

Summary
Pulmonary arterial hypertension (PAH) is a complex disease with significant morbidity and mortality. Recent animal and human studies have highlighted abnormalities in regulation and metabolism of insulin, sex hormones, adipokines, and lipids that may play a role in disease development. Mouse studies suggest features of the metabolic syndrome including insulin resistance, deficiencies in PPARγ and apolipoprotein E, and low adiponectin are linked to development of PAH. In humans, insulin resistance, the metabolic syndrome, and low levels of high-density lipoprotein have been associated with PAH. In addition, abnormal metabolism of estrogens has been demonstrated in human and animal models of PAH, suggesting an important relationship of sex hormones and pulmonary vascular disease. Improved understanding of how metabolic and hormonal derangements relate to development and progression of pulmonary hypertension may lead to better disease therapies and understanding of potential risk factors. This review will focus on the animal and human data regarding metabolic and sex hormone derangements in PAH.
Keywords: Metabolic syndrome, pulmonary hypertension, pulmonary arterial hypertension, sex hormones
Introduction
Despite major advances in understanding of the pathobiology of pulmonary arterial hypertension (PAH) and its survival in the last decade, PAH remains associated with substantial morbidity and mortality and a cure remains elusive1–4. The diverse group of diseases described to cause PAH5 highlights the complex pathobiology of this disease, and suggests involvement of multiple inflammatory and signaling pathways ultimately leading to endothelial damage, increased pulmonary vascular resistance, and right heart failure. Recent work in animal models of PAH and human disease have demonstrated a relationship of the metabolic syndrome and PAH, specifically highlighting the features of insulin resistance 6,7, dyslipidemia 8, and adiponectin deficiency9 in the development of pulmonary vascular disease. The well-known female predominance of PAH 10,11 and mouse and human data showing differences in sex hormone metabolism 12,13 in PAH underscore the influence of sex hormones as another aspect of PAH pathobiology.
As PAH is a relatively rare disease, our understanding of the epidemiology and clinical characteristics of PAH have been advanced through large registries of PAH patients. The REVEAL registry7 is a prospective study of nearly 3000 incident and prevalent cases of PAH in the United States (U.S.), and it demonstrates important potential associations of PAH and metabolic and hormonal influence. There is a strong female predominance in PAH patients (80% female in this registry), and co-morbid conditions including features of the metabolic syndrome were common: hypertension in 40%, obesity in 33%, diabetes mellitus in 12%, and ischemic cardiovascular disease in 9%7. Thus PAH is commonly accompanied by features of the metabolic syndrome. Improved understanding of these associations of metabolic derangements and sex hormones in PAH is critical to our understanding of PAH pathogenesis, and further study in these areas may uncover novel therapeutic targets and more effective treatment for this devastating disease.
Metabolic Syndrome
The metabolic syndrome (MS) is a group of metabolic and biochemical risk factors for developing cardiovascular disease and diabetes mellitus. The core features of MS include hypertension, elevated triglyceride levels, low high-density lipoprotein, obesity (particularly central obesity), and dysglycemia. Differing definitions of MS have lead to controversy over the last decade, however a new consensus definition proposed by the International Diabetes Federation and the American Heart Association/National Heart, Lung, and Blood Institute was recently published14. Table 1 shows the clinical criteria for diagnosis of the metabolic syndrome under this new consensus definition. The clinical cutoffs for central obesity (waist circumference) differ based on country-specific definitions as shown.
Table 1.
Criteria for diagnosis of the metabolic syndrome*
| Feature | Cutoffs |
|---|---|
| Elevated waist circumference | Population and country-specific definitions** |
| Elevated triglycerides or drug treatment for elevated triglycerides |
≥150 mg/dL (1.7 mmol/L) |
| Reduced high-density lipoprotein cholesterol (HDL-C) or drug treatment for reduced HDL-C |
< 40 mg/dL (1.0 mmol/L) in males < 50 mg/dL (1.3 mmol/L) in females |
| Hypertension (or antihypertensive drug therapy in patient with history of hypertension) |
Systolic ≥ 130 mm Hg and/or Diastolic ≥ 85 mm Hg |
| Elevated fasting glucose (or drug treatment for elevated glucose) |
≥ 100 mg/dL |
| Presence of 3 of the 5 criteria denote presence of metabolic syndrome | |
Adapted from Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009; 120:1640–164510.
For U.S.: ≥ 102cm for men, ≥ 88 cm for women. National Institutes of Health. Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults: the evidence report. Obes Res 1998; 6(suppl 2):51S–209S.
Metabolic syndrome is common and the prevalence is increasing worldwide14, likely related to increasingly sedentary lifestyles and obesity. MS is particularly common in the U.S., where an estimated 34% of adults over the age of 20 years meet criteria for MS15. The prevalence increases with age, and MS is found in over 54% of adults over the age of 60, compared to only 20% of adults in the 20–39 age group15. Thus MS will continue to grow as a clinical and public health concern as the global population ages.
The clustering of the components of MS has been described for decades, but the underlying causative factors of MS remain unknown. It is believed that insulin resistance plays a central role in development of MS, and may underlie all features of MS14,16. Central obesity is the most common feature of the MS in US population studies, with a crude prevalence of 53.2%15 and central adiposity may in fact precede development of other features of MS17.
