

Summary
Deletion of gene Rv3676 in Mycobacterium tuberculosis coding for a transcription factor belonging to the cAMP receptor protein (CRP) family caused growth defects in laboratory medium, in bone marrow-derived macrophages and in a mouse model of tuberculosis. Transcript profiling of M. tuberculosis grown in vitro identified 16 genes with significantly altered expression in the mutant compared with the wild type. Analysis of the DNA sequences upstream of the corresponding open reading frames revealed that 12 possessed sequences related to a consensus CRP binding site that could represent the sites of action of Rv3676. These included rpfA, lprQ, whiB1 and ahpC among genes with enhanced expression in the wild type, and Rv3616c-Rv3613c, Rv0188 and lipQ among genes exhibiting enhanced expression in the mutant. The activity of an rpfA::lacZ promoter fusion was lowered in the Rv3676 mutant and by mutation of the predicted Rv3676 binding site. Moreover, the product of Rv3676 (isolated as a TrxA fusion protein) interacted specifically with the rpfA promoter, and binding was inhibited by mutation of the Rv3676 site. Although Rv3676 retains four of the six amino acid residues that bind cAMP in Escherichia coli CRP addition of cAMP did not enhance Rv3676 binding at the rpfA promoter in vitro. In summary, it has been shown that Rv3676 is a direct regulator of rpfA expression, and because rpfA codes for a resuscitation promoting factor this may implicate Rv3676 in reactivation of dormant M. tuberculosis infections.
Introduction
Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), is one of the most successful of human pathogens, currently contributing to the deaths of ~2 million people every year (Dye et al., 1999). A characteristic feature of tuberculosis is that much disease actually occurs through reactivation of existing dormant infections; the lifetime risk of reactivation TB is estimated at 5–10% (Bloom and Murray, 1992). For M. tuberculosis, the preferred environment is the interior of the macrophage (Russell, 2001). The interplay between the bacterium and the eukaryotic cell, together with the interaction with the immune system, defines the outcome of the infection (Kaufmann, 2001). Following an initial infection, M. tuberculosis can persist for long periods of time in a dormant or non-replicating persistent state from which it may emerge several years later when conditions are more favourable, such as when the infected individual becomes immunocompromised (reviewed by Stewart et al., 2003). As about a third of the world’s population is thought to have a latent M. tuberculosis infection, the potential for reactivation TB is very large (Dye et al., 1999). Importantly, dormant bacteria are able to resist the effects of the human immune response and chemotherapy (reviewed by Stewart et al., 2003). The nature of the latent state is therefore of much interest and an understanding of mycobacterial persistence is critical to the control of TB.
Sequencing of the M. tuberculosis genome has revealed a number of putative regulatory genes, some identified as orthologues of well-known genes in other bacteria (Cole et al., 1998). Defining the functions of these regulators will provide new insights into the mechanisms by which M. tuberculosis controls gene expression in response to environmental and metabolic signals when it is present as an intracellular pathogen.
Members of the cAMP receptor protein (CRP)-FNR superfamily of transcription factors regulate a diversity of physiological functions in a wide range of bacteria. Different members of the family control aspects of carbon, sulphur and nitrogen metabolism, denitrification, nitrogen fixation, aerobic and anaerobic respiration, virulence gene expression and bacterial luminescence in response to a variety of environmental and metabolic signals (Green et al., 2001). Members of this large family of transcription regulators are all predicted to be structurally related to CRP. The archetypal CRP fold (Schultz et al., 1991) is a versatile structure that has evolved to accommodate different functional specificities in signal perception, DNA binding and interaction with RNA polymerase to allow different family members to respond to a wide range of signals (Green et al., 2001; Korner et al., 2003). The best-characterized member of the family is CRP itself. Escherichia coli CRP controls the expression of numerous genes in response to changes in the intracellular concentration of cAMP (Gosset et al., 2004). Upon binding cAMP, the cAMP:CRP complex binds to promoters containing DNA sequences related to the consensus TGTGANNNNNNT CACA (Berg and von Hippel, 1988). Once bound to promoter DNA CRP recruits RNA polymerase and specific protein:protein contacts are established that promote transcription of target genes (reviewed by Busby and Ebright, 1999). Alternatively, at some promoters CRP represses transcription by promoter occlusion.
The M. tuberculosis Rv3676 gene is annotated on TubercuList (http://genolist.pasteur.fr/TubercuList/) as a ‘probable transcriptional regulatory protein (probably CRP/FNR-family)’. Here an Rv3676 null mutant of M. tuberculosis is shown to be attenuated in macrophages and in a mouse model of tuberculosis infection. Transcriptome analysis in vitro reveals significant transcriptional changes in the Rv3676 mutant compared with the wild type, including altered expression the rpfA gene coding for one of five resuscitation promoting factor proteins (Rpfs), which could be involved in the regulation of persistence and reactivation (Mukamolova et al., 2002a). These proteins, originally discovered in Micrococcus luteus (Mukamolova et al., 1998), are potential regulators of persistence and reactivation and picomolar concentrations of Rpfs are able to stimulate the growth of extended stationary-phase cultures of Mycobacterium bovis BCG (Mukamolova et al., 2002a). They may therefore play a role in reactivation of latent infection. Little is yet known about how these genes are regulated. Here it is shown that the upstream region of rpfA contains a site similar to the CRP binding site in E. coli that is required for Rv3676-mediated regulation of rpfA expression. This therefore suggests that Rv3676 is an important transcriptional regulator in the control of M. tuberculosis infection and by regulating the expression of rpfA is implicated in reactivation during latency.
Results
Rv3676 is a member of the CRP family of transcription regulators
Escherichia coli CRP is the founder member of a superfamily of structurally related transcription regulators and the protein encoded by M. tuberculosis Rv3676 is a member of this family. Rv3676 is a deeply branching member related to the CRP and CooA groups of the family (Green et al., 2001; Korner et al., 2003). Thus, Rv3676 is 32% identical (53% similar) to E. coli CRP over 189 amino acids (residues 31–213 for Rv3676 and 24–206 for CRP) with most divergence within the N-terminal regions of the proteins. The N-terminal region of E. coli CRP contains two residues (His19 and His21) that interact with RNA polymerase and contribute to the activation of transcription from class II promoters (reviewed by Busby and Ebright, 1999). The lack of conserved amino acids in this region might suggest Rv3676 interacts differently with its cognate polymerase. Four of six residues that are involved in cAMP binding in E. coli CRP (Schultz et al., 1991) are conserved in Rv3676 (Fig. 1). Moreover, the amino acid residues (Arg180, Glu181 and Arg185) that are directly involved in DNA recognition in E. coli CRP (Schultz et al., 1991) are well conserved in Rv3676 with only Arg185 of CRP being replaced by Lys at the corresponding position in Rv3676 (Fig. 1). In contrast to the sustained similarities between CRP and Rv3676, most similarities to CooA are restricted to the C-terminal region (amino acids 140–209 of Rv3676 and 129–198 of CRP) and the amino acids (Pro2 and His77) that act as ligands for the CO-sensing haem cofactor of CooA (Lanzilotta et al., 2000) are not conserved in Rv3676. Therefore, these observations suggest that Rv3676 might share at least some of the functional characteristics of E. coli CRP, such as cyclic nucleotide binding and recognition of a similar DNA sequence.
Fig. 1.

Amino acid sequence alignments of the product of gene Rv3676 of M. tuberculosis with the CRP of E. coli. The highlighted residues are those involved in cyclic nucleotide binding in E. coli CRP and those that are conserved in M. tuberculosis Rv3676. There is no conservation between the other two cyclic nucleotide binding residues, R83 in E. coli (T90 in M. tuberculosis) or S129 (N135 in M. tuberculosis). The amino acids (Arg180, Glu181 and Arg185) of E. coli CRP directly involved in DNA recognition are indicated by underlining.
The Rv3676 deletion mutant is attenuated for growth in vitro and in vivo
To investigate the requirement of Rv3676 for normal in vitro growth, the Rv3676 gene was deleted in M. tuberculosis H37Rv, and the optical densities of both the Rv3676 mutant and wild-type strains were monitored in rolling culture bottles from inoculation until the cultures reached stationary phase. The Rv3676 mutant grew more slowly than the wild type, the doubling time being approximately 30 h compared with 16 h in the wild type (Fig. 2). In addition, the mutant strain grew very slowly on solid 7H11 agar plates, taking approximately 5 weeks to form colonies, compared with 2–3 weeks for the parental strain.
