

Abstract
The potential threat of a pandemic caused by H5N1 influenza A viruses has stimulated increased research on developing new antivirals against influenza A viruses. Current antivirals are directed against the M2 protein (named adamantanes) and the neuraminidase (named zanamivir and oseltamivir). However, both seasonal and H5N1 influenza A viruses have developed resistance to adamantanes and oseltamivir. Accordingly, new antivirals directed at the M2 and neuraminidase proteins, and against the hemagglutinin protein, are being developed. In addition, elucidation of the structural basis for several crucial functions of other viral proteins (specifically the non-structural NS1A protein, the nucleoprotein and the viral polymerase) has identified novel targets for the development of new antivirals. Here, we describe how functional and structural studies led to the discovery of these novel targets and also how structural information is facilitating the rational design of new drugs against previously identified targets.
Introduction
Influenza A and B viruses are enveloped RNA viruses, of which the genomes are in the form of eight segments [1] (Box 1). These viruses cause a highly contagious respiratory disease in humans, resulting in ~36 000 deaths and >100 000 hospitalizations in the USA annually (www.cdc.gov/flu). Influenza A viruses, but not influenza B viruses, are responsible for the periodic widespread epidemics, or pandemics, that have caused high mortality rates [2]. The most devastating pandemic occurred in 1918, which caused an estimated 40 million deaths worldwide [3]. Less devastating pandemics occurred in 1957 and 1968. H5N1 influenza A viruses (commonly known as ‘avian flu’), which are highly pathogenic in humans, are candidates for the next pandemic influenza virus [4,5] (Box 2). H5N1 viruses were first transmitted from chickens to humans in 1997 in Hong Kong, killing 6 out of 18 infected people [6,7]. After all the chickens in the poultry markets in Hong Kong were culled, H5N1 viruses continued to circulate in poultry markets in China, but transmission to humans was not documented until 2003 when fatal transmissions to humans resumed [8]. The human mortality rate has been high (~65%), resulting in >240 deaths. H5N1 viruses in avian species have now spread from Asia to Europe and Africa (www.who.int/csr/disease/avian_influenza/en/index.html). Fortunately, H5N1 viruses have not yet acquired the ability for efficient human-to-human transmission.
Box 1. Influenza virus genome and proteins.
Influenza A and B viruses are enveloped viruses that contain negative-stranded RNA genomes comprising eight RNA segments [1]. Because the viral genomes are of negative-strand polarity, the genome RNA segments must be transcribed by a virion-associated RNA-dependent RNA polymerase to produce the plus-strand viral mRNAs needed to initiate virus replication. Figure I shows a schematic representation of influenza A virus and its genome. The three largest genome segments encode the three subunits of the polymerase: PB1, PB2 and PA. The segment encoding PB1 also encodes a small nonstructural protein, PB1-F2, which has apoptotic functions. The intermediate-sized segments encode the hemagglutinin (HA), the neuraminidase (NA) and the nucleocapsid (NP) proteins. HA, the major surface protein of the virus, binds to sialic-acid-containing receptors on host cells, and is the protein against which neutralizing antibodies are produced. The NA viral surface protein removes sialic acid from glycoproteins. One of its major functions is to remove sialic acid during virus budding from the cell surface and from the HA and NA of the newly assembled virions, thereby eliminating aggregation of budding virions on the cell surface. NP protein molecules are bound at regular intervals along the entire length of each of the genomic RNAs to form viral ribonucleoproteins (vRNPs) and also have essential functions in viral RNA replication. The seventh genomic RNA segment encodes two proteins: M1 (matrix protein) and M2. The M1 protein underlies the viral lipid membrane and is thought to interact with the genomic vRNPs and with the inner (cytoplasmic) tails of HA and NA. The M2 protein is an ion channel protein that is essential for the uncoating of the virus. The smallest segment encodes two proteins, NS1A and NS2. The NS2 protein, also called NEP, mediates the export of newly synthesized viral RNPs from the nucleus to the cytoplasm. The NS1A protein, which is not incorporated into virions, is a multi-functional protein that has an integral role in suppressing host antiviral responses. We denote the NS1 protein of influenza A virus as the NS1A protein to distinguish it from the NS1 protein of influenza B virus, which can then be denoted as the NS1B protein. The sizes of both the genome segments and their encoded proteins differ between A and B viruses [1]. There are also other differences; for example, influenza B viruses do not have a M2 protein, and the NS1B protein does not bind the host proteins that bind to the NS1A protein (see main text).
Box 2. Influenza virus strains.
Influenza A and B viruses are enveloped RNA viruses in the Orthomyxoviridae family [1]. Influenza A viruses infect a wide number of avian and mammalian species, whereas influenza B viruses infect only humans. Influenza A viruses are organized into subtypes based on their surface hemagglutinin (HA) and neuraminidase (NA). Sixteen HA (H1 to H16) and nine NA (N1 to N9) subtypes are currently identified. The H3N2 and H1N1 influenza A subtypes (along with influenza B viruses) are responsible for recent seasonal influenza epidemics. The pathogenic influenza A virus that has been transmitted from avian species to humans since 1997 (commonly known as ‘avian flu’) is a H5N1 strain. A specific influenza strain is named according to a standard convention in the following order: virus type (the source organism of avian strains is also often specified), geographic origin, strain number, year and virus subtype; for example, influenza A/Udorn/307/1972 (H3N2) virus.
