

Abstract
Activator protein-1 (AP-1) and nuclear factor of activated T cells (NFAT) are two important transcription factors responsible for the regulation of cytokines, which are involved in cell proliferation and inflammation. Coal workers’ pneumoconiosis (CWP) is an occupational lung disease that may be related to chronic inflammation caused by coal dust exposure. In the present study, we demonstrate that coal from the Pennsylvania (PA) coalmine region, which has a high prevalence of CWP, can activate both AP-1 and NFAT in JB6 mouse epidermal cells. In contrast, coal from the Utah (UT) coalmine region, which has a low prevalence of CWP, has no such effects. The PA coal stimulates mitogen-activated protein kinase (MAPK) family members of extracellular signal-regulated kinases (ERKs) and p38 MAPK but not c-Jun-NH2-terminal kinases, as determined by the phosphorylation assay. The increase in AP-1 by the PA coal was completely eliminated by the pretreatment of cells with PD98059, a specific MAPK kinase inhibitor, and SB202190, a p38 kinase inhibitor, further confirming that the PA coal-induced AP-1 activation is mediated through ERKs and p38 MAPK pathways. Deferoxamine (DFO), an iron chelator, synergistically enhanced the PA coal-induced AP-1 activity, but inhibited NFAT activity. For comparison, cells were treated with ferrous sulfate and/or DFO. We have found that iron transactivated both AP-1 and NFAT, and DFO further enhanced iron-induced AP-1 activation but inhibited NFAT. These results indicate that activation of AP-1 and NFAT by the PA coal is through bioavailable iron present in the coal. These data are in agreement with our previous findings that the prevalence of CWP correlates well with levels of bioavailable iron in coals from various mining regions.
Coal remains a very significant part of U.S. energy needs. Nationwide, 55% of electricity is generated in coal-fired plants. With the current energy crisis in California, coal is more appealing now than it has been in years. Coal mining, however, causes health problems in coal workers. Among the respiratory diseases, coal workers’ pneumoconiosis (CWP) has received the most attention in the past decades because of its clear occupational association. Epidemiologic studies of the relationship between the prevalence of CWP and environmental measurements have consistently revealed that the predominant adverse exposure factor is respirable mixed coal dusts (1). Since CWP can become progressive after cessation of dust exposure, extensive at-risk populations consist of retired or ex-coal miners and those currently active in a workplace.
Despite comparable dust exposure levels, marked regional differences in the prevalence of CWP are well documented (2, 3). For example, there was a decline in prevalence from East to West in the United States, the disease being most common in Pennsylvania (PA) coalminers and least common in miners from Utah (UT) (4, 5). It has been found that quartz was a minor contributor to CWP development in general (6). Working on a large number of coal samples (a total of 28) representative of three coal mining regions, we have found that coals from the PA coal mine regions with a high prevalence of CWP release large amounts of bioavailable iron (BAI) in cells (7, 8). Coals from West Virginia (WV), with an intermediate prevalence of CWP, release moderate levels of BAI. In contrast, coals from UT, with a low prevalence of CWP, release little BAI. Iron is a well-known transition metal, capable of catalyzing oxidant formation by interacting with oxygen and/or hydrogen peroxide. These results suggest that BAI may play an important role in coal dust-induced CWP through oxidative stress pathways. Although CWP is considered to be one of the human lung pathologies related to oxidative stress (9), little evidence exists that an oxidant burden, which originates from the inhalation of high levels of iron-containing coal dusts, is an intrinsic mechanism in the development of CWP. The goal of the present study was to determine the early biologic responses of cells to BAI-induced oxidative stress caused by inhalation of coal dust, which may contribute to cell injury in vitro, and possibly lead to the development of CWP. Therefore, differences in levels of BAI in coals may be one of the important contributors to the observed regional differences in the prevalence of CWP. If this is the case, levels of BAI in coals could be used as a marker for predicting coal’s toxicity, even before its large-scale mining.