Fig. 2.
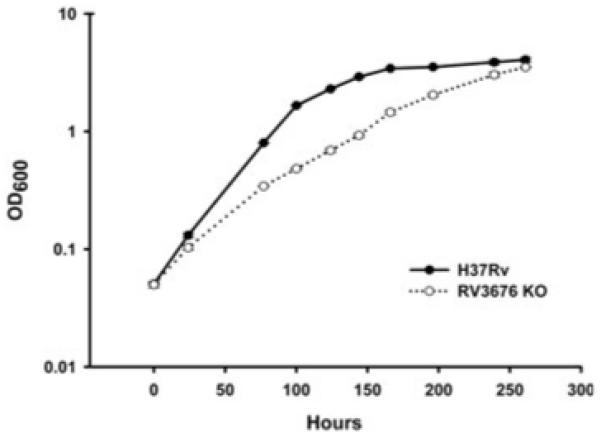
Growth curves for the Rv3676 null mutant (Rv3676 KO) and the parental strain M. tuberculosis H37Rv during in vitro liquid culture in rolling bottles at 37°C in Dubos medium. Each strain was grown to an optical density at 600 nm of 0.5, diluted 10-fold into fresh medium, and each strain split into two cultures and growth monitored for each. Each point represents the mean ± the standard deviation of the mean of the OD600 of the two samples of each strain. The growth of the complemented strain containing either the pRv3676long or the pRv3676short plasmid was indistinguishable from that of the wild type (data not shown).
The observation that the Rv3676 mutant exhibited impaired growth in vitro prompted experiments to investigate growth in unactivated mouse bone marrow-derived macrophages. These showed that growth of the Rv3676 knockout was severely impaired compared with the H37Rv wild type (Fig. 3). To ascertain whether the Rv3676 mutant was attenuated in vivo, growth of the mutant and wild-type strains in the lungs and spleens of BALB/c mice was determined following intravenous injection of the bacteria (Fig. 4). The Rv3676 mutant exhibited greatly reduced bacterial loads in the mouse lungs and spleen compared with infection by the wild type. It therefore falls within the growth in vivo (giv) type of mutant defined by Hingley-Wilson et al. (2003). The Rv3676 regulatory system therefore appears to be very important for the normal growth of the bacterium during the multiplication phase in macrophages and in mice, but did not appear necessary for the persistence phase of growth.
Fig. 3.
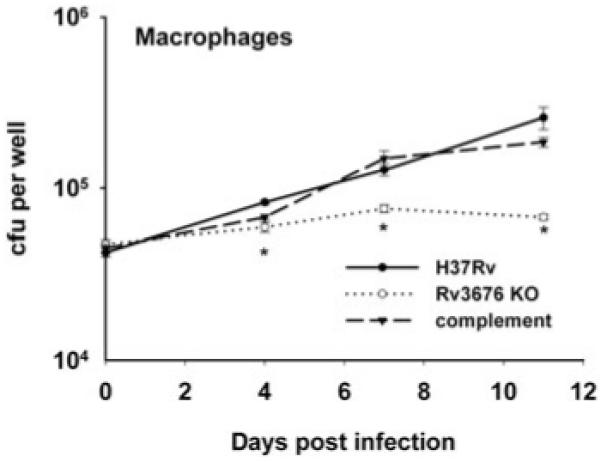
Growth of the Rv3676 deletion mutant (Rv3676 KO) is impaired in in vitro infections of mouse bone marrow-derived macrophages cells. Macrophages were isolated from BALB/c mice as described in the text and infected at a multiplicity of infection of one bacterium to two macrophages with each strain. The survival and multiplication of the M. tuberculosis strains were determined by cfu counts and are shown for the H37Rv wild type, the Rv3676 mutant and the complemented mutant strain. The results shown are with the pRv3676long complement; similar results were obtained with pRv3676short. The experiment was performed twice with similar results; the results of a representative experiment are shown. The results for each time point are the means of cfu determinations performed on triplicate infections, and the error bars show the standard deviations. The asterisk indicates that the result is significantly different from that of the wild type by the two-tailed Student’s t-test for groups of unequal variance (P < 0.01), as well as by single-factor analysis of variance (P < 0.01).
Fig. 4.
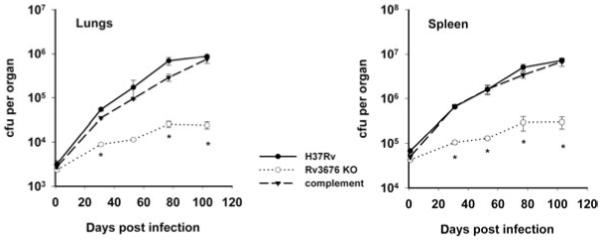
Growth of the Rv3676 null mutant (Rv3676 KO) is impaired in a mouse intravenous infection. BALB/c mice were inoculated intravenously with approximately 5 × 105 cfu of each strain. The survival and multiplication of the M. tuberculosis strains in the lungs and the spleen were determined by cfu counts and are shown for the wild-type strain H37Rv, the Rv3676 mutant and the complemented mutant strain. The results shown are with the pRv3676long complement; similar results were obtained with pRv3676short. The experiment was performed three times with similar results; the results of a representative experiment are shown. The results for each time point are the means of cfu determinations performed on organs from three mice, and the error bars show the standard deviations. The asterisk indicates that the result is significantly different from that of the wild type by the two-tailed Student’s t-test for groups of unequal variance (P < 0.01), as well as by single-factor analysis of variance (P < 0.01).
To ensure that the phenotypes observed were in fact due to deletion of the Rv3676 gene, the mutation was complemented with the wild-type allele using either a short (pRv3676short) or a longer construct (pRv3676long). Using either construct, the phenotype of the complemented strain in in vitro (data not shown), in macrophages (Fig. 3) and in mice (Fig. 4) was very similar to that of the wild-type strain and thus it was concluded that mutation of Rv3676 is the cause of the impaired growth phenotype in vitro and in vivo.
The Rv3676 mutation causes many transcriptional changes during in vitro growth
Whole genome DNA microarrays of M. tuberculosis H37Rv were used to identify genes and operons with altered expression in the Rv3676 mutant when grown in vitro. A total of seven hybridizations were performed from three replicate exponential phase RNA samples. This identified eight genes whose transcripts were >2.4-fold more abundant in the wild type than the Rv3676 mutant, and eight that were >2.4-fold more abundant in the mutant (Table 1). Among those transcripts that were more abundant in the wild type was that of the resuscitation promoting factor, Rv0867c (rpfA). This difference in rpfA expression between the wild type and Rv3676 mutant was confirmed by measuring the amount of rpfA mRNA using real-time polymerase chain reaction (PCR), standardizing to the amount of mRNA of a normalizer gene (sigA, the primary sigma factor), the expression of which did not change in the array analysis under these conditions and which has previously been shown to exhibit constant expression during exponential growth and so can be used as an internal standard for mRNA quantification (Dubnau et al., 2002). Using primers from the coding region of rpfA, 3.86-fold more rpfA transcript was detected in the wild type than in the Rv3676 mutant (not shown). From the DNA microarray analysis, other notable genes whose transcription was enhanced in the wild type were whiB1, a probable transcriptional regulatory protein, ahpC, coding for alkyl hydroperoxide reductase, lprQ, a probable conserved lipoprotein LPRQ, and fadD26, coding for acyl CoA synthase, and among genes enhanced in the Rv3676 mutant was fadD10, a possible fatty acid-CoA ligase, and lipQ, a probable carboxyesterase. Therefore, it was concluded that mutation of Rv3676 causes pleiotropic changes in the M. tuberculosis transcriptome.
Table 1.