MS is characterized by an inflammatory biochemical phenotype. This milieu is thought to contribute to endothelial dysfunction with activation of mediators resulting in oxidative injury, platelet activation, thrombus formation and development of atherosclerotic plaques and systemic microvascular disease18. The inflammatory biomarkers C-reactive protein (CRP) and myeloperoxidase (MPO) are elevated in MS19–21, and adiponectin, an adipokine with anti-inflammatory and insulin-sensitizing properties, is decreased in MS22,23. Insulin resistance has been linked to endothelial dysfunction and altered nitric oxide response24,25. Emerging literature summarized in this review points toward similar abnormalities of inflammatory mediators, adiponectin, and insulin resistance in pulmonary vascular disease and PAH in particular, thus the link between MS and PAH warrants further investigation.
Sex Hormones and the Metabolic Syndrome
There are important gender differences in the metabolic syndrome, and sex hormones have been recognized as important mediators in cardiovascular risk and systemic vascular disease. While there is no gender difference in age-adjusted prevalence estimates of MS in the U.S. using the NHANES 2003–2006 data, there are differences in the prevalence of each individual risk factor by sex with males having higher prevalence of hypertension, hyperglycemia, and elevated triglycerides than females15. Females had higher age-adjusted prevalence of central obesity and low high-density lipoprotein than males15. A review of gender differences in insulin resistance, central obesity, dyslipidemia and other features of MS is beyond the scope of this paper, but there is a large body of literature describing estrogen and testosterone effects on the components of MS [for a review, see Regitz-Zagrosek Clin Res Cardiol 200626]. Androgen deprivation therapy in males with prostate cancer has been shown to decrease insulin sensitivity, decrease lean body mass, elevate triglyceride and low-density lipoprotein levels, and increase fat mass [for a review see Saylor and Smith, J Urology 200927]. Estrogen and testosterone levels have been shown to associate with adiponectin levels in post-menopausal women28. Several studies have shown greater rates of insulin resistance in adolescent females29,30. Thus sex hormones may underlie the features of MS seen in PAH and represent a common disease modifier.
Metabolic and Hormonal Derangements in PAH: Animal Models
Substantial evidence has accumulated recently from a basic science perspective linking hormonal and metabolic derangements to pulmonary vascular disease. Insulin resistance, adipokines, lipids and estrogen metabolites are all potentially linked to development of pulmonary vascular disease in animal models and cell culture. Preclinical data will be explored below, in addition to a proposed mechanism for synthesis of these data.
Insulin Resistance
Altered BMPR2 signaling is a common feature of many types of PAH: mutations in BMPR2 have been well-described in heritable PAH, and decreased expression of BMPR2 has been found in other types of PAH not known to be associated with BMPR2 mutation31–33. One downstream target of this pathway is the transcription factor peroxisome proliferator-activated receptor γ (PPARγ) and its downstream effecter, ApoE34. PPARγ is best known for its role in glucose metabolism and adipogenesis35–37, but is an attractive target in PAH as it is highly expressed in the lungs, has been shown to have vasoprotective effects in systemic vascular disease38, and also regulates multiple other pathways known to be important in PAH including endothelin-1, MCP-1, and eNOS39,40. Moreover, there are several PPARγ ligands currently FDA-approved for the treatment of diabetes mellitus, thus making this molecule of potential therapeutic importance in PAH.
Lung tissue from PAH patients has been shown to have decreased expression of both PPARγ and ApoE41,42. ApoE plays a role in reduction of circulating oxidized low-density lipids and is thought to be protective in vascular disease43. Thus reductions in PPARγ and ApoE were hypothesized to potentially be causative in the development of pulmonary vascular disease. To study this potential relationship, Hansmann and colleagues evaluated ApoE deficient (ApoE −/−) mice fed a high fat diet and found that they develop pulmonary hypertension that could be reversed by administration of the PPARγ ligand rosiglitazone6. These findings have been recapitulated in normal and PAH patient-derived BMPR2 mutant pulmonary artery smooth muscle cells, where BMP-2 signaling had an antiproliferative effect that was PPARγ and ApoE-dependent44. Furthermore, transgenic mice with smooth muscle cell deletion of PPARγ spontaneously develop PAH44. Taken together, these findings unify dysfunction in BMPR2 signaling to downstream changes in nuclear transcription mediated by PPARγ and subsequently ApoE. It is attractive to postulate that use of PPARγ ligands in human disease may bypass the abnormal BMPR2 signaling and potentially be a potent antiproliferative treatment for PAH.
Adipokines
A critical feature of MS is obesity, particularly central adiposity17. Adipose cells are increasingly recognized not simply as innocent bystanders but as active participants in the hormonal milieu through production of cytokines (so called adipokines) and other factors45. The endocrine function of adipose tissue includes secretion of hormones that may have pulmonary vascular effects such as adiponectin, ghrelin and leptin9,46,47.
Adiponectin is a protein exclusively produced by fat cells that has known anti-inflammatory and insulin sensitizing properties48,49. Interestingly, in obese patients and those with type 2 diabetes, adiponectin levels are low and can be upregulated by PPARγ50. In the obese ApoE deficient mouse2, adiponectin levels were higher in female mice, and these mice also were also less likely to develop PAH compared to their male counterparts. Treatment of male transgenic mice with rosiglitazone increases adiponectin levels and results in reduced pulmonary artery pressures6,44. An adiponectin deficient mouse has also been studied and shown to develop pulmonary hypertension9,51. Adiponectin is known to inhibit PDGF, a potent mitogen for pulmonary artery smooth muscle cells, and this might be the mechanistic link for development of PAH52. Thus adiponectin appears to be intricately linked to insulin resistance and development of pulmonary vascular disease in animal models.
