

Abstract
Individuals infected with HIV-1 progress to AIDS at different rates. Rapid progressors develop AIDS within 2–5 years of initial infection, compared with approximately 10 years in typical progressors. Progression to AIDS is associated with impaired humoral and cellular immunity. In this issue of the JCI, Titanji and colleagues report that activated memory B (mBAct) cells are depleted in SIV-infected macaques defined as rapid progressors. Depletion was mediated by programmed death-1 (PD-1) and resulted in reduction of antibody titers specific for SIV and bacterial antigens. Interestingly, blockade of PD-1 in infected animals protected B cells from apoptosis and increased levels of SIV-specific antibodies in blood. These findings pave the way for a new therapeutic strategy aimed at improving humoral immunity in HIV-1 infection.
The use of antiretroviral therapy (ART) has dramatically reduced the number of individuals infected with HIV-1 that rapidly progress to AIDS in developed countries. However, rapid progression remains a problem in developing countries, where the majority of individuals infected with HIV-1 live, because use of ART is limited. Patients are clinically determined to have progressed to AIDS when their CD4+ T cell counts drop below 200 cells per microliter of blood. Although CD4+ T cell counts remain the gold standard to predict immunological impairment upon disease progression and to monitor immunological recovery during ART, the quality of responses mediated by CD4+ T cells during HIV-1 infection does not always correlate with their numbers (1). Thus, the identification of immunological biomarkers that reflect pathogenic events occurring during HIV-1 infection and that are predictive of progression to AIDS is an important research goal.
Progression to AIDS is associated with increased susceptibility to opportunistic infections as a result of both impaired cellular immune responses and dysfunctional humoral responses. In this regard, loss of memory B cells has been shown to lead to impaired HIV-specific and non–HIV-specific humoral immune responses (2). Understanding more clearly the mechanisms underlying B cell depletion during HIV-1 infection and the role of B cell defects in disease pathogenesis and progression could prove informative for developing approaches to improve humoral immunity in patients infected with HIV-1.
Depletion of memory B cells during SIV infection
In this issue of the JCI, Titanji and colleagues present new results on depletion of activated memory B (mBAct) cells during the early phases of SIV infection in rhesus macaques rapidly progressing to AIDS (3). In their study, depletion of this B cell subset was found to be associated with failure to produce SIV-specific antibodies. Alteration in B cell phenotype and in the percentages of different B cell subsets has also been described during acute and chronic phases of HIV-1 infection, with depletion of memory B cells (4–6). Accordingly, it is likely that the impaired capacity to mount an efficient neutralizing antibody response to HIV-1 may be directly correlated to the damage occurring to B cells devoted to antibody production.
SIV infection in macaques defined as rapid progressors not only affected SIV-specific memory B cells, but randomly eradicated memory B cells specific for other previously encountered antigens. Accordingly, the SIV-infected macaques in which depletion of mBAct cells was manifest showed a decline in antibody titers against intestinal bacteria (3). These data parallel similar findings on loss of antibody titers in response to viral and bacterial antigens in HIV-1–infected children and adults (2, 7, 8). As loss of mBAct cells was associated with rapid disease progression, Titanji et al. suggest that loss of mBAct cells should be evaluated as an early predictor of HIV-1 disease progression (3). If this holds true in individuals infected with HIV-1, this would be extremely important, as immunological biomarkers other than CD4+ T cell counts are needed to predict disease progression.
Altered homing of B cells to lymphoid tissues
Titanji and colleagues showed that following SIV infection of macaques, the percentages of total B cells and mBAct cells among blood lymphocytes rapidly decreased at 2 weeks after infection in both rapid and typical progressors (3). At 12 weeks after SIV infection, the total B cell numbers in blood rebounded in all animals, although the frequency of mBAct cells remained significantly lower than baseline levels in rapid progressors. Thus, the authors speculate that B cells, including mBAct cells, home away from the circulation to lymphoid tissues early after infection (at 2 weeks) and return to the circulation at 12 weeks after infection. This possibility is consistent with a previously published study in which a decline in the number of circulating total B cells was reported in SIVmac251-infected cynomolgus macaques 2 weeks after infection as a result of B cell trafficking to lymphoid organs, with preferential accumulation in spleen and intestine (9). Furthermore, altered expression of the chemokine receptor/ligand pair CXCR5/CXCL13, important for homing of B cells, has been reported during HIV-1 infection, especially in patients with low CD4+ T cell counts (10).