Genes with altered expression in the Rv3676 mutant compared with the parent.
| ORF | Gene | Gene product | Fold changea | P-value | Possible Rv3676 binding siteb |
|---|---|---|---|---|---|
| Rv0099 | fadD10 | Possible fatty-acid-CoA ligase | −3.00 | 0.033 | |
| Rv0188 | Possible transmembrane protein (upregulated at high temperature) | −6.11 | 0.047 | GGTGGCAGCGAGCCCA | |
| Rv0483 | lprQ | Probable conserved lipoprotein LPRQ | 3.04 | <0.001 | TGTGTTTGGTATCACA |
| Rv0867c | rpfA | Probable resuscitation promoting factor | 6.36 | <0.001 | TGTGACATTACCCACA |
| Rv1387 | PPE20 (repressed by low pH) | 2.43 | <0.001 | TGGCAATGGCCGCTCA | |
| Rv1592c | Conserved hypothetical (induced by hypoxia) | 2.53 | 0.009 | TCAGGGACCGATCACG | |
| Rv1623c | cydA | Probable cytochrome d ubiquinol oxidase subunit | −2.55 | 0.004 | |
| Rv2428 | ahpC | Alkyl hydroperoxide reductase | 3.24 | 0.01 | GGTGTGATATATCACC |
| Rv2485c | lipQ | Probable carboxylesterase | −2.46 | 0.001 | TGTGATCCTCGACACA |
| Rv2930 | fadD26 | Acyl CoA synthase | 2.91 | <0.001 | GATGGCTTCAGACACA |
| Rv3136 | PPE | 2.79 | 0.005 | TGCCATCGTCGTCGAA | |
| Rv3219 | whiB1 | Putative regulatory protein | 3.41 | 0.005 | AGTGAGATAGCCCACG |
| Rv3613c | Hypothetical protein (upregulated after starvation) | −3.14 | 0.023 | ||
| Rv3614c | Hypothetical (upregulated at low pH) | −5.76 | 0.029 | ||
| Rv3615c | Conserved hypothetical protein (upregulated at low pH) | −6.34 | 0.025 | ||
| Rv3616c | Conserved hypothetical protein (upregulated at low pH) | −7.26 | 0.058 | AGTGACACCGGTCACA |
Positive values represent genes with enhanced expression in the wild type, negative values represent genes with enhanced expression in the Rv3676 mutant.
Putative Rv3676 binding sites located upstream of indicated genes or operons. Conserved bases are highlighted in bold.
Determination of a possible Rv3676 binding site consensus sequence in M. tuberculosis
The intergenic DNA sequences of genes with altered expression profiles in the Rv3676 mutant (Table 1) were analysed to identify the likely binding site for Rv3676 in M. tuberculosis. The analysis was guided by the prediction that Rv3676 would recognize a site related to the E. coli CRP consensus (TGTGANNNNNNTCACA) (reviewed by Busby and Ebright, 1999). This assumption was based upon the conservation of amino acids directly involved in recognizing DNA, and the observation that expression of Rv3676 in E. coli inhibits transcription from a CRP-activated promoter, suggesting that CRP and Rv3676 compete for the same binding site (not shown). Possible Rv3676 binding sites associated with the genes differentially expressed in the Rv3676 mutant are listed in Table 1. Only for Rv0099 and Rv1623 was no obvious site resembling the search sequence identified; note that Rv3616c-Rv3613c probably form an operon transcribed from a Rv3676-regulated promoter located upstream of Rv3616c. Alignment of the putative Rv3676 binding sites revealed a binding site consensus consisting of GTGNNANNNNNCACA. Analysis of the intergenic regions of the M. tuberculosis genome using the TubercuList (http://genolist.pasteur.fr/TubercuList/) website revealed 15 matches to the putative Rv3676 consensus sequence and 242 sequences with one mismatch, suggesting that Rv3676 is a global regulator in M. tuberculosis.
Rv3676 directly regulates rpfA expression
The transcription profiling experiments suggested that rpfA expression is positively controlled by Rv3676 and analysis of the upstream DNA sequence revealed the presence of a putative Rv3676 binding site (Table 1). The site is located ~440 bp upstream of the rfpA translational start close to the moaD2 gene (Fig. 5A). The presence of an abundant rpfA transcript was detected using reverse transcription polymerase chain reaction (RT-PCR) with primers located in the rpfA coding region (Myc477) and downstream of the predicted Rv3676 binding site (Myc478) (Fig. 5A and B, lane 1). Much less abundant transcripts were detected using Myc477 and primers Myc479, located just upstream of the predicted Rv3676 binding site, or Myc486, located within moaD2 (Fig. 5A and B, lanes 3 and 5). This suggests that the major rpfA transcript initiates downstream of the putative Rv3676 binding site and also that some moa transcripts extend into rpfA. Accordingly, the activity of an rpfA::lacZ promoter fusion consisting of 597 bp DNA from immediately upstream of the rpfA translational start (details of co-ordinates in Experimental Procedures) was approximately 7.5-fold lower in the Rv3676 mutant strain compared with the parent (Fig. 6A). Moreover, mutation of the predicted RV3676 binding site in the rpfA promoter region (TGTGAcattacCCACA to TGTTAcattacCAACA) lowered in vivo transcription of the corresponding rpfA::lacZ fusion approximately ninefold (Fig. 6A). These differences are similar to those observed in the DNA microarray (approximately sixfold ) and real-time PCR experiments (approximately fourfold ) described above. A smaller construct of 335 bp gave similar results although the overall levels of expression were lower (Fig. 6A). The residual β-galactosidase activities observed with Rv3676 mutant, and the reporter with the mutated Rv3676 binding site, could result from the activity of the upstream promoter suggested by the RT-PCR experiments (Fig. 5B, lanes 3 and 5).
Fig. 5.
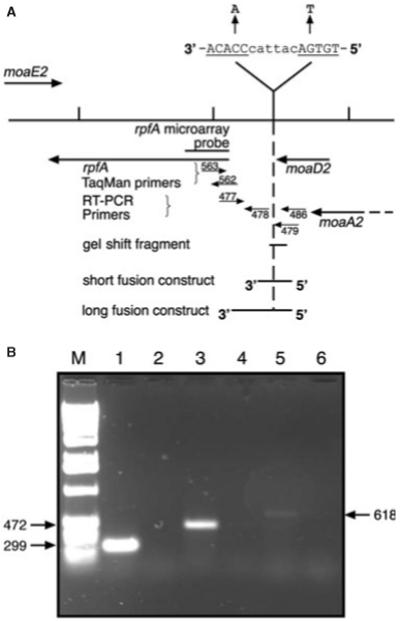
A. Diagram of the genomic region of M. tuberculosis containing the rpfA gene and adjacent genes, showing the position of the probe used for rpfA on the DNA microarray, the extent of the DNA fragment used for gel shift assays, and the position of the DNA fragments inserted into the lacZ reporter gene vector pEJ414. The numbers refer to the Myc oligonucleotides whose sequences are described in Experimental procedures. Also shown is the sequence of the putative Rv3676 binding site and the mutated version.
B. RT-PCR on the region upstream of the rpfA gene. This was performed on total RNA using the following primer pairs: lanes 1 and 2: Myc477 and Myc478; lanes 3 and 4: Myc477 and Myc479; lanes 5 and 6: Myc477 and Myc486. The reactions for lanes 2, 4 and 6 lacked reverse transcriptase. Lane M is a 1 kb ladder of molecular weight markers. The sizes of the RT-PCR products are shown in bp.
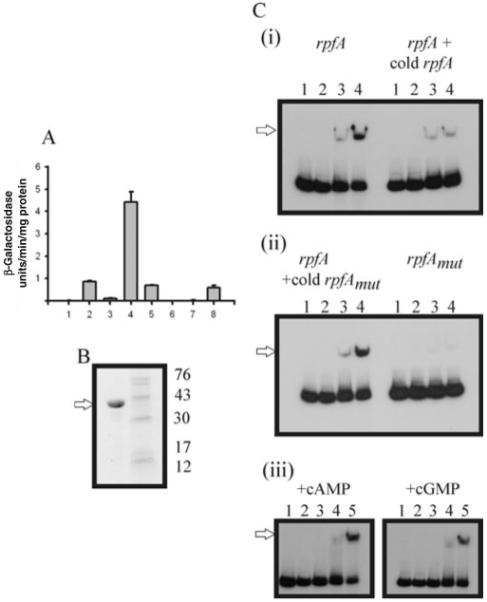
Fig. 6. Interaction of Rv3676 with the M. tuberculosis rpfA promoter.
A. In vivo transcription of rpfA. β-Galactosidase assays were performed on cell extracts from M. tuberculosis strains containing constructs with the rpfA promoter linked to the lacZ reporter gene. The plasmid constructs were, in a wild-type strain: (1) pEJ414 vector only; (2) pRB125 (prpfAPshort); (3) pRB127 (prpfAPshort: CRP site mutated); (4) pRB126 (prpfAPlong); (5) pRB128 (prpfAPlong: CRP site mutated); and in the Rv3676 mutant: (6) pEJ414 empty control plasmid only; (7) pRB125 (prpfAPshort); (8) pRB126 (prpfAPlong).