Dyslipidemia
Although hyperlipidemia is a key feature of MS, there is little basic science data to suggest that disordered lipids alone cause pulmonary vascular disease. All ApoE deficient mice fed a high fat diet develop elevated triglycerides and total cholesterol, though only a portion develop PAH, which correlates with the presence of insulin resistance, not degree of lipid derangement. Thus it is insulin resistance not abnormal lipids that promotes pulmonary vascular disease6,.
Sex Hormones
The compelling epidemiologic data showing marked female predominance in PAH makes study of the effects of sex hormones on the pulmonary vasculature of obvious importance10,11,53. There are several potential hypotheses as to why women are more prone to develop PAH than men. First, estrogens may promote pulmonary vascular disease, second, testosterone may prevent development of pulmonary vascular disease or, third, there may be some other factor associated with being a woman that predisposes to PAH. This “other factor” may be an environmental exposure or, perhaps, circulating fetal cells from pregnancy or some other unknown factor. Very little is known about the effects of testosterone or progesterone on the pulmonary vasculature, so we will focus on estrogens here.
In isolated pulmonary vessel studies, estrogen has been shown to have a salutary effect in preventing hypoxic or phenylephrine-induced vasoconstriction 54–56. Similarly, animal models of pulmonary hypertension such as monocrotaline and chronic hypoxia have shown a protective effect of estrogen administration and worsening with ovariectomy and subsequent estrogen deprivation57–61. These animal data are clearly in contrast to observations in humans, which has led to the hypothesis that altered estrogen metabolism may underlie phenotypic differences in animals and humans. Tofovic and colleagues have shown that the estrogen metabolite 2-ME is the primary mediator of the beneficial effects of estrogens in rodent PAH models57,62,63.
Interestingly, only male ApoE deficient mice fed a high fat diet develop pulmonary hypertension. Female ApoE deficient mice have normal pulmonary artery pressures when fed this specialized diet and this is associated with significantly higher adiponectin levels6. Testosterone inhibits secretion of adiponectin by adipocytes64 and thus may explain the male predominance in this model. The implications of this finding for humans are presently not clear. More study of the interplay between sex and metabolic hormones would greatly increase our understanding of these critical factors in pulmonary vascular disease development and progression.
Summary of Animal Data
How might all these various hormonal factors convene to cause pulmonary vascular disease? The data suggest that altered BMPR2 signaling in PAH changes metabolic phenotype through PPARγ-regulated transcription targets. As a downstream consequence, insulin resistance develops and protective adiopokines such as adiponectin are diminished, thus resulting in pulmonary vascular damage and proliferation (Figures 1, 2). It is possible that a shift occurs in the metabolic state of the pulmonary artery smooth muscles cells, perhaps triggered by insulin resistance and PPARγ-mediated altered transcription, that drives the cells into the so-called Warburg effect, resulting in increased proliferation, oxidant stress and further damage65,66. These hypotheses need to be further explored from both a basic science and translational perspective to identify novel treatment modalities for this deadly disease.
Figure 1. Metabolism and normal pulmonary vasculature.
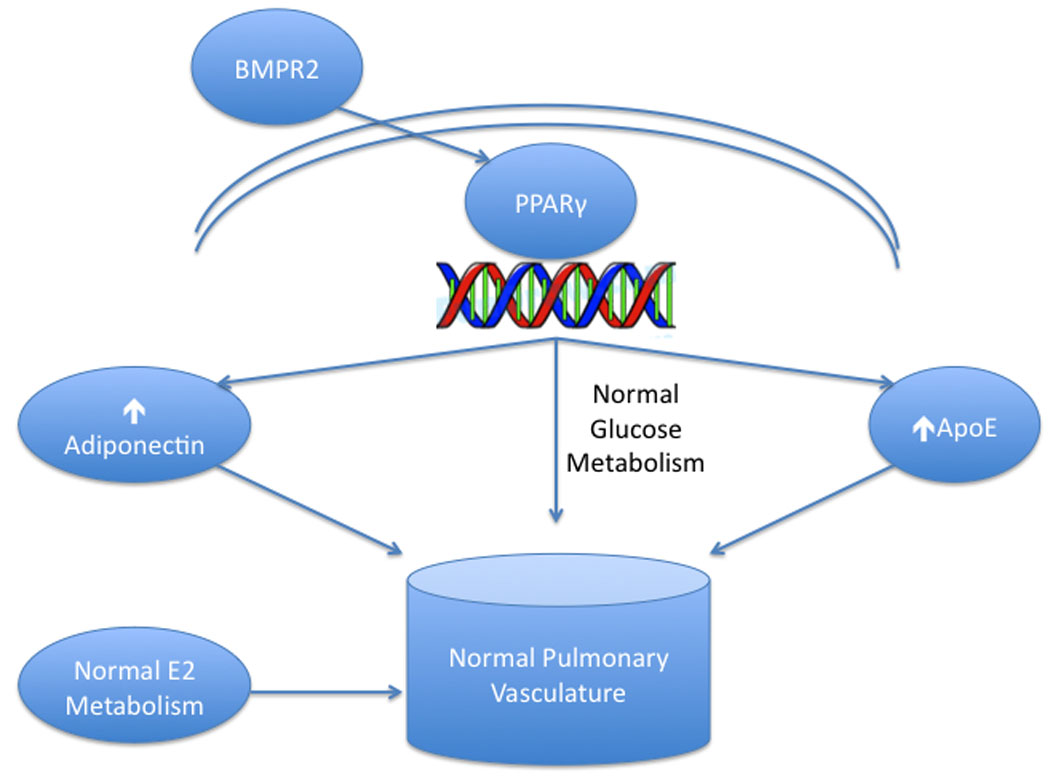
Under normal conditions, BMP-2 signaling stimulates the nuclear transcription factor PPARγ to increase expression of adiponectin and ApoE, which in concert with normal estrogen metabolism, promote normal pulmonary vascular function.