It is conceivable that the reduced numbers of total B cells and mBAct cells detected by Titanji and colleagues early during SIV infection (3) may be due to the transitory altered homing of B cells (Figure 1), occurring in all animals, combined in rapid progressors with the depletion of memory B cells, occurring by apoptosis in lymphoid tissues and leading to cumulative loss of B cells. Thus, the acute period of SIV infection appears to be characterized by intense fluctuation in levels of B cell chemoattractants in blood and affected tissues.
Figure 1. Two-phase model for depletion of mBAct from circulation.

The model outlined here is based on data generated by Titanji et al. (3) and Peruchon et al. (9). During the early stages of SIV infection (2 weeks), mBAct cells leave the blood to home to lymphoid tissues. At 12 weeks after SIV infection, mBAct cells migrate from the lymphoid tissues and rebound in the blood of typical progressor macaques and, to a much lesser extent, in the blood of rapid progressors. This model implies that mBAct in rapid progressors die through apoptosis in lymphoid tissues, according to a mechanism related to PD-1 signaling on B cells (see Figure 2).
Role of PD-1 in B cell survival and differentiation
All subsets of resting, activated, and tissue memory B cells in noninfected and SIV-infected animals analyzed by Titanji et al. expressed high levels of PD-1, with mBAct cells expressing the highest level of PD-1 and accounting for the largest number of PD-1+ cells (3). The depletion of mBAct cells shown to occur in rapid progressors related to high PD-1 expression. However, more studies are needed to clarify why signaling through PD-1 leads to depletion of mBAct cells only in rapid progressors, despite similar preinfection PD-1 expression levels on mBAct cells of rapid and typical progressors. The involvement of other apoptotic pathways, including Fas (3), and differential expression of the PD-1 ligands PD-L1 and PD-L2 in rapid and typical progressors may account for this difference.
PD-1 was first described as a negative regulatory molecule, the expression of which increases on T cells during activation (11). PD-1 expression during chronic viral infections has been linked to an exhausted phenotype, in which T cells are burned out and do not exert their effector functions. PD-L1 and PD-L2 are broadly expressed on dendritic cells, macrophages, B cells, and T cells. A role for PD-1 in negative regulation of B cell responses has been suggested by the findings that PD-1 inhibits BCR signaling through recruitment of src homology 2 domain–containing tyrosine phosphatase 2 (SHP-2) to its phosphotyrosine (12) and that PD-1–deficient mice develop antibody-mediated autoimmune diseases (13). A new view has recently emerged on the contribution of PD-1 signaling to the regulation of humoral immunity: interaction of PD-1 ligands on B cells with PD-1 on T cells was shown to exert a positive effect on antibody production by affecting the quality of germinal center (GC) responses and by fine-tuning the regulation of long-lived plasma cell formation (14). In the absence of PD-1 signals, augmented B cell death in GCs was reported to occur, with lower numbers of long-lived plasma cells formed (14).
Limited information is currently available on the autocrine role of PD-1 and PD-1 ligands in B cell responses and on the function of PD-1 on B cells in GC formation (15). Titanji and colleagues postulate a role for PD-1 signaling in regulating the survival/apoptosis of mBAct cells during SIV infection (Figure 2A), since in vitro exposure of B cells from SIV-infected macaques to PD-L1 expressed on a cell line increased mBAct cell apoptosis (3). Noticeably, upon these conditions, mBAct cells from control, noninfected animals did not die, even though they expressed similar levels of PD-1, which suggests that expression of proapoptotic factors prevails over that of antiapoptotic factors in mBAct cells of infected animals, especially fast progressors, and that differential expression of PD-L1 and PD-L2 may distinguish infected from noninfected animals. The cellular source of PD-L1 and PD-L2 in the SIV-infected macaques was not investigated. A previous study indicated that PD-L1 upregulation occurs in several cell types, including B cells, in progressive HIV-1 infection (16); however, more studies in SIV-infected animals are needed to define to what extent B cells express PD-L1 and/or PD-L2 and whether these ligands induce apoptosis of mBAct cells.
Figure 2. Biological action of PD-1 blockade on mBAct cells and antibody production during SIV infection.
