrial cyanobacterium cultured in a freshwater medium (Z). Z medium, like many freshwater, algal culture media, is composed of various inorganic salts dissolved in deionized H2O. Often many of these salts will have a chloride counter ion for the desired nutrient, e.g. FeCl3 for Fe3+ or MgCl2 for Mg2+. An inspection of Z medium protocol used by the UIC culture collection27 did not reveal any major components with a chloride counter ion. It was determined that the main source of chloride was due to the use of HCl to adjust the pH of the medium prior to autoclaving. It is estimated that a typical batch of Z medium, using Tris/HCl, has approximately 10-2 M Cl- as compared to seawater with a Cl- concentration near 0.6 M.
Previous reports of biological halogenation have shown that many of the halogenases have a low specificity for the halide substrate and the halogen preference is typically driven by the relative concentrations of each anion (Cl-, Br- and I-).28, 29 With this information in mind, Nostoc sp. (UIC 10022A) was cultured in varying concentrations of KBr (0.8, 8.4 and 84 mM) in Z medium devoid of Cl-. The cultures were grown under the same environmental conditions as the original 2 L cultures, i.e. 20 °C with 16/8 h light/dark cycle. After eight weeks of growth, the resulting biomass was harvested via centrifugation then freeze-dried. The lyophilized biomass for the KBr cultures were extracted with CH2Cl2:CH3OH (1:1). Each extract was subjected to HPLC-UV-MS analysis and the LC-MS data for all three of the extracts revealed the presence of several brominated cylindrocyclophanes. In particular, the extracted ion chromatogram for m/z 899 revealed a compound with a retention time of 12.82 min. This ion (m/z 899) was selected because it represents the major [M-H]- ion for tetrabrominated cylindrocyclophane A (C36H52Br4O6). The mass spectrum acquired at tR 12.82 min displayed a cluster of ion signals from m/z 895.0454 to 903.0418 with intensities that were consistent with a tetrabrominated molecule. The LC-MS data also indicated the presence of dibrominated cylindrocyclophane A ([M-H]-, m/z 739.2121) and tribrominated cylindrocyclophane A ([M-H]-, m/z 817.1344) at tR 12.43 min as well as dibrominated ([M-H]-, m/z 723.2265) and tribrominated cylindrocyclophane C ([M-H]-, m/z 803.1473) at tR 13.92 min.
All three extracts displayed the presence of brominated cylindrocyclophanes and the extracts were combined to yield 272.0 mg. This combined extract was subjected to C18 solid-phase extraction with a MeOH mobile phase to prepare the material for preparative HPLC. The MeOH extract (170.4 mg) was subjected to multiple rounds of semi-preparative reversed-phase (C18) HPLC to yield 0.4 mg of tetrabrominated cylindrocyclophane A, which we have named cylindrocyclophane AB4 (10).
Cylindrocyclophane AB4 (10) was obtained as white, amorphous solid. The negative mode HRESI-TOF-MS data for 10 indicated a molecular formula of C36H52Br4O6 ([M-H]-, m/z 895.0461) with an isotopic distribution pattern that was consistent with tetrabromination. The 1H NMR spectrum (Table 3) for 10 displayed signals indicaing that this compound followed the same halogenation pattern as 1. In particular, the 1H NMR spectrum for 10 displayed a triplet at δH 5.80 that correlated with δC 48.0 in the HSQC spectrum for 10. These signals were consistent with a dibromomethyl moiety and, considering that all other NMR signals were similar to those for 1, it was deduced that 10 is 30,30,34,34-tetrabromocylindrocyclophane A. The proton and carbon chemical shifts were assigned by comparing the NMR data for 1 with the NMR data for 10. Considering that 10 was isolated from the same organism, and thus produced by the same biosynthetic pathway as 1-4, we submit that 10 would have the same absolute configuration as 1-4.
Structure-activity Relationships
Compounds 1-5,7,9-13 were evaluated to determine inhibition of the 20S proteasome. These compounds displayed a wide range of activity with 1-3 being the most potent (IC50 = 2.55-3.93 μM) (Table 4). The structurally related 11 and 4 displayed IC50 values that were at least seven-fold less potent than 1-3. A common structural feature of 1-3 is the presence of a dichloromethyl moiety (C-30) at the end of the side chain (C-27 to C-30), while both 11 and 4 lack this moiety on either side chain (C-27/31 to C-30/34). These results indicate that the dichloromethyl moiety of 1-3 is important for the biological activity.
Table 4.
