

Abstract
Great skepticism has surrounded the question of whether modulation of voltage-gated Ca2+ channels (VGCCs) by the polyunsaturated free fatty acid arachidonic acid (AA) has any physiological basis. Here we synthesize findings from studies of both native and recombinant channels where micromolar concentrations of AA consistently inhibit both native and recombinant activity by stabilizing VGCCs in one or more closed states. Structural requirements for these inhibitory actions include a chain length of at least 18 carbons and multiple double bonds located near the fatty acid’s carboxy terminus. Acting at a second site, AA increases the rate of VGCC activation kinetics, and in CaV2.2 channels, increases current amplitude. We present evidence that phosphatidylinositol 4,5-bisphosphate (PIP2), a palmitoylated accessory subunit (β2a) of VGCCs and AA appear to have overlapping sites of action giving rise to complex channel behavior. Their actions converge in a physiologically relevant manner during muscarinic modulation of VGCCs. We speculate that M1 muscarinic receptors may stimulate multiple lipases to break down the PIP2 associated with VGCCs and leave PIP2’s freed fatty acid tails bound to the channels to confer modulation. This unexpectedly simple scheme gives rise to unanticipated predictions and redirects thinking about lipid regulation of VGCCs.
Keywords: CaV, cPLA2, DAG liapse, Gq, muscarinic receptors, ischemia, fatty acid
1. Introduction
Voltage-gated ion channel activity is exquisitely regulated to maintain accurate flow of information throughout the body allowing us to breathe, run, talk, learn, and remember. There is increasing appreciation that lipid molecules regulate and modulate ion channels that interact directly with them in the plasma membrane. Particular interest surrounds lipid regulation of voltage-gated Ca2+ channels (VGCCs) since changes in their activity affect not only membrane excitability, but also neurotransmitter release, Ca2+-dependent biochemical events, and activity-dependent gene transcription [1]. Thus, a change in VGCC activity may precipitate acute (msec-sec), short-term (sec-min), and long-term changes (hrs-days). Lipid-induced regulation of VGCC activity is incompletely understood; however there is growing appreciation that phosphatidylinositol 4,5-bis phosphate (PIP2) and free fatty acids (Fig. 1) play important roles in regulating VGCC activity. For example, PIP2 increases channel availability to open and promotes slowed gating kinetics, whereas fatty acids exert the opposite actions. In particular, growing evidence indicates that arachidonic acid (AA), a 20 carbon, polyunsaturated fatty acid is liberated from the sn-2 position of phospholipids, such as PIP2 (Fig. 1A), by phospholipases to modulate the activity of pore-forming VGCC subunits of the CaVα1 superfamily.
Figure 1.
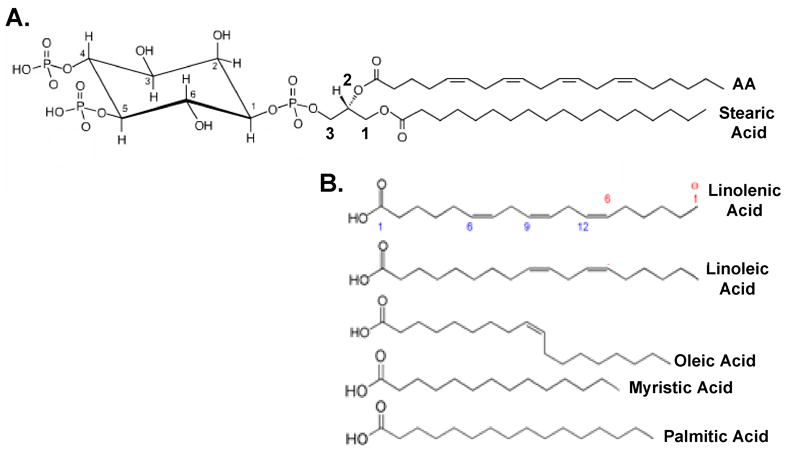
PIP2 is formed of a glycerol backbone with a phosphoinositol head group and two fatty acid tails. A. Stearic acid normally resides in the sn-1 position of PIP2, while AA is found in the sn-2 position. The three carbons of the glycerol backbone are numbered in red. The inositol headgroup of PIP2 attaches to the sn-3 carbon via a phosphoester linkage. The carbons of the inositol head group are numbered in black. Additional phosphate groups are located at carbons 4 and 5 of the inositol ring. B. Other fatty acids that can modulate VGCC activity. Carbon numbering starts from the carboxy end of fatty acids (shown in blue for linolenic acid) while defining the location of the first double-bond is determined from the amino terminus (shown in red).
The following review synthesizes current knowledge of the mechanisms used by PIP2 and AA to regulate VGCC currents. Basic chemistry about PIP2 and AA in the plasma membrane is presented when deemed helpful. A number of recent, comprehensive reviews thoughtfully address many of the physiological implications of phospholipid [2–4] and fatty acid [5–7] regulation of VGCCs. Where useful, AA’s actions will be compared to other fatty acids (Fig. 1B) but not its metabolites. For a thorough account of the effects of fatty acid metabolites on ion channels, see excellent reviews by Spector and Norris and by Meves [6, 8, 9]. Enzymes bind AA with Kms in the μM range, indicating that μM concentrations of AA are physiological [10]. At or above 20 μM, AA micelles can form though the exact value varies depending on ionic concentrations [11]. Above the critical micelle concentration, lipids will aggregate into spherical structures in solution where they can exert detergent effects on the lipid bilayer. Therefore we discuss studies where fatty acid concentrations used are less than 20 μM. A number of neurotransmitters that stimulate release of endogenous AA in a variety of cell types also modulate VGCCs. Some controversy exists as to whether released AA participates in transmitter modulation of VGCCs or whether simple dissociation of PIP2 is sufficient for decreased channel activity. Therefore we discuss the controversy surrounding the putative roles that PIP2 and AA may play in mediating VGCC modulation by Gq-coupled receptors (GqPCRs). A unifying model for VGCC modulation is presented that takes into account data from PIP2 and AA studies. Lastly we discuss the molecular significance of lipids in regulating VGCC activity.
2. Exogenously applied AA inhibits VGCC currents by stabilizing closed or inactivated conformations
The pore-forming CaVα1 subunits arise from a 4-fold repeat of 6 transmembrane segments with a pore-loop inserted between S5 and S6 (Fig 2A). Cytoplasmic loops link the four domains into a single polypeptide. Recent crystal structures of voltage-gated Kv channels serve as a rough template for interpreting very basic structure-function studies of VGCCs. As with Kv channels, S1-S4 form a voltage sensor unit with S4 containing multiple positively charged residues that move in response to changes in membrane potential. S5 and S6 form the pore of the channel with S6 serving as its inner lining. S6 contains amino acid residues responsible for channel opening, closing and certain forms of inactivation [12]. However, members within the VGCC family exhibit diverse gating behavior making it difficult to generalize about how their different structures affect activity.
Figure 2.

Schematics of AA’s effects on holding potential-dependent inactivation of Ca2+ channels. A. Topological orientation of a VGCC pore-forming subunit. The subunit is formed by four repeating domains (I–IV) each made up of six transmembrane segments (S1–S6) with each S4 containing at least four charged residues (+) that participate in voltage-sensing. Arrow indicates location of a high affinity binding site, called the alpha interaction domain (AID), for the β-subunit. Colored symbols approximate the location of critical residues that alter lipid regulation of identified voltage-gated ion channels. Yellow highlights identify regions critical for dihydropyridine (DHP) binding. B. In SCG neurons, AA (5 μM) increases the amount of inactivation as the holding potential becomes more positive. No shift in the voltage-sensitivity of inactivation is observed. C. For certain L- and T-channels, AA increases the voltage-sensitivity of inactivation, causing a leftward shift in the inactivation curve.
Nevertheless, from mutagenesis and kinetic studies there is general agreement that by mechanisms not yet well understood, movements of the voltage sensor somehow cause each S6 to flex away from one another, to open VGCCs. Closed times, measured from single channel patch-clamp recordings, distribute around several time points indicating that VGCCs have multiple closed states. Some combination of each voltage sensor moving through the membrane and S6 flexing may account for the multiple closed states [13]. Moreover when cells are held very negative, the four voltage sensors reside in their most internal position within the membrane. Upon depolarization, channels take a measurable amount of time to transition through these multiple closed conformations to open; this time is referred to as the first latency. An altered first latency indicates changes in stability of these closed conformations. As VGCCs transition through the partially activated closed states during membrane depolarization, they will undergo slow forms of voltage-dependent inactivation that develop over 100’s to thousands of msec. These slow forms of inactivation, often referred to as C-inactivation for K+ channels, are thought to occur following reorientation of residues within or near the outer pore region. VGCCs also undergo faster forms of inactivation, developing over tens to several hundreds of msec, where the mechanism underlying inactivation appears to vary depending on the type of VGCC. For some VGCCS, cytoplasmic loops appear to interact with the inner pore to block the current, giving rise to fast inactivation. Additionally fast forms of inactivation for some VGCCs can exhibit characteristics of C-type inactivation, underscoring the complexity of VGCCs’ biophysical properties. For more in depth discussion of VGCC gating see reviews by [12–16].
Exogenously applied AA inhibits currents of both native and recombinant VGCCs from all three families of α1 pore-forming subunits CaV1-CaV3. Where examined, all VGCCs exhibit sensitivity to AA with an IC50 within 1–10 μM [17–25]. CaV1 (L-channel) and CaV2 (N-, P/Q-and R-channels) pore-forming subunits exist in macromolecular complexes with accessory subunits α2δ and β, which help increase functional expression and tune the channels’ biophysical properties [26]. The β-subunit also aids in trafficking these channels to the cell membrane [27]. Despite the fact that multiple subunits form these VGCCs, they are referred to by the name of their CaVα1 pore-forming subunit. In contrast, CaV3 (T-channel) subunits give rise to recombinant current without a requirement for accessory subunits [28]. This latter finding suggests that whether direct or indirect, AA acts on CaV pore-forming subunits. Moreover, AA appears to alter the gating of VGCCs but not channel permeation. Details of AA’s actions on each VGCC family are presented below.
2.1 T-type Ca2+ Channels
AA inhibits T-current in adrenal zona fasciculate cells [29], rat osteoblast cells [30], NG108-15 cells [31] and currents from all three recombinant T-channels (CaV3.1, CaV3.2 and CaV3.3) [18, 20, 21] though CaV3.2 exhibits a greater magnitude of inhibition than CaV3.1 or CaV3.3 [18]. AA itself appears to modulate CaV3 channels since blocking AA metabolism has no effect on the magnitude of current inhibition. Additionally, AA can robustly inhibit T-currents from inside-out, ripped-off patches consistent with a direct effect [18, 21]. No change in unitary conductance was found, but rather, an increase in sweeps with no activity occurred following AA [21]. Consistent with this finding, at the whole-cell level AA could inhibit channels in the closed state. A Hill coefficient of 1.6 best describes inhibition of Cav3.1 by AA suggesting that at least two AA molecules cooperatively interact with one channel [18, 20, 21]. A detailed study by Chemin et al. demonstrated that other unsaturated free fatty acids can suppress CaV3 currents while saturated free fatty acids have no effect [18]. Fatty acid inhibition of T-current has a structural requirement of at least 18 carbons in chain length. Potency of inhibition increases with increasing numbers of cis double bonds and with placement of the double bonds near the carboxyl group [18].
Further kinetic studies examining the mechanism of inhibition indicate AA also increases the voltage-sensitivity of slow forms of inactivation of all three T-channels [18, 20, 21]. Talavera et al. (2004) found that the onset of current inhibition and changes in slow inactivation were different indicating AA has two different actions on CaV3.1. Changes in fast forms of inactivation do not appear to underlie inhibition. Talavera et al. came to this conclusion through studies using mutant CaV3.1 channels with decreased fast inactivation. Most notably, a point mutation in IIIS6 of CaV3.1 (M1510I) leads to currents that inactivate with a time constant 4-fold greater than for wt channels [32]. Despite the slower inactivation kinetics, AA inhibits M1510I currents more rapidly and with an approximately 10-fold lower IC50 (~0.3 μM) than wt currents (Fig. 2A) [21]. AA has little effect on the voltage-dependence of activation [18, 20, 21]; however, a small, but significant negative shift in the foot of the activation curve in CaV3.1 currents and a small change in the slope of the activation curve for CaV3.2 occurs following AA that can be accounted for by changes in the rate constants between closed states [20, 21]. Thus, AA appears to decrease channel availability by stabilizing closed conformations and also shifts the steady-state inactivation curve more negative.