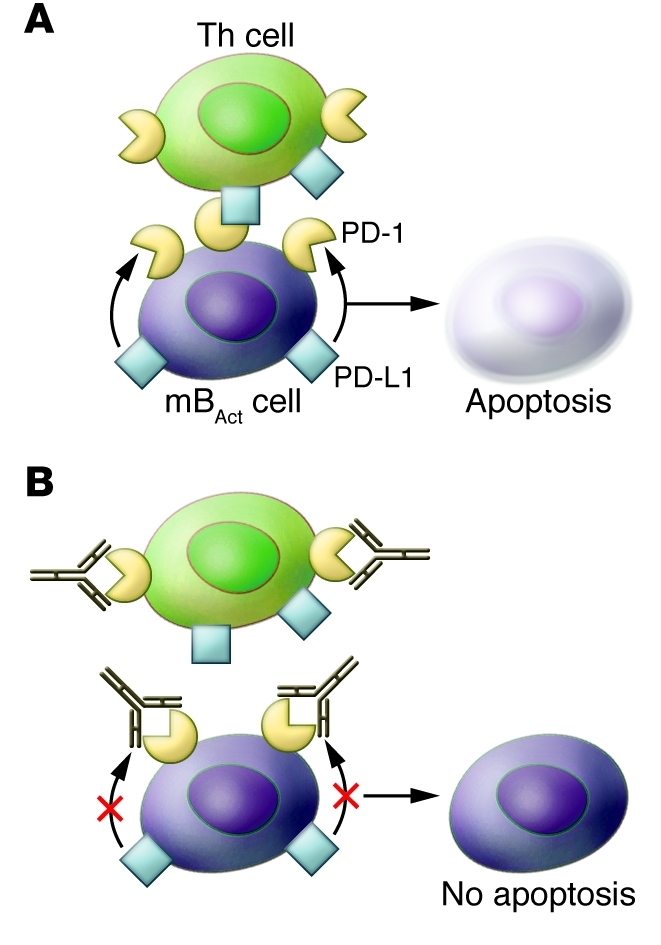
(A) As shown by Titanji and colleagues, PD-1 on mBAct of rapid progressors leads to apoptosis of these cells (3). The signal to undergo PD-1–mediated apoptosis can be provided in an autocrine fashion from PD-1 ligands present on B cells, or from PD-1 ligands on Th cells. In this context, it is likely that PD-1 present on Th cells does not receive signaling from PD-1 ligands present on B cells, since they are engaged in autocrine binding to PD-1 on B cells. (B) One possible interpretation of the data presented by Titanji et al. (3) is that upon PD-1 blockade, the autocrine binding of PD-1 to PD-1 ligands is inhibited, thus preventing apoptosis of B cells. Thus, blocking PD-1 on B cells can improve the possibility for these cells to produce specific antibodies to SIV and other microbial antigens, by channeling T cell help toward B cells in GCs.
PD-1 blockade contributes to production of SIV-specific antibodies
With an aim to unravel the role of the PD-1 pathway during SIV infection of macaques, Titanji et al. treated the SIV-infected macaques with PD-1–specific blocking antibodies (3). This approach turned into a successful therapeutic strategy to enhance the survival and proliferation of mBAct cells and the production of SIV-specific and non–SIV-specific antibodies. This is consistent with previous data from the same group suggesting that titers of SIV-specific antibodies could be enhanced as a consequence of PD-1 blockade (17).
Hyperactivation of immune cells, likely as a result of persistent antigenic stimulation, has been suggested as an important component of SIV and HIV-1 immunopathology. This abnormal activation leads to altered expression of mediators of cell survival/proliferation. Of particular interest is the suggestion by Titanji and colleagues of a connection between the PD-1 pathway and the Fas death receptor that leads to a reduced level of Fas-mediated apoptosis in B cells during PD-1 blockade; this would indicate that survival of memory B cells is negatively regulated by molecular interactions between Fas and PD-1 (3).
Blockade of PD-1 during SIV infection (3) may operate to control two pathogenic components linked to SIV infection (Figure 2B). First, PD-1 blockade on Th cells may lead to recovery of T cell exhaustion by interfering with PD-1 (15) and, indirectly, with additional molecules linked to T cell exhaustion. Second, the masking of PD-1 on B cells may result in increased survival of B cells, mBAct cells, and newly formed plasma cells in the GCs (Figure 2B). These two events may synergize and lead to higher levels of SIV-specific antibodies. A detailed analysis of the cellular components expressing PD-1 and its ligands needs to be performed in the GCs of SIV-infected animals to clarify the cellular components involved in PD-1 blockade.
