

Abstract
Normal, age-related depletion of the androgen testosterone is a risk factor for Alzheimer’s disease (AD) in men. Previously, we reported that experimental androgen depletion significantly accelerates development of AD-like neuropathology in the 3xTg-AD triple-transgenic mouse model of AD, an effect prevented by androgen treatment. Because testosterone is metabolized in brain into both the androgen dihydrotestosterone (DHT) and the estrogen 17β-estradiol (E2), testosterone can mediate its effects through androgen and or estrogen pathways. To define the role of androgen and estrogen pathways in regulation of AD-like neuropathology, we compared the effects of testosterone (T) and its metabolites DHT and E2 in male 3xTg-AD mice depleted of endogenous sex steroid hormones by gonadectomy (GDX). Male 3xTg-AD mice were sham GDX or GDX, immediately treated with vehicle, T, DHT, or E2, and 4 months later evaluated for two indices of AD-like neuropathology, β-amyloid (Aβ) accumulation and tau hyperphosphorylation. In comparison to sham GDX mice, we observed a significant increase in Aβ accumulation in GDX mice in subiculum, hippocampus, and amygdala. Treatment of GDX mice with T prevented the increased Aβ accumulation in all three brain regions. DHT treatment yielded similar results, significantly reducing Aβ accumulation across brain regions. Interestingly, E2 prevented Aβ accumulation in hippocampus but exerted only partial effects in subiculum and amygdala. Levels of tau hyperphosphorylation in sham GDX male 3xTg-AD mice were modest and only slightly increased by GDX. Treatment of GDX mice with T or E2 but not DHT reduced tau hyperphosphorylation to levels lower than observed in sham animals. These data suggest that testosterone regulates Aβ pathology through androgen and estrogen pathways and reduces tau pathology largely through estrogen pathways. These findings further define hormone pathways involved in regulation of AD-related pathology, information that is important for understanding disease etiology and developing pathway-specific hormone interventions.
Keywords: Alzheimer’s disease, β-amyloid, sex steroid hormones, tau hyperphosphorylation, transgenic mouse
1. INTRODUCTION
Advancing age is the most significant risk factor for the development of Alzheimer’s disease (AD) (Evans et al., 1989; Jorm et al., 1987; Rocca et al., 1986). One age-related change in men that has been identified as a risk factor for the development of AD is significant depletion in the primary male androgen, testosterone (T) (Pike et al., 2009; Rosario and Pike, 2008). The association between low testosterone and increased risk for AD likely reflects the loss of beneficial androgen-mediated actions in the brain. There are several neural androgen actions potentially relevant to AD, including promotion of neuron viability (Pike, 2001; Ramsden et al., 2003b), synaptic plasticity (Leranth et al., 2003), select aspects of cognition (Cherrier et al., 2005; Janowsky, 2006b; Moffat, 2005), and reduction of tau phosphorylation (Papasozomenos, 1997). Of particular importance is androgen regulation of Aβ, an hypothesized casual factor in the development of AD (Hardy, 1997). Elevated levels of Aβ have been observed in men with low levels of testosterone (Almeida and Papadopoulos, 2003; Gandy et al., 2001; Gillett et al., 2003; Rosario et al., 2009), suggesting that testosterone may function to decrease Aβ levels. Consistent with this possibility, cell culture studies have shown that T treatment reduces Aβ levels (Gouras et al., 2000; Yao et al., 2008). Importantly, findings in animal models also demonstrate that androgens are negative regulators of Aβ. Depletion of endogenous androgen levels by gonadectomy (GDX) resulted in a significant increase in soluble Aβ in wild-type male rats (Ramsden et al., 2003a) and elevated Aβ accumulation in male 3xTg-AD mice (Rosario et al., 2006), effects that were prevented by androgen treatment. Thus, regulation of Aβ is a neural androgen function that is compromised as a consequence of age-related T depletion and may contribute to AD pathogenesis.
An important issue that remains to be resolved is the role of androgen versus estrogen pathways in androgen regulation of AD pathology. Testosterone not only directly interacts with the androgen receptor (AR), but also acts as a prohormone, undergoing metabolism in brain to a variety of hormones that act by several mechanisms including activation of AR and estrogen receptors (ERs) (Attardi et al., 1976; Souttou et al., 1993). In particular, T is converted by aromatase to the estrogen 17β-estradiol (E2) and by 5α-reductase to the potent androgen dihydrotestosterone (DHT), hormones that specifically activate ERs and AR, respectively. Available cell culture evidence suggests that both estrogen and androgen pathways may be involved in androgen regulation of Aβ. First, testosterone has been shown to reduce Aβ levels in cultured neural cells by altering amyloid precursor protein processing (Gouras et al., 2000), however this action was inhibited by an aromatase inhibitor that prevents conversion of T to E2 (Goodenough et al., 2000). Second, we recently demonstrated that androgens reduce Aβ levels in culture by an AR-dependent mechanism involving increased expression of the Aβ-degrading enzyme neprilysin (Yao et al., 2008). To evaluate the potential involvement of androgen and estrogen pathways in regulation of AD-like neuropathology, we compared the effects of T, DHT, and E2 on levels of Aβ accumulation and tau hyperphosphorylation in male 3xTg-AD mice. Hormone levels were manipulated between ages 3–7 months, a time frame which corresponds to the early phases of pathology in male 3xTg-AD mice (Oddo et al., 2003a; Rosario et al., 2006), and thus allows for evaluation of androgens and estrogens on the development of AD-like pathology.
2. RESULTS
2.1 Efficacy of hormone manipulations
To evaluate the contribution of androgen and estrogen pathways in the regulation of AD-like pathology, young adult male 3xTg-AD mice were GDX to deplete endogenous sex steroid hormones, and then treated with T, DHT or E2 at previously established doses known to increase androgen or estrogen activity (Carroll et al., 2007; Rosario et al., 2006). In order to confirm the efficacy and specificity of the hormone manipulations, we measured seminal vesicle weight, a sensitive bioassay of androgen levels (Yamane et al., 1986). We observed a significant decrease in seminal vesicle weight in GDX mice relative to sham GDX mice (sham GDX, 63.5 ± 9.2 mg; GDX, 13.3 ± 1.4 mg, F (2,27) = 6.8, p <0.05). In comparison, the GDX +DHT group showed seminal vesicle weights (128.2 ± 20.1 mg) significantly higher than both the GDX and sham GDX groups (p <0.05), indicating that DHT treatment resulted in high physiological or supraphysiological DHT levels. Because the T pellets were depleted before their designed 60 day period (data not shown), we assessed T pellet efficacy in GDX mice 30 days after pellet implantation and found seminal vesicle weights significantly higher than GDX mice but not sham GDX mice, suggesting physiological replacement of T (GDX+T, 49.5 ± 7.6 mg F (3,15) = 9.1, p <0.05). E2 had no significant effect on seminal vesicle weight as compared to GDX or Sham GDX mice (GDX+E2, 24.2 ± 3.4 mg, p <0.05).