The primary means for controlling influenza virus epidemics has been vaccination with inactivated or live-attenuated virus [2]. The hemagglutinin (HA) of the virus in the vaccine elicits an immune response that neutralizes virus infectivity. The antigenic structures of the HAs of both influenza A and B viruses undergo antigenic drift, which results from the selection of mutant viruses that evade antibodies directed against the predominant antigenic type of the HA circulating in the human population. Mutant viruses are readily generated because the viral RNA polymerase has no proof-reading function. Because of antigenic drift, the vaccine has to be reformulated each year.
Pandemic influenza A viruses can apparently arise by two different mechanisms. In one mechanism, influenza A viruses undergo a process called antigenic shift, the reassortment of genomic RNA segments between human and avian influenza A virus strains, resulting in a new (potentially pandemic) virus encoding a novel avian-type HA that is immunologically distinct [2]. The influenza A viruses containing the H2 and H3 HA subtypes that caused pandemics in 1957 and 1968, respectively, resulted from the reassortment of avian and human genomic RNA segments. Influenza B viruses cannot undergo antigenic shift because they infect only humans. A second mechanism for generating pandemic influenza A viruses has been proposed based on the finding that all eight genomic RNAs of the 1918 pandemic virus contain avian-like sequences [9]. This finding suggested that all eight genomic RNAs were derived from an avian virus and that such a progenitor virus then underwent multiple mutations in the process of adapting to the mammalian host. H5N1 viruses, which contain only avian genes, seem to be undergoing this route for acquiring pandemic capability in humans [4,5]. As is the case for other avian HAs, humans have no immunological protection against the H5 HA subtype.
Antivirals have been used for both prophylactic and therapeutic treatments during seasonal epidemics [10]. For example, prophylactic treatment has been used for individuals at high risk who have not been vaccinated, such as the elderly in nursing homes and hospital personnel who could spread infection to patients. The most effective therapeutic treatment of patients requires early diagnosis because antivirals are most effective in alleviating symptoms if given within the first 30 hours after infection. If a H5N1 pandemic occurs, effective control of such a pandemic will require the use of antiviral drugs because it is not likely that sufficient amounts of an effective vaccine will be available, particularly in the early phase of a fast-spreading pandemic [11–13]. Antivirals can be stockpiled and, if used appropriately, should limit the spread of pandemic influenza virus. Importantly, the strategies that have been proposed for the use of antivirals to stem a H5N1 pandemic [11–13] could lead to more common and effective uses of antivirals during annual epidemics.
Current antivirals are directed against the M2 protein (these antivirals are known as adamantanes) and neuraminidase (NA; antivirals known as zanamivir and oseltamivir) [14–18]. However, both seasonal and H5N1 influenza A viruses have developed resistance to adamantanes and oseltamivir [19–26]. Consequently, new antivirals directed at these two viral proteins, and against HA, are being developed. In addition, novel targets for the development of new antivirals have been identified in the non-structural NS1A protein, the nucleoprotein (NP) and the viral polymerase. Here, we describe how functional and structural studies led to the discovery of these novel targets, and also how structural information is facilitating the rational design of new drugs against previously identified targets.
Current antiviral targets
M2 protein
Adamantanes (amantadine and rimantadine, marketed under the trade names Symmetrel and Flumadine, respectively) are directed against the M2 protein, a 97-amino-acid protein that is assembled into a tetramer that spans the viral membrane [14,15] (Figure 1). The tetramer possesses low pH-activated (H+)-ion-channel activity that is crucial for uncoating of influenza A virions, which enter cells via endosomes [14,15]. The M2 protein channel promotes the influx of H+ ions into the interior of the virions, thereby disrupting protein–protein interactions, specifically including those between the M1 protein and viral ribonucleoproteins (vRNPs). Two key residues (His37 and Trp41) within this tetrameric helical channel act as a pH sensor and a proton (H+ ion) gate, respectively. After the vRNPs are released from the endosomes into the cytoplasm, they are transported to the nucleus where they catalyze the initial transcription of the viral genome. Adamantanes inhibit M2-ion-channel activity and, as a result, the M1 protein remains associated with the released vRNPs, thereby inhibiting their nuclear import.
Figure 1.

Current structures of the influenza A virus M2 protein channel in complex with either amantadine or rimantadine. (a) X-ray structure (3.5 Å resolution) of the TM domain of M2 (residues 22–46) tetramer in complex (1:1) with amantadine in octyl-β-D-glucopyranoside detergent, pH 5.3 [27]. (b) Solution NMR structure of the TM domain and additional C-terminal region of M2 (residues 18–60) tetramer in complex (1:4) with rimantadine in dihexanoyl phosphatidylcholine detergent micelles, pH 7.5 [28]. The four rimantadine molecules are outside the channel. (c) Solid-state NMR structure of the TM domain of M2 (residues 22–46) tetramer in complex (1:1) with amantadine in dilauroyl phosphatidylcholine bilayers, pH 7.5 [29]. For each structure, side chains of Ser31 (red), His37 (cyan) and Trp41 (yellow) are shown, along with bound amanatadine or rimantadine (grey with nitrogen in blue; circled).
Virus mutants resistant to adamantanes readily emerge and are as fit as the wild-type virus [19,20]. In the 2005–2006 influenza season, 90.5% of the H3N2 viruses and 15.5% of the H1N1 viruses analyzed were resistant to adamantanes [21]; many human isolates of H5N1 viruses are also resistant [22]. A single substitution of any one of five amino acids in the M2 transmembrane (TM) domain can confer resistance [19,20]. In the 2005–2006 influenza season, the substitution of Asn for Ser at position 31 in the M2 protein was the predominant change that conferred resistance [21].