Concluding remarks
There is a need to develop innovative therapeutic strategies to ameliorate responses to common vaccination antigens in HIV-1–infected children and adults (3, 6), since limited access to ART and fast progression to AIDS upon HIV-1 infection in developing countries impairs responses to vaccination with bacterial and viral antigens in HIV-1–infected subjects. The present study from Titanji and colleagues (3) paves the way for a new treatment intervention, based on PD-1 blockade, that aims to ameliorate B cell responses. By reducing T cell exhaustion (17) and by improving B cell responses (3), this PD-1–based approach may provide a platform to accelerate immune reconstitution during HIV-1 infection.
Acknowledgments
The author’s work is supported through grants from the Swedish MRC, the Swedish International Development Agency (SIDA-SAREC), the Karolinska Institutet, the Fp6 EU Europrise network of excellence, the Fp7 EU NGIN Collaborative project Health-F3-2007-201433, and the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet.
Footnotes
Conflict of interest: F. Chiodi is a founder of and shareholder in Imed AB, a start-up company at the Karolinska Institutet Science Park.
Citation for this article: J Clin Invest. 2010;120(11):3810–3813. doi:10.1172/JCI44872.
See the related article beginning on page 3878.
References
- 1.INSIGHT-ESPRIT Study Group, et al. Interleukin-2 therapy in patients with HIV infection. N Engl J Med. 2009;361(16):1548–1559. doi: 10.1056/NEJMoa0903175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Milito A, et al. Mechanisms of hypergammaglobulinemia and impaired antigen-specific humoral immunity in HIV-1 infection. Blood. 2004;103(6):2180–2186. doi: 10.1182/blood-2003-07-2375. [DOI] [PubMed] [Google Scholar]
- 3.Titanji K, et al. Acute depletion of activated memory B cells involves the PD-1 pathway in rapidly progressing SIV-infected macaques. J Clin Invest. 2010;120(11):3878–3890. doi: 10.1172/JCI43271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moir S, Fauci AS. B cells in HIV infection and disease. . Nat Rev Immunol. 2009;9(4):235–245. doi: 10.1038/nri2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cagigi A, Nilsson A, Pensieroso S, Chiodi F. Dysfunctional B-cell responses during HIV-1 infection: implication for influenza vaccination and highly active antiretroviral therapy. Lancet Infect Dis. 2010;10(7):499–503. doi: 10.1016/S1473-3099(10)70117-1. [DOI] [PubMed] [Google Scholar]
- 6.Titanji K, et al. Primary HIV-1 infection sets the stage for important B lymphocyte dysfunctions. AIDS. 2005;19(17):1947–1955. doi: 10.1097/01.aids.0000191231.54170.89. [DOI] [PubMed] [Google Scholar]
- 7.Titanji K, et al. Loss of memory B cells impairs maintenance of long-term serologic memory during HIV-1 infection. Blood. 2006;108(5):1580–1587. doi: 10.1182/blood-2005-11-013383. [DOI] [PubMed] [Google Scholar]
- 8.Pensieroso S, et al. Timing of HAART defines the integrity of memory B cells and the longevity of humoral responses in HIV-1 vertically-infected children. Proc Natl Acad Sci U S A. 2009;106(19):7939–7944. doi: 10.1073/pnas.0901702106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peruchon S, et al. Tissue-specific B-cell dysfunction and generalized memory B-cell loss during acute SIV infection. PLoS One. 2009;4(6):e5966. doi: 10.1371/journal.pone.0005966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cagigi A, et al. Altered expression of the receptor-ligand pair CXCR5/CXCL13 in B cells during chronic HIV-1 infection. Blood. 2008;112(12):4401–4410. doi: 10.1182/blood-2008-02-140426. [DOI] [PubMed] [Google Scholar]
- 11.Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006;27(4):195–201. doi: 10.1016/j.it.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A. 2001;98(24):13866–13871. doi: 10.1073/pnas.231486598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okazaki T, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. 2003;9(12):1477–1483. doi: 10.1038/nm955. [DOI] [PubMed] [Google Scholar]
- 14.Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol. 2010;11(6):535–542. doi: 10.1038/ni.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nutt SL, Tarlinton DM. Give and take in the germinal center. Nat Immunol. 2010;11(6):464–466. doi: 10.1038/ni0610-464. [DOI] [PubMed] [Google Scholar]
- 16.Trabattoni D, et al. B7-H1 is up-regulated in HIV infection and is a novel surrogate marker of disease progression. Blood. 2003;101(7):2514–2520. doi: 10.1182/blood-2002-10-3065. [DOI] [PubMed] [Google Scholar]
- 17.Velu V, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458(7235):206–210. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]