2.2 L-type Ca2+ Channels
AA inhibits L-current in skeletal (CaV1.1) [22, 36], cardiac (CaV1.2) [17, 25, 37] and smooth muscle myocytes (CaV1.2) [38], SCG neurons (primarily CaV1.3) [23, 39–41], recombinant CaV1.3 current expressed in HEK 293 cells [42]; and in photoreceptors (primarily CaV1.4) [19]. In hippocampal neurons, which express both CaV1.2 and CaV1.3 channels, AA exerts similar inhibitory actions on the whole-cell L-current [43]. At the single channel level, AA inhibits unitary L-channel activity in SCG neurons by increasing mean first latency and mean closed time [41]. Additionally increased numbers of sweeps with no openings are observed, raising the possibility that AA also stabilizes inactivated states. Conductance and mean open time are unaffected by AA [41].
At the whole-cell level, AA increases the amount of CaV1.3b current inactivation at positive test potentials, but does not alter the voltage profile of inactivation [35]. This form of CaV1.3b has a truncated C-terminus ruling out AA binding to G-protein and calmodulin binding sites. Findings from three additional experiments, designed to minimize L-current inactivation, indicate AA decreases current independently of CaV1.3b inactivation [35]. 1) When CaV1.3b is held at −90 mV, few channels inactivate; most reside in the closed state whereas at −60 mV channels reside in a partially activated closed state become susceptible to slow forms of inactivation. Holding the membrane potential more negative (−90 mV versus −60 mV) approximately doubles CaV1.3b current inhibition by AA when cells were stepped to −10 mV every four seconds, the potential where maximal inward current normally occurs [35]. 2) In a similar experiment, current inhibition still occurs when L-channels are held closed during application of AA [19, 35, 38, 42], consistent with the hypothesis that AA stabilizes closed states. 3) Shortening the test potential duration from 40 ms to 10 ms decreases the number of channels that will inactivate. Under these conditions the magnitude of inhibition increased. In each of these three experiments the voltage protocol was manipulated to increase the time the channel spent in a closed state while minimizing the probability that channels would transition to inactivated conformations. In each case, inhibition remained the same or increased in magnitude.
These data from recombinant channels best fit a model where AA stabilizes channels in a deep closed state, rather than an inactivated state, and match whole-cell and single channel findings of native L-currents in SCG neurons [23, 41]. However, in other cell types, AA shifts the voltage-sensitivity of inactivation for L-current more negative [25, 37, 38, 44] similar to the schematic shown in Figure 2C. Amino acid residues found in transmembrane S5 and S6 segments regulate opening, closing and slow forms of inactivation [14, 32, 45–48]. Sequence variation in these regions among L-channels may explain these differences in response to AA.
2.3 N-type Ca2+ Channels
AA inhibits native N-current in SCG neurons [23, 39, 41, 49] and recombinant N-current (CaV2.2) expressed in HEK 293 cells [39, 50]. Other members of the CaV2 family [CaV2.1 (P/Q) and CaV2.3 (R)] have not been tested for sensitivity to AA. Native N-channels display distinct patterns of endogenous, heterogeneous activity called modes [51, 52]. AA alters the frequency of occurrence of particular modes to decrease unitary N-channel activity in cell-attached patches that contain one channel (Fig. 3A). AA decreases open probability (Po) by increasing the percentage of sweeps with no openings (nulls) and the number of null sweeps that cluster together [41]. AA stabilizes a null mode but does not alter modes with amplitude or open time changes. However, truncated closings (the last closing during the test pulse) increase 2–3-fold in duration, indicating channels enter a non-conducting state (closed or inactivated). Increases in first latency usually occur when channels transition through closed conformations to the open state more slowly than normal. First latency increases approximately 3-fold, indicating that AA stabilizes one or more intermediate closed states rather than an inactivated state.
Figure 3.

Schematic illustrating AA’s inhibitory actions on N-channel gating in SCG neurons. A. AA shifts the pattern of N-channel gating elicited with 750 ms test pulses. Eight idealized sweeps in the absence (CON) and presence of AA illustrate how AA shifts the pattern of unitary N-channel gating. Two minutes after puffing on micromolar concentrations of AA (red), the frequency of noninactivating sweeps decreases compared to control (CON), whereas of the numbers of null sweeps increase and are found clustered together. Additionally, in some sweeps, first latency increases (*) and some end with a long truncated closing (T). B. Noninactivating unitary N-channel activity is lost following AA. C. Idealized whole-cell current traces before (CON), and 2 min after puffing on AA. D. The whole-cell current inhibited by AA shows no fast inactivation consistent with a loss of unitary channel activity that is noninactivating (after Roberts-Crowley & Rittenhouse, 2008).
N-current comprises 80–90% of the whole-cell current in rat SCG neurons [53]. Kinetic analyses of these whole-cell currents revealed AA selectively decreases the frequency of non-inactivating gating with little change in fast inactivation (Figs. 3C–D). These findings are consistent with the single channel results where AA stabilizes one pattern of activity, the null mode, while specifically decreasing another, the noninactivating mode (Figs. 3A–B). Additionally whole-cell studies showed that AA increased the amount of holding potential-dependent inactivation at positive voltages [23], similar to that observed with recombinant CaV1.3b currents (Fig. 2B). This increase in inactivation may also contribute to the observed increased incidence of the null mode at the single channel level. However, no shift in voltage-sensitivity of inactivation was observed [23]. This lack of voltage dependency may be due to AA stabilizing a closed state that is susceptible to undergoing transitions to inactivated conformations [54]. Taken together, these findings support a model where AA may stabilize a closed conformation of the N-channel that normally promotes inactivation. Alternatively, if the rate of recovery from inactivation is also slowed, channels may accumulate in an inactivated conformation. Future biophysical studies combined with mutagenesis should allow more precise characterization of the mechanism of N-channel inhibition by AA.
In summary, the similar actions of AA across VGCC families suggest a common mechanism for inhibition. N-channels, as with T- and L-channels, exhibit changes in closings following exposure to AA. Additionally, AA appears to have a second though more varied effect on all three channels where kinetic changes in inactivation occur. Consistent with the idea of common mechanism, fatty acid inhibition of VGCC activity has similar structural requirements: 1) a minimum carbon chain length of 18 carbons; 2) the magnitude of inhibition increases with number of carbons 3) polyunsaturated bonds; 4) and double bond locations close to the carboxy terminus [18, 19, 23, 33–35, 39].
3. AA exerts biphasic actions on VGGCs
Despite the attention given to inhibition, reports of biphasic effects suggest AA modulates VGCCs at more than one site. In addition to inhibiting VGCC currents (Fig. 3C), AA acts at a second, distinct site to enhance N-current at negative test potentials [23, 39, 49]. Enhanced N-current exhibits an increased rate of activation that correlates in time with a leftward shift in voltage-sensitivity of activation [39]. N-current enhancement by AA is manifest in the current-voltage relationship at negative test potentials (such as depolarizations to −10 mV), whereas N-current inhibition dominates the modulation observed at positive test potentials (such as depolarizations to +10 mV). ETYA, myristic acid (Fig. 1B), and palmitic acid (5 μM) enhance, but do not inhibit N-current, demonstrating that the two effects have different fatty acid specificities [23, 39]. When alternating test pulses to −10 mV to monitor enhancement and to +10 mV to monitor inhibition, the opposing effects can be observed in the same time courses. The onset of enhancement develops significantly faster than inhibition, suggesting that the opposing effects of AA occur by separate mechanisms [23, 39].
Consistent with this possibility, the two actions separate with pharmacological manipulation [39]; inhibition appears to occur intracellularly or in the inner membrane leaflet while enhancement may occur extracellularly or in the outer membrane leaflet. When included in the pipette solution, bovine serum albumin (BSA) dialyzes into cells and serves as an AA scavenger since it binds AA quite effectively [55]. Dialyzing SCG neurons with BSA has no effect of its own on control currents nor does it alter enhancement following exposure to AA. However its presence minimizes N-current inhibition by AA, suggesting that the opposing effects have different sites of action. This hypothesis was further tested with bath application of AA covalently bound to coenzyme A. Very little of AA-coenzyme A (AA-CoA) crosses cell membranes because of its hydrophilic nature. When introduced into the bath, AA-CoA enhanced N-current at negative potentials but had no inhibitory effects at positive test potentials. Thus, intracellular regions of N-channels appear to confer inhibition, whereas enhancement by AA may be mediated extracellularly [23, 39]. Alternatively, enhancement may occur intracellularly but exhibit a higher affinity for free AA than the site of inhibition [23].
Two sites of action may exist for other types of VGCCs. In cardiac myocytes, AA produces both a hyperpolarizing shift in L-current activation and inactivation as well as a reduction in the maximum slope conductance [25]. In smooth muscle, myristic acid (Fig. 1B) enhances L-current at negative potentials [38], with a similar current-voltage profile as N-current modulation by myristic acid [39]. Other fatty acids such as palmitic acid stimulate L-current in pancreatic α-cells [56] while palmitic and oleic acids stimulates L-current in pancreatic β-cells [57, 58]. In contrast, only inhibition by AA has been observed for L-current in skeletal muscle myocytes (CaV1.1), smooth muscle myocytes (primarily CaV1.2) and SCG neurons (CaV1.3) [23, 38, 49]. Similarly, an increased rate of activation occurs in T-currents following AA with no increase in current amplitude [21, 42]. Thus, inhibition by AA may dominate VGCC activity, obscuring any appearance of the ongoing enhancing effects of AA [39]. When matched with conditions used in earlier studies, these results taken together provide an explanation for AA’s seemingly paradoxical actions on VGCC currents [38, 44, 59–62]. Future mutagenesis studies should resolve whether homologous, distinct sites exist across the three VGCC families that mediate enhancement and inhibition by AA.
4. Evidence for direct AA binding to channels
Once liberated, AA binds to a number of cytosolic proteins indicating that it can interact directly with specific proteins in either a lipid environment or in the more hydrophilic cytosolic environment [11, 44, 63–65]. Specific binding of AA to VGCCs remains difficult to demonstrate because of the overall size of channel proteins and the large number of hydrophobic residues that comprise the transmembrane domains that normally contact the lipid bilayer. Examples of proteins that bind AA include the cystic fibrosis antigen complex S100A8/S100A9 [66], certain forms of protein kinase C (PKC) [67], the catalytic subunit of protein phosphatase 2B (PP2B or calcineurin) [68], PP5 [69], fatty acid binding protein [70], and BSA [71]. In the C-terminal tail of S100A9, three consecutive histidine residues were identified for AA binding [66]. AA binds to BSA at multiple sites with high affinity (Kd, 62 nM) [71]. Surprisingly no sequence homology or obvious three dimensional structure is shared among the sites [72–74]. AA binding sites of other voltage-gated ion channels also remain mostly undefined; however, mutational studies have identified key residues in the pore region of the Na+ channel NaV1.5. A single point mutation (N406K) in IS6 (Fig. 2A) minimizes fatty acid inhibition of channel activity [75]. In contrast, AA inhibits a double mutant (L409C/A410W in the S6 of domain I), suggesting that multiple amino acids contribute to channel sensitivity to fatty acids. An asparagine is found in a homologous site in S6 of domains I–IV in all CaV channels, suggesting a possible common site of interaction with AA. In contrast to the possible common site of interaction for VGCCs and NaV1.5, three different potential sites of interaction with AA have been identified for K+ channels. Hamilton et al. (2003) identified two amino acids (Thr250 and Val275) in the pore-lining region of the human intermediate conductance, Ca2+-activated K+ channel (hIK1) as key residues for current inhibition by AA [76] while the amino terminus of ROMK1 and the carboxy terminus of Kir3 confer inhibition [77, 78]. In contrast, the carboxy terminus of TREK-2 confers enhancement by AA [79].