Figure 2. Metabolic Derangements in PAH.
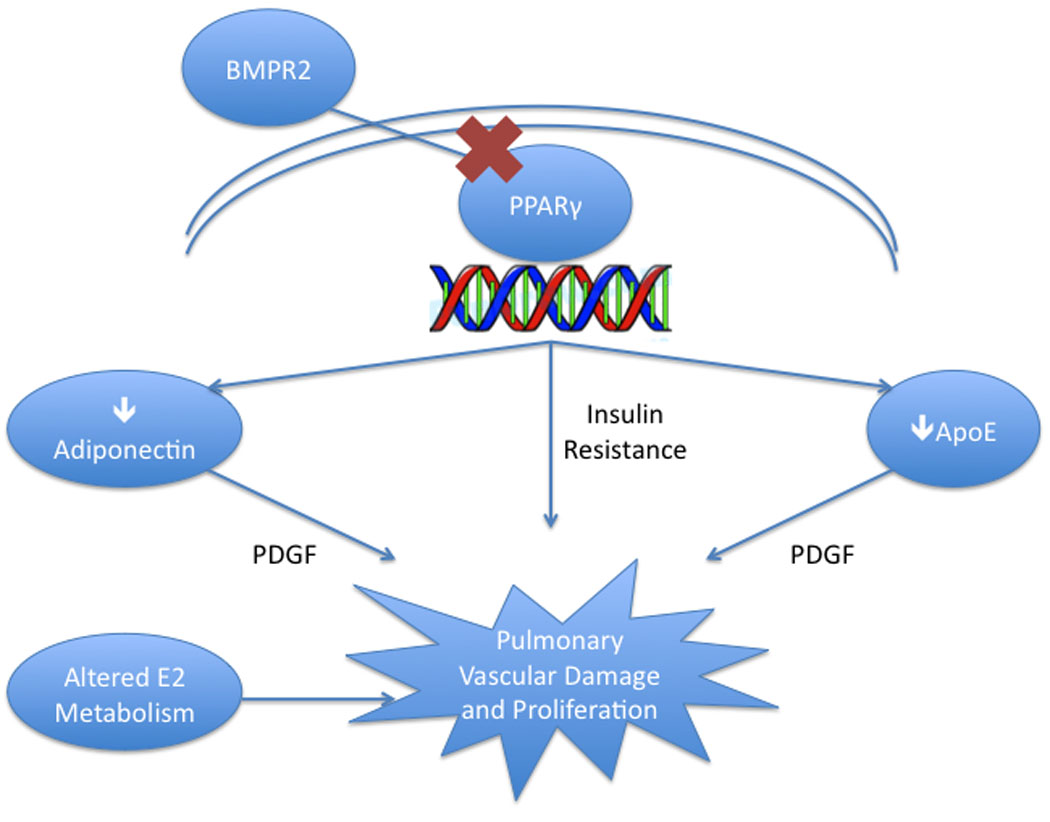
Abnormal BMPR2 signaling in many forms of PAH drives altered PPARγ-induced transcript production that results in three important derangements: insulin resistance, low levels of adiponectin and low ApoE. These alterations, likely with altered estrogen metabolism as well, may lead to the phenotype of PAH.
Metabolic and Hormonal Derangements in PAH: Humans
With emerging preclinical data showing a relationship of features of MS with pulmonary hypertension, there have been several small analyses of MS and its component features in humans. In a review of 122 consecutive patients referred for evaluation of pulmonary hypertension, Robbins et al found features of the MS were especially common in patients with pulmonary venous hypertension (World Health Organization Group II)67. Over 90% of patients with pulmonary venous hypertension had two or more features MS, with obesity (defined as BMI ≥ 30 kg/m2) and systemic hypertension as the most common features. While features of MS were less common in PAH, over one-third of PAH subjects had two or more features of MS, raising awareness of this syndrome in patients with PAH as well. While the study was limited by its retrospective and single-center design and lack of formal evaluation for features of MS, it was the first to draw attention to the important association of MS and pulmonary vascular disease.
Insulin Resistance
Insulin resistance is a core feature of the metabolic syndrome. Given the association of insulin resistance with pulmonary hypertension in animal models, several groups have evaluated insulin resistance or dysregulated glucose metabolism in humans with PAH. Using a validated, surrogate marker of insulin resistance (the triglyceride to high-density lipoprotein ratio), the prevalence of insulin resistance in a cohort of female PAH patients was found to be nearly 46%, significantly higher than the 22% prevalence by the same measure in a population cohort7. PAH patients with insulin resistance had similar age, body mass index, six-minute walk distance and NYHA functional class to those who were insulin sensitive. Despite these clinical similarities, insulin resistant PAH patients had a worse six-month event-free survival compared with insulin sensitive PAH patients7.