B. SDS-PAGE analysis of overproduced Trx–His(6)–Rv3676 fusion protein. Coomassie blue-stained gel showing the isolated Trx–His(6)–Rv3676 fusion protein (5 μg; arrowed) following affinity chromatography. The molecular mass markers and their sizes (kDa) are indicated.
C. Interaction of Rv3676 with the rpfA promoter. Radiolabelled unaltered rpfA promoter or rpfAmut promoter with mutations in the putative Rv3676 binding site (TGTGAcattacCCACA → TGTTAcattacCAACA) was incubated with Trx–His(6)–Rv3676 fusion protein before separation of Rv3676:rpfA complexes by gel electrophoresis. Where indicated unlabelled (cold) competitor DNA was present in 40-fold molar excess. When present cAMP or cGMP were added to the binding reactions and the polyacrylamide gels and buffers to a final concentration of 100 μM. Panels (i) and (ii): lanes 1, no Trx–His(6)–Rv3676 fusion; lanes 2, 0.5 μM; lanes 3, 5 μM; lanes 4, 10 μM. Panel (iii): lane 1, no Trx–His(6)–Rv3676 fusion; lane 2, 0.1 μM; lane 3, 0.5 μM; lane 4, 5 μM; lane 5, 10 μM. The Rv3676:rpfA complexes are arrowed.
To test whether the observed changes in rpfA expression are mediated directly by Rv3676, the protein was isolated as a TrxA–His(6)–Rv3676 fusion after overproduction in E. coli (Fig. 6B). The presence of the N-terminal His-tag facilitated affinity chromatography on HiTrap Chelating Sepharose to yield a fusion protein with the expected subunit Mr of ~37 000. Band shift assays showed that the Rv3676 fusion protein bound at the native rpfA promoter, and that addition of excess unlabelled rpfA promoter DNA effectively competed for the labelled promoter (Fig. 6C). The mutation of the predicted rpfA Rv3676 binding site (TGTGAcattacCCACA to TGTTAcattacCAACA) that led to lower expression in vivo (see above) also abolished Rv3676 binding in vitro (Fig. 6C). Furthermore, the mutant rpfA promoter DNA was unable to effectively compete with the unaltered promoter (Fig. 6C). Thus, it was concluded that Rv3676-mediated activation of rpfA expression is direct and requires interaction with the predicted binding site. For E. coli CRP DNA binding is greatly enhanced in the presence of its cognate signal molecule, cAMP. Despite retaining four of the six amino acid residues of E. coli CRP that directly contact cAMP, DNA binding of Rv3676 was not enhanced in the presence of cAMP or cGMP (Fig. 6C).
Discussion
Understanding the way in which M. tuberculosis regulates the expression of its genome is crucial in analysing the interaction with its environment. One way to do this is through the production and analysis of mutants of regulatory genes. Here it has been shown that M. tuberculosis Rv3676 is a member of the CRP family of transcription factors that is required for virulence in a mouse model of tuberculosis. Transcription profiling experiments comparing the wild type and Rv3676 mutant grown in vitro identified some of the genes that are regulated, directly or indirectly, by Rv3676 in M. tuberculosis. Further analysis of the regulation of one of these genes, rpfA coding for a resuscitation promoting factor thought to be involved in the extremely important processes of persistence and reactivation in M. tuberculosis infections, indicated that rpfA expression was directly regulated by Rv3676. The rpfA gene is one of five genes in M. tuberculosis encoding resuscitation promoting factor proteins (Mukamolova et al., 2002a). The Rpf proteins are secreted growth factors that stimulate the growth of aged cultures of M. tuberculosis, increasing the viable counts on solid agar by one or two orders of magnitude (Shleeva et al., 2004). Also a subpopulation of either injured or dormant M. tuberculosis cells obtained from macrophages, which were unculturable, could be recovered or resuscitated by addition of Rpf proteins in liquid medium (Biketov et al., 2000). The five Rpf proteins in M. tuberculosis appear to show functional redundancy, because deletion of any individual gene does not compromise growth (Downing et al., 2004; Tufariello et al., 2004), whereas the single rpf gene in M. luteus is an essential growth factor (Mukamolova et al., 2002b). Furthermore, there are differences in global gene expression between strains with different rpf mutations, so it is likely that they perform different functions and that their expression responds to different effector molecules (Downing et al., 2004). The Rv3676-regulated RpfA protein is the most potent Rpf in M. tuberculosis (Mukamolova et al., 2002a) and is among the most highly expressed genes in vitro and in infected macrophages (Triccas et al., 2001) and thus Rv3676 may play a key role in regulating the persistence and reactivation of M. tuberculosis.
The transcriptional profiling experiments showed that expression of some genes is lower in the mutant compared with the wild type whereas the expression of others is enhanced. This suggests that Rv3676 acts positively on genes such as rpfA, but negatively on others, although some of the changes in transcription may result from indirect effects, such as the slow growth of the mutant. Analysis of the M. tuberculosis genome sequence using an Rv3676 binding site consensus derived from comparison of the DNA sequences of the upstream regions of the differentially expressed genes listed in Table 1 suggests that all of the genes that are controlled by Rv3676 have not been identified and many may be co-dependent on additional transcription factors that are only activated during an infection. Thus, there are potentially numerous other interesting genes with Rv3676 binding sites that are not represented in the array experiments. For example, the icl gene coding for isocitrate lyase has been implicated in persistence (McKinney et al., 2000) and has an Rv3676 binding site upstream of the coding region with only one mismatch from the consensus sequence. Interestingly, in Corynebacterium glutamicum a CRP orthologue (GlxR) has been shown to bind to the promoter region of aceB (coding for isocitrate lyase) in the presence of cAMP to repress the glyoxylate bypass genes (Kim et al., 2004). Also potential Rv3676 binding sites are present in some genes, e.g. frdA, ndh and bioF2, that are regulated by FNR in E. coli (Guest et al., 1996).
The presence of a member of the CRP family of transcription factors in M. tuberculosis suggested that Rv3676 might respond to intracellular cAMP concentrations and it seems clear that cyclic nucleotides play a significant role in a number of metabolic processes in mycobacteria. Thus, a decrease in cAMP levels when glucose was added, the hallmark of catabolite repression, has been described for M. tuberculosis (Padh and Venkitasubramanian, 1976). It is also interesting to note that it has been reported that cAMP levels are much higher in mycobacteria, including M. tuberculosis, than in E. coli, and that extracellular cAMP goes on increasing even when stationary phase is reached suggesting that cAMP is continuously excreted into the medium (Padh and Venkitasubramanian, 1976). Certainly it is noteworthy that M. tuberculosis has 15 putative adenylate cyclases, implying that signal transduction involving cAMP is important in this bacterium (McCue et al., 2000). Indeed, cAMP has been shown to regulate the expression of galactokinase in Mycobacterium smegmatis (Raychaudhuri et al., 1998) and has been implicated in phospholipid synthesis in the same organism (Kaur and Khuller, 1995) and in the present study the expression of a number of genes involved in lipid metabolism was altered in the Rv3676 mutant. Interestingly in terms of infection, it has been reported that ingestion of live but not dead mycobacteria results in elevated cAMP levels which it was suggested could be the cause of the inhibition of phagosome–lysosome fusion (Lowrie et al., 1975) although evidence for this has not been forthcoming (Lowrie et al., 1979).
Despite the conservation of four of the six residues involved in cAMP binding in E. coli CRP (Fig. 1) thus far there is no evidence that cAMP has any affect on the DNA binding activity of Rv3676 in vitro. This suggests that the two non-conserved cAMP binding amino acids are crucial for mediating enhanced DNA binding in the presence of cAMP. In E. coli CRP Ser129 interacts with N6 of the adenine ring of cAMP and Arg83 interacts with O2P of the cAMP phosphate. These observations do not unequivocally show that Rv3676 does not bind cAMP, as it may do so in a way that does not improve the affinity of Rv3676 for target DNA. This could perhaps explain why a cAMP-binding protein was not found in a previous study (Padh and Venkitasubramanian, 1980). An alternative signal molecule cGMP has been demonstrated in M. smegmatis (Bhatnagar et al., 1984) but, like cAMP, cGMP did not improve Rv3676 DNA binding in vitro. Thus, the signal perceived by Rv3676 remains mysterious and future work should be directed towards identifying the environmental or metabolic signal perceived by Rv3676.