Thus, a common consensus sequence for AA binding may not exist across different protein families. Nevertheless, since AA elicits similar changes in L-, N- and T-channel activity, AA may act at one or more shared homologous sites. AA inhibits T-current recorded in ripped off, inside-out patches [18] where most second messenger systems are absent, ruling out a number of signaling molecules and suggesting direct binding. Moreover, T-channels require no accessory subunits for current expression, yet AA inhibits T-current similarly to native and recombinant Land N-currents [23, 35, 39], consistent with AA binding to homologous sites on pore-forming subunits. Future mutagenesis studies should reveal whether VGCCs have a common consensus site for AA binding and/or conferring modulation.
In the meantime, biochemical studies provide tantalizing clues as to regions of CaV1 that may interact with AA. Three amino acids in the pore-region of L-channels have been identified by extensive mutagenesis and chimera studies as critical for binding of dihydropyridines (DHPs) [80, 81], a class of L-channel agonists and antagonists (Fig. 2A). Docosahexaenoic acid (C22:6), but not AA, inhibits the binding of the DHP antagonist nitrendipine in cardiac myocytes [82, 83]. AA displaces the specific binding of the radiolabelled DHP antagonist PN200-110 with an IC50 of approximately 6 μM in rabbit skeletal muscle membrane assays [84] and decreases PN200-110 binding and 45Ca2+ flux by 50% in skeletal muscle T-tubules [22]. Blocking lipoxygenase activity has no significant effect on displacement of PN200-110, consistent with AA, rather than a metabolite, competing with DHP antagonist for binding to CaV1.1. Additionally, DHPs augment the binding of other CaV1 channel antagonists through allosteric interactions [81], a profile similar to that observed with AA where AA augments the binding of radiolabelled diltiazem and D888, L-channel antagonists with binding sites distinct from DHPs [84]. These findings identify the DHP binding site as one potential site for AA interaction with CaV1 channels. Whether AA interacts with analogous sites in the pore region of CaV2 and CaV3 channels awaits testing; however it is an attractive idea since high concentrations of DHPs will inhibit native N-current [85].
The canabinoids anandamide and 2-AG share structural similarities to AA and their binding to cannabinoid GPCRs results in modulation of VGCC activity [86]. Additionally, anandamide inhibits P-type Ca2+ currents in the cerebellum [87], L-current in T-tubule membranes [36], N-current in SCG neurons [88] and recombinant T-current [18, 89] independently of receptor activation. Anandamide produces a greater inhibition of all three (CaV3.1, CaV3.2, CaV3.3) T-currents than AA [18]. As reported for AA, anandamide competes with DHP agonist/antagonist binding to L-channels [82, 83], possibly by direct competition [84]. Together these findings identify the pore-region as a potential binding site for AA or alternatively as important for conferring Ca2+ channel modulation by AA [34].
5. Palmitoylation of the β2a subunit alters VGCC gating and modulation by AA
Studies using recombinant L- or N-channels revealed that the magnitude of current inhibition by AA varied with different β-subunits [50, 90]. Whole-cell currents from CaV1.3b coexpressed with β2a showed approximately half the inhibition by AA than when coexpressed with either β1, β3 or β4 [90]. Moreover, following AA the time to peak current decreased in channels coexpressed with β1, β3 or β4 whereas no change in activation kinetics was observed with β2a. Heterogeneous expression of β-subunits may explain the varied responses to AA by native CaV1.2 channels. Interestingly, AA inhibited currents from CaV2.2, coexpressed with β1, β3 or β4, whereas coexpression with β2a resulted in sustained enhancement of approximately 2-fold at negative test potentials [50]. Heterogeneous association of β-subunits with CaV2.2 may explain the biphasic action of AA in SCG neurons of enhancement of N-current at negative test potentials and inhibition at positive test potentials [23, 39]. Thus, depending on the β-subunit expressed with either L- or N-channels in a cell type, the profile of AA modulation may differ.
Notably β2a differs from β1, β3 and β4 in that it is uniquely palmitoylated at cysteine 3 and 4 of its N-terminus [91]. Channels coexpressed with palmitoylated β2a exhibit noninactivating currents and a rightward shift in the voltage sensitivity of slow forms of inactivation [35, 92–100]. By tethering β2a to the membrane, its movements as well as those of the intracellular loops that interact with β2a are impeded. Restricting β2a’s movements results in noninactivating CaV1 and CaV2 currents [94, 101]. Coexpression of a mutant depalmitoyated βa, which has had cysteine 3 and 4 substituted with serine (βaC3,4S), results in N-current inhibition rather than enhancement by AA [50]. This finding raises the possibility that the fatty acid palmitoyl groups of βa disrupt N-current inhibition by exogenously applied AA. When palmitoylation was transferred to βb by substituting the 16 amino terminus of βa for βb’s amino terminus while the rest of β1 remained unchanged, AA no longer inhibits currents of CaV2.2 coexpressed with the palmitoylated βa β1 chimera. Instead, these currents exhibit enhancement at negative test potentials similar to wt βa. This result suggests that the palmitoyl groups confer the block of inhibition.
One consequence of tethering β2a is that the currents from CaV2.2 coexpressed with β2a exhibit little inactivation compared to currents from CaV2.2 coexpressed with β1, β3 or β4. Since both inactivation and modulation are different in β2a-containing channels, the question arises of whether the dramatic slowing of channel inactivation rather than from palmitoylation underlies β2a’s effects. Indeed, depalmitoylated β2aC3,4S containing L- and N-channels exhibit increased inactivation as well as inhibition by AA [35, 102]. In other words, the palmitoyl groups may block inhibition – not by competing for the same putative binding site as AA – but rather by keeping channels from inactivating. Though AA’s primary action appears to stabilize channels in a closed conformation, AA also promotes channel inactivation. Since the palmitoyl groups restrain channels from inactivating, perhaps no inhibition can occur. However, several pieces of data indicate that differences in channel inactivation cannot account for the ability of palmitoylated β2a to block inhibition [35]. First, when cells expressing CaV1.3b and depalmitoylated β2aC3,4S are preincubated with free palmitic acid, inhibition by AA is lost while fast inactivation remains. Second, attaching the transmembrane protein segment CD8 to β2aC3,4S rescues noninactivating kinetics [97, 98]. Despite the rescued non-inactivating kinetics, coexpression of CD8-β2aC3,4S with CaV1.3b exhibits robust inhibition by AA. Third, the β2e splice variant contains a unique hydrophobic N-terminus tail that may insert into the membrane. When coexpressed with either CaV1.2 or CaV1.3b, the L-current exhibits little inactivation [35, 103], yet is robustly inhibited by AA similar to CD8-β2aC3,4S. These studies indicate that free palmitic acid or the palmitoyl groups of β2a may competitively block inhibition of CaV1.3 by AA independently of a channel’s ability to undergo fast inactivation. Taken together, these L- and N-channel studies with β-subunits reveal a potentially novel role for a palmitoylated protein. The palmitoyl groups ofβ2a don’t simply tether the subunit to the membrane but rather may interact with VGCCs to block or interfere with AA’s binding site that confers current inhibition (Fig. 5A). Whether other proteins such palmitoylated syntaxins, G-proteins, or scaffolding proteins might also interact with VGCCs to alter their gating remains to be tested. Critical to developing this model will be further experiments that test whether certain channel residues change the response to AA and to palmitoylation.
Figure 5.

PIP2–AA model of VGCC inhibition by GqPCRs. A. Model comparing CaV1.3 channels with palmitoylated β2a to inhibited channels with β3. B. PIP2–AA model proposes that M1R coupling to Gq activates PLC to remove the inositol head group from PIP2 associated with the channel. Both DAG lipase (DAGL) and cPLA2 must cleave the two fatty acid tails from the remaining glycerol backbone in order to observe current inhibition. Once freed from the glycerol backbone the two fatty acids confer inhibition. C. VGCC current modulation by 10 μM Oxo-M of a cortical neuron in the absence (left) or presence of the PLA2 antagonist OPC (right). D. VGCC current modulation of a SCG neuron in the absence (left) or presence of the DAG lipase antagonist RHC 80267 (right).
6. PIP2 exerts actions that oppose AA on VGCC Currents
Currents from CaV1 and CaV2 channels decrease or “run down” irreversibly over time from ripped off patches of membrane. This observation has led to the hypothesis that critical intracellular components facilitate channel gating, but are lost upon ripping off the patch. What that critical component might be remained elusive until a study by Wu et al., [104] found that application of exogenous PIP2 slowed rundown of currents from giant, inside-out patches of Xenopus oocyte membrane expressing CaV2.1 (P/Q), α2δ, and β4 subunits. In contrast antibodies against PIP2 accelerated rundown. Similar decreases in rundown were obtained with CaV1.2 and CaV2.2 channels, expressed in inside-out oocyte membrane patches following PIP2 application [105, 106] or with whole-cell N-current when dialyzing the PIP2 analog diC8-PIP2 into SCG neurons [106]. When conditions were manipulated to increase endogenous PIP2 levels, currents were maintained or enhanced. Similarly in portal vein myocytes whole-cell L-current increased when dialyzed with 1 μM PIP3 or PI(3,4)P2 but not PI(4,5)P2 suggesting more specific structural requirements for enhancement [107]. Additional testing may provide a more complete profile of specificity for each VGCC by different phosphatidylinositols.
In contrast, conditions that decreased endogenous PIP2 levels caused currents to run down more rapidly than observed under control conditions [104, 108]. Interestingly, the presence of active catalytic subunit of protein kinase A (PKA) also decreased rundown of CaV2.1 currents. PKA’s effects were not additive with exogenously applied PIP2 [104]. These findings suggested that PIP2 may directly associate with recombinant channel protein preventing rundown by maintaining channel availability. Wu et al [104] further found that PIP2 has a second action where it decreases CaV2.1 currents by promoting reluctant gating. Positive test pulses minimize this effect of PIP2. Slowed activation kinetics accompanies the voltage-dependence of PIP2’s actions similar to the modulation profile of CaV2.1 currents by Gβγ of pertussis-toxin sensitive G-proteins [109]. Wu et al. [104] hypothesized that one binding site stabilized CaV2.1 channels, increasing their availability to open and thus called it the “S” site, whereas they named the second site that confers reluctant gating the “R” site. The actions of PIP2 appear the opposite of AA: AA decreases availability of channels to open and also acts at a second site to increase activation kinetics promoting willing gating. This mirror image of action though antagonistic suggests that PIP2 and AA may act at similar sites.
The Yang lab has performed over 85 point mutations in the four S6 segments of CaV2.1 and characterized current amplitude and kinetic activity of these mutants [46, 105]. Mutating one particular residue, an isoleucine located at the intracellular end of IIIS6 to histidine (I1520H) or aspartate (I1502D), significantly decreased rundown whereas point mutations in homologous positions in the other three domains had so significant effect [105]. Similarly homologous point mutations in domain III of L- and N-channels to histidine or aspartate also slowed rundown (Fig. 2A). Exposing mutant channels to PIP2 had no further effect; however the mutant channels still required PIP2 since their currents rapidly ran down when endogenous PIP2 was sequestered by exposing the membrane patches to a positively charged peptide sequence from myristolated alanine-rich protein kinase C substrate (MARCKS). MARCKS sequesters PIP2 through nonspecific electrostatic interactions between the negative charges of the phosphorylated inositol headgroup [110]. These findings raise the possibility that PIP2 may normally interact with positively charged amino acids that are spatially near IIIS6. The C-terminus of CaV2.1 interacts with PIP2 as well as other phospholipids such as phosphatidic acid in lipid-protein overlay assays consistent with direct interaction between pore-forming subunits and phospholipids [111]. Determining the precise locations of PIP2 interaction sites remains an open and challenging question since complex interactions among the intracellular loops appears to occur [112–114]. Potential PIP2 interactions with Ca2+ channel auxillary subunits have not been tested.