Activator protein-1 (AP-1) and nuclear factor of activated T cells (NFAT) are important transcription factors controlling the upregulation of proinflammatory cytokines, which contribute to the development of fibrosis, an advanced disease of pneumoconiosis (9, 10). AP-1 consists of a family of Jun/Fos dimers that include different Jun proteins (c-Jun, JunB, and JunD) and Fos proteins (c-Fos, FosB, Fra-1, Fra-2, and FosB2) (11). AP-1 activation is regulated at multiple levels by the activation of mitogen-activated protein kinases (MAPK), involving three major pathways of extracellular signal-related kinases (ERKs), stress-activated protein kinases/c-Jun NH2-terminal kinases (JNKs), and p38 MAPK (12). The ERKs pathway is considered responsive to the growth/differentiation signals, while the JNKs and p38 pathways are responsive to environmental stresses such as heat shock. A variety of environmental pollutants, which include arsenic, vanadium, and quartz, activate all or part of the ERKs and JNKs/p38 pathways (13–17). The NFAT was originally described as a transcription factor expressed in activated but not in resting T cells. Growing evidence indicates that NFAT is also expressed in a variety of lymphoid tissues, including heart, ovary, lung, skin, as well as liver (18). In contrast to AP-1, dephosphorylation activates NFAT protein. This is achieved through the activation of signaling pathways that produce a rise in intracellular free calcium levels. An increase in intracellular free calcium induces activation of the phosphatase calcineurin, which dephosphorylates NFAT proteins. In the present study, we have found that the coals from PA coal-mine regions transactivate both AP-1 and NFAT transcription factors, whereas those from UT do not, and that this activation is mediated by the BAI present in the coals. This finding may provide an explanation for the known regional differences in the prevalence of CWP.
Materials and Methods
Chemical Agents
Fetal bovine serum (FBS) was from Life Technologies, Inc. (Grand Island, NY); Eagle’s minimal essential medium (MEM), PD98059, and SB202190 were purchased from Calbiochem (San Diego, CA); Dulbecco’s modified Eagle’s medium were from BioWhittaker (Walkersville, MD); luciferase assay substrate was from Promega (Madison, WI); phospho-MAPK antibody kits were from Cell Signaling (Beverly, MA); Deferoxamine (DFO) and ferrous sulfate heptahydrate (FeSO4·7H2O) were obtained from Sigma (St. Louis, MO).
Selection of Coal Samples
Two bituminous coal samples, one from the PA coal mine region (PSOC no. 1,198), with a prevalence of 26% CWP, the other from UT (PSOC no. 459), with a prevalence of 4% CWP, were selected from the 28 coal samples pool that we have studied in relation to coal’s toxicity (7, 8). The time periods during which the coals were sampled ranged from the 1970s to the 1980s, the same period during which the first and second rounds of a national study of CWP were performed (4, 5). In our opinion, these two coal samples are representative of the two coalmine regions. They were purchased from the Penn State Coal Sample Bank (Pennsylvania State University, University Park, PA). The coal samples for the present study were selected based on the published epidemiologic data, which indicated a different prevalence of CWP for coal seams in specific counties and states, as previously described (8). Physico-chemical characteristics of the coal that are thought to play an important role include coal rank (carbon content or molar ratio of carbon to hydrogen), quartz content, and BAI and are presented in Table 1.
TABLE 1.
Characteristics of two bituminous coals from the Pennsylvania and Utah coal mine regions
| Coal | CWP (%)* | Carbon (%)† | Atomic C/H (Molar Ratio)† | Quartz (%)† | Bioavailable Iron (ppm)‡ |
|
|---|---|---|---|---|---|---|
| Fe2+ | Fe3+ | |||||
| PSOC-1198 (PA) | 26 | 72.44 | 1.39 | 3.50 | 15,010 | 8,588.0 |
| PSOC-459 (UT) | 4 | 71.32 | 1.16 | 3.63 | 0 | 4.6 |
Definition of abbreviations: carbon to hydrogen ratio, C/H; coal workers’ pneumoconiosis, CWP; Pennsylvania, PA; Utah, UT.
Prevalence of CWP was obtained from Morgan and coworkers (4).
Data on carbon content or molar ratio of carbon/hydrogen as indicators of coal rank and quartz content in the coals are provided by the Penn State Coal Sample Bank and Database.
Levels of bioavailable iron (both Fe2+ and Fe3+) were determined spectrophotometrically by 2,2-dipyrydil-Fe2+ and deferoxamine-Fe3+ complexes at 520 nm and 430 nm, respectively. Bioavailable iron is defined as iron released in 10 mM phosphate solution, pH 4.5, which mimics the phagolysosomes of cells (7).