Several recent 3D structures of the M2 channel in the presence of one of the adamantane drugs have been determined using X-ray crystallography and NMR spectroscopy [27–29] (Figure 1). The X-ray structure [27] and one NMR structure [29] indicate that the M2 ion channel TM domain binds to a single amantadine molecule plugging the pore in the vicinity of Ser31 (Figure 1a,c). The position of amantadine in the pore suggested that new drugs that target different regions of the pore would be less susceptible to the generation of resistant mutants. In contrast, another NMR structure [28] of the closed state of the M2 ion channel suggested a totally different, allosteric mechanism for inhibition by rimantadine. In this model, the drug binds at four equivalent sites in the vicinity of the Trp41 proton gate, but on the outside of the TM helices, thereby stabilizing the closed state of the channel and inhibiting its opening (Figure 1b). Subsequent functional results have favored the first model [30]. The key question is whether these new structures taken together will enable investigators to develop new antiviral drugs against the M2 protein that are less susceptible to the development of resistant virus mutants. To this end, researchers are screening for chemicals that bind to mutant forms of M2 that are not inhibited by adamantanes.
NA
The first antivirals directed against NA were rationally designed on the basis of the X-ray crystal structure of N2-type NAs (i.e. N2 and N9) [16,31,32]. NA is a tetramer, and each monomer has a sialic-acid-binding pocket lined by amino acids that are conserved in all influenza A and B virus strains. A sialic acid derivative in which the 4-hydroxyl group of sialic acid is replaced with a guanidinyl group binds to this pocket tighter than sialic acid itself. The resulting compound, zanamivir (commercially called Relenza), effectively inhibits the NA of influenza A and B viruses and is orders of magnitude less active against non-influenza neuraminidases. Zanamivir must be delivered by an oral inhaler.
Subsequently, a different compound containing a lipophilic side chain was synthesized (oseltamivir, commercially called Tamiflu), which is administered orally as the prodrug for oseltamivir carboxylate, the actual NA inhibitor [17,18]. Pathogenic H5N1 viruses resistant to oseltamivir were isolated from patients undergoing treatment with this drug [23–25]. In addition, oseltamivir-resistant H1N1 viruses emerged globally in the 2007–2009 influenza seasons in the absence of exposure of patients to the drug [26]. All these resistant viruses had the same amino acid substitution, His274Tyr, in their NA protein, which had no apparent effect on the fitness of the virus. The H5N1 and H1N1 viruses with the His274Tyr mutant NA remained susceptible to zanamivir, demonstrating that even if such mutant viruses became predominant at least one NA inhibitor would still be potent. In addition, a recent structural analysis has shown that type 1 NAs (i.e. N1, N4 and N8), unlike the N2 NA that was used to design oseltamivir and zanamivir, have a large cavity adjacent to the sialic-acid-binding site [33]. This cavity can potentially be exploited for the design of new antivirals directed at type 1 NAs by adding additional chemical groups to existing NA inhibitors. Such antivirals would be expected to be effective both against currently circulating human H1N1 viruses and pathogenic H5N1 viruses.
HA
The HA of influenza A viruses is a homotrimer that binds to sialic receptors on cells [1,34]. The low pH in the endosomes causes irreversible conformational changes of the HA [35] that lead to the fusion of the viral and cellular membranes and hence uncoating of the virus. Chemicals that inhibit these conformational changes inhibit virus replication [36–38]. Insight into ways to improve the activity of such inhibitors was provided by the crystal structures of two HAs in complex with one such inhibitor, tert-butyl hydroquinone (TBHQ) [39]. This compound binds in a hydrophobic pocket at the interface between HA monomers, thereby stabilizing the neutral pH conformation of HA. The way that TBHQ fits into this pocket suggests that specific modifications of this chemical would improve binding affinity, potentially leading to an effective antiviral drug. The HAs used for this crystal structure fall into the phylogenetic group of HAs that includes the H3 HA of viruses currently circulating in humans. However, the TBHQ-binding pocket is absent from the HAs of the second phylogenetic HA group that includes both the H1 HA of viruses currently circulating in humans and the H5 HA of pathogenic H5N1 viruses. Other chemicals inhibit membrane fusion by the HAs of this second phylogenetic group [38], and crystal structures of these HAs in complex with their inhibitory compounds might lead to the design of antiviral drugs against viruses containing these HAs.
Emerging antiviral targets
NS1A protein
The NS1 protein from influenza A (NS1A) is a small (230–237 residue), dimeric, multi-functional protein that has a crucial role in the ability of the virus to evade the antiviral response of the infected cell [40,41]. The protein comprises two domains: (i) the N-terminal (residues 1–73) RNA-binding domain (RBD), and (ii) a C-terminal (residues 74–230/237) effector domain (ED) [40,41]. The NS1A RBD binds to double-stranded RNA (dsRNA) in a non-sequence-specific manner [42–45]. The primary, if not only, role of NS1A dsRNA-binding activity is the inhibition of the 2′–5′ oligo A synthetase/RNase L pathway that is induced by interferon-α/β (IFN-α/β) [46]. The C-terminal ED of NS1A interacts with several host cellular proteins, including: (i) the cellular 30-kDa subunit of the cleavage and polyadenylation specificity factor (CPSF30), thereby inhibiting the 3′ end processing of cellular pre-mRNAs and, hence, the production of all cellular mRNAs including IFN-β mRNA; (ii) the p85β subunit of phosphatidylinositol-3 (PI3) kinase, resulting in the activation of PI3 kinase signaling; and (iii) protein kinase R (PKR), resulting in the inhibition of PKR activation that would otherwise inhibit virus replication [47–52].