7. Physiological implications of VGCC modulation by lipids
A number of neurotransmitters including acetylcholine, dopamine, and serotonin, hormones and growth factors bind to GPCRs that stimulate phospholipases to liberate fatty acids, such as AA, from autonomic end organs and from nerve cell membranes [115–120]. AA release following muscarinic stimulation has been examined most extensively where AA is primarily liberated from inositol phospholipids. This relationship raises the question of whether neurotransmitters regulate PIP2 and AA levels to modulate VGCC activity. Muscarinic modulation of L- and N-current by a slow signaling pathway [121–124], first described in SCG neurons (see Hille 1994[125] and Suh & Hille 2005 [126] for review), mimics current enhancement and inhibition by exogenously applied AA [23, 39, 49, 127, 128]. However, whether endogenously released AA plays a role in modulating VGCC currents following muscarinic receptor activation is controversial. Here, we discuss the evidence for and against endogenously liberated AA downstream of GqPCRs mediating VGCC current modulation.
7.1 AA participation in Ca2+ current modulation by GqPCRs is controversial
The muscarinic agonist oxotremorine-M (Oxo-M) inhibits L- and N-current in SCG neurons by the slow pathway via M1 muscarinic receptors (M1Rs), Gq and metabolism of PIP2 by phospholipase C (PLC) [49, 104, 106, 129–131]. Additional phospholipid breakdown appears to participate in current modulation since decreasing AA release by pharmacologically antagonizing PLA2 activity with oleyloxyethyl phosphorylcholine (OPC) or 7,7-dimethyl-5,8-eicosadienoic acid (DEDA), minimized VGCC current inhibition in SCG and cortical (Fig. 5C) neurons [49, 127, 128, 132]. This slow pathway contrasts the fast, voltage-dependent N-current inhibition by direct G-protein binding to the pore-forming subunit [113]. OPC and DEDA have no inhibitory effects on this fast pathway [133, 134].
Little free AA is found in cell membranes. Instead AA is situated in phospholipids of the inner leaflet of the cell membrane. Acyl transferases covalently link AA to the C2 (also referred to as the sn-2) carbon of the glycerol backbone of phospholipids, whereas a saturated or mono-saturated fatty acid normally occupies the sn-1 position [135]. 80% of PIP2 has AA in the sn-2 position [136] (Fig. 1A). Phospholipase A2 (PLA2) cleaves fatty acids from the sn-2 position of phospholipids, with lysophospholipid arising as the other by-product. Group IVa PLA2, a cytoplasmic, Ca2+-sensitive PLA2, also called cPLA2, exhibits selectivity specifically for AA in the sn-2 position of phospholipids [137]. cPLA2 associates with membranes by binding to PIP2 via a C2 domain with an effective increase in enzyme activity [137]. Phosphorylation by ERK1/2 acutely activates cPLA2 following GqPCR stimulation. These same receptors are associated with phosphatidylinositol metabolism, suggesting that cPLA2 in vivo may exhibit some preference for cleaving AA from PIP2. However careful studies with purified or recombinant cPLA2 have not been performed to fully understand its substrate specificity. PLA2 will also cleave AA from phosphatidic acid. When antibodies were dialyzed into SCG neurons as functional antagonists of slow pathway modulation, cPLA2 antibodies but not non-immunized IgG or iPLA2 or sPLA2 antibodies minimized inhibition by Oxo-M raising the intriguing possibility that in neurons, cPLA2 may act to liberate AA from PIP2 to inhibit VGCC activity.
The finding that cPLA2 may participate in M1R inhibition of L- and N-current challenged a model, illustrated in Figure 4, where dissociation of PIP2 from VGCCs and subsequent breakdown by PLC is not only necessary but also sufficient for observing inhibition [2, 104, 106]. The Hille lab originally proposed this “PIP2” model for M-current modulation by M1Rs. In this model, PIP2 molecules cycle between binding and dissociating from the KCNQ proteins that give rise to M-current. PIP2 association increases channel availability to open. Following stimulation of M1Rs, activated PLC cleaves the inositol head group from unbound PIP2 [138–141]. Consequently less PIP2 is present to rebind to channels resulting in decreased M-current. Whether PLA2 participates in M-current modulation was examined by pharmacologically antagonizing PLA2 but no adverse effect was found [127, 142, 143]. Additionally small molecules and various kinases were tested but were also found to play no role in current inhibition [125, 126] supporting the idea that PIP2 breakdown by PLC is sufficient for its modulation by M1Rs. Because L-, M- and N-current inhibition by the slow pathway in both primary neurons and recombinant systems requires M1Rs, Gq and PLC [49, 104, 106, 126, 127, 138, 140–142, 144], the Hille lab and others proposed that the same signal transduction pathway similarly modulates all three currents.
Figure 4.

The PIP2 model of VGCC inhibition by GqPCRs. Top; PIP2 exists in a steady-state relationship with VGCCs, where it binds and unbinds with particular rate constants for each VGCC. When PLC is activated, it will metabolize PIP2 that has dissociated from VGCCs.
Consistent with the PIP2 model, Gamper et al. [106] reported no effect of OPC following a 2 min incubation period on N-current inhibition by Oxo-M, when recording from perforated patches of SCG neurons. These findings contrasted whole-cell studies where a 2 minute preincubation with the same OPC concentration resulted in a loss of inhibition by Oxo-M [49]. Gamper et al. [106] suggested that the differences observed with OPC, most likely reflect variations in experimental design rather than in the biology of the system. Similarly, Bannister et al. [145] found that the PLA2 antagonist quinacrine had no effect on slow pathway inhibition of recombinant L-current (CaV1.2); however this compound poorly antagonizes cPLA2 [146]. Lastly, Lechner et al. [147] found that inhibition of presynaptic currents by bradykinin was blocked when PIP2 breakdown was antagonized, yet remained normal when PLA2 was antagonized with DEDA. In each study, no control experiments were presented that documented selective cPLA2 antagonism, nevertheless these studies raised questions surrounding cPLA2’s role in the slow pathway of VGCC currents.
In a fourth study the PIP2 analog, diC8-PIP2 was dialyzed into SCG neurons to effectively expose the VGCCS to unlimited amounts of PIP2 agonist. Under these conditions, minimal Ca2+ current inhibition by Oxo-M occurred, suggesting that if enough endogenous PIP2 or exogenously applied diC8-PIP2 remains available to bind to VGCCs, no inhibition will occur [106]. DiC8-PIP2 lacks the normal fatty acid chains associated with PIP2, e.g., AA and stearic acid in the sn-1 and sn-2 positions respectively. Thus diC8-PIP2 might also act as a substrate competitor of cPLA2, antagonizing AA release from PIP2 and as a consequence, minimal current inhibition would occur. While this experiment highlighted the importance of PIP2 in VGCC gating, it did not rule out a requirement for PLA2 during slow pathway stimulation, leaving cPLA2’s role uncertain.
Subsequent biochemical, imaging, and genetic approaches pointed to a prominent role for cPLA2 activity during L- and N-current inhibition by the slow pathway. First, cPLA2 protein was acutely phosphorylated in SCG neurons following exposure to muscarinic agonist, but blocked in the presence of the MT-7 toxin, a M1R antagonist, demonstrating its acute activation by M1Rs [127]. Second, including BSA either in the pipette or bath solution to limit AA levels, antagonized L- and N-current inhibition [49, 127]. Moreover, when the BSA-containing bath solution was analyzed for fatty acid content by GC-MS, free AA levels were found to increase approximately 2-fold following ganglion exposure to Oxo-M documenting muscarinic stimulation of AA release from SCG. Most notably, L- and N-current inhibition by Oxo-M, was lost in cPLA2−/− SCG neurons [127, 128]. No significant difference in control current amplitude or magnitude of current inhibition by AA was observed between cPLA2+/+ versus cPLA2−/− neurons, indicating normal VGCC activity in cPLA2−/− neurons. In contrast to VGCCs, no change in M-current inhibition occurred in cPLA2−/− SCG neurons, demonstrating that breakdown of PIP2 by PLC still must occur under these experimental conditions despite loss of L- and N-current inhibition.
Taken together the data indicate that L- and N-current inhibition occurs by a signal transduction cascade diverging from that mediating M-current inhibition [49, 127, 128, 132, 133]. This conclusion is supported by recent work using palmitoylated charged peptides to sequester PIP2. Dialyzing low peptide concentrations into neurons disrupted M-current but not N-current modulation by M1Rs [148]. Free fatty acids, most likely AA, liberated from the sn-2 position during phospholipid metabolism mediate current inhibition. AA itself, rather than a metabolite, serves as the effector since antagonizing cyclo-oxygenases, lipoxygenases or P-450 epoxygenases individually or all together had no effect on AA or Oxo-M’s ability to inhibit current [23, 39, 49, 133].
7.2 PLC, cPLA2, and DAG lipase participate in M1R modulation of Ca2+ currents
Recent findings help advance our understanding in how PIP2 and AA regulate VGCC activity. In addition to PIP2 breakdown by PLC and AA release by acutely activated cPLA2, a third lipase called diacylglycerol (DAG) lipase appears to antagonize inhibition of native and recombinant L- and N-current (Fig. 5D) following M1R stimulation [149]. DAG lipase α and β cleave fatty acids (normally stearic acid) preferentially from the sn-1 position of PIP2 or from DAG [150, 151]. This conclusion of DAG lipase involvement in the slow pathway is based on findings that pharmacologically antagonizing DAG lipases with the selective compound RHC-80267 minimizes L- and N-current inhibition. In contrast RHC-80267 has no effect on M-current inhibition by the slow pathway, N-current inhibition by direct G-protein binding to CaV2.2, or L-and N-current inhibition by exogenous AA [134], indicating its actions selectively target slow pathway modulation of L- and N-currents [108, 149].
That three lipases are required for VGCC inhibition may seem implausible and excessively complicated. However, the need for all three lipases suggests a linear signal transduction cascade that generates a signaling molecule, such as AA, may not be responsible for Ca2+ current inhibition [127]. Similarly, the PIP2 model where removal of the phosphoinositol head group by PLC is necessary and sufficient for observing Ca2+ current inhibition [104, 106] also cannot explain why cPLA2−/− neurons exhibit little L- and N-current inhibition, while M-current inhibition proceeds normally, indicating that PLC is active [127, 134]. Thus both models fail in explaining all the results; however a remarkably simple “PIP2-AA” model shown in Figure 5B successfully combines the essence of each model to explain slow pathway modulation of Ca2+ currents.
Moreover, the PIP2-AA model successfully incorporates virtually all previously published findings from a number of research groups into an unexpectedly simple scheme. Oxo-M binds to M1Rs which couple to Gq to activate PLC. Activated PLC directly cleaves the inositol head group in the sn-3 from PIP2 molecules associated with channels Additionally PLC stimulates phosphorylation of cPLA2. Activated cPLA2 and DAG lipase liberate the two fatty acid tails of PIP2 in situ by acting specifically at the sn-2 and sn-1 positions respectively of the glycerol backbone. Consequently, the released IP3 and glycerol will enter the cytoplasm while the two freed fatty acids remain bound to channels. Alternatively the phosphoinositide headgroup could remain bound to the channel. Disassembling PIP2 may uncouple a hydrophobic region of the channel that binds the fatty acid tails from the predicted charged pocket that interacts with the phosphoinositol head group. This separation may impede coordinated conformational changes that closed channels undergo as they reconfigure into an open conformation, thus changing the availability of channels to open. The bound free fatty acid tails antagonize binding of a PIP2 to the site. Thus it is the loss of the glycerol backbone uncoupling the fatty acid tails from the head group that causes a decrease in channel opening, stabilizing a closed conformation. Three findings hint that a similar mechanism may occur for some K+ channels: 1) the antagonistic actions of PIP2 and AA on certain K+ channels [78, 152]; 2) overlapping binding sites for PIP2 and AA at the proximal end of the C-terminus of Kir3 channels [78]; and 3) the importance of the head group and fatty acid tails for coupling voltage sensing to opening [153, 154].