Preparation of Coal Samples for Cell Treatment
Coal samples were ground and size-classified using the Mercer Impactor (Intox, Albuquerque, NM). Coal samples were ground in a ball mill with 5-mm diameter glass beads for 24 h. A 30 g aliquot of the ground coal sample was resuspended in a 1-L glass jar with 5 L of nitrogen (N2) per minute. Coal dusts larger than 10 μm were removed by passing them through a cyclone (BGI, Waltham, MA) with a 10-μm cut-off size, and the respirable fraction of the coal dusts were collected on teflon filters (0.45 μm pore size; Millipore, Bedford, MA). The size distribution of the coal dusts collected on the teflon filter was determined using the Mercer Impactor, and the diameter of over 80% of the coal particles was less than 5 μm (data not shown).
Cell Culture
AP-1-luciferase reporter stably-transfected mouse epidermal JB6 P+1–1 cells were cultured in monolayers in MEM containing 5% FBS, 2 mM L-glutamine, and 25 μg of gentamicin/ml (19, 20); Mouse embryo fibroblast PW cells and its transfectant, PW NFAT mass1 cells, were cultured in Dulbecco’s modified Eagle’s medium with 10% FBS, 2 mM L-glutamine, and 25 μg gentamicin/ml (21).
AP-1 Activity Assay
Confluent monolayers of JB6 P+1–1 cells were trypsinized, and 8 × 103 viable cells suspended in 100 μl of 5% FBS MEM were added to each well of a 96-well plate, and incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air for 12 to 24 h. Cells were starved by culturing them in 0.1% FBS MEM for 12–24 h and were then exposed to the PA (PSOC-1198) or UT (PSOC-459) coals for AP-1 induction. The reason for using 0.1% FBS was to ensure that BAI present in the coals would not be chelated by transferrin in serum. As a negative control, each experiment included plates with cells to which nothing had been added. After different periods of culture times, cells were extracted with lysis buffer, and luciferase activity was measured using a luminometer (Monolight 2,010; Monolight, Gaithersburg, MD). The results are expressed as relative AP-1 activity (15, 22).
Protein Kinase Phosphorylation Assay
Immunoblotting for the phosphorylation of ERKs, JNKs, and p38 kinase was performed as described in the protocol from Cell Signaling Technology using phosphospecific antibodies against phosphorylated sites of ERKs, JNKs, and p38 kinase, respectively. Non-phosphospecific antibodies against ERKs, JNKs, and p38 kinase proteins provided in each assay kit were used to normalize the phosphorylation assay using the same transferred membrane blots.
Assay for Transactivation of NFAT
Confluent monolayers of PW NFAT mass1 cells were trypsinized, and 5 × 103 viable cells suspended in 100 μl of medium were added to each well of a 96-well plate. Plates were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air. These cells were exposed to the PA coal (PSOC-1198) or UT coal (PSOC-459) for the indicated times and doses. The control and coal-treated cells were extracted with lysis buffer and luciferase activity was measured as previously described (21). The results are expressed as NFAT activity relative to untreated controls (21).
Results
Induction of AP-1 in JB6 Cells by the PA but Not the UT Coal Dusts
To study the regulation of AP-1 transcription activity by coals from different coal mine regions, we used stable AP-1–luciferase reporter plasmid-transfected mouse epidermal JB6 P+ cells. The results observed from this stable transfectant show that AP-1 activity was markedly induced by exposure of the cells to the PA coal dust but not to the UT coal dust. At a dose of 40 μg/cm2 (or 160 μg/ml), relative luciferase activity was increased 8-fold by the PA coal sample (Figure 1). In contrast, luciferase activity in cells treated with the UT coal remained unchanged, as compared with the control cells. The activation of AP-1 by the PA coal appears to be time- and dose-dependent (Figures 2A and 2B). The maximum induction of AP-1 by the PA coal occurred at 40 μg/cm2 for a 36-h incubation.
Figure 1.