2.2 Hormone regulation of Aβ deposition
To determine whether androgen regulation of Aβ pathology is mediated by androgen and or estrogen pathways, we examined the effects of the hormone manipulations on Aβ deposition in three brain regions known to be affected by Aβ pathology in 3xTg-AD mice and human AD, the subiculum, hippocampus CA1, and amygdala. Statistical analysis with repeated measures ANOVA indicated a significant effect of condition on Aβ load across brain regions [F = 2.95 (8,62), p < 0.01]. Specifically, we observed a significant increase in the intensity and area of Aβ immunoreactivity in androgen depleted GDX mice in comparison to sham GDX animals in the subiculum (Fig. 1), CA1 region of hippocampus (Fig. 2), and amygdala (Fig. 3). Treatment of GDX mice with DHT prevented the increased Aβ accumulation in all three brain regions that is observed in GDX mice, yielding Aβ load values at or below the levels observed in the sham GDX group (Figs. 1–3). Testosterone also significantly prevented Aβ accumulation in comparison to GDX mice with the same regional efficacy and a similar magnitude as observed in the GDX+DHT group. The GDX+E2 group showed mixed effects when compared with GDX mice. In hippocampus CA1, E2 treatment significantly attenuated Aβ accumulation with an efficacy similar to that of T and DHT (Fig. 2). However, in both subiculum and amygdala, E2 treatment resulted in Aβ load values that were not significantly different from either the sham GDX or GDX groups (Figs. 1, 3), suggesting partial efficacy.
Fig. 1.
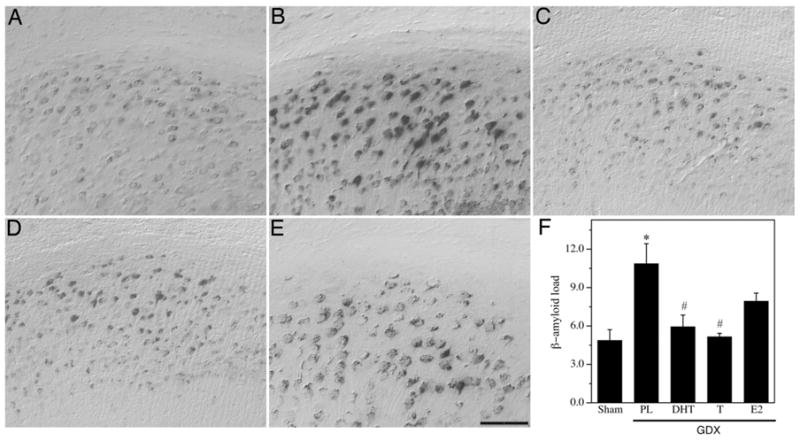
Hormone regulation of Aβ in subiculum of male 3xTg-AD mice. Representative high magnification photomicrographs show Aβ immunoreactivity in male 3xTg-AD mice in the following treatment groups: Sham GDX (A), GDX+PL (B), GDX+DHT (C), GDX+T (D), and GDX +E2 (E). Scale bar = 100μm. (F) Levels of Aβ immunoreactive load in subiculum were quantified across groups. Data show mean Aβ load values (± SEM). * Denotes p < 0.05 in comparison to Sham GDX (Sham) group, # denotes p < 0.05 versus GDX+PL group.
Fig. 2.

Hormone regulation of Aβ in hippocampus CA1 of male 3xTg-AD mice. Representative high magnification photomicrographs show Aβ immunoreactivity in male 3xTg-AD mice in the following treatment groups: Sham GDX (A), GDX+PL (B), GDX+DHT (C), GDX+T (D), and GDX +E2 (E). Scale bar = 100μm. (F) Levels of Aβ immunoreactive load in CA1 of hippocampus were quantified across groups. Data show mean Aβ load values (± SEM). * Denotes p < 0.05 in comparison to Sham GDX (Sham) group, # denotes p < 0.05 versus GDX+PL group.
Fig. 3.

Hormone regulation of Aβ in amygdala of male 3xTg-AD mice. Representative high magnification photomicrographs show Aβ immunoreactivity in male 3xTg-AD mice in the following treatment groups: Sham GDX (A), GDX+PL (B), GDX+DHT (C), GDX+T (D), and GDX +E2 (E). Scale bar = 100μm. (F) Levels of Aβ immunoreactive load in amygdala were quantified across groups. Data show mean Aβ load values (± SEM). * Denotes p < 0.05 in comparison to Sham GDX (Sham) group, # denotes p < 0.05 versus GDX+PL group.
In addition to Aβ load, we also counted numbers of extracellular plaques as a measure of Aβ deposition. Although we observed very few plaques in male 3xTg-AD mice at age 7 mo, there was a significant hormone-related effect on extracellular plaque number [F (5,45) = 3.32, p <0.05]. Androgen depletion significantly increased the number of extracellular plaques (sham GDX, 0.09 ± 0.091; GDX, 1.0 ± 0.35), an effect prevented by both T and DHT treatment (GDX +T, 0.14 ± 0.14; GDX+DHT, 0.0 ± 0.0). E2 had no effect on the number of extracellular plaques in GDX mice (GDX+ E2, 1.0 ± 0.45).