The dsRNA-binding surface of the RBD is an attractive target for antiviral drug development. Solution NMR and X-ray crystal structures of the isolated RBD of the NS1A protein of influenza A/Udorn/307//72 (Ud) virus (H3N2) revealed a unique six-helical symmetric head-to-tail homodimer structure [53,54] (Figure 2a). Only one amino acid, Arg38/Arg38′, is absolutely required for dsRNA binding [44]. A mutant Ud virus expressing a NS1A protein in which Arg at position 38 was replaced with an Ala residue is highly attenuated [46], demonstrating that dsRNA-binding activity is required for efficient virus replication. Several basic residues including Arg38/Arg38′, are located in symmetry-related, antiparallel helices (α2 and α2′) on one face of the protein [53,54]. An analogous six-helical homodimer motif was observed in the X-ray crystal structure of the RBD of the NS1B protein of an influenza B virus [55]. The dsRNA-binding surfaces in both NS1A and NS1B are defined by tracks of highly conserved basic and hydrophilic residues that are complementary to the phosphate backbone of canonical A-form dsRNA, as supported by NMR chemical shift perturbation data for the Ud NS1A(1–73):dsRNA complex [55] (Figure 2b). This binding mode was confirmed by an X-ray crystal structure of the complex of a NS1A RBD and 19-base-pair dsRNA [56] (Figure 2c). This structure shows that the NS1A RBD interacts with backbone atoms and 2′-OH moieties along the major groove of the RNA duplex. Numerous hydrophilic or charged residues on the binding face of NS1A contribute to dsRNA binding, particularly Arg38/Arg38′, which penetrates into the helix, forming hydrogen bonds with phosphates on both strands. An important feature of the NS1A dsRNA-binding face is a pocket consisting of numerous hydrophilic and hydrophobic residues. Chemicals that specifically bind to this pocket and interfere with the interaction of NS1A residues with dsRNA would inhibit virus replication, making this pocket an inviting target for antiviral drug design.
Figure 2.

Potential antiviral target in the NS1A dsRNA-binding domain. (a) Ribbon representation of the solution NMR structure of Ud NS1A(1–73) [53]. NS1A and NS1A′ subunits are shown in green and cyan, respectively, and the side chains of Arg38 and Arg38′ (R38 and R38′) are in red. α2 and α2′ denote the antiparallel helices; C stands for C terminus. (b) Space-filling model of the Ud NS1A(1–73), showing tracks of conserved residues on the RNA-binding face of the protein [55]. Residues are colored according to their degree of conservation across the entire NS1A family, ranging from highest (purple) to lowest (cyan). The separation across the basic edges of the conserved tracks corresponds to 10 Å, the width of the major groove in dsRNA. The pocket on the RNA-binding face is indicated. Reproduced, with permission, from Ref. [55]. (c) The crystal structure of NS1A(1–70) from influenza A/Puerto Rico/8/34 (H1N1) in complex with a 19-bp dsRNA duplex [56]. Ribbon representation of NS1A(1–70)–dsRNA showing the major grove mode of binding in the complex. NS1A RBD subunits are colored as in panel (a); the dsRNA backbone is orange. Reproduced, with permission, from Ref. [56].
The CPSF30-binding pocket in the ED is also an attractive target for antiviral drug development. Several lines of evidence established the importance of this binding site for virus replication. One set of experiments utilized the CPSF30 domain comprising its second and third zinc finger (F2F3) that binds efficiently to the NS1A ED [57]. In cells that constitutively express F2F3, replication of influenza A viruses was inhibited coupled with increased production of IFN-β mRNA, presumably as a result of F2F3 occupying the CPSF30 binding site on the NS1A protein and, hence, blocking the binding of endogenous CPSF30 to this site. These results show that the CPSF30-binding site of the NS1A protein can be blocked without any apparent effect on the ability of CPSF30 to carry out its required functions in the processing of cellular pre-mRNAs. The X-ray crystal structure of Ud NS1A(85–215) in complex with the F2F3 fragment revealed that the ED undergoes a dramatic structural rearrangement from its dimer structure [58–60] to form a tetrameric complex comprising two F2F3 units wrapped around two NS1A EDs arranged in a head-to-head manner [61] (Figure 3, inset). The CPSF30-binding pocket identified in this crystal structure is largely hydrophobic and contains several highly conserved residues that interact with aromatic side chains from the F3 zinc finger [61] (Figure 3). A recombinant Ud virus expressing a NS1A mutant protein that does not bind to F2F3 of CPSF30 is attenuated and does not inhibit IFN-β pre-mRNA processing [61]. Hence, disruption of the NS1A:CPSF30 complex, which suppresses this crucial host antiviral response and is required for efficient virus replication, makes this conserved binding pocket in NS1A ED an attractive target for antiviral drug development.
Figure 3.