Biological Activity of Isolated Cylindrocyclophanes (1-5,7,9-13).
| Compound | R1 | R2 | R3 | R4 | 20S Proteasome IC50 (μM)a |
HT-29 EC50 (μM)b,c |
|---|---|---|---|---|---|---|
| 1 | OH | CHCl2 | OH | CHCl2 | 3.93 ± 0.18 | 2.0 |
| 2 | OH | CH2Cl | OH | CHCl2 | 2.75 ± 0.31 | 0.5 |
| 3 | OH | CH3 | OH | CHCl2 | 2.55 ± 0.11 | 1.7 |
| 4 | OH | CH3 | OH | CH2Cl | 27.6 ± 0.6 | - |
| 5 | OH | CHCl2 | H | CHCl2 | 11.2 ± 1.2 | 2.8 |
| 7 | OH | CH3 | H | CHCl2 | 22.8 ± 2.7 | 0.9 |
| 9 | H | CHCl2 | H | CHCl2 | 44.8 ± 2.0 | - |
| 10 | OH | CHBr2 | OH | CHBr2 | 2.23 ± 0.48 | 0.5 |
| 11 | OH | CH3 | OH | CH3 | 33.9 ± 3.0 | - |
| 12 | OH | CH3 | H | CH3 | 59.3 ± 8.2 | - |
| 13 | H | CH3 | H | CH3 | > 100 | - |
Positive controls:
bortezomib (IC50 2.5 nM) and
camptothecin (EC50 60 nM).
A dash (-) denotes the compound was not evaluated.
A comparison of the biological activity of 11-13 revealed that 11 was twice as potent as 12, while 13 was inactive at a concentration greater than 100 μM. The only structural difference among these molecules is the presence or absence of a hydroxy moiety at C-1 and C-14. The most potent of these three compounds, i.e. 11, has hydroxy moieties at both C-1 and C-14, while the inactive compound 13 lacks both hydroxy moieties. Based upon these results, we submit that the hydroxy moieties at C-1 and C-14 are important for inhibition of the proteasome. This hypothesis is also supported by the fact that 12, which has only one hydroxy moiety at C-1, displayed half the potency of 11. Given that 11 is symmetrical it is possible that both hydroxy moieties (C-1 and C-14) are not required for binding but rather that the C2 axis of symmetry of the molecule would decrease the entropy of binding.
The initial comparison of the structures and related activities for 4 and 3 revealed the importance of the dichloromethyl moiety (C-30/34) and the comparison of 11-13 revealed the importance of the benzylic hydroxy moiety (C-1/14) for the biological activities of these compounds. A comparison of the structures and activities of 3, 5, and 7 was used to join these two “pharmacophores”. The structure of 7 was important to this study since the dichloromethyl (C-30) and the benzylic hydroxy moiety (C-1) were “distal” (Figure 2, dashed line), while the benzylic carbon (C-14) that was “adjacent” (Figure 2, solid line) lacked a hydroxy group. In the structure of 3, both benzylic carbons (C-1 and C-14) possessed an attached hydroxy group. Biological evaluation of 7 (IC50 = 22.8 ± 2.7 μM) revealed that it was more than eight-fold less potent as proteasome inhibitor than 3 (IC50 = 2.55 ± 0.11 μM). Based upon these results, it was apparent that the “adjacent” relationship of the benzylic hydroxy and the dichloromethyl moiety was important for inhibition of the proteasome. The importance of the “adjacent” relationship was also confirmed by the activity of 5 (Figure 2). The difference between the structures of 7 and 5 is that the methyl group (C-34) that is “adjacent” to the benzylic hydroxy moiety (C-1) is dichlorinated in 5, while C-34 is not modified in 7. As with the comparison of 3 and 5, the molecule containing a dichloromethyl moiety “adjacent” to a benzylic hydroxy (5), was more potent (IC50 = 11.2 ± 1.23 μM) than the molecule that lacked this relationship (7, IC50 = 22.8 ± 2.7 μM).
Figure 2.
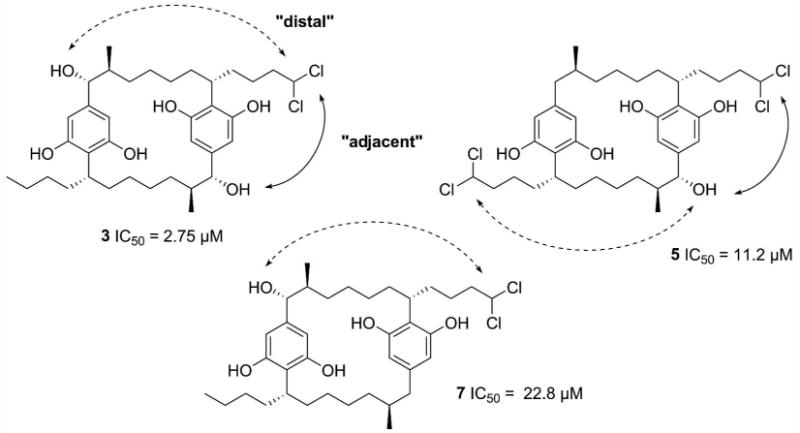
Chemical structures of 3, 5, and 7 with corresponding IC50 values for the 20S proteasome bioassay. The key “adjacent” relationship is shown with a solid double arrow.