Using hemoglobin A1c (HbA1c), a marker of chronic glycemia and a recommended test for diagnosis of diabetes and those at high-risk for diabetes, we found that 56% of PAH patients without a prior diagnosis of diabetes had glucose intolerance (defined as HbA1c ≥ 6.0%)68. Age and BMI were similar in PAH patients with glucose intolerance to those with normal HbA1c, however PAH patients with glucose intolerance had more severe heart failure by NYHA classification and significantly lower mean six-minute walk distance68.
Thus insulin resistance and dysregulated glucose metabolism may be modifiers of pulmonary vascular disease. One hypothesis is that insulin resistance and the accompanying inflammatory milieu, dysregulated nitric oxide response, and endothelial damage may worsen pulmonary vascular injury in a susceptible host, contributing to more severe disease. Another hypothesis is that PAH leads to insulin resistance, perhaps through increased sedentary lifestyle in the setting of vascular disease and heart failure. Further studies in humans and animal models will hopefully begin to answer this important mechanistic question.
Dyslipidemia
Reduced levels of high-density lipoprotein cholesterol (HDL-C) are another important feature of the metabolic syndrome, and clinically, elevating HDL-C is often a target of drug and lifestyle therapy to reduce the risk of cardiovascular disease69. In a recent study, PAH patients were found to have a significantly lower HDL-C than control patients with cardiovascular risk factors, and an HDL-C of 35 mg/dL discriminated PAH survivors from nonsurvivors with a sensitivity of 100% and specificity of 60%8. Low HDL-C in PAH has been reported in other studies as well7. HDL-C is known to have anti-inflammatory properties 70–72 and thus abnormalities in HDL-C found in MS and PAH may be contributing to pulmonary vascular injury, or low HDL-C may simply represent altered lipid metabolism resulting from sedentary lifestyle and altered metabolism in the setting of pulmonary vascular disease.
Currently, screening for and aggressive interventions for dyslipidemia are not part of standard clinical practice in pulmonary arterial hypertension. It’s unknown whether interventions to raise HDL-C in PAH have an effect on disease severity or outcome, and further study in this area is needed. But growing evidence supporting another link of PAH and the metabolic syndrome stresses the potential importance of this fertile area of research.
Sex hormones
Early reports of primary pulmonary hypertension, what we now call idiopathic PAH, demonstrated a female predominance53,73 and recent large registries of PAH in France and the U.S. continue to show this gender bias10,11. In the U.S. Chicago registry, of 578 total PAH patients with more than half classified as IPAH, HPAH or anorexigen-associated, the female to male ratio was 3:174. In the national REVEAL U.S. registry, the female to male ratio of PAH was reported 4.3 to 1, significantly higher than the 1.9:1 female to male ratio in the French cohort. Female predominance is seen in heritable forms of PAH as well: PAH caused by mutation in BMPR2 has a female to male ratio of 2.7 to 153. Portopulmonary and scleroderma-associated PAH both affect females more than males75–77.
While there have been multiple studies of the effects of estrogens and testosterone in PAH in animal models, there is relatively little data in humans. There are no human studies of testosterone effects on the pulmonary vasculature or right ventricle in PAH, although emerging data suggests this may be a factor in normal right ventricular structure78. There have been a few studies evaluating the role of estrogen signaling and metabolism in human PAH, and interestingly, most of these studies suggest that higher levels of estrogens or perhaps altered metabolism of estrogens contribute to the PAH75,76.
Gene array studies in affected and unaffected BMPR2 mutation carriers have pointed toward abnormalities in the cytochrome P450 enzyme CYP1B1 as a potential explanation for the gender bias seen in hereditary PAH. CYP1B1 is an enzyme that catalyzes the oxidation of estrogens. Alterations in CYP1B1 expression could lead to alterations in the types of estrogen metabolites produced. Austin and colleagues found that affected BMPR2 mutation carriers more commonly had wild-type CYP1B1 than unaffected mutation carriers, and that affected patients had altered levels of estrogen metabolite ratios (2-OHE to 16α-OHE) in the urine than unaffected individuals as would be predicted by altered CYP1B1 function12. This study suggests that alterations in CYP1B1 may account for aberrant estrogen metabolism and perhaps explain the female predominance in heritable PAH.
In another study of gene arrays in idiopathic PAH (IPAH) it was shown that patients with IPAH have higher levels of estrogen receptor α than normal female controls79. A study of portopulmonary hypertension has demonstrated that single nucleotide polymorphisms in aromatase and estrogen receptor 1, critical enzymes in the pathway of estrogen signaling and metabolism, are associated with a risk of developing portopulmonary hypertension76. Polymorphisms in these enzymes correlated with higher estradiol levels, which seem to implicate estrogen as a critical actor in the development of portopulmonary hypertension. Further studies to better understand how estrogen affects the pulmonary vasculature and interacts with the metabolic syndrome, and which estrogen metabolites are most involved is critical to our understanding of these observations.
Future Directions
Although tremendous advances have been made in the last two decades with the advent of multiple effective drug therapies for PAH, the underlying complex pathobiology is incompletely understood, and there are no curative therapies available apart from lung transplantation. Multiple studies in humans and animal models of PAH now point toward metabolic and sex hormone disturbances that may underlie disease development or progression. Understanding how sex hormones and the metabolic syndrome features of obesity, dyslipidemia, systemic hypertension, and insulin resistance affect the pulmonary vasculature may uncover novel therapeutic targets or clinical management strategies. Further studies of metabolic derangements in heritable PAH in humans and animal models of BMPR2 mutation are ongoing, and it is likely that in the next decade we may understand why these metabolic disturbances are so prevalent in PAH. The strong epidemiologic, basic science, and human data supporting a role for sex hormones in PAH development support estrogen as another possible risk factor for pulmonary endothelial damage and development of clinical PAH. Whether manipulation of sex hormone metabolism could be helpful in PAH requires further study. More preclinical and human data is needed to understand whether modification of obesity, insulin resistance, and dyslipidemia is favorable in PAH or at risk groups.