Another way to think of the consequences of PIP2 breakdown may be that somehow voltage sensing uncouples from channel opening. Though sheer speculation, this idea brings to mind, intriguing crystal data from the MacKinnon lab of phospholipid placement in the inner regions of a voltage-dependent K+ channel where just one phospholipid crystallized with each subunit [154]. The lipid tails sandwich between the voltage sensor and the inner pore with the head group pointing towards the cytoplasm. One could image that the phospholipid acts like Velcro between the two regions perhaps slipping as S6 flexes during opening only to re-attach as S6 straightens on closing. Whether VGCCs have a phospholipid in a similar region remains to be determined though a similar placement of PIP2 in a VGCC would be an ideal site to explain PIP2 and AA’s actions at the “S” site. Few clues exist that hint as to whether the PIP2 “R” site (which we hypothesizes confers enhancement by AA) is also located at the inner pore. It is possible that 4 phospholipids wedge between each voltage sensor and inner pore helix. Whether one or more of these wedged PIP2s may confer reluctant gating kinetics is an attractive possibility. Unlike the crystallized K+ channels, VGCCs do not have four-fold symmetry, so that homologous PIP2 interaction sites may influence channel opening differently. Alternatively recent findings with K+ channel structures reveal the importance of S1 for exerting the force on the outer pore region by the voltage sensor paddle [155]. Whether lipid packing in the crevices around S1 and the pore is important for channel opening no doubt will be an important question to answer.
Oxo-M also enhances N-current in SCG neurons similar to AA by increasing the voltage-sensitivity and kinetics of activation. Moreover muscarinic enhancement of N-current also involves PLA2 activity [49]. In recombinant studies M1Rs stimulation, as with exogenously applied AA, resulted in sustained enhancement of N-current only when CaV2.2 was expressed with the palmitoylated β2a [102]. We imagine that by forming multiple binding sites with cytoplasmic domains of CaV2.2, the CaVβ2a protein effectively “docks” its two palmitic acids at a site extremely close to CaV2.2 promoting their interaction with the channel. This constraint creates a high local concentration of palmitic acid that promotes competition with AA and possibly PIP2 for the “S” site acting there as a phospholipid mimic. No change in channel availability is observed with muscarinic stimulation, only the “R” site undergoes modulation. Thus the PIP2-AA model successfully incorporates the actions of three lipid moities: PIP2, AA and palmitoylated β2a. No doubt, lipid regulation of VGCCs does not occur exactly this way. This thinking however, creates a starting point for testing this molecular image of lipids competing for sites on VGCCs that effect channel opening. Critical to developing this model will be further experiments that examine whether mutating certain channel residues change the response to AA and to palmitoylation.
7.3 Control of PIP2 interaction with VGCCs by three lipases provides a highly regulated mechanism for modulation
The PIP2-AA model is appealing in that it provides more regulatory control of lipid interaction with channels compared to a “naked” channel with no bound lipid (Fig. 4). The local breakdown of PIP2 should give rise to variety of byproducts that each alters VGCC gating in unique ways. Moreover, this new model resolves previous conceptual conflicts in mechanism and provides a framework for raising new predictions and pursuing a new direction of questioning around how lipids regulate VGCCs in normal and pathological situations. First, this model predicts that the lipid tails of PIP2 and free AA interact with CaVα1 though whether PIP2 or free AA act by directly interacting with the channel remains uncertain. The PIP2-AA model is compatible with the idea that loss of PIP2 from channels occurs following M1R stimulation [104, 106]. If we assume that PIP2 directly interacts with the channel, then the simplest model would predict that cPLA2 liberates AA on location and exogenously applied AA may dislodge PIP2 by competing with its fatty acid tails for binding sites. Metabolizing PIP2 still bound to channels can account for why inhibiting either PLA2 or DAG lipase will antagonize channel modulation. However whether these enzymes are able to access PIP2 molecules while bound to the channel needs to be tested more directly.
Though increases in free AA in the bath solution are detected following muscarinic stimulation [127], this new model predicts that free AA generated locally confers inhibition. In support of this idea, AA inhibits recombinant T-current, exhibiting similar inhibition as L- and N-currents [20, 21]. Since T-current arises from channels with no accessory subunits, AA’s actions are predicted to occur on the pore-forming subunit. Whether the Hill coefficient of ~2 for AA binding to T-channels [21] suggests that AA acts at two sites on the channel or that the two tails of PIP2 act together at one site remains to be tested. This question of whether two tails participate in current inhibition raise an additional question of whether two AAs mediate inhibition or whether inhibition occurs from AA and the fatty acid, most likely stearic acid, from the sn-1 position; or just AA from the sn-2 position. The actions of stearic acid have not yet been tested on L- or N-current though other saturated fatty acids can enhance N-current (Barrett et al., 2001). Once liberated, AA may diffuse a short distance to its site of action. Whether other GqPCRs require both cPLA2 and DAG lipase to observe VGCC modulation has not been examined. Notably M3Rs couple to Gq to activate an endogenous AA-dependent noncapacitative Ca2+ current in HEK cells that also requires cPLA2 [156]. However, other GqPCRs require both PLA2 and DAGL activity and AA release to regulate a variety of cellular processes [156–163], documenting widespread association between DAGL and cPLA2. These questions and many others arise from contemplating the predictions of this simple new model; the answers of which may be as surprising as what the field has discovered so far on how lipids regulate VGCC activity.
Acknowledgments
The authors would like to acknowledge the efforts of former lab members in developing the ideas on lipid regulation of channels and support by funds from UMMS. We also would like to acknowledge the help from past and present collaborators. Research from the Rittenhouse lab discussed in this review was funded by a Grant-In-Aid and and an Established Investigator Award from the American Heart Association and grants from the NIH (NS057473, NS07366, and NS34195).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci U S A. 2001;98:11024–31. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delmas P, Coste B, Gamper N, Shapiro MS. Phosphoinositide lipid second messengers: new paradigms for calcium channel modulation. Neuron. 2005;47:179–82. doi: 10.1016/j.neuron.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Gamper N, Shapiro MS. Regulation of ion transport proteins by membrane phosphoinositides. Nat Rev Neurosci. 2007;8:921–34. doi: 10.1038/nrn2257. [DOI] [PubMed] [Google Scholar]
- 4.Michailidis IE, Zhang Y, Yang J. The lipid connection-regulation of voltage-gated Ca(2+) channels by phosphoinositides. Pflugers Arch. 2007;455:147–55. doi: 10.1007/s00424-007-0272-9. [DOI] [PubMed] [Google Scholar]
- 5.Boland LM, Drzewiecki MM. Polyunsaturated fatty acid modulation of voltage-gated ion channels. Cell Biochem Biophys. 2008;52:59–84. doi: 10.1007/s12013-008-9027-2. [DOI] [PubMed] [Google Scholar]
- 6.Meves H. Arachidonic acid and ion channels: an update. Br J Pharmacol. 2008;155:4–16. doi: 10.1038/bjp.2008.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts-Crowley ML, Rittenhouse AR. Biophysics of Ion Channels and Diseases. Transworld Research Network 2008 [Google Scholar]
- 8.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- 9.Meves H. The action of prostaglandins on ion channels. Curr Neuropharmacol. 2006;4:41–57. doi: 10.2174/157015906775203048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villarroel A, Schwarz TL. Inhibition of the Kv4 (Shal) family of transient K+ currents by arachidonic acid. J Neurosci. 1996;16:2522–32. doi: 10.1523/JNEUROSCI.16-08-02522.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meves H. Modulation of ion channels by arachidonic acid. Prog Neurobiol. 1994;43:175–86. doi: 10.1016/0301-0082(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 12.Hille B. Ion Channels of Excitable Membranes 2001 [Google Scholar]
- 13.Tombola F, Pathak MM, Isacoff EY. How does voltage open an ion channel? Annu Rev Cell Dev Biol. 2006;22:23–52. doi: 10.1146/annurev.cellbio.21.020404.145837. [DOI] [PubMed] [Google Scholar]
- 14.Hering S, Berjukow S, Sokolov S, Marksteiner R, Weiss RG, Kraus R, Timin EN. Molecular determinants of inactivation in voltage-gated Ca2+ channels. J Physiol, 528 Pt. 2000;2:237–49. doi: 10.1111/j.1469-7793.2000.t01-1-00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones SW. Calcium channels: unanswered questions. J Bioenerg Biomembr. 2003;35:461–75. doi: 10.1023/b:jobb.0000008020.86004.28. [DOI] [PubMed] [Google Scholar]
- 16.Stotz SC, Jarvis SE, Zamponi GW. Functional roles of cytoplasmic loops and pore lining transmembrane helices in the voltage-dependent inactivation of HVA calcium channels. J Physiol. 2004;554:263–73. doi: 10.1113/jphysiol.2003.047068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao YF, Gomez AM, Morgan JP, Lederer WJ, Leaf A. Suppression of voltage-gated L-type Ca2+ currents by polyunsaturated fatty acids in adult and neonatal rat ventricular myocytes. Proc Natl Acad Sci U S A. 1997;94:4182–7. doi: 10.1073/pnas.94.8.4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chemin J, Nargeot J, Lory P. Chemical determinants involved in anandamide-induced inhibition of T-type calcium channels. J Biol Chem. 2007;282:2314–23. doi: 10.1074/jbc.M610033200. [DOI] [PubMed] [Google Scholar]
- 19.Vellani V, Reynolds AM, McNaughton PA. Modulation of the synaptic Ca2+ current in salamander photoreceptors by polyunsaturated fatty acids and retinoids. J Physiol, 529 Pt. 2000;2:333–44. doi: 10.1111/j.1469-7793.2000.00333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Cribbs LL, Satin J. Arachidonic acid modulation of alpha1H, a cloned human T-type calcium channel. Am J Physiol Heart Circ Physiol. 2000;278:H184–93. doi: 10.1152/ajpheart.2000.278.1.H184. [DOI] [PubMed] [Google Scholar]
- 21.Talavera K, Staes M, Janssens A, Droogmans G, Nilius B. Mechanism of arachidonic acid modulation of the T-type Ca2+ channel alpha1G. J Gen Physiol. 2004;124:225–38. doi: 10.1085/jgp.200409050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oz M, Tchugunova Y, Dinc M. Differential effects of endogenous and synthetic cannabinoids on voltage-dependent calcium fluxes in rabbit T-tubule membranes: comparison with fatty acids. Eur J Pharmacol. 2004;502:47–58. doi: 10.1016/j.ejphar.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 23.Liu L, Barrett CF, Rittenhouse AR. Arachidonic acid both inhibits and enhances whole cell calcium currents in rat sympathetic neurons. Am J Physiol Cell Physiol. 2001;280:C1293–305. doi: 10.1152/ajpcell.2001.280.5.C1293. [DOI] [PubMed] [Google Scholar]