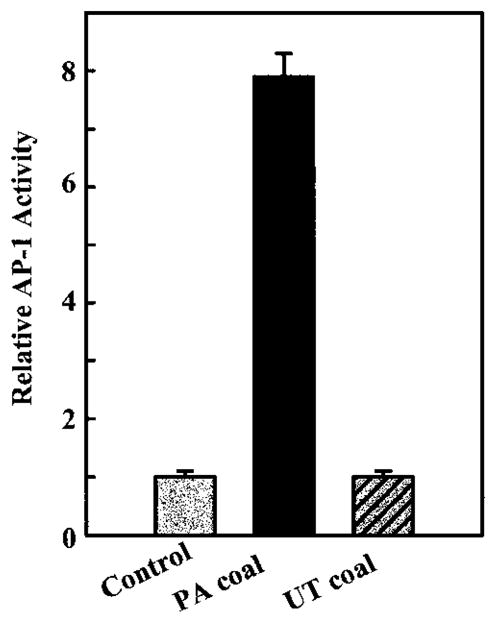
Effects of coals from PA (PSOC-1198) and UT (PSOC-459) on AP-1 activation in mouse epidermal JB6 cells. A total of 8 × 103 P+1-1 cells were seeded into each well of 96-well plates. After being cultured at 373C overnight, the cells were starved for 12 h by replacing medium with 0.1% FBS MEM. Then, the cells were treated with the PSOC-1198 or PSOC-459 coals at a dose of 40 μg/cm2 for 36 h. The luciferase activity was then measured and the results are presented as relative AP-1 activity. Each bar indicates the mean and standard deviation of four identically treated assay wells.
Figure 2.

Time course and dose response studies on AP-1 trans-activation by coals from PA (PSOC-1198; closed circles) and UT (PSOC-459; open circles). A total of 8 × 103 P+1-1 were seeded, cultured, and processed as described in the legend for Figure 1. (a) For the dose response study, cells were treated with different amounts of PSOC-1198 or PSOC-459. After being cultured for 36 h, the luciferase activity was measured. (b) For the time course study, cells were treated with the PSOC-1198 or PSOC-459 coals at 40 μg/cm2 and harvested at time points indicated for the luciferase activity assay. The results are presented as relative AP-1 activity. Each bar indicates the mean and standard deviation of four identically treated assay wells.
Activation of ERKs and p38 MPAK but Not JNKs by the PA Coal
AP-1 activation is regulated at multiple levels by the activation of MAPK, involving ERKs, JNKs, and p38 MAPK. Using antibodies specific for the MAPKs and phosphospecific for the phosphorylated MAPKs, we studied ERKs, JNKs, and p38 kinase proteins and their phosphorylation. Figure 3 shows that exposure to the PA coal with a high prevalence of CWP significantly stimulated the phosphorylation of ERKs and p38 kinase, but not JNKs. Interestingly, the coal from the UT coal mine region with a low prevalence of CWP did not induce phosphorylation of ERKs or JNKs, although it activated the phosphorylation of p38 kinase as the PA coal did, but to a lesser extent. The positive control, 2 μM benzo(a)pyrene diol-epoxide did activate the phosphorylation of JNKs, indicating that coals from the PA and UT coal mine region did not induce phosphorylation of JNKs under our experimental conditions. Phosphorylation of ERKs was maximal at 30 min after exposure to the PA coal, while that of p38 kinase was maximal at 90 min after exposure.
Figure 3.

Effects of the PA (PSOC-1198) and UT (PSOC-459) coals on the phosphorylation of ERKs, JNKs, and p38 MAPK. 8 × 103 P+1-1 were seeded, cultured, and processed as described in the legend of Figure 1. Cells were then exposed to the PA (PSOC-1198) and UT (PSOC-459) coals for different periods of time as indicated, or various amounts of coal. After lysis, phosphorylated and nonphosphorylated ERKs, JNKs, and p38 kinase proteins were assayed using the corresponding specific antibodies. The phosphorylated and nonphosphorylated proteins were analyzed using the same transferred membrane blots. Benzo(a)pyrene diol-epoxide was used as a positive control for JNKs.
Inhibition of the PA Coal-induced AP-1 Activation by PD98059 and SB202190
To further confirm that ERKs and p38 kinase pathways are activated by the PA coal, specific inhibitors of each pathway were added to the tissue culture media 30 min before coal treatment. Inhibitors remained in the culture medium for the duration of experiments. PD 98059, a selective and cell-permeable inhibitor of MAPK kinase, and SB202190, a potent and cell-permeable inhibitor of p38 MAPK with no effect on the activity of the ERKs or JNKs subgroups, were used in these experiments. We have found that the pretreatment of cells with PD98059 and SB202190 resulted in the inhibition of the PA coal-induced AP-1 luciferase activity (Figures 4A and 4B). These results suggest that AP-1 activation by the PA coal occurs through at least two major pathways of ERKs and p38 MAPK.