REFERENCES
- 1.Thenappan T, Shah SJ, Rich S, et al. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010;35:1079–1087. doi: 10.1183/09031936.00072709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Humbert M, Sitbon O, Yaici A, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010 doi: 10.1183/09031936.00057010. [DOI] [PubMed] [Google Scholar]
- 3.Humbert MSO, et al. Survival in patients with idiopathic, familial and anorexigen associated pulmonary arterial hypertension in the modern management era. Circulation. 2010 doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 4.Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting Survival in Pulmonary Arterial Hypertension. Insights From the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010 doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 5.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 6.Hansmann G, Wagner RA, Schellong S, et al. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2007;115:1275–1284. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- 7.Zamanian RT, Hansmann G, Snook S, et al. Insulin resistance in pulmonary arterial hypertension. Eur Respir J. 2009;33:318–324. doi: 10.1183/09031936.00000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heresi GA, Aytekin M, Newman J, et al. Plasma Levels of High-density Lipoprotein Cholesterol and Outcomes in Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2010 doi: 10.1164/rccm.201001-0007OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Summer R, Fiack CA, Ikeda Y, et al. Adiponectin deficiency: a model of pulmonary hypertension associated with pulmonary vascular disease. Am J Physiol Lung Cell Mol Physiol. 2009;297:L432–L438. doi: 10.1152/ajplung.90599.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–1030. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 11.Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137:376–387. doi: 10.1378/chest.09-1140. [DOI] [PubMed] [Google Scholar]
- 12.Austin ED, Cogan JD, West JD, et al. Alterations in estrogen metabolism: Implications for higher penetrance of FPAH in females. Eur Respir J. 2009 doi: 10.1183/09031936.00010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pugh ME, Hemnes AR. Development of pulmonary arterial hypertension in women: interplay of sex hormones and pulmonary vascular disease. Womens Health (Lond Engl) 2010;6:285–296. doi: 10.2217/whe.09.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 15.Ervin RB. Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race and ethnicity, and body mass index: United States, 2003–2006. Natl Health Stat Report. 2009:1–7. [PubMed] [Google Scholar]
- 16.Eckel RH, Alberti KG, Grundy SM, et al. The metabolic syndrome. Lancet. 2010;375:181–183. doi: 10.1016/S0140-6736(09)61794-3. [DOI] [PubMed] [Google Scholar]
- 17.Cameron AJ, Boyko EJ, Sicree RA, et al. Central obesity as a precursor to the metabolic syndrome in the AusDiab study and Mauritius. Obesity (Silver Spring) 2008;16:2707–2716. doi: 10.1038/oby.2008.412. [DOI] [PubMed] [Google Scholar]
- 18.Koh KK, Han SH, Quon MJ. Inflammatory markers and the metabolic syndrome: insights from therapeutic interventions. J Am Coll Cardiol. 2005;46:1978–1985. doi: 10.1016/j.jacc.2005.06.082. [DOI] [PubMed] [Google Scholar]
- 19.Koh ET, Lee P, Gladman DD, et al. Pulmonary hypertension in systemic sclerosis: an analysis of 17 patients. Br J Rheumatol. 1996;35:989–993. doi: 10.1093/rheumatology/35.10.989. [DOI] [PubMed] [Google Scholar]
- 20.Zhang R, Brennan ML, Fu X, et al. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286:2136–2142. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]
- 21.Torres JL, Ridker PM. High sensitivity C-reactive protein in clinical practice. Am Heart Hosp J. 2003;1:207–211. doi: 10.1111/j.1541-9215.2003.02109.x. [DOI] [PubMed] [Google Scholar]
- 22.Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. 2010;314:1–16. doi: 10.1016/j.mce.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 23.Arita Y, Kihara S, Ouchi N, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 24.Cubbon RM, Kahn MB, Wheatcroft SB. Effects of insulin resistance on endothelial progenitor cells and vascular repair. Clin Sci (Lond) 2009;117:173–190. doi: 10.1042/CS20080263. [DOI] [PubMed] [Google Scholar]
- 25.Kim JA, Montagnani M, Koh KK, et al. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 26.Regitz-Zagrosek V, Lehmkuhl E, Weickert MO. Gender differences in the metabolic syndrome and their role for cardiovascular disease. Clin Res Cardiol. 2006;95:136–147. doi: 10.1007/s00392-006-0351-5. [DOI] [PubMed] [Google Scholar]
- 27.Saylor PJ, Smith MR. Metabolic complications of androgen deprivation therapy for prostate cancer. J Urol. 2009;181:1998–2006. doi: 10.1016/j.juro.2009.01.047. discussion 2007–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merki-Feld GS, Imthurn B, Rosselli M, et al. Serum concentrations of high-molecular weight adiponectin and their association with sex steroids in premenopausal women. Metabolism. 2010 doi: 10.1016/j.metabol.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 29.Travers SH, Jeffers BW, Eckel RH. Insulin resistance during puberty and future fat accumulation. J Clin Endocrinol Metab. 2002;87:3814–3818. doi: 10.1210/jcem.87.8.8765. [DOI] [PubMed] [Google Scholar]