- 24.Bringmann A, Schopf S, Faude F, Reichenbach A. Arachidonic acid-induced inhibition of Ca2+ channel currents in retinal glial (Muller) cells. Graefes Arch Clin Exp Ophthalmol. 2001;239:859–64. doi: 10.1007/s004170100372. [DOI] [PubMed] [Google Scholar]
- 25.Liu SJ. Inhibition of L-type Ca2+ channel current and negative inotropy induced by arachidonic acid in adult rat ventricular myocytes. Am J Physiol Cell Physiol. 2007 doi: 10.1152/ajpcell.00284.2007. [DOI] [PubMed] [Google Scholar]
- 26.Singer D, Biel M, Lotan I, Flockerzi V, Hofmann F, Dascal N. The roles of the subunits in the function of the calcium channel. Science. 1991;253:1553–7. doi: 10.1126/science.1716787. [DOI] [PubMed] [Google Scholar]
- 27.Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T, Mori Y, De Waard M. The I–II loop of the Ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron. 2000;25:177–90. doi: 10.1016/s0896-6273(00)80881-8. [DOI] [PubMed] [Google Scholar]
- 28.Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee JH. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- 29.Danthi SJ, Enyeart JA, Enyeart JJ. Modulation of native T-type calcium channels by omega-3 fatty acids. Biochem Biophys Res Commun. 2005;327:485–93. doi: 10.1016/j.bbrc.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 30.Chesnoy-Marchais D, Fritsch J. Concentration-dependent modulations of potassium and calcium currents of rat osteoblastic cells by arachidonic acid. J Membr Biol. 1994;138:159–70. doi: 10.1007/BF00232644. [DOI] [PubMed] [Google Scholar]
- 31.Schmitt H, Meves H. Model experiments on squid axons and NG108-15 mouse neuroblastoma × rat glioma hybrid cells. J Physiol Paris. 1995;89:181–93. doi: 10.1016/0928-4257(96)83635-7. [DOI] [PubMed] [Google Scholar]
- 32.Marksteiner R, Schurr P, Berjukow S, Margreiter E, Perez-Reyes E, Hering S. Inactivation determinants in segment IIIS6 of Ca(v)3.1. J Physiol. 2001;537:27–34. doi: 10.1111/j.1469-7793.2001.0027k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balazy M, Poff CD. Biological nitration of arachidonic acid. Curr Vasc Pharmacol. 2004;2:81–93. doi: 10.2174/1570161043476465. [DOI] [PubMed] [Google Scholar]
- 34.Jarrahian A, Hillard CJ. Arachidonylethanolamide (anandamide) binds with low affinity to dihydropyridine binding sites in brain membranes. Prostaglandins Leukot Essent Fatty Acids. 1997;57:551–4. doi: 10.1016/s0952-3278(97)90559-7. [DOI] [PubMed] [Google Scholar]
- 35.Roberts-Crowley ML, Rittenhouse AR. Arachidonic Acid Inhibition of L-type Calcium (CaV1.3) Channels Varies with Accessory Beta Subunits. Journal of General Physiology. 2009 doi: 10.1085/jgp.200810047. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oz M, Tchugunova YB, Dunn SM. Endogenous cannabinoid anandamide directly inhibits voltage-dependent Ca(2+) fluxes in rabbit T-tubule membranes. Eur J Pharmacol. 2000;404:13–20. doi: 10.1016/s0014-2999(00)00396-4. [DOI] [PubMed] [Google Scholar]
- 37.Petit-Jacques J, Hartzell HC. Effect of arachidonic acid on the L-type calcium current in frog cardiac myocytes. J Physiol. 1996;493 (Pt 1):67–81. doi: 10.1113/jphysiol.1996.sp021365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimada T, Somlyo AP. Modulation of voltage-dependent Ca channel current by arachidonic acid and other long-chain fatty acids in rabbit intestinal smooth muscle. J Gen Physiol. 1992;100:27–44. doi: 10.1085/jgp.100.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrett CF, Liu L, Rittenhouse AR. Arachidonic acid reversibly enhances N-type calcium current at an extracellular site. Am J Physiol Cell Physiol. 2001;280:C1306–18. doi: 10.1152/ajpcell.2001.280.5.C1306. [DOI] [PubMed] [Google Scholar]
- 40.Liu L, Gonzalez PK, Barrett CF, Rittenhouse AR. The calcium channel ligand FPL 64176 enhances L-type but inhibits N-type neuronal calcium currents. Neuropharmacology. 2003;45:281–92. doi: 10.1016/s0028-3908(03)00153-9. [DOI] [PubMed] [Google Scholar]
- 41.Liu L, Rittenhouse AR. Effects of arachidonic acid on unitary calcium currents in rat sympathetic neurons. J Physiol. 2000;525(Pt 2):391–404. doi: 10.1111/j.1469-7793.2000.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roberts-Crowley ML, Rittenhouse AR. Arachidonic Acid Inhibition of L-type Calcium (CaV1.3) Channels Varies with Accessory Beta Subunits. Journal of General Physiology. 2008 doi: 10.1085/jgp.200810047. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keyser DO, Alger BE. Arachidonic acid modulates hippocampal calcium current via protein kinase C and oxygen radicals. Neuron. 1990;5:545–53. doi: 10.1016/0896-6273(90)90092-t. [DOI] [PubMed] [Google Scholar]
- 44.Schmitt H, Meves H. Modulation of neuronal calcium channels by arachidonic acid and related substances. J Membr Biol. 1995;145:233–44. doi: 10.1007/BF00232715. [DOI] [PubMed] [Google Scholar]
- 45.Xie C, Zhen XG, Yang J. Localization of the activation gate of a voltage-gated Ca2+ channel. J Gen Physiol. 2005;126:205–12. doi: 10.1085/jgp.200509293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhen XG, Xie C, Fitzmaurice A, Schoonover CE, Orenstein ET, Yang J. Functional architecture of the inner pore of a voltage-gated Ca2+ channel. J Gen Physiol. 2005;126:193–204. doi: 10.1085/jgp.200509292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berjukow S, Marksteiner R, Sokolov S, Weiss RG, Margreiter E, Hering S. Amino acids in segment IVS6 and beta-subunit interaction support distinct conformational changes during Ca(v)2.1 inactivation. J Biol Chem. 2001;276:17076–82. doi: 10.1074/jbc.M010491200. [DOI] [PubMed] [Google Scholar]
- 48.Shi C, Soldatov NM. Molecular determinants of voltage-dependent slow inactivation of the Ca2+ channel. J Biol Chem. 2002;277:6813–21. doi: 10.1074/jbc.M110524200. [DOI] [PubMed] [Google Scholar]
- 49.Liu L, Rittenhouse AR. Arachidonic acid mediates muscarinic inhibition and enhancement of N-type Ca2+ current in sympathetic neurons. Proc Natl Acad Sci U S A. 2003;100:295–300. doi: 10.1073/pnas.0136826100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heneghan JF, Mitra-Ganguli T, Stanish LF, Liu L, Rittenhouse AR. Cavβ2a toggles N-current modulation by Gq-coupled receptors from inhibition to enhancement. 2008 Submitted. [Google Scholar]
- 51.Plummer MR, Hess P. Reversible uncoupling of inactivation in N-type calcium channels. Nature. 1991;351:657–9. doi: 10.1038/351657a0. [DOI] [PubMed] [Google Scholar]
- 52.Rittenhouse AR, Hess P. Microscopic heterogeneity in unitary N-type calcium currents in rat sympathetic neurons. J Physiol. 1994;474:87–99. doi: 10.1113/jphysiol.1994.sp020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Plummer MR, Logothetis DE, Hess P. Elementary properties and pharmacological sensitivities of calcium channels in mammalian peripheral neurons. Neuron. 1989;2:1453–63. doi: 10.1016/0896-6273(89)90191-8. [DOI] [PubMed] [Google Scholar]
- 54.Patil PG, Brody DL, Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–38. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- 55.Spector A. Aggregation of a-crystallin and its possible relationship to cataract formation. Isr J Med Sci. 1972;8:1577–82. [PubMed] [Google Scholar]
- 56.Olofsson CS, Salehi A, Gopel SO, Holm C, Rorsman P. Palmitate stimulation of glucagon secretion in mouse pancreatic alpha-cells results from activation of L-type calcium channels and elevation of cytoplasmic calcium. Diabetes. 2004;53:2836–43. doi: 10.2337/diabetes.53.11.2836. [DOI] [PubMed] [Google Scholar]
- 57.Tian Y, Corkey RF, Yaney GC, Goforth PB, Satin LS, Moitoso de Vargas L. Differential modulation of L-type calcium channel subunits by oleate. Am J Physiol Endocrinol Metab. 2008;294:E1178–86. doi: 10.1152/ajpendo.90237.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olofsson CS, Salehi A, Holm C, Rorsman P. Palmitate increases L-type Ca2+ currents and the size of the readily releasable granule pool in mouse pancreatic beta-cells. J Physiol. 2004;557:935–48. doi: 10.1113/jphysiol.2004.066258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khurana G, Bennett MR. Nitric oxide and arachidonic acid modulation of calcium currents in postganglionic neurones of avian cultured ciliary ganglia. Br J Pharmacol. 1993;109:480–5. doi: 10.1111/j.1476-5381.1993.tb13594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hatton CJ, Peers C. Arachidonic acid inhibits both K+ and Ca2+ currents in isolated type I cells of the rat carotid body. Brain Res. 1998;787:315–20. doi: 10.1016/s0006-8993(97)01486-8. [DOI] [PubMed] [Google Scholar]
- 61.Bug W, Williams JT, North RA. Membrane potential measured during potassium-evoked release of noradrenaline from rat brain neurons: effects of normorphine. J Neurochem. 1986;47:652–5. doi: 10.1111/j.1471-4159.1986.tb04549.x. [DOI] [PubMed] [Google Scholar]
- 62.Piomelli D, Shapiro E, Feinmark SJ, Schwartz JH. Metabolites of arachidonic acid in the nervous system of Aplysia: possible mediators of synaptic modulation. J Neurosci. 1987;7:3675–86. doi: 10.1523/JNEUROSCI.07-11-03675.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakanishi H, Exton JH. Purification and characterization of the zeta isoform of protein kinase C from bovine kidney. J Biol Chem. 1992;267:16347–54. [PubMed] [Google Scholar]
- 64.Gailly P, Gong MC, Somlyo AV, Somlyo AP. Possible role of atypical protein kinase C activated by arachidonic acid in Ca2+ sensitization of rabbit smooth muscle. J Physiol. 1997;500 (Pt 1):95–109. doi: 10.1113/jphysiol.1997.sp022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mukai H, Kitagawa M, Shibata H, Takanaga H, Mori K, Shimakawa M, Miyahara M, Hirao K, Ono Y. Activation of PKN, a novel 120-kDa protein kinase with leucine zipper-like sequences, by unsaturated fatty acids and by limited proteolysis. Biochem Biophys Res Commun. 1994;204:348–56. doi: 10.1006/bbrc.1994.2466. [DOI] [PubMed] [Google Scholar]
- 66.Sopalla C, Leukert N, Sorg C, Kerkhoff C. Evidence for the involvement of the unique C-tail of S100A9 in the binding of arachidonic acid to the heterocomplex S100A8/A9. Biol Chem. 2002;383:1895–905. doi: 10.1515/BC.2002.213. [DOI] [PubMed] [Google Scholar]
- 67.McPhail LC, Clayton CC, Snyderman R. A potential second messenger role for arachidonic acid: activation of Ca2+-dependent protein kinase. Trans Assoc Am Physicians. 1984;97:222–31. [PubMed] [Google Scholar]
- 68.Kessen U, Schaloske R, Aichem A, Mutzel R. Ca(2+)/calmodulin-independent activation of calcineurin from Dictyostelium by unsaturated long chain fatty acids. J Biol Chem. 1999;274:37821–6. doi: 10.1074/jbc.274.53.37821. [DOI] [PubMed] [Google Scholar]
- 69.Skinner J, Sinclair C, Romeo C, Armstrong D, Charbonneau H, Rossie S. Purification of a fatty acid-stimulated protein-serine/threonine phosphatase from bovine brain and its identification as a homolog of protein phosphatase 5. J Biol Chem. 1997;272:22464–71. doi: 10.1074/jbc.272.36.22464. [DOI] [PubMed] [Google Scholar]