Experimental procedures
Strains and plasmids
Mycobacterium tuberculosis and E. coli strains and plasmids used are listed in Table 2.
Table 2.
Bacterial strains and plasmids used in this study
| Relevant genotype or characteristics | Reference or source | |
|---|---|---|
| Bacterial strain | ||
| M. tuberculosis H37Rv | Wild-type virulent strain | Kubica et al. (1972) |
| M. tuberculosis Rv3676 | H37Rv, deletion of Rv3676 | This work |
| M. tuberculosis Rv3676 short complement |
M. tuberculosis Rv3676, pRB131 | This work |
| M. tuberculosis Rv3676 long complement |
M. tuberculosis Rv3676, pRB132 | This work |
| E. coli BL21λDE3 | Lysogen of λDE3 carrying a copy of the T7 RNA polymerase under the control of the IPTG-inducible lacUV5 promoter; ApR |
Novagen |
| E. coli JRG5250 | BL21λDE3 pGS1698 | This work |
| M. tuberculosis shuttle plasmids | ||
| p2NIL | Suicide gene delivery vector, oriE, kan | T. Parish (Parish and Stoker, 2000) |
| pLR2 | Final Rv3676 knock out p2NIL derivative (see Experimental procedures) | This work |
| pKP186 | Integrase-negative derivative of the integrating vector pMV306 | K.G. Papavinasasundaram (unpublished); Stover et al. (1992) |
| pBS-int | Integrase-positive plasmid lacking a M. tuberculosis origin of replication | B. Springer (unpublished) |
| pRB131 (pRv3676short) | Rv3676 complementing derivative of pKP186, short construct | This work |
| pRB132 (pRV3676long) | Rv3676 complementing derivative of pKP186, long construct | This work |
| pEJ414 | lacZ transcriptional reporter plasmid | E.O. Davis (Papavinasasundaram et al., 2001) |
| E. coli plasmids | ||
| pET32a | An IPTG-inducible expression vector | Novagen |
| pCR4Blunt-TOPO | General cloning vector for blunt-ended PCR products; ApR, KanR | Invitrogen |
| pGS1698 | A pET32a derivative encoding M. tuberculosis Rv3676 as a Trx–His(6)–Rv3676 fusion protein; ApR |
This work |
| pRB129 | A pCR4Blunt-TOPO derivative containing the M. tuberculosis rpfA upstream region between Myc657 and Myc658 |
This work |
| pRB130 | As pRB129, with a mutated CRP binding site | This work |
| pGOAL19 | hyg, Phsp60–sacB, PAg85a–lacZ marker gene cassette, amp, oriE | T. Parish (Parish and Stoker, 2000) |
| pRB125 (prpfAPshort) | pEJ414 derivative containing the short rpfA upstream region | This work |
| pRB126 (prpfAPIong) | pEJ414 derivative containing the long rpfA upstream region | This work |
| pRB127 | As pRB125, with a mutated CRP binding site | This work |
| pRB128 | As pRB126, with a mutated CRP binding site | This work |
Construction of a deletion mutant of Rv3676
Approximately 1.6 kb of DNA sequence from each side of the Rv3676 gene were amplified from M. tuberculosis genomic DNA with HotStarTaq (Qiagen) using the following primer pairs: for the 5′ side Myc19 (5′-CTGGATCCTGATATTCGGC CATCAC-3′) and Myc20 (5′-CTGGATCC-GTGTCGAGCCG TTCTAA-3′) (bp 4114738–4116363), and for the 3′ side Myc27 (5′-GCGGCCGCTACCGAGTCCGCAGATAGTC-3′) and Myc28 (5′-GCGGCCGCTTGCCGGTGACAAGGTG CAG-3′) (bp 4117258–4118860). The 5′ fragments were cloned into the BamHI site and 3′ fragments into the NotI site of the suicide gene delivery vector p2NIL which is capable of replication within E. coli but lacks a mycobacterial origin of replication (Parish and Stoker, 2000). A hygromycin cassette was cloned into the KpnI site of p2NIL to replace the Rv3676 gene. A PacI fragment from pGOAL19 containing the lacZ and sacB genes was inserted to give the completed suicide delivery vector pLR2 (Table 2). This vector contains the sacB gene from Bacillus subtilis that causes lethality when expressed in M. tuberculosis making it an effective counterselectable marker, and a lacZ gene. The virulent M. tuberculosis strain H37Rv was transformed with pLR2 and selection was made for potential single cross-overs as kanamycin- and hygromycin-resistant colonies expressing the lacZ gene. These colonies were streaked onto sucrose plates containing hygromycin only. Twelve colonies with the expected phenotype (kanamycin-sensitive, sucrose-resistant and white due to the loss of the lacZ gene) were screened for potential double cross-overs by PCR using the following pairs of primers: for the 5′ side, Myc166 (5′-TCGTTCTGGC CCTGATG-3′) and Myc167 (5′-ACTTCGACGAGTAGGAC-3′), and for the 3′ side, Myc168 (5′-CCACGCCTTGGAA GATT-3′) and Myc169 (5′-TCACGCGAGACGGTGAA-3′). Southern blot analysis on genomic DNA was performed with probes derived using the following primer pairs: for the 5′ side, Myc61 (5′-AATCACCTTGCTCCACA-3′) and Myc62 (5′-TTTGGAGCCGTTCAGCC-3′), and for the 3′ side, Myc63 (5′-CAGATGGAACTGCCGAT-3′) and Myc64 (5′-CCAATGG CACGCGTTGT-3′). DNA sequencing was performed using the primers Myc36 (5′-ACGTCTGGAGCAGATAC-3′) in gene Rv3677 and Myc40 (5′-CCTGCATCGACGTCAAG-3′) in gene Rv3675.
Complementation of the Rv3676 deletion mutation
Two complementing constructs were prepared by PCR of genomic DNA, a longer (pRv3676Plong: bp 4115784–4117351) one using primers Myc432 (5′-GAATTCAAGTCC AGCTCGCAGTA-3′) located in gene Rv3674c and Myc433 (5′-ATCGATTCGTCGAACACGTCTAC-3′) in gene Rv3677c, and a shorter (pRv3676Pshort: bp 4115784–4116181) one using Myc430 (5′-GAATTCTACGCTCGCTCGGAATG-3′) located in gene Rv3675, and Myc433. Fragments were cloned into EcoRI and ClaI sites of pKP186, an integrase-negative derivative of the integrating vector pMV306 (Stover et al., 1992) kindly provided by Dr K.G. Papavinasasundaram and sequenced completely to verify the absence of PCR-derived mutations. The plasmids were co-transformed into the Rv3676 null mutant along with plasmid pBS-int (kindly provided by Dr B. Springer) carrying the integrase gene necessary to achieve integration of the plasmid into the chromosome. The pBS-int plasmid lacks a mycobacterial origin of replication and is therefore lost from the bacterium.
In vitro growth determination
This was carried out in 1 l polycarbonate culture bottles (Techmate) in a Bellco roll-in incubator (2 r.p.m.) at 37°C in Dubos broth containing 0.05% (v/v) Tween supplemented with 0.2% (v/v) glycerol and 4% (v/v) Dubos medium albumin. Optical density readings on aliquots removed were taken at 600 nm.
Growth of bacteria in mouse macrophage cell culture
Bone marrow cells were extracted from the hind legs of 8-week-old adult female BALB/c mice as described previously (Tascon et al., 2000). The cells were resuspended in IMDM + 5% FCS, 2 mM l-glutamine, 80μM β-mercaptoethanol to which was added 10% by volume of supernatant from L929 cells that produce M-CSF. The cells were plated in six-well plates and incubated at 37°C and 5% CO2 atmosphere. At day 2 the cells were washed with fresh prewarmed medium. Fresh medium was added to the plates containing the adherent bone marrow macrophage cells. The cells were cultured for a further 3 days and at day 5 they were infected for 6 h at a ratio of 2:1 cells:acid fast bacilli. After infection the medium was removed and replaced with fresh medium. The bone marrow macrophages were lysed with 2% saponin and incubated for 1 h at 37°C. Growth was determined by viable counts on Middlebrook 7H11 agar plates containing Middlebrook OADC supplement at days 0 (6 h post infection), 4, 7 and 11 post infection. The numbers of colony forming units (cfu) were determined after the plates had been incubated at 37°C for approximately 20 days for the wild type and after approximately 35 days for the Rv3676 mutant.