The final part of this preliminary SAR investigation was the comparison of 1 and 10. The difference between these two molecules is the halogen present in the dihalogenated methyl moiety, Cl for 1 and Br for 10. Both compounds displayed similar potencies (IC50 = 3.93 ± 0.18 and 2.23 ± 0.48 μM for 1 and 10, respectively), indicating that the type of halogen, i.e. Cl or Br, is not as important to the level of activity as the other structural features described previously.
The cytotoxicity EC50 values (Table 4) for 1-3, 5, 7, and 10 did not reveal any variation that could be used to elucidate any structure-activity relationships. While evaluation of all of the cylindrocyclophanes could produce some variation that could be used for SAR studies, it is important to note that the EC50 values acquired during this project (0.9-2.8 μM) were similar to the cytotoxic potencies reported by Moore et al. (0.5-5 μg/mL) for cylindrocyclophanes A-F and Bui et al. (3.3-5.1 μM) for carbamidocyclophanes A-C.21, 22 Based upon these reports and the data acquired here, we do not anticipate any significant difference in the cytotoxic activity of the chlorinated cylindrocyclophanes as compared to 11-13, thus making any SAR studies difficult. Also, it is important to note that 7, which displayed proteasome inhibition that was more than seven-fold less potent than 3, displayed similar cytotoxicity (EC50 = 0.9 μM and EC50= 1.7 μM, respectively) as 3. Based upon these results, it is unlikely that inhibition of the proteasome is the primary mode of action for the cytotoxicity of these compounds.
Taxonomic Identification
The initial taxonomic identification of UIC 10022A was conducted using traditional techniques, i.e. morphological analysis. The morphological features (see Supporting Information) suggested that this strain was a member of the genus Nostoc,30-32 however the genus Trichormus could not be definitively ruled out solely on morphological analysis. Therefore, it was important to compare genetic material of UIC 10022A with reference strains of the genera Nostoc and Trichormus.
The genetic analysis of UIC 10022A was performed using a 1.2 kb sequence of the 16S rRNA gene. For the phylogenetic comparison, reference sequences were first selected using the list of reference strains found in Bergey's Manual of Systematic Bacteriology.33 Only sequences of at least 1 kb were retrieved from GenBank. For the genus Nostoc, the reference strains were PCC73102, PCC6720, PCC7120, and PCC7423. Strains for the genus Trichormus were also added to allow a phylogenetic comparison of UIC 10022A with that genus.34 In addition, the cryptophycin producing Nostoc strains GSV224 and ATCC53789 and the cylindrocyclophane A-F producing Cylindrospermum lichenforme UTEX2014 were also added.
A BLAST search of the UIC 10022A 16S rRNA sequence was employed to retrieve sequence data for the phylogenetic analysis. Top scoring sequences included in the phylogenetic analysis were from the strains Nostoc sp. PCC7423, Nostoc sp. KK-01, Nostoc muscorum CENA61, Anabaena flos-aquae UTCC64, and Anabaena variabilis ATCC29413. Phylogenetic trees were constructed using neighbor joining (NJ), maximum parsimony (MP), and minimum evolution (ME) methods. In all three cases, the resulting phylogenetic trees had similar topology near the node for UIC 10022A and the Nostoc cluster containing UIC 10022A was identical for all trees with the same bootstrap value. The minimum evolution tree (ME) (Figure 3) revealed that the 16S rRNA sequence for UIC 10022A was a member of a monophyletic clade that included the Nostoc sp. strains PCC7423, KK-01, and CENA61. According to Bergey's Manual of Systematic Bacteriology, PCC7423 is the reference strain for Nostoc cluster 3.3, thus we gave that clade the same designation.33 The phylogenetic tree also revealed that reference strains for the genus Trichromus did form a monophyletic clade, but this cluster was quite distant from Nostoc cluster 3.3. These results support the taxonomic assignment of Nostoc sp. to UIC 10022A.
Figure 3.