- 30.Moran A, Jacobs DR, Jr, Steinberger J, et al. Insulin resistance during puberty: results from clamp studies in 357 children. Diabetes. 1999;48:2039–2044. doi: 10.2337/diabetes.48.10.2039. [DOI] [PubMed] [Google Scholar]
- 31.Hamid R, Cogan JD, Hedges LK, et al. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum Mutat. 2009;30:649–654. doi: 10.1002/humu.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Machado RD, Pauciulo MW, Thomson JR, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du L, Sullivan CC, Chu D, et al. Signaling molecules in nonfamilial pulmonary hypertension. N Engl J Med. 2003;348:500–509. doi: 10.1056/NEJMoa021650. [DOI] [PubMed] [Google Scholar]
- 34.Galetto R, Albajar M, Polanco JI, et al. Identification of a peroxisome-proliferator-activated-receptor response element in the apolipoprotein E gene control region. Biochem J. 2001;357:521–527. doi: 10.1042/0264-6021:3570521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He W, Barak Y, Hevener A, et al. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A. 2003;100:15712–15717. doi: 10.1073/pnas.2536828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hevener AL, He W, Barak Y, et al. Muscle-specific Pparg deletion causes insulin resistance. Nat Med. 2003;9:1491–1497. doi: 10.1038/nm956. [DOI] [PubMed] [Google Scholar]
- 37.Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 38.Duan SZ, Ivashchenko CY, Usher MG, et al. PPAR-gamma in the Cardiovascular System. PPAR Res. 2008;2008:745–804. doi: 10.1155/2008/745804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Itoh T, Nagaya N, Ishibashi-Ueda H, et al. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology. 2006;11:158–163. doi: 10.1111/j.1440-1843.2006.00821.x. [DOI] [PubMed] [Google Scholar]
- 40.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 41.Ameshima S, Golpon H, Cool CD, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res. 2003;92:1162–1169. doi: 10.1161/01.RES.0000073585.50092.14. [DOI] [PubMed] [Google Scholar]
- 42.Geraci MW, Moore M, Gesell T, et al. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88:555–562. doi: 10.1161/01.res.88.6.555. [DOI] [PubMed] [Google Scholar]
- 43.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 44.Hansmann G, de Jesus Perez VA, Alastalo TP, et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118:1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trayhurn P, Bing C, Wood IS. Adipose tissue and adipokines--energy regulation from the human perspective. J Nutr. 2006;136:1935S–1939S. doi: 10.1093/jn/136.7.1935S. [DOI] [PubMed] [Google Scholar]
- 46.Hansmann G, Rabinovitch M. The protective role of adiponectin in pulmonary vascular disease. Am J Physiol Lung Cell Mol Physiol. 2010;298:L1–L2. doi: 10.1152/ajplung.00367.2009. [DOI] [PubMed] [Google Scholar]
- 47.Maruna P, Lindner J, Kubzova KM. Leptin and soluble leptin receptor changes after pulmonary endarterectomy: relations to cortisol and cytokine network. Physiol Res. 2009;58:569–576. doi: 10.33549/physiolres.931523. [DOI] [PubMed] [Google Scholar]
- 48.Spranger J, Kroke A, Mohlig M, et al. Adiponectin and protection against type 2 diabetes mellitus. Lancet. 2003;361:226–228. doi: 10.1016/S0140-6736(03)12255-6. [DOI] [PubMed] [Google Scholar]
- 49.Spranger J, Kroke A, Mohlig M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812–817. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- 50.Swarbrick MM, Havel PJ. Physiological, pharmacological, and nutritional regulation of circulating adiponectin concentrations in humans. Metab Syndr Relat Disord. 2008;6:87–102. doi: 10.1089/met.2007.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Medoff BD, Okamoto Y, Leyton P, et al. Adiponectin deficiency increases allergic airway inflammation and pulmonary vascular remodeling. Am J Respir Cell Mol Biol. 2009;41:397–406. doi: 10.1165/rcmb.2008-0415OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guignabert C, Alvira CM, Alastalo TP, et al. Tie2-mediated loss of peroxisome proliferator-activated receptor-gamma in mice causes PDGF receptor-beta-dependent pulmonary arterial muscularization. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1082–L1090. doi: 10.1152/ajplung.00199.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loyd JE, Butler MG, Foroud TM, et al. Genetic anticipation and abnormal gender ratio at birth in familial primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;152:93–97. doi: 10.1164/ajrccm.152.1.7599869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lahm T, Crisostomo PR, Markel TA, et al. Selective Estrogen Receptor-Alpha And Estrogen Receptor-Beta Agonists Rapidly Decrease Pulmonary Artery Vasoconstriction By A Nitric Oxide Dependent Mechanism. Am J Physiol Regul Integr Comp Physiol. 2008 doi: 10.1152/ajpregu.90667.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lahm T, Crisostomo PR, Markel TA, et al. The effects of estrogen on pulmonary artery vasoreactivity and hypoxic pulmonary vasoconstriction: potential new clinical implications for an old hormone. Crit Care Med. 2008;36:2174–2183. doi: 10.1097/CCM.0b013e31817d1a92. [DOI] [PubMed] [Google Scholar]