In vivo growth characteristics of the Rv3676 mutant in a mouse model of TB infection
The wild type and Rv3676 mutant of M. tuberculosis H37Rv were grown as rolling cultures in Dubos broth (as above) to mid-exponential phase. Each strain was diluted in phosphate-buffered saline to give a suspension of approximately 106 cfu ml−1 and 0.2 ml of these suspensions were inoculated intravenously into 6-to 8-week-old female BALB/c mice. The infection was monitored by removing the lungs and spleens of three to five infected mice at intervals. The tissues were homogenized by shaking with 2 mm diameter glass beads in chilled saline with a Mini-Bead Beater (Biospec Products, Bartlesville, Oklahoma). Serial 10-fold dilutions of the resultant suspensions were plated onto Dubos 7H11 agar with Dubos oleic albumin complex supplement (Difco Laboratories, Surrey, UK). The numbers of cfu were determined after the plates had been incubated at 37°C for approximately 20 days for the wild type and after approximately 35 days for the Rv3676 mutant.
Transcription analysis using DNA microarrays
Bacteria were grown in Dubos medium (supplemented with 0.2% v/v glycerol, 4% v/v Dubos medium albumin) in roller bottles at 37°C. Growth was measured at OD600 and bacteria were harvested at mid-exponential phase. Total RNA was extracted using a Hybaid RNA isolation kit (Hybaid). RNA was treated twice with RNase-free DNase (Roche) in the presence of 1 unit ml−1 RNasin ribonuclease inhibitor (Promega), 20 mM sodium acetate, 10 mM magnesium chloride and 10 mM sodium chloride, and purified using RNeasy columns (Qiagen) between treatments. RNA was checked for DNA contamination by PCR. DNA microarray experiments were performed using a whole genome microarray of M. tuberculosis H37Rv prepared in the laboratory of Dr P. Butcher (St. George’s Hospital Medical School, London). Fluorescently labelled cDNA copies of total RNA (5–10 μg) were generated by direct incorporation of Cy3- or Cy5-labelled dCTP (Amersham) using 6 μg of random hexamers together with 500 units Superscript RNase H− reverse transcriptase (GibcoBRL), 1x first-strand buffer, 10 mM DTT, dNTPs (40 μM dCTP, 100 μM each of dATP, dGTP, dTTP) and 1.5 nmol Cy3 or Cy5 labelled dCTP. RNA and random primers were heated at 95°C for 5 min in a final volume of 11 μl, snap cooled before adding the remaining components. Incubation was at 25°C for 10 min, followed by 42°C for 90 min. The two labelled samples to be compared (Cy3 and Cy5) were combined and purified using MinElute columns (Qiagen) and the pellets resuspended in 12.5 μl of water, 4x SSC, 0.3% SDS, heated at 95°C for 2 min. Hybridization of microarray slides was performed under a coverslip as described (Wilson et al., 1999) in hybridization chambers at 60°C for 16–24 h and washed in 1x SSC, 0.005% SDS for 2 min and in 0.06x SSC for 4 min. Microarray slides were scanned for fluorescence with an Axon 4000A microarray scanner, local background subtracted using the GenePix software and normalized equivalent readings were plotted using GeneSpring software (Silicon Genetics). Data were obtained from at least six slides, including dye swaps. To account for dye swap, the signal channel and control channel measurements for dye swap samples were reversed. Each gene’s measured intensity (wild type) was divided by its control channel value (mutant) in each sample to give the ratio of expression; if the control channel were below 10 then 10 was used instead. If the control channel and the signal channel were both below 10 then no data was reported. The 50th percentile of all measurements was used as a positive control for each sample; each measurement for each gene was divided by this synthetic positive control. The percentile was calculated using only genes marked present. This normalizes each array so that the 50th percentile/median measurement (ratio) for each array is equal to 1 and ensures that any systematic effects are removed (caused by scanning, labelling, hybridization, etc.), enabling valid comparison between different arrays as each array is scaled/transformed to have the same median value. Genes were defined as being differentially regulated where there was a greater than twofold change in at least four of the hybridizations and where P < 0.05 by Student’s t-test. The t-test was conducted on the six experiments as a group. To limit the list of genes it was arbitrarily limited to those with a 2.4-fold increase or decrease in expression.
Real-time quantitative PCR
Real-time quantitative PCR was carried out on the ABI Prism 7700 Sequence Detection system using the Taqman Universal PCR Master Mix (PE Applied Biosystems). The primers and the Taqman probes (carrying both a fluorophore and a quencher) were designed using the Primer Express software (PE Applied Biosystems). The forward rpfA primer was Myc562 (5′-CGTAAGCCCACCACATCCA-3′), the reverse primer was Myc563 (5′-CGAGTACTGCGCCGGTAAA-3′) and the probe primer was Myc564 (5′-CGTCAGCGTCGCCAAG ATCGC-3′). The forward primer from sigA, the normalizer gene, was Myc598 (5′-TCGGTTCGCGCCTACCT-3′), the reverse primer Myc599 (5′-TGGCTAGCTCGACCTCTTCCT-3′) and the probe primer Myc600 (5′-TTGAGCAGCGCTAC CTTGCCG-3′).
RT-PCR
Total RNA was used for RT-PCR as described by Papavinasasundaram et al. (1997) but using Superscript II reverse transcriptase (Invitrogen). The primers used for the region upstream of rpfA were Myc477 (5′-TTACGGTGGCGTCCA CTCATA-3′), Myc478 (5′-CTCGCCGATAGCACGCCGAAC-3′) and Myc486 (5′-GATAGACGGCCTGTCGGT-3′) (see Fig. 5A).
Construction of reporter gene plasmids using the upstream region of rpfA
Two regions of the DNA sequence of the region upstream of rpfA were generated by PCR from M. tuberculosis genomic DNA using the primer pairs Myc486 (5′-TCTAGAGATAGA CGGCCTGTCGGT-3′) and Myc487 (5′-AAGCTTGACGAGC CTCCCGAGACG-3′) for the short promoter construct (bp 966133–965798), and Myc486 and Myc488 (5′-AAGCTTA CGTTAGGTAATTCCTCT-3′) for the long promoter construct (bp 966133–965536). These fragments were cloned into the XbaI and HindIII sites of the polylinker in the lacZ transcriptional reporter plasmid pEJ414 (Papavinasasundaram et al., 2001) to make pRB125 (prpfAPshort) and pRB126 (prpfAPlong). These were verified by DNA sequencing.
Mutagenesis of CRP binding site in plasmids pRB125 and pRB126
This was performed using the Stratagene Quik Change mutagenesis kit using the primers Myc474 (5′-GCTAAG GCCATGTTACATTACCAACATAACGGAACGATAAC-3′) and Myc475 (5′-GTTATCGTTCCGTTATGTTGGTAATGTAACAT GGCCTTAGC-3′) to generate pRB127 (from pRB125, prpfAPshort mutated construct) and pRB128 (from pRB126, prpfAPlong mutated construct).
Transformation of plasmids into M. tuberculosis
Bacteria were grown in rolling bottles at 37°C and electro-competent cells were prepared as described by Papavinasasundaram et al. (1998). Electroporation was performed as described by Jacobs et al. (1991) and colonies were selected on 7H11 agar plates containing kanamycin.
Preparation of cell-free extracts and assay for β-galactosidase reporter enzyme
Mycobacterium tuberculosis cultures were grown in Dubos medium in rolling bottles at 37°C and cell-free extracts and β-galactosidase assays were carried out as described by Papavinasasundaram et al. (2001). For E. coli cultures grown at 37°C in L broth (tryptone, 10 g l−1; yeast extract, 5 g l−1, NaCl, 5 g l−1) for 16 h, β-galactosidase activities were determined as described by Miller (1972).