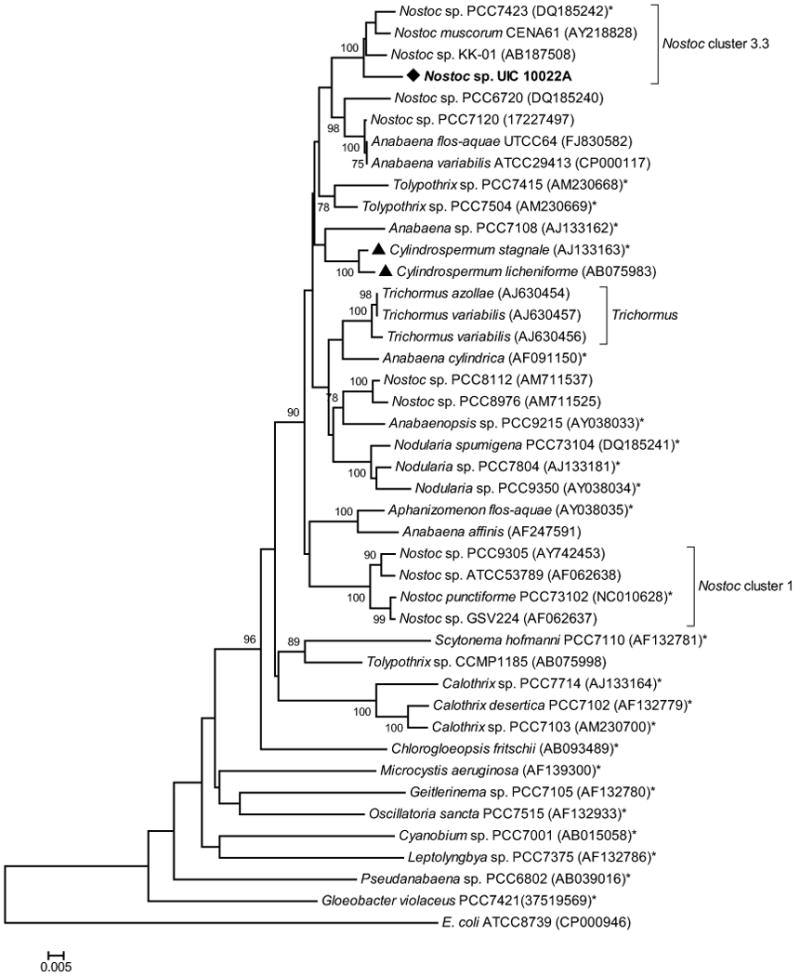
Phylogenetic relationships of 16S rDNA from cyanobacteria. The tree was constructed using maximum evolution (ME) method with a bootstrap consensus of 1000 replicates. Strains denoted with an asterisk (*) are “Bergey's” reference strains.33 Cyanobacterial strains previously reported to produce cylindrocyclophanes are denoted by a triangle (▲). Only bootstrap values greater than or equal to 75% are displayed.
Quite surprisingly, the phylogenetic tree also revealed that Nostoc cluster 3.3 was not part of a strong, monophyletic clade with other “Bergey's” reference Nostoc strains, i.e. PCC73102, PCC6720, and PCC7120. In particular, the node for Nostoc punctiforme PCC73102, reference strain for Nostoc cluster 1,33 was a member of a monophyetic clade that included the cryptophycin producing strains GSV224 and ATCC29413. However, the clade that included both Nostoc clusters 1 and 3.3 also included reference strains from other genera, specifically Tolypothrix spp. PCC7504 and PCC7415, Anabaena sp. PCC7108, and Cylindrospermum stagnale PCC7417. Further study of these organisms, e.g. the phylogenetic analysis of additional genes such as ITS, rpoB, rpoC, and rbcL/rbcX, would be required to determine the taxonomic implications of the heterogeneity found in this clade.
It is also important to note that that both Cylindrospermum strains previously reported to produce cylindrocyclophanes formed a monophyletic clade that was not closely associated with the clade that included Nostoc sp. UIC 10022A. Considering the evolutionary distance between these organisms and the heterogeneity of the group that includes all three cylindrocyclophane producing cyanobacteria, it is possible that the biosynthetic genes were shared via horizontal gene transfer between the Cylindrospermum and Nostoc species. Further genetic analysis of members of both genera would be needed to validate this hypothesis.
Experimental Section
General Experimental Procedures
Optical rotations were determined on a Perkin-Elmer 421 polarimeter. UV spectra were obtained on a Varian Cary 50 Bio spectrophotometer. CD spectra were obtained on a Jasco J-710 spectropolarimeter. IR spectra were obtained on a Jasco FTIR-40 Fourier transform infrared spectrometer. The NMR spectra were acquired at 600 MHz for 1H and 226 MHz for 13C on Bruker DRX600 MHz and Bruker AVII900 MHz NMR spectrometers, respectively. The TOF-MS was acquired on a Shimadzu IT-TOF spectrometer. HPLC separations were performed on a Thermo HPLC system consisting of a P4000 pump and UV1000 UV absorbance detector. HPLC-UV-MS analyses were performed on a Shimadzu Prominence liquid chromatograph with UV-Vis photodiode array detector and the above mentioned IT-TOF mass spectrometer.
Biological Material
A Nostoc sp. (10022A) was isolated from an algal assemblage growing on the soil in a parkway in the 400 block of West Melrose Avenue in Chicago, Illinois (N 41° 56.46′ W 87° 38.39′). The unialgal strain (10022A) was produced through a combination of streak plate and micropipette isolation techniques.16 The strain was cultured in 2 L of Z medium27 in 2.8 L Fernbach flasks and 20 L of Z medium in a 22 L glass carboy, both with sterile aeration. After 6-8 weeks of growth, the cultures were harvested by centrifugation and then freeze dried.