2.3 Hormone regulation of tau hyperphosphorylation
To evaluate tau hyperphosphorylation across the previously described treatment groups in the male 3xTg-AD mice, we counted numbers of cells strongly immunoreactive for the antibody AT8, which recognizes phosphorylation of tau at Ser202 and Thr205. Consistent with previous reports (Oddo et al., 2003a; Oddo et al., 2003b), we observed a significant age-related increase in AT8-immunreactive neurons, which were predominantly localized in subiculum and to lesser extent hippocampus CA1 (Fig. 4A–E). At age 7 mo, we observed modest numbers of AT8-labeled cells which showed a statistically non-significant increase in the GDX group. Interestingly, the GDX+T and GDX+E2 groups exhibited significantly fewer numbers of AT8-immunoreactive neurons than both the sham GDX and GDX groups (Fig. 4F). DHT had no significant effect on the number of AT8-immunoreactive neurons.
Fig. 4.

Age and regulation of tau hyperphosphorylation in male 3xTg-AD mice. (A–C) Representative photomicrographs show tau immunoreactivity with the phospho-specific antibody AT8 in hippocampus CA1 of Sham GDX male 3xTg-AD mice at (A) age 3 mo, (B) age 7 mo, and (C) age 13 mo old. Scale bar = 100 μm. (D) High magnification photomicrograph of a cell strongly immunoreactive for AT8 antibody. Scale bar = 25μm. (E) Levels of tau hyperphosphorylation were quantified by counts of cells strongly immunoreactive for AT8. Data show mean numbers (± SEM) of AT8-immunoreactive cells in Sham GDX males at ages 3 mo, 7 mo, and 13 mo. * Denotes p < 0.05 in comparison to 3 mo group. (F) Data show mean numbers (± SEM) of AT8-immunoreactive cells in Sham GDX (Sham), GDX+PL, GDX+DHT, GDX+T, and GDX+E2 mice at age 7 mo. * Denotes p < 0.05 in comparison to Sham group, and # denotes p < 0.05 in comparison to GDX+PL group.
3. DISCUSSION
In this study, we examined whether the recently established androgen regulation of AD neuropathology is mediated through androgen and or estrogen pathways. Our findings indicate sex steroid regulation of Aβ, evidenced by a significant increase in intraneuronal and extracellular Aβ pathology following GDX-induced androgen depletion. Further, the data suggest that hormonal regulation of Aβ is mediated largely via activation of androgen pathways although E2 also exhibited Aβ-lowering actions. In contrast, regulation of tau hyperphosphorylation appeared to be regulated predominantly by estrogen pathways as numbers of pre-tangle neurons were reduced by T and E2 but not DHT. These data indicate that T utilizes both androgen and estrogen pathways to reduce indices of AD pathology in male 3xTg-AD mice.
3.1 Roles of androgen and estrogen pathways in androgen regulation of Aβ
Consistent with our previous work (Rosario et al., 2006), we found that androgen depletion accelerates the development of Aβ pathology in the 3xTg-AD mouse model of AD, findings which implicate androgens as endogenous regulators of Aβ. Our observation that T prevented the GDX-induced increase in Aβ pathology confirms androgen regulation of Aβ but does not define the relative involvement of androgen and estrogen pathways. This is because T functions in brain as a prohormone, converted by aromatase into the estrogen 17β-estradiol and by 5∞-reductase into the potent androgen DHT. Like T, DHT is also a substrate in brain for enzymes whose actions generate active hormones. Of particular interest, DHT can be converted to 5α-androstan-3β,17β-diol, which is an agonist for estrogen receptor β (ERβ) (Weihua et al., 2001; Weihua et al., 2002) and mediates some neural effects of DHT (Handa et al., 2008; Handa et al., 2009; Lund et al., 2006; Pak et al., 2005). Thus, both the T metabolite 17β-estradiol and the DHT metabolite 5α-androstan-3β,17β-diol are ER agonists that activate estrogen pathways and may contribute to observed Aβ-lowering effects of T and DHT.
Supporting a role of androgen pathways in Aβ regulation, we observed that T and DHT treatments similarly impacted Aβ accumulation in a manner that significantly differed from E2 treatment. Further comparisons on the relative efficacies of T and DHT are not possible from our study since these androgens differ in their affinities for AR and the absolute levels of the hormones were not assessed. It is also important to note that the utilized method of hormone delivery has limitations. Subcutaneous pellets are advantageous in that they allow continuous hormone treatment for extended periods without repeated invasive procedures, however they can also yield uneven hormone delivery with very high initial levels and low final levels (Strom et al., 2008, 2009).
Whereas both T and DHT strongly inhibited the elevated Aβ accumulation induced by GDX across all three examined brain regions, E2 treatment was associated with more modest Aβ reduction that was statistically significant only in hippocampus CA1. Thus, we conclude that androgens can lower Aβ by mechanism(s) independent of estrogen pathways. Although the experimental design does not exclude the possibility that DHT may indirectly activate estrogen pathways via metabolism to 5α-androstan-3β,17β-diol which acts on ERβ, we have previously reported that Aβ levels in hippocampus CA1 and subiculum in female 3xTg-AD mice were reduced by an ER∞ agonist but not an ERβ agonist indicating ER∞ is more important in Aβ regulation (Carroll and Pike, 2008). Further, we previously found that GDX-induced elevation of soluble Aβ in male Sprague-Dawley rats was prevented by DHT but not E2 treatment (Ramsden et al., 2003a). Also consistent with an androgen pathway of Aβ regulation is recent work in guinea pigs that demonstrates an inverse relationship between levels of soluble Aβ in cerebrospinal fluid and serum levels of T (Wahjoepramono et al., 2008). Similarly, brain levels of Aβ in aged men are inversely associated with brain levels of T but not E2 (Rosario et al., 2009).
While the data suggest that androgens utilize an estrogen-independent mechanism to reduce Aβ levels, they also indicate involvement of estrogen pathway(s). Specifically, in GDX male 3xTg-AD mice, we found that E2 treatment resulted in significantly lower Aβ levels in hippocampus CA1 and intermediate Aβ levels in subiculum and amygdala that were between sham GDX and GDX groups. Since E2 is not an effective agonist for AR, these findings implicate an AR-independent estrogen pathway in regulation of Aβ levels in male brain. Interestingly, we observe that the same dose of E2 delivered to ovariectomized female 3xTg-AD mice significantly reduces Aβ accumulation not only in hippocampus CA1 but also in subiculum, amygdala, and frontal cortex (Carroll et al., 2007; Carroll and Pike, 2008). Unclear is why E2 was only partially effective in reducing Aβ in males. Although activation of estrogen pathways has several important functions in the male brain including regulation of aspects of development and sexual differentiation (Cooke et al., 1998; Forger, 2006), some established E2 actions in female brain are not observed in male brain. For example, E2 is an established regulator of spine density in female hippocampus (Woolley and McEwen, 1992; Woolley and McEwen, 1994) whereas DHT but not E2 increases spine density in male hippocampus (Leranth et al., 2003; Leranth et al., 2004). Thus, our data suggest that estrogen pathways contribute to androgen regulation of Aβ but that such pathways may be less effective in males than in females.