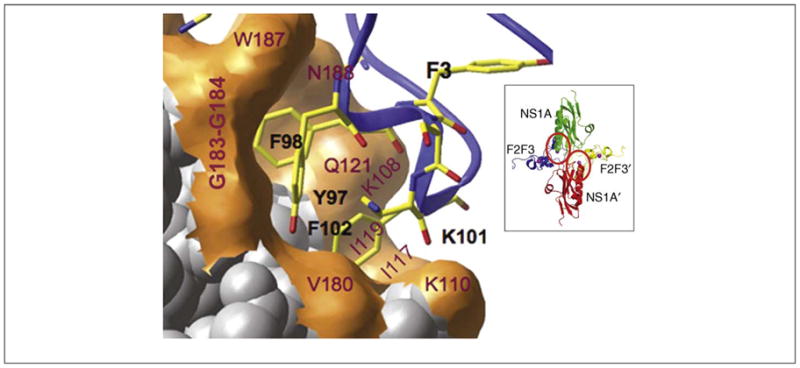
Potential antiviral target in the NS1A effector domain. Expanded view of the F2F3-binding pocket of the Ud NS1A ED [61]. NS1A residues labeled red interact with aromatic side chains in the F3 zinc finger. Inset: ribbon representation of the Ud NS1A(85–215)–F2F3 complex, comprising two NS1A effector domains (green and red) and two F2F3 zinc finger domains (blue and yellow). The F2F3-binding pockets in the two NS1A subunits of the tetramer are circled. Modified, with permission, from Ref. [61].
A recent study showed that several chemicals that bind to undetermined regions of the NS1A protein inhibit virus replication [62], verifying that the NS1A protein is a viable target for the generation of antivirals against influenza A viruses. Other investigators are specifically targeting the dsRNA- and CPSF30-binding sites for the development of antivirals.
NP
The viral NP, which has RNA-binding activity with no apparent specificity, has several crucial functions during infection. It binds along the entire length of the viral RNA segments to form the vRNPs that are packaged into the virus [63] and is required for the switch from viral mRNA synthesis to viral RNA replication [64]. Viral mRNA synthesis is initiated by capped RNA fragments of 10–13 bases that are excised from cellular pre-mRNAs by the intrinsic endonuclease of the viral polymerase (a process called cap-snatching), whereas viral RNA replication is initiated without a primer [64]. A recent study has shown how NP mediates this switch: it interacts with one or more subunits of the viral polymerase, resulting in modification of the polymerase in favor of unprimed initiation [65].
The X-ray crystal structures of NP from both H1N1 and H5N1 viruses identified two potential targets for antiviral development [66,67]. NP (which forms trimers in the crystal) folds into a crescent shape with a head and body domain (Figure 4a). Each NP monomer has a tail loop at the back of the molecule that mediates oligomerization via its insertion into a pocket in the body domain of a neighboring molecule. The tail loop interactions between subunits are almost identical in the H1N1 and H5N1 NP structures. Single amino acid substitutions in the tail loop eliminate oligomerization, and the resulting monomeric NP is not active in viral RNA replication. Consequently, the tail-loop-binding pocket is a potential target for the development of antivirals. Another possible antiviral target in NP is its RNA-binding site [66] (Figure 4b). The RNA-binding groove is found between the head and body domains at the exterior surface of the trimer. It contains a large number of basic residues, and some of the residues required for RNA-binding have been identified.
Figure 4.

Potential antiviral targets in NP. Crystal structure of the NP from HINI influenza A/WSN/33 [66]. (a) Interactions between subunit A (green) and the tail loop of subunit B (red) from the trimeric structure of NP shown on the left. For each denoted amino acid, the subunit in which it is located (A or B) is shown in a parenthesis. (b) Electrostatic surface potential (blue, positive; red, negative) of subunit B from NP, identifying a putative RNA-binding site. Reproduced, with permission, from Ref. [66].
The viral RNA-dependent RNA polymerase
The RNA-dependent RNA polymerase (RdRp) of influenza A virus is a large (250 kDa) heterotrimer complex comprising three subunits: PB1, PB2 and PA. All three subunits are required for both capped RNA-primed viral mRNA synthesis and for unprimed viral RNA replication [65]. Proper formation of the trimeric polymerase requires a specific interaction between the C-terminal domain of PA (PAC) and the N terminus of PB1 (PB1N). The 3D structure of this binding surface was revealed in crystal structures of PAC in complex with PB1N, using both H5N1 and H1N1 viral proteins. PAC forms a clamp around PB1N [68,69] (Figure 5). Binding is mediated by hydrophobic interactions and hydrogen bonds. The PA and PB1 residues involved in this interaction are conserved across all influenza A virus strains. Mutation of key residues in the PB1-binding site of PAC and in PB1N lead to a loss of polymerase activity and a reduction in virus replication. Because the PB1N peptide has been shown to inhibit viral replication, presumably by blocking trimer assembly [70], the PAC–PB1N binding surface provides a potential target for anti-influenza drug development.
Figure 5.

Potential antiviral target in the viral polymerase. Structure of the PAC–PB1N complex. Surface image showing the hydrophobic contacts between PA (green) and the N terminus of PB1 (orange) in the crystal structure of PA(239–716)–PB1(1–81) from influenza A/Puerto Rico/8/1934 (H1N1) [68]. All other residues in PA are colored blue. Reproduced, with permission, from Ref. [68].
The crystal structures of the two functional domains of the cap-dependent endonuclease, its cap-binding site and endonuclease active site have recently been determined [71–73]. These two polymerase domains might also be suitable targets for influenza antiviral drug development.