Strain Identification
Cultured material was used for both morphological studies and phylogenetic analysis. Microscopic observation was performed using a Zeiss Axiostar Plus light microscope. The following parameters were utilized for identification: thallus morphology, relative size and shape of vegetative cells, presence and arrangement of heterocytes, presence, relative size of akinetes, trichome polarity, end cell morphology, and colony structure (see Supporting Information). Identification by phenotype was performed according to the system by Komárek et al.30
DNA Extraction, 16S rDNA PCR Amplification, and Sequencing
Prior to DNA extraction, 8 mL of a static culture of Nostoc sp. (UIC 10022A) was combined with 1.5 mL of 100 mM HEPES/NaOH, pH 6.8, 10 mM EDTA and 0.5 mL of 10 mg/mL lysozyme then incubated in a 35 °C water bath for 1 h. After incubation, the cell material was recovered via centrifugation and transferred to a clean 2 mL microcentrifuge tube. DNA was extracted from the resulting cell material using the Wizard Genomic DNA purification kit (Promega). The manufacturer's protocol was modified to include mechanical disruption after the addition of the nuclei lysis solution (step 6). A portion of the 16S rDNA gene was amplified by PCR from the DNA using the cyanobacterial specific primers 106F and 1509R previously described by Nubel et al.35 Reaction mixtures consisted of 1 μL of DNA (100 ng), 4 μL of 5× Phusion HF buffer, 0.4 μL of dNTP mix (10 μM of dATP, dCTP, dGTP, and dTTP), 1 μL of each primer (10 μM), 0.2 μL of Phusion high-fidelity DNA polymerase (New England Biolabs cat. F-530S), and 12.6 μL of H2O for a total volume of 25 μL. The reaction was performed using a Bio-Rad C1000 Thermal Cycler with the following reaction program: initial denaturation for 130 s at 98 °C, 24 cycles of amplification with 10 s at 95 °C, 30 s at 50 °C, and 60 s at 72 °C, and a final elongation of 3 min at 72 °C. PCR products were purified using the MinElute PCR purification kit from Qiagen and sequenced in both directions using the previously described cyanobacterial primers (106F and 1509R) as well as internal primers 359F and 781R.35 The resulting sequence was deposited with NCBI Genbank (acc. no. HM359085).
Phylogenetic Analysis
Phylogenetic analyses were conducted in MEGA4.36 The resulting sequence chromatograms were visually inspected and the total sequence of 1215 nucleotides was aligned with 41 cyanobacterial species obtained from GenBank (http://www.ncbi.nlm.nih.gov), as well as the evolutionary distant E. coli ATCC8739. Multiple sequence alignment was performed via the ClustalW interface of MEGA4 using default gap opening (15) and extension (6.66) penalties. The aligned sequences were used to construct phylogenetic trees in MEGA4. The evolutionary history was inferred using the neighbor-joining (NJ), minimum evolution (ME), and maximum parsimony (MP) methods. For each method, the bootstrap consensus tree inferred from 1000 replicates was taken to represent the evolutionary history of the taxa analyzed.
Extraction and Dereplication
Two 2 L cultures of 10022A (0.92 g lyophilized biomass) were extracted with 1:1 CH2Cl2:CH3OH to yield 110.9 mg of extract. The extract (83.5 mg) was subjected to flash chromatography utilizing HP20SS Diaion resin and a H2O:IPA step gradient (1:0, 4:1, 3:2, 2:3, 3:7, 1:4, 1:9, 0:1) to yield eight fractions. A portion (5.0 mg) of fraction 4 (8.50 mg) was subjected to semi-preparative HPLC (Agilent Series 1100, Phenomenex Onyx C18, 4.6 × 100 mm, H2O:MeCN) to yield 32 fractions collected in a 96 deep well plate. The collected material was dried in vacuo and resuspended in MeOH to create daughter plates for biological evaluation and ESI-TOF-MS analysis. The remaining material was again dried in vacuo and select fractions were resuspended in MeOH- d4 for 1H NMR analysis.
Isolation of Chlorinated Cylindrocyclophanes
A 20 L culture of 10022A (12.76 g lyophilized biomass) yielded 557.8 mg of extract (CH2Cl2:CH3OH 1:1). This extract was subjected to flash chromatography utilizing HP20SS Diaion resin and a H2O:IPA gradient to yield eight fractions. These fractions were subjected to HPLC-ESI-TOF-MS analysis to determine the presence of cylindrocyclophanes. Multiple rounds of semi-preparative reversed-phase HPLC of F4 (Varian Dynamax C18, 10×250 mm, H2O:MeOH) yielded 4.7, 0.5, 1.6, 0.3, 0.8, 1.1, 2.0, 0.5, 0.1, 0.3, 0.3, and 0.1 mg of 1-9 and 11-13, respectively. The elution order of the cylindrocyclophanes was 4, 11, 2, 3, 1, 8, 12, 6, 7, 5, 13, and 9.