- 70.Norris AW, Spector AA. Very long chain n-3 and n-6 polyunsaturated fatty acids bind strongly to liver fatty acid-binding protein. J Lipid Res. 2002;43:646–53. [PubMed] [Google Scholar]
- 71.Demant EJ. Bovine serum albumin-(7-hydroxycoumarin-4-acetic acid) complex: applications to the fluorometric measurement of fatty acid concentrations. Anal Biochem. 1999;267:366–72. doi: 10.1006/abio.1998.3019. [DOI] [PubMed] [Google Scholar]
- 72.Spector AA. Fatty acid binding to plasma albumin. J Lipid Res. 1975;16:165–79. [PubMed] [Google Scholar]
- 73.Petitpas I, Grune T, Bhattacharya AA, Curry S. Crystal structures of human serum albumin complexed with monounsaturated and polyunsaturated fatty acids. J Mol Biol. 2001;314:955–60. doi: 10.1006/jmbi.2000.5208. [DOI] [PubMed] [Google Scholar]
- 74.Huang BX, Dass C, Kim HY. Probing conformational changes of human serum albumin due to unsaturated fatty acid binding by chemical cross-linking and mass spectrometry. Biochem J. 2005;387:695–702. doi: 10.1042/BJ20041624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiao YF, Ke Q, Wang SY, Auktor K, Yang Y, Wang GK, Morgan JP, Leaf A. Single point mutations affect fatty acid block of human myocardial sodium channel alpha subunit Na+ channels. Proc Natl Acad Sci U S A. 2001;98:3606–11. doi: 10.1073/pnas.061003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hamilton KL, Syme CA, Devor DC. Molecular localization of the inhibitory arachidonic acid binding site to the pore of hIK1. J Biol Chem. 2003;278:16690–7. doi: 10.1074/jbc.M212959200. [DOI] [PubMed] [Google Scholar]
- 77.Macica CM, Yang Y, Hebert SC, Wang WH. Arachidonic acid inhibits activity of cloned renal K+ channel, ROMK1. Am J Physiol. 1996;271:F588–94. doi: 10.1152/ajprenal.1996.271.3.F588. [DOI] [PubMed] [Google Scholar]
- 78.Rogalski SL, Chavkin C. Eicosanoids inhibit the G-protein-gated inwardly rectifying potassium channel (Kir3) at the Na+/PIP2 gating site. J Biol Chem. 2001;276:14855–60. doi: 10.1074/jbc.M010097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim Y, Gnatenco C, Bang H, Kim D. Localization of TREK-2 K+ channel domains that regulate channel kinetics and sensitivity to pressure, fatty acids and pHi. Pflugers Arch. 2001;442:952–60. doi: 10.1007/s004240100626. [DOI] [PubMed] [Google Scholar]
- 80.Sinnegger MJ, Wang Z, Grabner M, Hering S, Striessnig J, Glossmann H, Mitterdorfer J. Nine L-type amino acid residues confer full 1,4-dihydropyridine sensitivity to the neuronal calcium channel alpha1A subunit. Role of L-type Met1188. J Biol Chem. 1997;272:27686–93. doi: 10.1074/jbc.272.44.27686. [DOI] [PubMed] [Google Scholar]
- 81.Striessnig J, Goll A, Moosburger K, Glossmann H. Purified calcium channels have three allosterically coupled drug receptors. FEBS Lett. 1986;197:204–10. doi: 10.1016/0014-5793(86)80327-1. [DOI] [PubMed] [Google Scholar]
- 82.Hallaq H, Smith TW, Leaf A. Modulation of dihydropyridine-sensitive calcium channels in heart cells by fish oil fatty acids. Proc Natl Acad Sci U S A. 1992;89:1760–4. doi: 10.1073/pnas.89.5.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pepe S, Bogdanov K, Hallaq H, Spurgeon H, Leaf A, Lakatta E. Omega 3 polyunsaturated fatty acid modulates dihydropyridine effects on L-type Ca2+ channels, cytosolic Ca2+, and contraction in adult rat cardiac myocytes. Proc Natl Acad Sci U S A. 1994;91:8832–6. doi: 10.1073/pnas.91.19.8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shimasue K, Urushidani T, Hagiwara M, Nagao T. Effects of anandamide and arachidonic acid on specific binding of (+)-PN200-110, diltiazem and (−) -desmethoxyverapamil to L-type Ca2+ channel. Eur J Pharmacol. 1996;296:347–50. doi: 10.1016/0014-2999(95)00826-8. [DOI] [PubMed] [Google Scholar]
- 85.Jones SW, Marks TN. Calcium currents in bullfrog sympathetic neurons. I. Activation kinetics and pharmacology. J Gen Physiol. 1989;94:151–67. doi: 10.1085/jgp.94.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brown SP, Safo PK, Regehr WG. Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci. 2004;24:5623–31. doi: 10.1523/JNEUROSCI.0918-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fisyunov A, Tsintsadze V, Min R, Burnashev N, Lozovaya N. Cannabinoids modulate the P-type high-voltage-activated calcium currents in purkinje neurons. J Neurophysiol. 2006;96:1267–77. doi: 10.1152/jn.01227.2005. [DOI] [PubMed] [Google Scholar]
- 88.Guo J, Ikeda SR. Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol Pharmacol. 2004;65:665–74. doi: 10.1124/mol.65.3.665. [DOI] [PubMed] [Google Scholar]
- 89.Chemin J, Monteil A, Perez-Reyes E, Nargeot J, Lory P. Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. Embo J. 2001;20:7033–40. doi: 10.1093/emboj/20.24.7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roberts-Crowley ML, Rittenhouse AR. Arachidonic Acid Inhibition of L-type Calcium (CaV1.3) Channels Varies with Accessory Beta Subunits. 2008 doi: 10.1085/jgp.200810047. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chien AJ, Carr KM, Shirokov RE, Rios E, Hosey MM. Identification of palmitoylation sites within the L-type calcium channel beta2a subunit and effects on channel function. J Biol Chem. 1996;271:26465–8. doi: 10.1074/jbc.271.43.26465. [DOI] [PubMed] [Google Scholar]
- 92.Olcese R, Qin N, Schneider T, Neely A, Wei X, Stefani E, Birnbaumer L. The amino terminus of a calcium channel beta subunit sets rates of channel inactivation independently of the subunit’s effect on activation. Neuron. 1994;13:1433–8. doi: 10.1016/0896-6273(94)90428-6. [DOI] [PubMed] [Google Scholar]
- 93.Cens T, Restituito S, Galas S, Charnet P. Voltage and calcium use the same molecular determinants to inactivate calcium channels. J Biol Chem. 1999;274:5483–90. doi: 10.1074/jbc.274.9.5483. [DOI] [PubMed] [Google Scholar]
- 94.Qin N, Platano D, Olcese R, Costantin JL, Stefani E, Birnbaumer L. Unique regulatory properties of the type 2a Ca2+ channel beta subunit caused by palmitoylation. Proc Natl Acad Sci U S A. 1998;95:4690–5. doi: 10.1073/pnas.95.8.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stephens GJ, Page KM, Bogdanov Y, Dolphin AC. The alpha1B Ca2+ channel amino terminus contributes determinants for beta subunit-mediated voltage-dependent inactivation properties. J Physiol. 2000;525(Pt 2):377–90. doi: 10.1111/j.1469-7793.2000.t01-1-00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Feng ZP, Hamid J, Doering C, Bosey GM, Snutch TP, Zamponi GW. Residue Gly1326 of the N-type calcium channel alpha 1B subunit controls reversibility of omega-conotoxin GVIA and MVIIA block. J Biol Chem. 2001;276:15728–35. doi: 10.1074/jbc.M100406200. [DOI] [PubMed] [Google Scholar]
- 97.Restituito S, Cens T, Barrere C, Geib S, Galas S, De Waard M, Charnet P. The [beta]2a subunit is a molecular groom for the Ca2+ channel inactivation gate. J Neurosci. 2000;20:9046–52. doi: 10.1523/JNEUROSCI.20-24-09046.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Restituito S, Cens T, Rousset M, Charnet P. Ca(2+) channel inactivation heterogeneity reveals physiological unbinding of auxiliary beta subunits. Biophys J. 2001;81:89–96. doi: 10.1016/S0006-3495(01)75682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Richards MW, Leroy J, Pratt WS, Dolphin AC. The HOOK-domain between the SH3 and the GK domains of Cavbeta subunits contains key determinants controlling calcium channel inactivation. Channels (Austin) 2007;1:92–101. doi: 10.4161/chan.4145. [DOI] [PubMed] [Google Scholar]
- 100.Chan AW, Owens S, Tung C, Stanley EF. Resistance of presynaptic CaV2.2 channels to voltage-dependent inactivation: dynamic palmitoylation and voltage sensitivity. Cell Calcium. 2007;42:419–25. doi: 10.1016/j.ceca.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 101.Chien AJ, Hosey MM. Post-translational modifications of beta subunits of voltage-dependent calcium channels. J Bioenerg Biomembr. 1998;30:377–86. doi: 10.1023/a:1021941706726. [DOI] [PubMed] [Google Scholar]
- 102.Heneghan JF, Mitra-Ganguli T, Stanish LF, Liu L, Rittenhouse AR. The Ca2+ Channel Beta Subunit Determines Whether Stimulation of M1 Muscarinic Receptors Enhance or Inhibit N-Current. Society for General Physiologists 62nd Annual Meeting and Symposium; 2008. Poster 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Takahashi SX, Mittman S, Colecraft HM. Distinctive modulatory effects of five human auxiliary beta2 subunit splice variants on L-type calcium channel gating. Biophys J. 2003;84:3007–21. doi: 10.1016/S0006-3495(03)70027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu L, Bauer CS, Zhen XG, Xie C, Yang J. Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature. 2002;419:947–52. doi: 10.1038/nature01118. [DOI] [PubMed] [Google Scholar]
- 105.Zhen XG, Xie C, Yamada Y, Zhang Y, Doyle C, Yang J. A single amino acid mutation attenuates rundown of voltage-gated calcium channels. FEBS Lett. 2006;580:5733–8. doi: 10.1016/j.febslet.2006.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS. Phosphatidylinositol [correction] 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci. 2004;24:10980–92. doi: 10.1523/JNEUROSCI.3869-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Le Blanc C, Mironneau C, Barbot C, Henaff M, Bondeva T, Wetzker R, Macrez N. Regulation of vascular L-type Ca2+ channels by phosphatidylinositol 3,4,5-trisphosphate. Circ Res. 2004;95:300–7. doi: 10.1161/01.RES.0000138017.76125.8b. [DOI] [PubMed] [Google Scholar]
- 108.Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–7. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–62. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 110.McLaughlin S, Wang J, Gambhir A, Murray D. PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–75. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 111.Rousset M, Cens T, Gouin-Charnet A, Scamps F, Charnet P. Ca2+ and phosphatidylinositol 4,5-bisphosphate stabilize a Gbeta gamma-sensitive state of Ca V2 Ca 2+ channels. J Biol Chem. 2004;279:14619–30. doi: 10.1074/jbc.M313284200. [DOI] [PubMed] [Google Scholar]
- 112.Agler HL, Evans J, Tay LH, Anderson MJ, Colecraft HM, Yue DT. G protein-gated inhibitory module of N-type (ca(v)2.2) ca2+ channels. Neuron. 2005;46:891–904. doi: 10.1016/j.neuron.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 113.De Waard M, Hering J, Weiss N, Feltz A. How do G proteins directly control neuronal Ca2+ channel function? Trends Pharmacol Sci. 2005;26:427–36. doi: 10.1016/j.tips.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 114.Stotz SC, Zamponi GW. Structural determinants of fast inactivation of high voltage-activated Ca(2+) channels. Trends Neurosci. 2001;24:176–81. doi: 10.1016/s0166-2236(00)01738-0. [DOI] [PubMed] [Google Scholar]
- 115.Biddlecome GH, Berstein G, Ross EM. Regulation of phospholipase C-beta1 by Gq and m1 muscarinic cholinergic receptor. Steady-state balance of receptor-mediated activation and GTPase-activating protein-promoted deactivation. J Biol Chem. 1996;271:7999–8007. doi: 10.1074/jbc.271.14.7999. [DOI] [PubMed] [Google Scholar]
- 116.Reichman M, Nen W, Hokin LE. Acetylcholine releases prostaglandins from brain slices incubated in vitro. J Neurochem. 1987;49:1216–21. doi: 10.1111/j.1471-4159.1987.tb10013.x. [DOI] [PubMed] [Google Scholar]