Overproduction and purification of Rv3676
The Rv3676 open reading frame was amplified by PCR with appropriate primers (Myc249: 5′-GAATTCGTGGACGAGAT CCTGGCCAG-3′ and Myc250: 5′-GAATTCCCTCGCTCGG CGGGCCAGTC-3′) containing 5′ EcoRI restriction sites. The amplified DNA was ligated into the corresponding sites in pGEX-6P-1 to create pGS1644. The pGS1644 plasmid was then used as a template to amplify the Rv3676 coding region using primers (5′-CTGGGATCCCCGGAATTCCATGGACGA GATCCTG-3′ and 5′-CCGTCATCACCGAACCTCGAGAAG CTTATCGTCAGTCAG-3′) containing engineered NcoI and XhoI restriction sites to allow ligation into the corresponding sites in pET32a (Novagen). The resulting plasmid (pGS1698; Table 2) encoded a TrxA–His(6)–Rv3676 fusion protein. Incorporation of the NcoI restriction site resulted in the alteration of codon 1 of Rv3676 from GTG (Val) to ATG (Met). Expression of the TrxA–His(6)–Rv3676 fusion protein was induced by the addition of IPTG (30 μg ml−1) to mid-exponential phase (OD600 of 0.4–0.6) aerobic cultures (25 ml) of E. coli strain JRG5250 (BL21λDE3 pGS1698; Table 2) in L broth at 37°C. Incubation was continued for a further 2 h at which point the bacteria were collected by centrifugation. The bacteria were suspended in a total volume of 5 ml of 10 mM Tris-HCl (pH 8.0) buffer containing 10 mM NaCl and then lysed by sonication. The lysate was clarified by centrifugation at 20 000 r.p.m. for 1 h and 2 ml aliquots were applied to nickel-loaded HiTrap Chelating Sepharose columns (1 ml) (Amersham). Bound fusion protein was eluted with a gradient of imidazole (0–500 mM, 20 ml in 20 mM sodium phosphate buffer, pH 7.4 containing 0.5 M NaCl).
Band shift assays
Myc657 (5′-GAATTCGACGGGATTGTCGTCCGA-3′) and Myc658 (5′-GGATCCCACAATCGCCGGCAAGAA-3′) were used for PCR with genomic DNA to generate the region upstream of the rpfA gene containing the putative Rv3676 binding site. This 200 bp DNA fragment (bp 966066–965867) was blunt-end cloned into plasmid pCR4Blunt-TOPO (Invitrogen) to produce plasmid pRB129 and verified by DNA sequencing. The Stratagene Quik Change mutagenesis kit with pRB129 as the template and the primers Myc474 (5′-GCTAAGGCCATGTTACATTACCAACATAACGGAACGAT AAC-3′) and Myc475 (5′-GTTATCGTTCCGTTATGTTGGTA ATGTAACATGGCCTTAGC-3′) was used to mutate the putative Rv3676 binding site in rpfA creating pRB130. DNA fragments containing the wild type and mutated rpfA promoter regions were generated by PCR amplification from plasmids pRB129 and pRB130, respectively, using oligonucleotide primers Myc657 and Myc658 (see above). The fragments were labelled by the inclusion of 0.5 μl (0.185 MBq) of [α-32P]-dATP in each reaction; unincorporated radionucleotides were removed using a Qiaquick PCR clean-up kit (Qiagen). Samples of purified TrxA–His(6)–Rv3676 fusion protein were incubated with the appropriate DNA fragment in binding buffer (20 mM Hepes, 5 mM MgCl2, 50 mM potassium glutamate, 1 mM dithiothreitol, 30 μg ml−1 herring sperm DNA, 500 μg ml−1 bovine serum albumin, pH 8.0) for 10 min before separating free DNA and DNA:protein complexes by electrophoresis on 6% polyacrylamide gels, buffered with TBE. Where indicated a 40-fold molar excess of cold competitor DNA was included in the incubations. Also where indicated cAMP or cGMP was included in the binding reactions and in the polyacrylamide gels and buffers at a final concentration of 100 μM.
Statistical analysis
For in vitro growth measurements each point represents the mean ± the standard deviation of the mean of the OD600 of the two samples of each strain. For the results from macrophage and mouse infections the results for each time point are the means of cfu determinations performed on organs from three macrophage infections or three mice infections, and the error bars show the standard deviations. The asterisk indicates that the result is statistically significantly different from that of the wild type by the two-tailed Student’s t-test for groups of unequal variance (P < 0.01), as well as by single-factor analysis of variance (P < 0.01). For the microararray analysis, genes were defined as being differentially regulated where there was a greater than twofold change in at least four of the hybridizations and where P < 0.05 by Student’s t-test. For the reporter gene assays each bar represent the mean of three assays on each sample ± the standard deviation of the mean.
Supplementary Material
Acknowledgements
We should like to thank John Guest for initial discussions, Mike Young for numerous consultations on rpf genes, Elaine Davis for providing the reporter gene vector pEJ414 and for help with the technique, Tanya Parish for providing plasmids p2NIL and pGOAL, K.G. Papavinasasundaram and Bernhard Springer for providing plasmids pKP186 and pBS-int, Peter Artymiuk for discussion on cAMP binding, Natalie Garton for discussion on lipid metabolism, and Vangelis Stavropoulos and John Brennan for their help with infection experiments. We acknowledge BμG@S (the Bacterial Microarray Group at St. George’s Hospital Medical School) and especially Philip Butcher, for the supply of the M. tuberculosis microarrays and advice, and The Wellcome Trust for funding the multicollaborative microbial pathogen microarray facility under its Functional Genomics Resources Initiative. Lisa Rickman was supported by a Medical Research Council research studentship and Carmen Menéndez by an EU Marie Curie fellowship (Contract No. HPMFCT-2002-016015). This work was supported at NIMR by the Medical Research Council and at Sheffield by the Biotechnology and Biological Science Research Council through a project grant (P19192).
Footnotes
Deceased 20 February 2003.
References
- Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins. II. The binding specificity of cyclic AMP receptor protein to recognition sites. J Mol Biol. 1988;200:709–723. doi: 10.1016/0022-2836(88)90482-2. [DOI] [PubMed] [Google Scholar]
- Bhatnagar NB, Bhatnagar R, Venkitasubramanian TA. Characterization and metabolism of cyclic guanosine 3′5′-monophosphate in Mycobacterium smegmatis. Biochem Biophys Res Commun. 1984;121:634–640. doi: 10.1016/0006-291x(84)90229-8. [DOI] [PubMed] [Google Scholar]
- Biketov S, Mukamolova GV, Potapov V, Gilenkov E, Vostroknutova G, Kell DB, et al. Culturability of Mycobacterium tuberculosis cells isolated from murine macrophages: a bacterial growth factor promotes recovery. FEMS Immunol Med Microbiol. 2000;29:233–240. doi: 10.1111/j.1574-695X.2000.tb01528.x. [DOI] [PubMed] [Google Scholar]
- Bloom BR, Murray CJ. Tuberculosis: commentary on a reemergent killer. Science. 1992;257:1055–1064. doi: 10.1126/science.257.5073.1055. [DOI] [PubMed] [Google Scholar]
- Busby S, Ebright RH. Transcription activation by catabolite activator protein (CAP) J Mol Biol. 1999;293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Downing KJ, Betts JC, Young DI, McAdam RA, Kelly F, Young M, Mizrahi V. Global expression profiling of strains harbouring null mutations reveals that the five rpf-like genes of Mycobacterium tuberculosis show functional redundancy. Tuberculosis. 2004;84:167–179. doi: 10.1016/j.tube.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Dubnau E, Fontan P, Manganelli R, Soares-Appel S, Smith I. Mycobacterium tuberculosis genes induced during infection of human macrophages. Infect Immun. 2002;70:2787–2795. doi: 10.1128/IAI.70.6.2787-2795.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA. 1999;282:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- Gosset G, Zhang ZG, Nayyar SN, Cuevas WA, Saier MH. Transcriptome analysis of Crpdependent catabolite control of gene expression in Escherichia coli. J Bacteriol. 2004;186:3516–3524. doi: 10.1128/JB.186.11.3516-3524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J, Scott C, Guest J. Functional versatility in the CRP-FNR superfamily of transcription factors: FNR and FLP. Adv Microb Physiol. 2001;44:1–34. doi: 10.1016/s0065-2911(01)44010-0. [DOI] [PubMed] [Google Scholar]