Culture in KBr Enriched Media
Three separate 2 L cultures of 10022A were grown in the presence of 0.1, 1.0 and 10.0 g/L KBr, respectively. The base Z medium for each culture was modified by substituting Tris/HCl with Tris/H2SO4 to reduce the concentration of chloride. The cultures were grown under the same environmental conditions as the original 2 L cultures. After eight weeks of growth, the resulting biomass was harvested via centrifugation and freeze dried. The lyophilized biomass for the 0.1, 1.0 and 10.0 g/L KBr cultures (1.03, 1.05 and 0.49 g, respectively) was extracted with CH2Cl2:CH3OH (1:1) to yield 95.4, 113.8 and 106.8 mg, respectively. For each extract, 5 μL of a 10 mg/mL MeOH solution was subjected to HPLC-UV-MS analysis (Shimadzu Prominence liquid chromatograph, Varian Microsorb 100-5 C18, 2.0 × 250 mm, 5% MeOH(aq) with 0.1% acetic acid:MeOH). Chromatographic data were acquired for UV-Vis absorbance (photodiode array detector, λ: 200-700 nm) and HRESI-TOF-MS (scan events: (+) m/z 100-500, (+) m/z 400-2000, (-) m/z 100-500, and (-) m/z 400-2000).
Isolation of Cylindrocyclophane AB4
The extracts of the three KBr cultures were combined to yield 272.0 mg. This combined extract was subjected to C18 solid phase extraction with a MeOH mobile phase to prepare the material for preparative HPLC. The MeOH extract (170.4 mg) was subjected to multiple rounds of semi-preparative reversed-phase HPLC (Varian Dynamax C18, 10×250 mm, H2O:MeOH) to yield 0.4 mg of 10.
Cylindrocyclophane A4 (1)
White amorphous powder; [α]D-26 (c 0.2, MeOH); UV(MeOH) λmax (log ε) 227 (3.88), 274 (3.17) nm; CD (c 0.006, MeOH) λmax (Δε) 207 (6.4), 218 (-3.2), 247 (-0.13), 261 (0.21), 281 (-1.0) nm; IR (neat) νmax 3266 (br), 2926, 1594, 1431, 1018, 951, 838, 804 cm-1; 1H and 13C NMR see Table 1; HR-ESI-TOF-MS (-) m/z 719.2392 [M-H]- (calcd for C36H51Cl4O6, 719.2445).
Cylindrocyclophane A3 (2)
White amorphous powder; [α]D -27 (c 0.06, MeOH); UV(MeOH) λmax (log ε) 217 (4.04), 279 (3.18) nm; IR (neat) νmax 3283 (br), 2928, 2856, 1594, 1431, 1016, 950, 842 cm-1; 1H and 13C NMR see Table 1; HR-ESI-TOF-MS (-) m/z 685.2877 [M-H]- (calcd for C36H52Cl3O6, 685.2835).
Cylindrocyclophane A2 (3)
White amorphous powder; [α]D -26 (c 0.07, MeOH); UV(MeOH) λmax (log ε) 227 (3.88), 274 (3.21) nm; IR (neat) νmax 3259 (br), 2927, 2855, 1594, 1431, 1372, 1017, 949, 843 cm-1; 1H and 13C NMR see Table 1; HR-ESI-TOF-MS (-) m/z 651.3257 [M-H]- (calcd for C36H53Cl2O6, 651.3225).
Cylindrocyclophane A1 (4)
White amorphous powder; UV(MeOH) λmax (log ε) 212 (3.79), 274 (2.62) nm; IR (neat) νmax 3401 (br), 2927, 2855, 1629, 1430, 1375, 1232, 1091, 1016, 986, 828 cm-1; 1H and 13C NMR see Table 1; HR-ESI-TOF-MS (-) m/z 617.3639 [M-H]- (calcd for C36H54ClO6, 617.3818).
Cylindrocyclophane C4 (5)
White amorphous powder; [α]D -40 (c 0.03, MeOH); UV(MeOH) λmax (log ε) 229 (3.66), 270 (2.53) nm; IR (neat) νmax cm-1; 1H and 13C NMR see Table 2; HR-ESI-TOF-MS (-) m/z 703.2469 [M-H]- (calcd for C36H51Cl4O5, 703.2496).
Cylindrocyclophane C3 (6)
White amorphous powder; [α]D -39 (c 0.05, MeOH); UV(MeOH) λmax (log ε) 216 (3.74), 274 (2.94) nm; IR (neat) νmax 3310 (br), 2930, 1629, 1429, 1374, 1011 cm-1; 1H and 13C NMR see Table 2; HR-ESI-TOF-MS (-) m/z 669.2858 [M-H]- (calcd for C36H52Cl3O5, 669.2886).
Cylindrocyclophane C2 (7)
White amorphous powder; [α]D -37 (c 0.15, MeOH); UV(MeOH) λmax (log ε) 227 (3.83), 274 (3.14) nm; IR (neat) νmax 3233 (br), 2925, 2855, 1512, 1429, 1015, 949, 854 cm-1; 1H and 13C NMR see Table 2; HR-ESI-TOF-MS (-) m/z 635.3247 [M-H]- (calcd for C36H53Cl2O5, 635.3276).