- 56.Lahm T, Patel KM, Crisostomo PR, et al. Endogenous estrogen attenuates pulmonary artery vasoreactivity and acute hypoxic pulmonary vasoconstriction: the effects of sex and menstrual cycle. Am J Physiol Endocrinol Metab. 2007;293:E865–E871. doi: 10.1152/ajpendo.00201.2007. [DOI] [PubMed] [Google Scholar]
- 57.Tofovic SP, Zhang X, Jackson EK, et al. 2-Methoxyestradiol mediates the protective effects of estradiol in monocrotaline-induced pulmonary hypertension. Vascul Pharmacol. 2006;45:358–367. doi: 10.1016/j.vph.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 58.Moore LG, McMurtry IF, Reeves JT. Effects of sex hormones on cardiovascular and hematologic responses to chronic hypoxia in rats. Proc Soc Exp Biol Med. 1978;158:658–662. doi: 10.3181/00379727-158-40268. [DOI] [PubMed] [Google Scholar]
- 59.Rabinovitch M, Gamble WJ, Miettinen OS, et al. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Physiol. 1981;240:H62–H72. doi: 10.1152/ajpheart.1981.240.1.H62. [DOI] [PubMed] [Google Scholar]
- 60.McMurtry IF, Frith CH, Will DH. Cardiopulmonary responses of male and female swine to simulated high altitude. J Appl Physiol. 1973;35:459–462. doi: 10.1152/jappl.1973.35.4.459. [DOI] [PubMed] [Google Scholar]
- 61.Resta TC, Kanagy NL, Walker BR. Estradiol-induced attenuation of pulmonary hypertension is not associated with altered eNOS expression. Am J Physiol Lung Cell Mol Physiol. 2001;280:L88–L97. doi: 10.1152/ajplung.2001.280.1.L88. [DOI] [PubMed] [Google Scholar]
- 62.Tofovic SP, Zhang X, Jackson EK, et al. 2-methoxyestradiol attenuates bleomycin-induced pulmonary hypertension and fibrosis in estrogen-deficient rats. Vascul Pharmacol. 2009 doi: 10.1016/j.vph.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tofovic SP, Zhang X, Jackson EK, et al. 2-methoxyestradiol attenuates bleomycin-induced pulmonary hypertension and fibrosis in estrogen-deficient rats. Vascul Pharmacol. 2009;51:190–197. doi: 10.1016/j.vph.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu A, Chan KW, Hoo RL, et al. Testosterone selectively reduces the high molecular weight form of adiponectin by inhibiting its secretion from adipocytes. J Biol Chem. 2005;280:18073–18080. doi: 10.1074/jbc.M414231200. [DOI] [PubMed] [Google Scholar]
- 65.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Warburg OPK, Negelein E. Ueber den Stoffwechsel der Tumor. Biochemische Zeitschrift. 1924;152:319–344. [Google Scholar]
- 67.Robbins IM, Newman JH, Johnson RF, et al. Association of the metabolic syndrome with pulmonary venous hypertension. Chest. 2009;136:31–36. doi: 10.1378/chest.08-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pugh M. Unrecognized glucose intolerance is common in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;181:A4813. doi: 10.1016/j.healun.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–3421. [PubMed] [Google Scholar]
- 70.Gonzalez-Diez M, Rodriguez C, Badimon L, et al. Prostacyclin induction by high-density lipoprotein (HDL) in vascular smooth muscle cells depends on sphingosine 1-phosphate receptors: effect of simvastatin. Thromb Haemost. 2008;100:119–126. doi: 10.1160/TH07-11-0675. [DOI] [PubMed] [Google Scholar]
- 71.Toikka JO, Ahotupa M, Viikari JS, et al. Constantly low HDL-cholesterol concentration relates to endothelial dysfunction and increased in vivo LDL-oxidation in healthy young men. Atherosclerosis. 1999;147:133–138. doi: 10.1016/s0021-9150(99)00186-0. [DOI] [PubMed] [Google Scholar]
- 72.O'Connell BJ, Genest J., Jr High-density lipoproteins and endothelial function. Circulation. 2001;104:1978–1983. doi: 10.1161/hc3901.096667. [DOI] [PubMed] [Google Scholar]
- 73.Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216–223. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- 74.Thenappan T, Shah SJ, Rich S, et al. A USA-based registry for pulmonary arterial hypertension: 1982–2006. Eur Respir J. 2007;30:1103–1110. doi: 10.1183/09031936.00042107. [DOI] [PubMed] [Google Scholar]
- 75.Kawut SM, Krowka MJ, Trotter JF, et al. Clinical risk factors for portopulmonary hypertension. Hepatology. 2008;48:196–203. doi: 10.1002/hep.22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Roberts KE, Fallon MB, Krowka MJ, et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med. 2009;179:835–842. doi: 10.1164/rccm.200809-1472OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chung L, Liu J, Parsons L, et al. Characterization of Connective Tissue Disease Associated Pulmonary Arterial Hypertension from the Reveal Registry: Identifying Systemic Sclerosis as a Unique Phenotype. Chest. 2010 doi: 10.1378/chest.10-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ventetuolo CEOP, Bluemke DA, Tandri H, Barr RG, Bagiella E, Bristow M, Johnson C, Kizer J, Lima JA, Kawut SM. Sex Hormones and the Right Ventricle: The MESA-Right Ventricle Study. Abstract Presentation, Proceedings of the American Thoracic Society. 2009 [Google Scholar]
- 79.Rajkumar R, Konishi K, Richards TJ, et al. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2010;298:H1235–H1248. doi: 10.1152/ajpheart.00254.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]