- Guest JR, Green J, Irvine AS, Spiro S. The FNR modulon and FNR-regulated gene expression. In: Lin EC, Lynch AS, editors. Regulation and Gene Expression in Escherichia coli. R.G. Landes & Co; Austin, TX: 1996. pp. 317–342. [Google Scholar]
- Hingley-Wilson SM, Sambandamurthy VK, Jacobs WR., Jr Survival perspectives from the world’s most successful pathogen, Mycobacterium tuberculosis. Nat Immunol. 2003;4:949–955. doi: 10.1038/ni981. [DOI] [PubMed] [Google Scholar]
- Jacobs WR, Jr, Kalpana GV, Cirillo JD, Pascopella L, Snapper SB, Udani RA, et al. Genetic systems for mycobacteria. Methods Enzymol. 1991;204:537–555. doi: 10.1016/0076-6879(91)04027-l. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH. How can immunology contribute to the control of tuberculosis? Nat Rev Immunol. 2001;1:20–30. doi: 10.1038/35095558. [DOI] [PubMed] [Google Scholar]
- Kaur H, Khuller GK. Role of cyclic adenosine monophosphate in phospholipid synthesis in Mycobacterium smegmatis ATCC 607. Lipids. 1995;30:345–349. doi: 10.1007/BF02536043. [DOI] [PubMed] [Google Scholar]
- Kim H-J, Kim T-H, Kim Y, Lee H-S. Identification and characterization of glxR, a gene involved in regulation of glyoxylate bypass in Corynebacterium glutamicum. J Bacteriol. 2004;186:3453–3460. doi: 10.1128/JB.186.11.3453-3460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korner H, Sofia HJ, Zumft WG. Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol Rev. 2003;27:559–592. doi: 10.1016/S0168-6445(03)00066-4. [DOI] [PubMed] [Google Scholar]
- Kubica GP, Kim TH, Dunbar FP. Designation of strain H37Rv as the neotype of Mycobacterium tuberculosis. Int J Syst Bacteriol. 1972;22:99–106. [Google Scholar]
- Lanzilotta WN, Schuller DJ, Thorsteinsson MV, Kerby RL, Roberts GP, Poulos TL. Structure of the CO sensing transcription activator CooA. Nat Struct Biol. 2000;7:876–880. doi: 10.1038/82820. [DOI] [PubMed] [Google Scholar]
- Lowrie DB, Jackett PS, Ratcliffe NA. Mycobacterium microti may protect itself from intracellular destruction by releasing cyclic AMP into phagosomes. Nature. 1975;254:600–602. doi: 10.1038/254600a0. [DOI] [PubMed] [Google Scholar]
- Lowrie DB, Aber VR, Jackett PS. Phagosome–lysosome fusion and cyclic adenosine 3′:5′-monophosphate in macrophages infected with Mycobacterium microti, Mycobacterium bovis BCG or Mycobacterium lepraemurium. J Gen Microbiol. 1979;110:431–441. doi: 10.1099/00221287-110-2-431. [DOI] [PubMed] [Google Scholar]
- McCue LA, McDonough KA, Lawrence CE. Functional classification of cNMP-binding proteins and nucleotide cyclases with implications for novel regulatory pathways in Mycobacterium tuberculosis. Genome Res. 2000;10:204–219. doi: 10.1101/gr.10.2.204. [DOI] [PubMed] [Google Scholar]
- McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1972. Assay of β-galactosidase; pp. 352–355. [Google Scholar]
- Mukamolova GV, Kaprelyants AS, Young DI, Young M, Kell DB. A bacterial cytokine. Proc Natl Acad Sci USA. 1998;95:8916–8921. doi: 10.1073/pnas.95.15.8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamolova GV, Turapov OA, Young DI, Kaprelyants AS, Kell DB, Young M. A family of autocrine growth factors in Mycobacterium tuberculosis. Mol Microbiol. 2002a;46:623–635. doi: 10.1046/j.1365-2958.2002.03184.x. [DOI] [PubMed] [Google Scholar]
- Mukamolova GV, Turapov OA, Kazarian K, Telkov M, Kaprelyants AS, Kell DB, Young M. The rpf gene of Micrococcus luteus encodes an essential secreted growth factor. Mol Microbiol. 2002b;46:611–621. doi: 10.1046/j.1365-2958.2002.03183.x. [DOI] [PubMed] [Google Scholar]
- Padh H, Venkitasubramanian TA. Adenosine 3′,5′-monophosphate in Mycobacterium phlei and Mycobacterium tuberculosis H37Ra. Microbios. 1976;16:183–189. [PubMed] [Google Scholar]
- Padh H, Venkitasubramanian TA. Lack of adenosine-3′,5′-monophosphate receptor protein and apparent lack of expression of adenosine-3′,5′-monophosphate functions in Mycobacterium smegmatis CDC 46. Microbios. 1980;27:69–78. [PubMed] [Google Scholar]
- Papavinasasundaram KG, Movahedzadeh F, Keer JT, Stoker NG, Colston MJ, Davis EO. Mycobacterial recA is cotranscribed with a potential regulatory gene called recX. Mol Microbiol. 1997;24:141–153. doi: 10.1046/j.1365-2958.1997.3441697.x. [DOI] [PubMed] [Google Scholar]
- Papavinasasundaram KG, Colston MJ, Davis EO. Construction and complementation of a recA deletion mutant of Mycobacterium smegmatis reveals that the intein in Mycobacterium tuberculosis recA does not affect RecA function. Mol Microbiol. 1998;30:525–534. doi: 10.1046/j.1365-2958.1998.01083.x. [DOI] [PubMed] [Google Scholar]
- Papavinasasundaram KG, Anderson C, Brooks PC, Thomas NA, Movahedzadeh F, Jenner PJ, et al. Slow induction of RecA by DNA damage in Mycobacterium tuberculosis. Microbiology. 2001;147:3271–3279. doi: 10.1099/00221287-147-12-3271. [DOI] [PubMed] [Google Scholar]
- Parish T, Stoker NG. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology. 2000;146:1969–1975. doi: 10.1099/00221287-146-8-1969. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Basu M, Mandal NC. Glutamate and cyclic AMP regulate the expression of galactokinase in Mycobacterium smegmatis. Microbiology. 1998;144:2131–2140. doi: 10.1099/00221287-144-8-2131. [DOI] [PubMed] [Google Scholar]
- Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2:569–577. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- Schultz SC, Shields GC, Steitz TA. Crystal structure of a CAP–DNA complex: the DNA is bent by 90 degrees. Science. 1991;253:1001–1007. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- Shleeva M, Mukamolova GV, Young M, Williams HD, Kaprelyants AS. Formation of ‘non-culturable’ cells of Mycobacterium smegmatis in stationary phase in response to growth under suboptimal conditions and their Rpf-mediated resuscitation. Microbiology. 2004;150:1687–1697. doi: 10.1099/mic.0.26893-0. [DOI] [PubMed] [Google Scholar]
- Stewart GR, Robertson BD, Young DB. Tuberculosis: a problem with persistence. Nat Rev Microbiol. 2003;1:97–105. doi: 10.1038/nrmicro749. [DOI] [PubMed] [Google Scholar]
- Stover CK, de la Cruz VF, Bansal GP, Hanson MS, Fuerst TR, Jacobs WR, Jr, Bloom BR. Use of recombinant BCG as a vaccine delivery vehicle. Adv Exp Med Biol. 1992;327:175–182. doi: 10.1007/978-1-4615-3410-5_19. [DOI] [PubMed] [Google Scholar]
- Tascon RE, Soares CS, Ragno S, Stavropoulos E, Hirst EM, Colston MJ. Mycobacterium tuberculosis-activated dendritic cells induce protective immunity in mice. Immunology. 2000;99:473–480. doi: 10.1046/j.1365-2567.2000.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triccas JA, Britton WJ, Gicquel B. Isolation of strong expression signals of Mycobacterium tuberculosis. Microbiology. 2001;147:1253–1258. doi: 10.1099/00221287-147-5-1253. [DOI] [PubMed] [Google Scholar]
- Tufariello JM, Jacobs WR, Jr, Chan J. Individual Mycobacterium tuberculosis resuscitationpromoting factor homologues are dispensable for growth in vitro and in vivo. Infect Immun. 2004;72:515–526. doi: 10.1128/IAI.72.1.515-526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M, DeRisi J, Kristensen H-H, Imboden P, Rane S, Brown PO, Schoolnik GK. Exploring drug-induced alterations in gene expression in Mycobacterium tuberculosis by microarray hybridization. Proc Natl Acad Sci USA. 1999;96:12833–12838. doi: 10.1073/pnas.96.22.12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.