Cylindrocyclophane C1 (8)
White amorphous powder; [α]D -44 (c 0.05, MeOH); UV(MeOH) λmax (log ε) 227 (3.33), 274 (2.78) nm; IR (neat) νmax 3319 (br), 2931, 2859, 1631, 1458, 1429, 1374, 1090, 1055, 1010, 831 cm-1; 1H and 13C NMR see Table 2; HR-ESI-TOF-MS (-) m/z 601.3643 [M-H]- (calcd for C36H54ClO5, 601.3665).
Cylindrocyclophane F4 (9)
White amorphous powder; UV(MeOH) λmax 219, 270 nm; IR (neat) νmax cm-1; 1H and 13C NMR see Table 3; HR-ESI-TOF-MS (-) m/z 687.2618 [M-H]- (calcd for C36H51Cl4O4, 687.2547).
Cylindrocyclophane AB4 (10)
White amorphous powder; [α]D -45 (c 0.04, MeOH); UV(MeOH) λmax (log ε) 227 (3.90), 274 (3.54) nm; IR (neat) νmax 3318 (br), 2923, 2854, 1595, 1430, 1374, 1013, 832 cm-1; 1H and 13C NMR see Table 3; HR-ESI-TOF-MS (-) m/z 895.0461 [M-H]- (calcd for C36H51Br4O4, 895.0425).
20S Proteasome Bioassay
Evaluation of inhibition of the 20S proteasome was performed as previously described.16
HT-29 assay
Cytotoxicity against the HT-29 cancer cell line was performed according to established protocols.37
Supplementary Material
Acknowledgments
This research was supported by P01 CA125066 from NCI/NIH. We thank the Research Resources Center (RRC) at UIC for high resolution mass spectrometry. We also thank Dr. H.-B. Chai from the Ohio State University for performing the HT-29 assay and Dr. Kroll from the Research Triangle Institute for performing the MCF7, H460, and SF268 assays. We are also grateful to Mr. N. Engene from Scripps Institute of Oceanography and the UIC DNA Services facility for assistance with the genetic sequencing and analysis.
Footnotes
Supporting Information Available: The 1H, gCOSY, gHSQC and gHMBC spectra of 1 - 10; The DEPTQ spectra of 1, 7 and 10; The gTOCSY spectrum of 7; Photomicrograph and morphological description of Nostoc sp. UIC 10022A. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Welker M. Cyanobacterial Hepatotoxins: Chemistry, Biosynthesis, and Occurrence. In: Botana LM, editor. Seafood and Freshwater Toxins. CRC Press; Boca Raton, FL: 2008. pp. 825–843. [Google Scholar]
- 2.Gademann K, Portmann C. Curr Org Chem. 2008;12:326–341. [Google Scholar]
- 3.Burja AM, Banaigs B, Abou-Mansour E, Burgess JG, Wright PC. Tetrahedron. 2001;57:9347–9377. [Google Scholar]
- 4.König GM, Kehraus S, Seibert SF, Abdel-Lateff A, Mueller D. ChemBioChem. 2006;7:229–238. doi: 10.1002/cbic.200500087. [DOI] [PubMed] [Google Scholar]
- 5.Tan LT. Phytochemistry. 2007;68:954–979. doi: 10.1016/j.phytochem.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz RE, Hirsch CF, Sesin DF, Flor JE, Chartrain M, Fromtling RE, Harris GH, Salvatore MJ, Liesch JM, Yudin K. J Ind Microbiol. 1990;5:113–123. [Google Scholar]
- 7.Wagner MM, Paul DC, Shih C, Jordan MA, Wilson L, Williams DC. Cancer Chemother Pharmacol. 1999;43:115–125. doi: 10.1007/s002800050871. [DOI] [PubMed] [Google Scholar]
- 8.Edelman MJ, Gandara DR, Hausner P, Israel V, Thornton D, DeSanto J, Doyle LA. Lung Cancer. 2003;39:197–199. doi: 10.1016/s0169-5002(02)00511-1. [DOI] [PubMed] [Google Scholar]
- 9.D'Agostino G, Del Campo J, Mellado B, Izquierdo MA, Minarik T, Cirri L, Marini L, Perez-Gracia JL, Scambia G. Int J Gynecol Cancer. 2006;16:71–76. doi: 10.1111/j.1525-1438.2006.00276.x. [DOI] [PubMed] [Google Scholar]
- 10.Golakoti T, Yoshida WY, Chaganty S, Moore RE. J Nat Prod. 2001;64:54–59. doi: 10.1021/np000316k. [DOI] [PubMed] [Google Scholar]
- 11.Ploutno A, Carmeli S. J Nat Prod. 2000;63:1524–1526. doi: 10.1021/np0002334. [DOI] [PubMed] [Google Scholar]
- 12.Mehner C, Muller D, Kehraus S, Hautmann S, Gutschow M, Konig GM. ChemBioChem. 2008;9:2692–2703. doi: 10.1002/cbic.200800415. [DOI] [PubMed] [Google Scholar]