Conclusions
The potential threat of a pandemic caused by H5N1 influenza A viruses has stimulated increased research on developing new antivirals directed against several viral proteins. An additional incentive has been the emergence of influenza A viruses, both seasonal and pathogenic H5N1 viruses, that are resistant to some of the current antivirals that are directed against the M2 and NA proteins. Accordingly, new antivirals against these two viral proteins are being developed relying largely on structure-based optimization of drug candidates. In addition, recent structural results have provided a more informed approach to the development of antivirals against a well-known target, HA. Furthermore, functional and structural studies have identified novel targets in other viral proteins, specifically the NS1A protein, NP and viral polymerase proteins. The new targets in these proteins are conserved structures that mediate crucial virus functions. Because many amino acid changes in most of these conserved functional sites are deleterious [44,46,61,66–69], it is anticipated that it will be difficult for influenza virus to develop resistance to compounds targeting these sites. However, this prediction will have to be verified, particularly considering that a similar prediction was made for NA antivirals. The development of antivirals targeted against several influenza proteins would make it possible to use combinations of antivirals to minimize the selection of resistant viruses, in light of the report that combined use of amantadine and oseltamivir reduced the emergence of drug-resistant influenza A viruses [74].
Figure I.
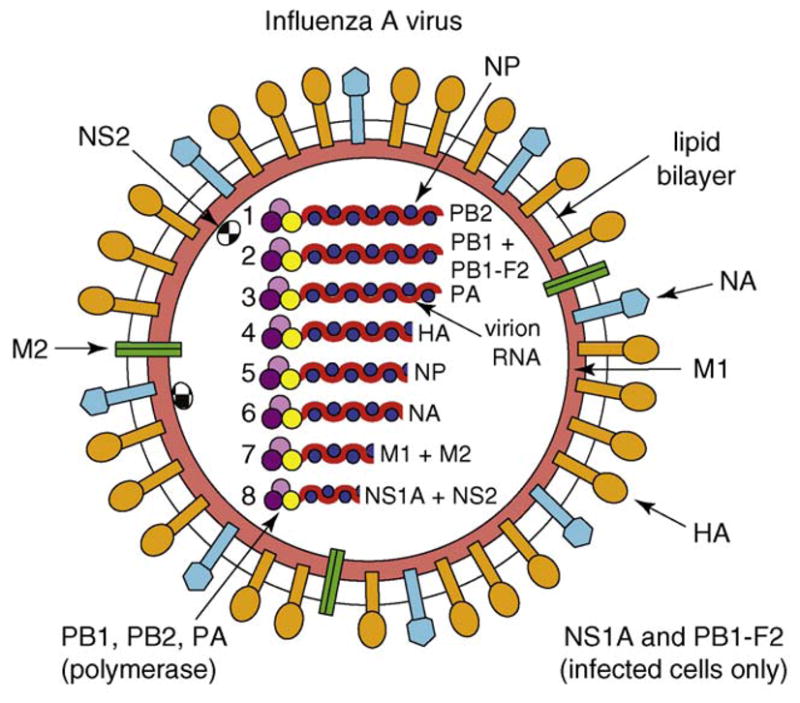
Schematic representation of influenza A virus.
Acknowledgments
Research in our laboratories is supported by grants from the National Institutes of Health USA (www.nih.gov).
Footnotes
Disclosure statement
The authors are funded by a grant from the National Institutes of Health USA to develop antivirals against the dsRNA- and CPSF30-binding surfaces in the NS1A protein. Applications for patents directed at targeting these two NS1A surfaces for the development of antivirals have been submitted.
References
- 1.Lamb RA, Krug RM. Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Lippincott Williams & Wilkins; 2001. pp. 1487–1532. [Google Scholar]
- 2.Wright PF, Webster RG. Orthomyxoviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Lippincott Williams & Wilkins; 2001. pp. 1533–1579. [Google Scholar]
- 3.Reid AH, et al. The 1918 Spanish influenza: integrating history and biology. Microbes Infect. 2001;3:81–87. doi: 10.1016/s1286-4579(00)01351-4. [DOI] [PubMed] [Google Scholar]
- 4.Horimoto T, Kawaoka Y. Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol. 2005;3:591–600. doi: 10.1038/nrmicro1208. [DOI] [PubMed] [Google Scholar]
- 5.Noah DL, Krug RM. Influenza virus virulence and its molecular determinants. In: Maramorsch K, Shatkin AJ, editors. Advances in Virus Research. Vol. 65. Elsevier; 2005. pp. 121–145. [DOI] [PubMed] [Google Scholar]
- 6.de Jong JC, et al. A pandemic warning? Nature. 1997;389:554. doi: 10.1038/39218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subbarao K, et al. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science. 1998;279:393–396. doi: 10.1126/science.279.5349.393. [DOI] [PubMed] [Google Scholar]
- 8.Peiris JS, et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet. 2004;363:617–619. doi: 10.1016/S0140-6736(04)15595-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taubenberger JK, et al. Characterization of the 1918 influenza virus polymerase genes. Nature. 2005;437:889–893. doi: 10.1038/nature04230. [DOI] [PubMed] [Google Scholar]
- 10.Hayden FG. Antivirals for influenza: historical perspectives and lessons learned. Antiviral Res. 2006;71:372–378. doi: 10.1016/j.antiviral.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 11.Longini IM, Jr, et al. Containing pandemic influenza at the source. Science. 2005;309:1083–1087. doi: 10.1126/science.1115717. [DOI] [PubMed] [Google Scholar]
- 12.Ferguson NM, et al. Strategies for mitigating an influenza pandemic. Nature. 2006;442:448–452. doi: 10.1038/nature04795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Germann TC, et al. Mitigation strategies for pandemic influenza in the United States. Proc Natl Acad Sci U S A. 2006;103:5935–5940. doi: 10.1073/pnas.0601266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hay AJ, et al. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985;4:3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamb RA, et al. The influenza A virus M2 ion channel protein and its role in the influenza virus life cycle. In: Wimmer D, editor. Receptor-Mediated Virus Entry Into Cells. Cold Spring Harbor Press; 1994. pp. 303–321. [Google Scholar]
- 16.von Itzstein M, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 17.Kim CU, et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis and structural analysis of carbocyclic sialiac acid analogues with potent anti-influenza activity. J Am Chem Soc. 1997;119:681–690. doi: 10.1021/ja963036t. [DOI] [PubMed] [Google Scholar]