Figure 4.
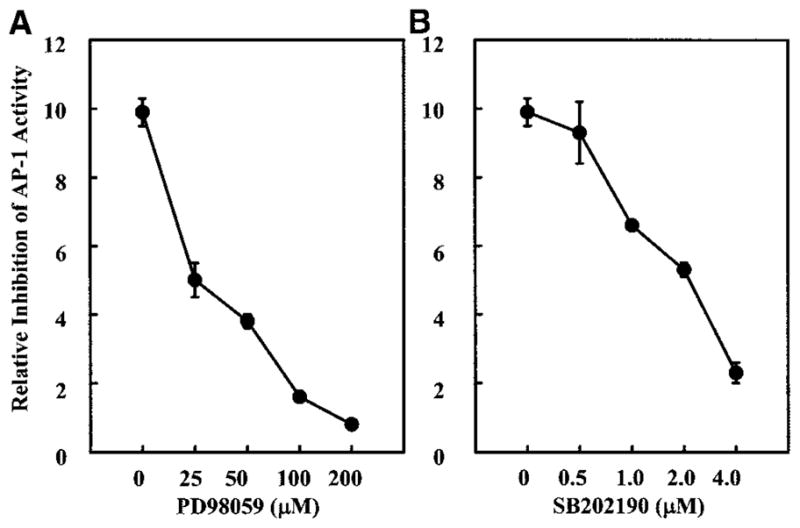
Effects of the pretreatment of cells with PD98059 or SB202190 on AP-1 activity induced by the PA coal (PSOC-1198). JB6 P+1-1 cells were seeded, cultured and processed as described in Figure 1. Cells were first treated for 30 min with different concentrations of (a) PD98059 or (b) SB202190, as indicated, and then were exposed to 40 μg/cm2 of PSOC-1198 coal for 36 h; luciferase activity was measured, and results are presented as relative AP-1 activity.
Opposite Effects of DFO on the PA Coal-induced NFAT and AP-1 Activation
The identification of active compound(s) in the complicated mixes of coal dusts is a key step for predicting coal’s toxicity and for monitoring early adverse effects on and health of coal workers. To prove that BAI in the coal is the likely active component in the activation of AP-1, possibly leading to the development of CWP, JB6 cells were pretreated with DFO, a chelator that specifically binds iron. To our surprise, DFO exhibited a synergistic augmentation of the PA coal-induced AP-1 activation (Figure 5A), whereas DFO alone did not induce AP-1 activity in the cells (data not shown). Preincubation of the PA coal with DFO also synergistically increased AP-1 activity (data not shown), similar to the pretreatment of cells with DFO followed by the coal. To test whether the synergistic effect of DFO is AP-1 specific, confluent monolayers of PW NFAT mass1 cells were also used for the PA coal and DFO treatments. Interestingly, the PA coal also transactivated NFAT in a dose-dependent manner, while pretreatment of cells with DFO inhibited the PA coal-induced NFAT activation (Figure 5B). The distinct effects of DFO on the PA coal-induced AP-1 and NFAT activation indicate that, although DFO is both effective and efficient in preventing iron-catalyzed oxidant formation (23), there may be some side-effects caused by DFO treatment, due to its contribution to AP-1 activation.
Figure 5.