Although these data define contributions of both androgen and estrogen pathways in regulation of Aβ, they do not elucidate the underlying signaling mechanisms. Sex steroid hormones function largely via binding to and activating their specific steroid receptors, AR for T and DHT and ERα and ERβ for E2. As with other steroid hormone receptors, activation of AR and ER can initiate both classic genomic and rapid cell signaling pathways (Falkenstein et al., 2000). AR and ERs are localized in many brain regions, including hippocampus and amygdala (Kerr et al., 1995; Simerly et al., 1990), which were examined in this study. Although both ERα and ERβ are present in the hippocampus and amygdala, ERβ has been shown to be expressed at greater levels than ERα in both the hippocampus and amygdala (Mehra et al., 2005; Shughrue et al., 1997). Previous reports indicate that both AR and ER are involved in regulation of Aβ. In the case of AR, we observed that T and DHT reduced soluble Aβ in a cell culture paradigm by a AR-dependent genomic mechanism involving increased expression of the Aβ-catabolizing enzyme neprilysin (Yao et al., 2008). Similarly, in vitro and in vivo evidence has identified activation of ERβ as important to E2 regulation of Aβ (Carroll and Pike, 2008) by a mechanism involving increased expression of insulin degrading enzyme (Zhao et al., 2010), an Aβ-catabolizing protein. Besides these established AR- and ER-dependent pathways, the observed effects of T, DHT and E2 may involve other non-receptor dependent pathways. For example, sex steroid hormone regulation of Aβ could involve lowering levels of luteinizing hormone (LH) via regulation of the hypothalamic-pituitary-gonadal axis. Elevated LH is associated with AD (Bowen et al., 2000) and may have a role in modulating Aβ levels (Meethal et al., 2005). In addition, some neural actions of T and DHT are mediated by an AR-independent pathway involving DHT metabolism to 5α-androstan-3β,17β-diol which acts on GABAA receptors (Edinger and Frye, 2004). One limitation of this study is that it does not specifically define the relative roles of AR and ER as mediators of observed androgen and estrogen pathways.
3.2 Roles of androgen and estrogen pathways in androgen regulation of tau hyperphosphorylation
In contrast to several studies describing a relationship between androgens and inhibition of Aβ accumulation (Pike et al., 2009), few studies have investigated the possibility that androgens may reduce AD-like tau pathology. Notably, work by Papasozomenos and colleagues found that androgens can reduce tau hyperphosphorylation induced by acute heat shock in male rats (Papasozomenos, 1997; Papasozomenos and Shanavas, 2002). In this study, we observed a statistically nonsignificant trend of elevated tau phosphorylation in GDX males that was not apparent in the DHT group. Interestingly, in middle-aged male 3xTg-AD mice that exhibit more advanced tau pathology, we do observe a significant relationship between androgen depletion and increased tau pathology that is prevented by DHT (unpublished observations). Perhaps unexpectedly, we found that the modest numbers of AT8-immunoreactive neurons observed in the relatively young sham GDX mice from this study were significantly lower in groups receiving T but not in those receiving DHT. Further, E2 treatment was also associated with a significant decrease in tau hyperphosphorylation. Together, these data suggest a potential role for estrogen pathways in regulation of tau phosphorylation since both E2 and T (via aromatization to E2) treatments are expected to increase brain levels of estrogens. A role of estrogen pathways in reducing tau phosphorylation is consistent with our observations in female 3xTg-AD mice (Carroll et al., 2007). Further, E2 has been found to decrease tau phosphorylation in cell culture (Liu et al., 2008; Zhang et al., 2008), an effect inhibited by ER antagonism (Alvarez-de-la-Rosa et al., 2005). Interestingly, in the heat shock model used by Papasozomenos and colleagues (1997), testosterone inhibition of tau hyperphosphorylation was estrogen independent.
An important consideration in the interpretation of tau hyperphosphorylation data in the 3xTg-AD mouse is the relationship between tau and Aβ pathologies. Previous work in this AD mouse model has found that reduction of Aβ by Aβ immunotherapy also results in decreased levels of tau phosphorylation, suggesting Aβ accumulation contributes to tau pathology in the 3xTg-AD model (Oddo et al., 2003a; Oddo et al., 2006). However, tau hyperphosphorylation in 3xTg-AD mice can be reduced in the absence of changes in Aβ levels by directly targeting phosphorylation pathways (Caccamo et al., 2007; Kitazawa et al., 2005). Our observations are more consistent with the latter finding in which Aβ and tau are not inextricably linked. First, although DHT reduced Aβ levels this androgen treatment did not significantly affect tau pathology. Second, E2 only modestly reduced Aβ yet strongly lowered numbers of neurons with hyperphosphorylated tau. Thus, our data are consistent with the hypothesis that estrogen pathways can reduce tau pathology by affecting phosphorylation pathways, as suggested by in vitro studies (Alvarez-de-la-Rosa et al., 2005; Liu et al., 2008; Zhang et al., 2008).