- 18.Mendel DB, et al. Oral administration of a prodrug of the influenza virus neuraminidase inhibitor GS 4071 protects mice and ferrets against influenza infection. Antimicrob Agents Chemother. 1998;42:640–646. doi: 10.1128/aac.42.3.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hay AJ, et al. Molecular basis of resistance of influenza A viruses to amantadine. J Antimicrob Chemother. 1986;18 (Suppl B):19–29. doi: 10.1093/jac/18.supplement_b.19. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki H, et al. Emergence of amantadine-resistant influenza A viruses: epidemiological study. J Infect Chemother. 2003;9:195–200. doi: 10.1007/s10156-003-0262-6. [DOI] [PubMed] [Google Scholar]
- 21.Deyde VM, et al. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis. 2007;196:249–257. doi: 10.1086/518936. [DOI] [PubMed] [Google Scholar]
- 22.Puthavathana P, et al. Molecular characterization of the complete genome of human influenza H5N1 virus isolates from Thailand. J Gen Virol. 2005;86:423–433. doi: 10.1099/vir.0.80368-0. [DOI] [PubMed] [Google Scholar]
- 23.de Jong MD, et al. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N Engl J Med. 2005;353:2667–2672. doi: 10.1056/NEJMoa054512. [DOI] [PubMed] [Google Scholar]
- 24.Le QM, et al. Avian flu: isolation of drug-resistant H5N1 virus. Nature. 2005;437:1108. doi: 10.1038/4371108a. [DOI] [PubMed] [Google Scholar]
- 25.Yen HL, et al. Neuraminidase inhibitor-resistant recombinant A/Vietnam/1203/04 (H5N1) influenza viruses retain their replication efficiency and pathogenicity in vitro and in vivo. J Virol. 2007;81:12418–12426. doi: 10.1128/JVI.01067-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lackenby A, et al. Emergence of resistance to oseltamivir among influenza A(H1N1) viruses in Europe. Euro Surveill. 2008;13:8026. doi: 10.2807/ese.13.05.08026-en. [DOI] [PubMed] [Google Scholar]
- 27.Stouffer AL, et al. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cady SD, et al. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding. J Mol Biol. 2009;385:1127–1141. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jing X, et al. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc Natl Acad Sci U S A. 2008;105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varghese JN, et al. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 Å resolution. Nature. 1983;303:35–40. doi: 10.1038/303035a0. [DOI] [PubMed] [Google Scholar]
- 32.Gubareva LV, et al. Inhibition of replication of avian influenza viruses by the neuraminidase inhibitor 4-guanidino-2,4-dideoxy-2,3-dehydro-N-acetylneuraminic acid. Virology. 1995;212:323–330. doi: 10.1006/viro.1995.1489. [DOI] [PubMed] [Google Scholar]
- 33.Russell RJ, et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45–49. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
- 34.Wilson IA, et al. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 35.Bullough PA, et al. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 36.Bodian DL, et al. Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry. 1993;32:2967–2978. doi: 10.1021/bi00063a007. [DOI] [PubMed] [Google Scholar]
- 37.Hoffman LR, et al. Structure-based identification of an inducer of the low-pH conformational change in the influenza virus hemagglutinin: irreversible inhibition of infectivity. J Virol. 1997;71:8808–8820. doi: 10.1128/jvi.71.11.8808-8820.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu KL, et al. Structure-activity relationships for a series of thiobenzamide influenza fusion inhibitors derived from 1,3,3-trimethyl-5-hydroxy-cyclohexylmethylamine. Bioorg Med Chem Lett. 2002;12:3379–3382. doi: 10.1016/s0960-894x(02)00761-8. [DOI] [PubMed] [Google Scholar]
- 39.Russell RJ, et al. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc Natl Acad Sci U S A. 2008;105:17736–17741. doi: 10.1073/pnas.0807142105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krug RM, et al. Intracellular warfare between human influenza viruses and human cells: the roles of the viral NS1 protein. Virology. 2003;309:181–189. doi: 10.1016/s0042-6822(03)00119-3. [DOI] [PubMed] [Google Scholar]
- 41.Hale BG, et al. The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008;89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- 42.Hatada E, Fukuda R. Binding of influenza A virus NS1 protein to dsRNA in vitro. J Gen Virol. 1992;73:3325–3329. doi: 10.1099/0022-1317-73-12-3325. [DOI] [PubMed] [Google Scholar]
- 43.Lu Y, et al. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology. 1995;214:222–228. doi: 10.1006/viro.1995.9937. [DOI] [PubMed] [Google Scholar]
- 44.Wang W, et al. RNA-binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic amino acids. RNA. 1999;5:195–205. doi: 10.1017/s1355838299981621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chien CY, et al. Biophysical characterization of the complex between double-stranded RNA and the N-terminal domain of the NS1 protein from influenza A virus: evidence for a novel RNA-binding mode. Biochemistry. 2004;43:1950–1962. doi: 10.1021/bi030176o. [DOI] [PubMed] [Google Scholar]
- 46.Min JY, Krug RM. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: inhibiting the 2′–5′ OAS/RNase L pathway. Proc Natl Acad Sci U S A. 2006;103:7100–7105. doi: 10.1073/pnas.0602184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nemeroff ME, et al. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′ end formation of cellular pre-mRNAs. Mol Cell. 1998;1:991–1000. doi: 10.1016/s1097-2765(00)80099-4. [DOI] [PubMed] [Google Scholar]