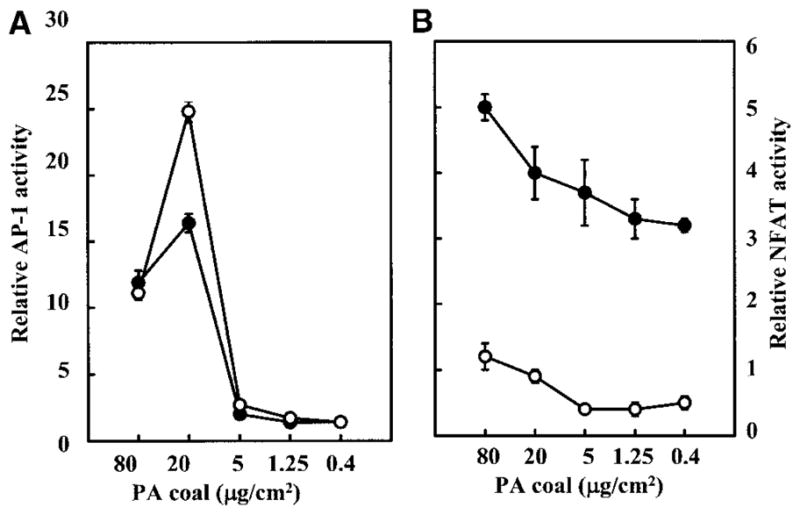
Effects of DFO on AP-1 and NFAT activities induced by the PA coal (PSOC-1198). (a) JB6 P+1-1 cells or (b) PW NFAT mass1 cells were seeded and cultured in 96-well plates. Cells were treated with 0.2 mM of DFO plus different amounts of PSOC-1198 coal as indicated for 36 h. The luciferase activity was measured and results are presented as relative AP-1 activity or relative NFAT activity, respectively.
Effects of FeSO4 on AP-1 and NFAT Activation
Our previous studies have indicated that FeSO4 is one of the major bioavailable iron compounds in the sampled coals, some of which originate from the oxidation of pyrite, a typical contaminant of coals (24). To confirm that iron is the active compound in the coal for the activation of the transcription factors, both stable transfectants of AP-1 and NFAT were treated with pure FeSO4 and/or DFO. We have found that iron can activate both AP-1 and NFAT (Figures 6A and 6B), and that pretreatment of cells with DFO synergistically enhances iron-induced AP-1 activation (Figure 6A), but inhibits iron-induced NFAT activation (Figure 6B). These results are in agreement with the effects observed in the PA coal-treated cells. Our results strongly suggest that BAI present in the PA coals is likely the active component responsible for the activation of AP-1 and NFAT transcription factors in the exposed cells.
Figure 6.

Activation of AP-1 and NFAT by FeSO4 and opposite effects of DFO on their activities. (a) JB6 P+1-1 cells or (b) PW NFAT mass1 cells were seeded and cultured in 96-well plates. Cells were treated with 0.2 mM of DFO plus 0.2 mM FeSO4, as indicated, for 36 h. Luciferase activity was measured and results are presented as relative AP-1 activity or relative NFAT activity, respectively.
Discussion
Occupational exposure to coal dust is associated with the development of pneumoconiosis. Pneumoconiosis can result in progressive massive fibrosis, even after cessation of coal dust exposure. Increasing evidence demonstrates that CWP is one of the human lung pathologies related to oxidative stress and chronic inflammation (9). For example, previous studies on symptomatic coal miners have shown that alveolar macrophages recovered from bronchoalveolar lavage released excessive amounts of oxidants and inflammatory cytokines (25–29). Our previous studies have shown that BAI present in the coals may be responsible for the oxidant formation and a subsequent cytokine release in the lung after inhalation (8, 30). Therefore, differences in the levels of BAI in the coals may contribute to the observed regional differences in the prevalence of CWP. However, there is little direct evidence that BAI in the coal is the active compound in inducing lung injury, and the molecular mechanisms involved in coal dust-induced pneumoconiosis and fibrosis are unclear.
In the present study, the results show that BAI in the PA coal with a high prevalence of CWP stimulates the AP-1 and NFAT transactivation in a time- and dose-dependent manner. The coal from UT with a low prevalence of CWP does not exhibit these effects. We realize that the mouse epidermal JB6 cell line used in the present study may not be the best cell model for studies on lung disease. However, this cell line is a unique cell model sensitive to oxidative stress (31). Coal dusts, which often elicit weak or no biologic response in cell or animal models as compared with that of quartz (32), triggered distinctive effects on AP-1 activation in the present study.