3.3 Clinical implications: androgen therapy and AD
Age-related T depletion in men is a recently identified risk factor for the development of AD (Hogervorst and Bandelow, 2004; Moffat, 2005; Pike et al., 2009; Rosario et al., 2004). Testosterone-based androgen therapy is increasingly utilized in aging men to treat clinical manifestations of hypogonadism, including sexual function, muscle mass and strength, bone mineral density, cognition, and overall state of well being or quality of life (Bhasin et al., 2006; Bhasin et al., 2007; Kaufman and Vermeulen, 2005). Androgens also promote cognitive function in both men and male rodents (Janowsky, 2006a; Janowsky, 2006b), actions that apparently involve both androgen and estrogen pathways as evidenced by similarities and differences in the cognitive and behavioral effects of T and DHT (Benice and Raber, 2009; Bimonte-Nelson et al., 2003; Cherrier et al., 2003; Edinger and Frye, 2004; Frye et al., 2004). Given the present findings as well as the recent literature suggesting neuroprotective effects of androgens against AD-related pathology (Pike et al., 2009; Raber, 2008; Rosario and Pike, 2008), further clinical evaluation of androgen therapy to reduce the risk of AD and improve neural health in aging men with low T appears warranted. Because our data demonstrate that androgens utilize both androgen and estrogen pathways to reduce Aβ and tau pathologies, the most effective androgen therapies for combating AD-related pathologies may be T and selective androgen receptor modulators that are capable of acting through both androgen and estrogen pathways.
4. EXPERIMENTAL PROCEDURES
4.1 Animals and hormone treatments
We utilized adult male 3xTg-AD mice, a triple transgenic model that expresses disease-related mutations in amyloid precursor protein, presenilin 1, and tau (Oddo et al., 2003b), to examine the effect of T, DHT, and E2 on regulation of AD pathology. All the mice used in this study were bred in our laboratory, had ad libitum access to food and water, and were housed individually under a 12h light/12h dark schedule. Mice were divided into 5 treatment groups (N = 6–7 per group): sham GDX (treated with placebo), GDX (treated with placebo), GDX+T, GDX +DHT, and GDX+E2 and were maintained under these hormone conditions for 4 months, from 3 months of age until 7 months of age. Under pentobarbital (50 mg/kg) anesthesia, 3 mo-old male 3xTg-AD mice were either sham GDX or GDX and immediately implanted with slow-release, subcutaneous delivery pellets (Innovative Research of America, Sarasota, FL) containing placebo (hormone-free, 90-day pellet), DHT (10 mg, 90-day pellet), T (10 mg 60-day pellet) or E2 (0.025mg 60-day pellet). To maintain treatments over a 4-month period, animals were delivered a second pellet 2 mo (T, E2) or 3 mo (Pl, DHT) after the initial pellet. Both 60- and 90-day placebo pellets were utilized in Sham GDX and GDX groups; no apparent effects on pathology were noted between 60- and 90-day placebo pellets (data not shown).
Two additional groups of sham GDX animals at ages 3 mo and 13 mo were also included for tau analyses to identify any age-related changes in tau. At the end of the treatment period, mice were anesthetized with pentobarbital (100 mg/kg), blood was collected for hormone analyses, and animals were perfused with PBS. Seminal vesicles were dissected, blotted, and weighed to determine effectiveness of androgen treatments (Yamane et al., 1986). The brain was dissected and immersion fixed in 4% paraformaldehyde for immunohistochemical analyses. Animal studies were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and under an institutionally approved animal protocol.
4.2 Immunohistochemistry
Paraformaldehyde-fixed hemi-brains were sectioned in the horizontal plane (40 μm) using a vibratome. Tissue sections were immunostained in a few large batches in which sections from the different treatment groups were evenly distributed to control for inter-batch variability. In each experiment, sections were immunolabeled following a previously described protocol (Pike, 1999). Briefly, sections were quenched of endogenous peroxidase by H2O2 rinse then blocked for non-specific binding by incubation in TBS-Triton-X buffer containing 2% serum. For Aβ immunolabeling, sections were also pretreated with an antigen unmasking step (5 min rinse in 99% formic acid) to increase Aβ staining (Cummings, 2002). Sections were incubated overnight with buffer containing a primary antibody specific for either hyperphosphorylated tau (AT8 monoclonal antibody, 1:1000; Pierce, Rockford, IL) or Aβ (rabbit anti-human Aβ1-43, 1:300 dilution; Zymed, San Fransisco, CA). Immunoreactivity was visualized using the Vector Elite ABC kit followed by diaminobenzidine reaction according to manufacturer’s instructions (Vector, Burlingame, CA). Immunolabeled sections were dehydrated by sequential rinses in solutions with increasing alcohol content and cover-slipped in permanent mounting medium.
4.3 Quantification of immunohistochemistry
Immunohistochemistry was quantified by two methods. First, Aβ levels in immunostained sections were determined using an immunoreactive load technique previously described (Carroll et al., 2007; Rosario et al., 2006). In brief, high magnification fields (420 μm × 330 μm) from Aβ-immunolabeled sections were collected and stored on computer by a video capture system (B/W CCD camera coupled to an Olympus BX40 upright microscope). For each brain, images were collected for each brain region (subiculum, hippocampus CA1, amygdala) from 5 separate sections. In a regular pattern, 2 non-overlapping fields for subiculum and amygdala and 3 non-overlapping fields for CA1 were captured per section. Using NIH Image 1.61 software, gray scale images were processed with a constant threshold value to create a binary separation between positive and negative immunoreactivity such that each pixel was either black or white. This conversion permits calculation of the percentage of section area occupied by immunoreactive label (i.e., load); it is not a measure of immunoreactive intensity.
Second, levels of hyperphosphorylated tau pathology and numbers of extracellular, plaque-like Aβ deposits were determined by a counting method based on our previously established exclusion and inclusion criteria (Carroll et al., 2007; Rosario et al., 2006). Tau pathology was quantified by counting the combined number of AT8-immunoreactive neurons, as defined by immunoreactive staining throughout the cell body, in hippocampus and subiculum from 11–12 sections per brain (each separated by 280 μm). The number of extracellular plaque-like Aβ deposits was counted in a similar manner in hippocampus and subiculum from 11–12 sections per brain from Aβ-immunolabeled sections in which the size of the immunolabeled plaque was at least twice the size of a neuron cell body. Although plaque number is related to Aβ IHC load, and both measures are affected by threshold parameters, they represent different stages of pathology with the plaque stage representing later, more advanced pathology in 3xTg-AD mice (Oddo et al., 2003; Rosario et al., 2006).
Raw data from Aβ load data was analyzed by repeated measures ANOVA with the Wilks’ lambda test. Raw data from Aβ load values, AT8 counts, and Aβ plaque counts were statistically analyzed by a one-way ANOVA, followed by between group comparisons using Fisher LSD. Effects with p < 0.05 were considered statistically significant.