- 13.Okino T, Qi S, Matsuda H, Murakami M, Yamaguchi K. J Nat Prod. 1997;60:158–161. doi: 10.1021/np970146k. [DOI] [PubMed] [Google Scholar]
- 14.Ploutno A, Carmeli S. Tetrahedron. 2002;58:9949–9957. [Google Scholar]
- 15.Murakami M, Sun Q, Ishida K, Matsuda H, Okino T, Yamaguchi K. Phytochemistry. 1997;45:1197–1202. [Google Scholar]
- 16.Chlipala G, Mo S, de Blanco EJC, Ito A, Bazarek S, Orjala J. Pharm Biol. 2009;47:53–60. doi: 10.1080/13880200802415483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burger AM, Seth AK. Eur J Cancer. 2004;40:2217–2229. doi: 10.1016/j.ejca.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Khan T, Stauffer JK, Williams R, Hixon JA, Salcedo R, Lincoln E, Back TC, Powell D, Lockett S, Arnold AC, Sayers TJ, Wigginton JM. J Immunol. 2006;176:6302–6312. doi: 10.4049/jimmunol.176.10.6302. [DOI] [PubMed] [Google Scholar]
- 19.Ciechanover A. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 20.Adams J. Cancer Cell. 2004;5:417–421. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 21.Moore BS, Chen J, Patterson GML, Moore RE. Tetrahedron. 1992;48:3001–3006. [Google Scholar]
- 22.Bui HTN, Jansen R, Pham HTL, Mundt S. J Nat Prod. 2007;70:499–503. doi: 10.1021/np060324m. [DOI] [PubMed] [Google Scholar]
- 23.Moore BS, Chen J, Patterson GML, Moore RE, Brinen LS, Kato Y, Clardy J. J Am Chem Soc. 1990;112:4061–4063. [Google Scholar]
- 24.Marquez BL, Watts KS, Yokochi A, Roberts MA, Verdier-Pinard P, Jimenez JI, Hamel E, Scheuer PJ, Gerwick WH. J Nat Prod. 2002;65:866–871. doi: 10.1021/np0106283. [DOI] [PubMed] [Google Scholar]
- 25.Orsini MA, Pannell LK, Erickson KL. J Nat Prod. 2001;64:572–577. doi: 10.1021/np000452p. [DOI] [PubMed] [Google Scholar]
- 26.Orjala J, Gerwick WH. J Nat Prod. 1996;59:427–430. doi: 10.1021/np960085a. [DOI] [PubMed] [Google Scholar]
- 27.Falch BS, König GM, Wright AD, Sticher O, Angerhofer CK, Pezzuto JM, Bachmann H. Planta Med. 1995;61:321–328. doi: 10.1055/s-2006-958092. [DOI] [PubMed] [Google Scholar]
- 28.Eustaquio AS, Pojer F, Noel JP, Moore BS. Nat Chem Biol. 2008;4:69–74. doi: 10.1038/nchembio.2007.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner C, El Omari M, Konig GM. J Nat Prod. 2009;72:540–553. doi: 10.1021/np800651m. [DOI] [PubMed] [Google Scholar]
- 30.Komárek J, Komárková J, Kling H. Filamentous Cyanobacteria. In: Wehr JD, Sheath RG, editors. Freshwater Algae of North America. Academic Press; San Diego: 2003. pp. 117–196. [Google Scholar]
- 31.Herdman M, Castenholz RW, Rippka R. Form-genus VIII Nostoc Vaucher 1803. In: Boone DR, Castenholz RW, editors. Bergey's Manual of Systematic Bacteriology. Vol. 1. Springer; New York: 2001. pp. 575–580. [Google Scholar]
- 32.Komárek J, Anagnostidis K. Arch Hydrobiol Suppl. 1989;82:247–345. [Google Scholar]
- 33.Castenholz RW. Phylum BX. Cyanobacteria. In: Boone DR, Castenholz RW, editors. Bergey's Manual of Systematic Bacteriology. Vol. 1. Springer; New York: 2001. pp. 473–599. [Google Scholar]
- 34.Rajaniemi P, Hrouzek P, Kastovska K, Willame R, Rantala A, Hoffmann L, Komárek J, Sivonen K. Int J Syst Evol Microbiol. 2005;55:11–26. doi: 10.1099/ijs.0.63276-0. [DOI] [PubMed] [Google Scholar]
- 35.Nubel U, GarciaPichel F, Muyzer G. Appl Environ Microbiol. 1997;63:3327–3332. doi: 10.1128/aem.63.8.3327-3332.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamura K, Dudley J, Nei M, Kumar S. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 37.Seo EK, Kim NC, Mi Q, Chai H, Wall ME, Wani MC, Navarro HA, Burgess JP, Graham JG, Cabieses F, Tan GT, Farnsworth NR, Pezzuto JM, Kinghorn AD. J Nat Prod. 2001;64:1483–1485. doi: 10.1021/np0103158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.