- 117.Gammon CM, Allen AC, Morell P. Bradykinin stimulates phosphoinositide hydrolysis and mobilization of arachidonic acid in dorsal root ganglion neurons. J Neurochem. 1989;53:95–101. doi: 10.1111/j.1471-4159.1989.tb07299.x. [DOI] [PubMed] [Google Scholar]
- 118.Felder CC, Kanterman RY, Ma AL, Axelrod J. Serotonin stimulates phospholipase A2 and the release of arachidonic acid in hippocampal neurons by a type 2 serotonin receptor that is independent of inositolphospholipid hydrolysis. Proc Natl Acad Sci U S A. 1990;87:2187–91. doi: 10.1073/pnas.87.6.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tence M, Cordier J, Premont J, Glowinski J. Muscarinic cholinergic agonists stimulate arachidonic acid release from mouse striatal neurons in primary culture. J Pharmacol Exp Ther. 1994;269:646–53. [PubMed] [Google Scholar]
- 120.Bernstein LS, Ramineni S, Hague C, Cladman W, Chidiac P, Levey AI, Hepler JR. RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11alpha signaling. J Biol Chem. 2004;279:21248–56. doi: 10.1074/jbc.M312407200. [DOI] [PubMed] [Google Scholar]
- 121.Beech DJ, Bernheim L, Mathie A, Hille B. Intracellular Ca2+ buffers disrupt muscarinic suppression of Ca2+ current and M current in rat sympathetic neurons. Proc Natl Acad Sci U S A. 1991;88:652–6. doi: 10.1073/pnas.88.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bernheim L, Beech DJ, Hille B. A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron. 1991;6:859–67. doi: 10.1016/0896-6273(91)90226-p. [DOI] [PubMed] [Google Scholar]
- 123.Bernheim L, Mathie A, Hille B. Characterization of muscarinic receptor subtypes inhibiting Ca2+ current and M current in rat sympathetic neurons. Proc Natl Acad Sci U S A. 1992;89:9544–8. doi: 10.1073/pnas.89.20.9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mathie A, Bernheim L, Hille B. Inhibition of N- and L-type calcium channels by muscarinic receptor activation in rat sympathetic neurons. Neuron. 1992;8:907–14. doi: 10.1016/0896-6273(92)90205-r. [DOI] [PubMed] [Google Scholar]
- 125.Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–6. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 126.Suh BC, Hille B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol. 2005;15:370–8. doi: 10.1016/j.conb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 127.Liu L, Zhao R, Bai Y, Stanish LF, Evans JE, Sanderson MJ, Bonventre JV, Rittenhouse AR. M1 muscarinic receptors inhibit L-type Ca2+ current and M-current by divergent signal transduction cascades. J Neurosci. 2006;26:11588–98. doi: 10.1523/JNEUROSCI.2102-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Grigg P, Liu L, Bonventre JV, Rittenhouse AR. Increased nerve cell firing in sympathetic neurons missing cytoplasmic phosholipase A2. Society for Neuroscience, 2008. 2008 Poster# 132.20/D45. [Google Scholar]
- 129.Delmas P, Abogadie FC, Dayrell M, Haley JE, Milligan G, Caulfield MP, Brown DA, Buckley NJ. G-proteins and G-protein subunits mediating cholinergic inhibition of N-type calcium currents in sympathetic neurons. Eur J Neurosci. 1998;10:1654–66. doi: 10.1046/j.1460-9568.1998.00170.x. [DOI] [PubMed] [Google Scholar]
- 130.Haley JE, Abogadie FC, Delmas P, Dayrell M, Vallis Y, Milligan G, Caulfield MP, Brown DA, Buckley NJ. The alpha subunit of Gq contributes to muscarinic inhibition of the M-type potassium current in sympathetic neurons. J Neurosci. 1998;18:4521–31. doi: 10.1523/JNEUROSCI.18-12-04521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shapiro MS, Wollmuth LP, Hille B. Modulation of Ca2+ channels by PTX-sensitive G-proteins is blocked by N-ethylmaleimide in rat sympathetic neurons. J Neurosci. 1994;14:7109–16. doi: 10.1523/JNEUROSCI.14-11-07109.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu L, Roberts ML, Rittenhouse AR. Phospholipid metabolism is required for M1 muscarinic inhibition of N-type calcium current in sympathetic neurons. Eur Biophys J. 2004;33:255–64. doi: 10.1007/s00249-003-0387-7. [DOI] [PubMed] [Google Scholar]
- 133.Liu L, Rittenhouse AR. Pharmacological discrimination between muscarinic receptor signal transduction cascades with bethanechol chloride. Br J Pharmacol. 2003;138:1259–70. doi: 10.1038/sj.bjp.0705157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liu L, Heneghan JF, Stanish LF, Michael GJ, Erganov M, Rittenhouse AR. L-and N-Current but not M-Current Inhibition by M1 Muscarinic Receptors Requires DAG Lipase Activity. 2008 doi: 10.1002/jcp.21378. Submitted. [DOI] [PubMed] [Google Scholar]
- 135.Bennett CN, Horrobin DF. Gene targets related to phospholipid and fatty acid metabolism in schizophrenia and other psychiatric disorders: an update. Prostaglandins Leukot Essent Fatty Acids. 2000;63:47–59. doi: 10.1054/plef.2000.0191. [DOI] [PubMed] [Google Scholar]
- 136.Wenk MR, Lucast L, Di Paolo G, Romanelli AJ, Suchy SF, Nussbaum RL, Cline GW, Shulman GI, McMurray W, De Camilli P. Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat Biotechnol. 2003;21:813–7. doi: 10.1038/nbt837. [DOI] [PubMed] [Google Scholar]
- 137.Leslie CC. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot Essent Fatty Acids. 2004;70:373–6. doi: 10.1016/j.plefa.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 138.Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35:507–20. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- 139.Zhang W, Crocker E, McLaughlin S, Smith SO. Binding of peptides with basic and aromatic residues to bilayer membranes: phenylalanine in the myristoylated alanine-rich C kinase substrate effector domain penetrates into the hydrophobic core of the bilayer. J Biol Chem. 2003;278:21459–66. doi: 10.1074/jbc.M301652200. [DOI] [PubMed] [Google Scholar]
- 140.Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–62. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- 141.Ford CP, Stemkowski PL, Light PE, Smith PA. Experiments to test the role of phosphatidylinositol 4,5-bisphosphate in neurotransmitter-induced M-channel closure in bullfrog sympathetic neurons. J Neurosci. 2003;23:4931–41. doi: 10.1523/JNEUROSCI.23-12-04931.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Robbins J, Marsh SJ, Brown DA. On the mechanism of M-current inhibition by muscarinic m1 receptors in DNA-transfected rodent neuroblastoma × glioma cells. J Physiol. 1993;469:153–78. doi: 10.1113/jphysiol.1993.sp019809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Villarroel A. On the role of arachidonic acid in M-current modulation by muscarine in bullfrog sympathetic neurons. J Neurosci. 1994;14:7053–66. doi: 10.1523/JNEUROSCI.14-11-07053.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, Logothetis DE. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003;37:963–75. doi: 10.1016/s0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]
- 145.Bannister RA, Melliti K, Adams BA. Reconstituted slow muscarinic inhibition of neuronal (Ca(v)1.2c) L-type Ca2+ channels. Biophys J. 2002;83:3256–67. doi: 10.1016/S0006-3495(02)75327-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kim DK, Bonventre JV. Purification of a 100 kDa phospholipase A2 from spleen, lung and kidney: antiserum raised to pig spleen phospholipase A2 recognizes a similar form in bovine lung, kidney and platelets, and immunoprecipitates phospholipase A2 activity. Biochem J. 1993;294 (Pt 1):261–70. doi: 10.1042/bj2940261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lechner SG, Hussl S, Schicker KW, Drobny H, Boehm S. Presynaptic inhibition via a phospholipase C- and phosphatidylinositol bisphosphate-dependent regulation of neuronal Ca2+ channels. Mol Pharmacol. 2005;68:1387–96. doi: 10.1124/mol.105.014886. [DOI] [PubMed] [Google Scholar]
- 148.Robbins J, Marsh SJ, Brown DA. Probing the regulation of M (Kv7) potassium channels in intact neurons with membrane-targeted peptides. J Neurosci. 2006;26:7950–61. doi: 10.1523/JNEUROSCI.2138-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Liu L, Heneghan JF, Michael GJ, Stanish LF, Egertova M, Rittenhouse AR. Land N-current but not M-current inhibition by M1 muscarinic receptors requires DAG lipase activity. J Cell Physiol. 2008;216:91–100. doi: 10.1002/jcp.21378. [DOI] [PubMed] [Google Scholar]
- 150.Dixon JF, Hokin LE. Secretogogue-stimulated phosphatidylinositol breakdown in the exocrine pancreas liberates arachidonic acid, stearic acid, and glycerol by sequential actions of phospholipase C and diglyceride lipase. J Biol Chem. 1984;259:14418–25. [PubMed] [Google Scholar]
- 151.Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–8. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004;304:265–70. doi: 10.1126/science.1094113. [DOI] [PubMed] [Google Scholar]
- 153.Xu Y, Ramu Y, Lu Z. Removal of phospho-head groups of membrane lipids immobilizes voltage sensors of K+ channels. Nature. 2008;451:826–9. doi: 10.1038/nature06618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–82. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 155.Lee SY, Banerjee A, Mackinnon R. Two Separate Interfaces between the Voltage Sensor and Pore Are Required for the Function of Voltage-Dependent K(+) Channels. PLoS Biol. 2009;7:e47. doi: 10.1371/journal.pbio.1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Osterhout JL, Shuttleworth TJ. A Ca(2+)-independent activation of a type IV cytosolic phospholipase A(2) underlies the receptor stimulation of arachidonic acid-dependent noncapacitative calcium entry. J Biol Chem. 2000;275:8248–54. doi: 10.1074/jbc.275.11.8248. [DOI] [PubMed] [Google Scholar]
- 157.Ambs P, Baccarini M, Fitzke E, Dieter P. Role of cytosolic phospholipase A2 in arachidonic acid release of rat-liver macrophages: regulation by Ca2+ and phosphorylation. Biochem J. 1995;311 (Pt 1):189–95. doi: 10.1042/bj3110189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Antoniotti S, Fiorio Pla A, Pregnolato S, Mottola A, Lovisolo D, Munaron L. Control of endothelial cell proliferation by calcium influx and arachidonic acid metabolism: a pharmacological approach. J Cell Physiol. 2003;197:370–8. doi: 10.1002/jcp.10359. [DOI] [PubMed] [Google Scholar]
- 159.Pandol SJ, Hsu YL, Kondratenko NF, Schoeffield-Payne MS, Steinbach JH. Dual pathways for agonist-stimulated arachidonic acid release in pancreatic acini: roles in secretion. Am J Physiol. 1991;260:G423–33. doi: 10.1152/ajpgi.1991.260.3.G423. [DOI] [PubMed] [Google Scholar]
- 160.Carraway RE, Hassan S, Cochrane DE. Regulation of neurotensin receptor function by the arachidonic acid-lipoxygenase pathway in prostate cancer PC3 cells. Prostaglandins Leukot Essent Fatty Acids. 2006;74:93–107. doi: 10.1016/j.plefa.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 161.Hou W, Arita Y, Morisset J. Endogenous arachidonic acid release and pancreatic amylase secretion. Pancreas. 1997;14:301–8. doi: 10.1097/00006676-199704000-00014. [DOI] [PubMed] [Google Scholar]
- 162.Mau SE, Vilhardt H. Cross talk between substance P and melittin-activated cellular signaling pathways in rat lactotroph-enriched cell cultures. J Neurochem. 1997;69:762–72. doi: 10.1046/j.1471-4159.1997.69020762.x. [DOI] [PubMed] [Google Scholar]
- 163.Liu L. Regulation of lung surfactant secretion by phospholipase A2. J Cell Biochem. 1999;72:103–10. [PubMed] [Google Scholar]