Research Highlights.
Testosterone reduces Alzheimer-related pathology in a transgenic mouse model
An androgen metabolite of testosterone reduces beta-amyloid but not tau pathology
An estrogen metabolite of testosterone is effective in reducing tau pathology
Testosterone uses both androgen and estrogen pathways to protect against Alzheimer
Acknowledgments
This study was supported by the National Institute of Aging (AG23739 to CJP).
Abbreviations
- Aβ
β-amyloid
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- AR
androgen receptor
- DHT
dihydrotestosterone
- E2
17β-estradiol
- ER
estrogen receptor
- GDX
gonadectomy
- T
testosterone
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Almeida TA, Papadopoulos N. Progression model of prostate cancer. Methods Mol Biol. 2003;222:211–22. doi: 10.1385/1-59259-328-3:211. [DOI] [PubMed] [Google Scholar]
- Alvarez-de-la-Rosa M, Silva I, Nilsen J, Perez MM, Garcia-Segura LM, Avila J, Naftolin F. Estradiol prevents neural tau hyperphosphorylation characteristic of Alzheimer’s disease. Ann N Y Acad Sci. 2005;1052:210–24. doi: 10.1196/annals.1347.016. [DOI] [PubMed] [Google Scholar]
- Attardi B, Geller LN, Ohno S. Androgen and estrogen receptors in brain cytosol from male, female, and testicular feminized (tfm/y hermaphrodite) mice. Endocrinology. 1976;98:864–74. doi: 10.1210/endo-98-4-864. [DOI] [PubMed] [Google Scholar]
- Benice TS, Raber J. Testosterone and dihydrotestosterone differentially improve cognition in aged female mice. Learn Mem. 2009;16:479–85. doi: 10.1101/lm.1428209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, Montori VM. Testosterone therapy in adult men with androgen deficiency syndromes: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2006;91:1995–2010. doi: 10.1210/jc.2005-2847. [DOI] [PubMed] [Google Scholar]
- Bhasin S, Parker RA, Sattler F, Haubrich R, Alston B, Umbleja T, Shikuma CM Team ACTGPAS. Effects of testosterone supplementation on whole body and regional fat mass and distribution in human immunodeficiency virus-infected men with abdominal obesity. J Clin Endocrinol Metab. 2007;92:1049–1057. doi: 10.1210/jc.2006-2060. [DOI] [PubMed] [Google Scholar]
- Bimonte-Nelson HA, et al. Testosterone, but not nonaromatizable dihydrotestosterone, improves working memory and alters nerve growth factor levels in aged male rats. Exp Neurol. 2003;181:301–12. doi: 10.1016/s0014-4886(03)00061-x. [DOI] [PubMed] [Google Scholar]
- Bowen RL, Isley JP, Atkinson RL. An association of elevated serum gonadotropin concentrations and Alzheimer disease? J Neuroendocrinol. 2000;12:351–4. doi: 10.1046/j.1365-2826.2000.00461.x. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Tran LX, LaFerla FM. Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am J Pathol. 2007;170:1669–75. doi: 10.2353/ajpath.2007.061178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JC, Rosario ER, Chang L, Stanczyk FZ, Oddo S, LaFerla FM, Pike CJ. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci. 2007;27:13357–65. doi: 10.1523/JNEUROSCI.2718-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JC, Pike CJ. Selective estrogen receptor modulators differentially regulate Alzheimer-like changes in female 3xTg-AD mice. Endocrinology. 2008 doi: 10.1210/en.2007-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherrier MM, Craft S, Matsumoto AM. Cognitive changes associated with supplementation of testosterone or dihydrotestosterone in mildly hypogonadal men: a preliminary report. J Androl. 2003;24:568–76. doi: 10.1002/j.1939-4640.2003.tb02708.x. [DOI] [PubMed] [Google Scholar]
- Cherrier MM, Matsumoto AM, Amory JK, Asthana S, Bremner W, Peskind ER, Raskind MA, Craft S. Testosterone improves spatial memory in men with Alzheimer disease and mild cognitive impairment. Neurology. 2005;64:2063–8. doi: 10.1212/01.WNL.0000165995.98986.F1. [DOI] [PubMed] [Google Scholar]
- Cooke B, Hegstrom CD, Villeneuve LS, Breedlove SM. Sexual differentiation of the vertebrate brain: principles and mechanisms. Front Neuroendocrinol. 1998;19:323–362. doi: 10.1006/frne.1998.0171. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Manson A, Kim R, Sheu P, Anderson A. Optimization of techniques for the maximal detection and quantification of Alzheimer’s-related neuropathology with digital imaging. Neurobiol Aging. 2002;23:161–170. doi: 10.1016/s0197-4580(01)00316-5. [DOI] [PubMed] [Google Scholar]
- Edinger KL, Frye CA. Testosterone’s analgesic, anxiolytic, and cognitive-enhancing effects may be due in part to actions of its 5alpha-reduced metabolites in the hippocampus. Behav Neurosci. 2004;118:1352–64. doi: 10.1037/0735-7044.118.6.1352. [DOI] [PubMed] [Google Scholar]
- Evans DA, Funkenstein HH, Albert MS, Scherr PA, Cook NR, Chown MJ, Hebert LE, Hennekens CH, Taylor JO. Prevalence of Alzheimer’s disease in a community population of older persons. Higher than previously reported. Jama. 1989;262:2551–6. [PubMed] [Google Scholar]
- Falkenstein E, Tillmann HC, Christ M, Feuring M, Wehling M. Multiple actions of steroid hormones--a focus on rapid, nongenomic effects. Pharmacol Rev. 2000;52:513–56. [PubMed] [Google Scholar]
- Forger NG. Cell death and sexual differentiation of the nervous system. Neuroscience. 2006;138:929–38. doi: 10.1016/j.neuroscience.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Frye CA, et al. 5alpha-reduced androgens may have actions in the hippocampus to enhance cognitive performance of male rats. Psychoneuroendocrinology. 2004;29:1019–27. doi: 10.1016/j.psyneuen.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Gandy S, Almeida OP, Fonte J, Lim D, Waterrus A, Spry N, Flicker L, Martins RN. Chemical andropause and amyloid-beta peptide. JAMA. 2001;285:2195–6. doi: 10.1001/jama.285.17.2195-a. [DOI] [PubMed] [Google Scholar]