- 48.Kim MJ, et al. Human influenza viruses activate an interferon-independent transcription of cellular antiviral genes: outcome with influenza A virus is unique. Proc Natl Acad Sci U S A. 2002;99:10096–10101. doi: 10.1073/pnas.152327499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Noah DL, et al. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAs. Virology. 2003;307:386–395. doi: 10.1016/s0042-6822(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 50.Hale BG, et al. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc Natl Acad Sci U S A. 2006;103:14194–14199. doi: 10.1073/pnas.0606109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Min JY, et al. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology. 2007;363:236–243. doi: 10.1016/j.virol.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 52.Shin YK, et al. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J Gen Virol. 2007;88:13–18. doi: 10.1099/vir.0.82419-0. [DOI] [PubMed] [Google Scholar]
- 53.Chien CY, et al. A novel RNA-binding motif in influenza A virus non-structural protein 1. Nat Struct Biol. 1997;4:891–895. doi: 10.1038/nsb1197-891. [DOI] [PubMed] [Google Scholar]
- 54.Liu J, et al. Crystal structure of the unique RNA-binding domain of the influenza virus NS1 protein. Nat Struct Biol. 1997;4:896–899. doi: 10.1038/nsb1197-896. [DOI] [PubMed] [Google Scholar]
- 55.Yin C, et al. Conserved surface features form the double-stranded RNA-binding site of non-structural protein 1 (NS1) from influenza A and B viruses. J Biol Chem. 2007;282:20584–20592. doi: 10.1074/jbc.M611619200. [DOI] [PubMed] [Google Scholar]
- 56.Cheng A, et al. Structural basis for dsRNA recognition by NS1 protein of influenza A virus. Cell Res. 2008;19:187–195. doi: 10.1038/cr.2008.288. [DOI] [PubMed] [Google Scholar]
- 57.Twu KY, et al. The CPSF30 binding site on the NS1A protein of influenza A virus is a potential antiviral target. J Virol. 2006;80:3957–3965. doi: 10.1128/JVI.80.8.3957-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bornholdt ZA, Prasad BV. X-ray structure of influenza virus NS1 effector domain. Nat Struct Mol Biol. 2006;13:559–560. doi: 10.1038/nsmb1099. [DOI] [PubMed] [Google Scholar]
- 59.Hale BG, et al. Structure of an avian influenza A virus NS1 protein effector domain. Virology. 2008;378:1–5. doi: 10.1016/j.virol.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 60.Xia S, et al. Structure of NS1A effector domain from the influenza A/Udorn/72 virus. Acta Crystallogr D Biol Crystallogr. 2009;65:11–17. doi: 10.1107/S0907444908032186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Das K, et al. Structural basis for suppression by influenza A virus of a host antiviral response. Proc Natl Acad Sci U S A. 2008;105:13093–13098. doi: 10.1073/pnas.0805213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Basu D, et al. Novel influenza virus NS1 antagonists block replication and restore innate immune function. J Virol. 2009;83:1881–1891. doi: 10.1128/JVI.01805-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Portela A, Digard P. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J Gen Virol. 2002;83:723–734. doi: 10.1099/0022-1317-83-4-723. [DOI] [PubMed] [Google Scholar]
- 64.Krug RM, et al. Expression and replication of the influenza virus genome. In: Krug RM, editor. The influenza viruses. Plenum Press; 1989. pp. 89–152. [Google Scholar]
- 65.Newcomb LL, et al. Interaction of the influenza a virus nucleocapsid protein with the viral RNA polymerase potentiates unprimed viral RNA replication. J Virol. 2009;83:29–36. doi: 10.1128/JVI.02293-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ye Q, et al. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature. 2006;444:1078–1082. doi: 10.1038/nature05379. [DOI] [PubMed] [Google Scholar]
- 67.Ng AK, et al. Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB J. 2008;22:3638–3647. doi: 10.1096/fj.08-112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Obayashi E, et al. The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature. 2008;454:1127–1131. doi: 10.1038/nature07225. [DOI] [PubMed] [Google Scholar]
- 69.He X, et al. Crystal structure of the polymerase PA(C)-PB1(N) complex from an avian influenza H5N1 virus. Nature. 2008;454:1123–1126. doi: 10.1038/nature07120. [DOI] [PubMed] [Google Scholar]
- 70.Ghanem A, et al. Peptide-mediated interference with influenza A virus polymerase. J Virol. 2007;81:7801–7804. doi: 10.1128/JVI.00724-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guilligay D, et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol. 2008;15:500–506. doi: 10.1038/nsmb.1421. [DOI] [PubMed] [Google Scholar]
- 72.Dias A, et al. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature. 2009;458:914–918. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 73.Yuan P, et al. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature. 2009;458:909–913. doi: 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
- 74.Ilyushina NA, et al. Combination therapy, a potential strategy for reducing the emergence of drug-resistant influenza a variants. Antiviral Res. 2006;70:121–131. doi: 10.1016/j.antiviral.2006.01.012. [DOI] [PubMed] [Google Scholar]