- Gillett MJ, Martins RN, Clarnette RM, Chubb SA, Bruce DG, Yeap BB. Relationship between testosterone, sex hormone binding globulin and plasma amyloid beta peptide 40 in older men with subjective memory loss or dementia. J Alzheimers Dis. 2003;5:267–9. doi: 10.3233/jad-2003-5401. [DOI] [PubMed] [Google Scholar]
- Goodenough S, Engert S, Behl C. Testosterone stimulates rapid secretory amyloid precursor protein release from rat hypothalamic cells via the activation of the mitogen-activated protein kinase pathway. Neuroscience Letters. 2000;296:49–52. doi: 10.1016/s0304-3940(00)01622-0. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Xu H, Gross RS, Greenfield JP, Hai B, Wang R, Greengard P. Testosterone reduces neuronal secretion of Alzheimer’s beta-amyloid peptides. PNAS. 2000;97:1202–5. doi: 10.1073/pnas.97.3.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa RJ, Pak TR, Kudwa AE, Lund TD, Hinds L. An alternate pathway for androgen regulation of brain function: activation of estrogen receptor beta by the metabolite of dihydrotestosterone, 5alpha-androstane-3beta,17beta-diol. Horm Behav. 2008;53:741–52. doi: 10.1016/j.yhbeh.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa RJ, Weiser MJ, Zuloaga DG. A role for the androgen metabolite, 5alpha-androstane-3beta,17beta-diol, in modulating oestrogen receptor beta-mediated regulation of hormonal stress reactivity. J Neuroendocrinol. 2009;21:351–8. doi: 10.1111/j.1365-2826.2009.01840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20:154–9. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- Hogervorst E, Bandelow S. The controversy over levels of sex steroids in cases with Alzheimer’s disease. Journal of Neuroendocrinology. 2004;16:93–4. doi: 10.1111/j.0953-8194.2004.01134.x. [DOI] [PubMed] [Google Scholar]
- Janowsky JS. The role of androgens in cognition and brain aging in men. Neuroscience. 2006a;138:1015–20. doi: 10.1016/j.neuroscience.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Janowsky JS. Thinking with your gonads: testosterone and cognition. Trends Cogn Sci. 2006b;10:77–82. doi: 10.1016/j.tics.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Jorm AF, Korten AE, Henderson AS. The prevalence of dementia: a quantitative integration of the literature. Acta Psychiatr Scand. 1987;76:465–79. doi: 10.1111/j.1600-0447.1987.tb02906.x. [DOI] [PubMed] [Google Scholar]
- Kaufman JM, Vermeulen A. The decline of androgen levels in elderly men and its clinical and therapeutic implications. Endocr Rev. 2005 doi: 10.1210/er.2004-0013. [DOI] [PubMed] [Google Scholar]
- Kerr JE, Allore RJ, Beck SG, Handa RJ. Distribution and hormonal regulation of androgen receptor (AR) and AR messenger ribonucleic acid in the rat hippocampus. Endocrinology. 1995;136:3213–21. doi: 10.1210/endo.136.8.7628354. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–53. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leranth C, Petnehazy O, MacLusky NJ. Gonadal hormones affect spine synaptic density in the CA1 hippocampal subfield of male rats. J Neurosci. 2003;23:1588–92. doi: 10.1523/JNEUROSCI.23-05-01588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leranth C, Hajszan T, MacLusky NJ. Androgens increase spine synapse density in the CA1 hippocampal subfield of ovariectomized female rats. J Neurosci. 2004;24:495–9. doi: 10.1523/JNEUROSCI.4516-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XA, Zhu LQ, Zhang Q, Shi HR, Wang SH, Wang Q, Wang JZ. Estradiol attenuates tau hyperphosphorylation induced by upregulation of protein kinase-A. Neurochem Res. 2008;33:1811–20. doi: 10.1007/s11064-008-9638-4. [DOI] [PubMed] [Google Scholar]
- Lund TD, Hinds LR, Handa RJ. The androgen 5alpha-dihydrotestosterone and its metabolite 5alpha-androstan-3beta, 17beta-diol inhibit the hypothalamo-pituitary-adrenal response to stress by acting through estrogen receptor beta-expressing neurons in the hypothalamus. J Neurosci. 2006;26:1448–56. doi: 10.1523/JNEUROSCI.3777-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meethal SV, Smith MA, Bowen RL, Atwood CS. The gonadotropin connection in Alzheimer’s disease. Endocrine. 2005;26:317–26. doi: 10.1385/ENDO:26:3:317. [DOI] [PubMed] [Google Scholar]
- Mehra RD, Sharma K, Nyakas C, Vij U. Estrogen receptor alpha and beta immunoreactive neurons in normal adult and aged female rat hippocampus: a qualitative and quantitative study. Brain Res. 2005;1056:22–35. doi: 10.1016/j.brainres.2005.06.073. [DOI] [PubMed] [Google Scholar]
- Moffat SD. Effects of testosterone on cognitive and brain aging in elderly men. Ann N Y Acad Sci. 2005;1055:80–92. doi: 10.1196/annals.1323.014. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiology of Aging. 2003a;24:1063–70. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003b;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, Laferla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006 doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- Pak TR, Chung WC, Lund TD, Hinds LR, Clay CM, Handa RJ. The androgen metabolite, 5alpha-androstane-3beta, 17beta-diol, is a potent modulator of estrogen receptor-beta1-mediated gene transcription in neuronal cells. Endocrinology. 2005;146:147–55. doi: 10.1210/en.2004-0871. [DOI] [PubMed] [Google Scholar]
- Papasozomenos SC. The heat shock-induced hyperphosphorylation of tau is estrogen-independent and prevented by androgens: implications for Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:6612–7. doi: 10.1073/pnas.94.13.6612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasozomenos SCh, Shanavas A. Testosterone prevents the heat shock-induced overactivation of glycogen synthase kinase-3 beta but not of cyclin-dependent kinase 5 and c-Jun NH2-terminal kinase and concomitantly abolishes hyperphosphorylation of tau: implications for Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:1140–5. doi: 10.1073/pnas.032646799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ. Estrogen modulates neuronal Bcl-xL expression and beta-amyloid-induced apoptosis: relevance to Alzheimer’s disease. J Neurochem. 1999;72:1552–63. doi: 10.1046/j.1471-4159.1999.721552.x. [DOI] [PubMed] [Google Scholar]
- Pike CJ. Testosterone attenuates beta-amyloid toxicity in cultured hippocampal neurons. Brain Research. 2001;919:160–5. doi: 10.1016/s0006-8993(01)03024-4. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Nguyen TV, Ramsden M, Yao M, Murphy MP, Rosario ER. Androgen cell signaling pathways involved in neuroprotective actions. Horm Behav. 2008;53:693–705. doi: 10.1016/j.yhbeh.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ, Carroll JC, Rosario ER, Barron AM. Protective actions of sex steroid hormones in Alzheimer’s disease. Front Neuroendocrinol. 2009;30:239–58. doi: 10.1016/j.yfrne.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber J. AR, apoE, and cognitive function. Horm Behav. 2008;53:706–15. doi: 10.1016/j.yhbeh.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden M, Nyborg AC, Murphy MP, Chang L, Stanczyk FZ, Golde TE, Pike CJ. Androgens modulate beta-amyloid levels in male rat brain. J Neurochem. 2003a;87:1052–5. doi: 10.1046/j.1471-4159.2003.02114.x. [DOI] [PubMed] [Google Scholar]
- Ramsden M, Shin TM, Pike CJ. Androgens modulate neuronal vulnerability to kainate lesion. Neuroscience. 2003b;122:573–8. doi: 10.1016/j.neuroscience.2003.08.048. [DOI] [PubMed] [Google Scholar]
- Rocca WA, Amaducci LA, Schoenberg BS. Epidemiology of clinically diagnosed Alzheimer’s disease. Ann Neurol. 1986;19:415–24. doi: 10.1002/ana.410190502. [DOI] [PubMed] [Google Scholar]
- Rosario ER, Pike CJ. Androgen regulation of beta-amyloid protein and the risk of Alzheimer’s disease. Brain Res Rev. 2008;57:444–53. doi: 10.1016/j.brainresrev.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario ER, Carroll JC, Oddo S, LaFerla FM, Pike CJ. Androgens regulate the development of neuropathology in a triple transgenic mouse model of Alzheimer’s disease. J Neurosci. 2006;26:13384–9. doi: 10.1523/JNEUROSCI.2514-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario ER, Chang L, Head EH, Stanczyk FZ, Pike CJ. Brain levels of sex steroid hormones in men and women during normal aging and in Alzheimer’s disease Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.04.008. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario ER, Chang L, Stanczyk FZ, Pike CJ. Age-related testosterone depletion and the development of Alzheimer disease. JAMA. 2004;292:1431–2. doi: 10.1001/jama.292.12.1431-b. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–525. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Simerly RB, Chang C, Muramatsu M, Swanson LW. Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. The Journal of Comparative Neurology. 1990;294:76–95. doi: 10.1002/cne.902940107. [DOI] [PubMed] [Google Scholar]
- Souttou B, Moretti JL, Gros J, Guilloteau D, Crepin M. Receptor binding and biological effects of three 125I-iodinated estrogen derivatives in human breast cancer cells (MCF-7) J Steroid Biochem Mol Biol. 1993;44:105–12. doi: 10.1016/0960-0760(93)90017-q. [DOI] [PubMed] [Google Scholar]
- Strom JO, Theodorsson A, Theodorsson E. Dose-related neuroprotective versus neurodamaging effects of estrogens in rat cerebral ischemia: a systematic analysis. J Cereb Blood Flow Metabol. 2009;29:1359–1372. doi: 10.1038/jcbfm.2009.66. [DOI] [PubMed] [Google Scholar]
- Strom JO, Theodorsson A, Theodorsson E. Order of magnitude differences between methods for maintaining physiological 17beta-oestradiol concentrations in ovariectomized rats. Scand J Clin Lab Invest. 2008;68:814–22. doi: 10.1080/00365510802409703. [DOI] [PubMed] [Google Scholar]
- Wahjoepramono EJ, Wijaya LK, Taddei K, Martins G, Howard M, de Ruyck K, Bates K, Dhaliwal SS, Verdile G, Carruthers M, Martins RN. Distinct effects of testosterone on plasma and cerebrospinal fluid amyloid-beta levels. J Alzheimers Dis. 2008;15:129–37. doi: 10.3233/jad-2008-15111. [DOI] [PubMed] [Google Scholar]
- Weihua Z, Makela S, Andersson LC, Salmi S, Saji S, Webster JI, Jensen EV, Nilsson S, Warner M, Gustafsson J. A role for estrogen receptor β in the regulation of growth of the ventral prostate. Proc Natl Acad Sci. 2001:6330–6335. doi: 10.1073/pnas.111150898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihua Z, Lathe R, Warner M, Gustafsson JA. An endocrine pathway in the prostate, ERbeta, AR, 5alpha-androstane-3beta,17beta-diol, and CYP7B1, regulates prostate growth. Proc Natl Acad Sci U S A. 2002;99:13589–94. doi: 10.1073/pnas.162477299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–2554. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Estradiol regulates hippocampal dendritic spine density via an N-methyl-D-aspartate receptor-dependent mechanism. J Neurosci. 1994;14:7680–7. doi: 10.1523/JNEUROSCI.14-12-07680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane T, Kitamura Y, Terada N, Matsumoto K. Proliferative response of seminal vesicle cells to androgen and estrogen in neonatally castrated mice. J Steroid Biochem. 1986;24:703–8. doi: 10.1016/0022-4731(86)90846-0. [DOI] [PubMed] [Google Scholar]
- Yao M, Nguyen TV, Rosario ER, Ramsden M, Pike CJ. Androgens regulate neprilysin expression: role in reducing beta-amyloid levels. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05341.x. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Wang R, Khan M, Mahesh V, Brann DW. Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J Neurosci. 2008;28:8430–41. doi: 10.1523/JNEUROSCI.2752-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Yao J, Mao Z, Chen S, Wang Y, Brinton RD. 17beta-Estradiol regulates insulin-degrading enzyme expression via an ERbeta/PI3-K pathway in hippocampus: Relevance to Alzheimer’s prevention. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]