Our results suggest that simultaneous activation of AP-1 and NFAT induced by the PA coal may be an important mechanism contributing to cytokine formation and possibly leading to the development of CWP, since a high prevalence of CWP was observed in workers of the PA coal mine region. The promoter/enhancer regions of many genes contain adjacent AP-1 and NFAT binding sites. The cooperative binding of AP-1 and NFAT proteins to these sites greatly enhances the DNA binding affinity and the transcriptional activity of the AP-1–NFAT ternary complex compared with either NFAT or AP-1 alone (33). Cooperation between AP-1 and NFAT transcription factors has been demonstrated in many genes, such as interleukin (IL)-2, IL-4, IL-5, IL-8, IL-13, granulocyte macrophage-colony stimulating factor (GM-CSF), and tumor necrosis factor-α (33–36). For example, upstream of the GM-CSF gene lies an enhancer element that controls GM-CSF expression and contains four NFAT sites, three of which support cooperative binding with AP-1 (36). Previous studies have indicated that the expression of IL-8, tumor necrosis factor-α, and other cytokines is associated with the initiation and control of effective immune and inflammatory responses and may be related to cancer development (31). Our results suggest that AP-1 and NFAT may be involved in the coal dust-induced lung inflammation following inhalation and may subsequently be involved in the fibrogenic effects of the coal.
Most importantly, we have found that the phosphorylation of ERKs and p38 kinase, but not of JNKs, is induced by the PA coal, as shown by Western immunoblotting. AP-1 is a downstream target of these three MAPK members. The signal transduction pathways leading to the AP-1 activation have been extensively studied in the last several years. It is known that stress-related signals such as ultraviolet light induce the activation of all three MAPK pathways (ERKs, JNKs, and p38). To further confirm our finding, we have shown that the pretreatment of cells with the p38 and ERK inhibitors SB203580 and PD98059 suppressed AP-1 trans-activation induced by the PA coal. Thus, these results suggest that the PA coal-induced AP-1 activation may occur through ERKs and p38 MAPK pathways, at least in the JB6 cells that we have tested.
Using DFO, a chelator binding iron, we found that DFO synergistically increased the PA coal-induced AP-1 activation. To test whether the synergistic effect of DFO is AP-1 specific, we then studied the effects of the PA coal with and without DFO on NFAT activities. Interestingly, the PA coal also transactivated NFAT. In contrast to its enhancing effect on AP-1, DFO inhibited the PA coal-induced NFAT activation, suggesting two different mechanisms of DFO on iron-induced AP-1 and NFAT activities. DFO can bind Fe3+, as well as Al3+ and Ga3+. However, neither Al3+ nor Ga3+ ions are likely to be found in aqueous coal suspensions at high concentrations. Al and Ga are trace metals in the parts-per-million or parts-per-billion range according to the data provided by the Penn State Coal Sample Bank. Therefore, the biologic responses inhibited by pretreatment with DFO are solely due to the presence of iron. To further support this conclusion, the pure iron compound FeSO4 was used. Similar to the PA coal, iron activated both AP-1 and NFAT, while DFO synergistically augmented the iron-induced AP-1 activation but inhibited the iron-induced NFAT activation. These data demonstrate for the first time that iron present in the coals induces AP-1 and NFAT activation. Our results suggest that the inhalation of iron-containing coals, such as those from the PA coal mine region, imposes a threat to the lungs of coal workers, leading to pulmonary oxidative stress and the development of CWP. These results are well correlated with BAI content in the coals, but not with coal rank or levels of quartz in the coals.
The mechanisms of synergistic effects of DFO on the PA coal or iron-induced AP-1 activation remain unclear. DFO alone had no effect on AP-1 activation. It is the DFO–Fe3+ complex (ferroxamine), which caused an increased activation of AP-1 in comparison with iron alone. This may be due to a reducing milieu within the cells when DFO was used to prevent iron from redox cycling. Redox factor 1, a Class II hydrolytic apurinic/apyrimidinic endonuclease, has been shown to stimulate the DNA-binding activity of AP-1 proteins by a redox-dependent mechanism in cooperation with thioredoxin (37, 38). Thioredoxin was shown to increase DNA binding activity of AP-1 by reducing Cys-154 in c-Fos and Cys-272 in c-Jun (39, 40). Whether ferroxamine reacts with thioredoxin is unknown and needs further investigation.
In conclusion, our studies have shown that the PA coal with a high prevalence of CWP transactivates both AP-1 and NFAT. These data support our hypothesis that BAI in the coal may be the active compound in inducing activation of AP-1 and NFAT. Since AP-1 and NFAT are important transcription factors that have been demonstrated in the promoter/enhancer region of genes of several cytokines and chemokines, such as IL-2, IL-6, IL-8, and GM-CSF, the biologic outcomes of AP-1 and NFAT activation by the coals may be a powerful mechanism in coal-induced lung disease.
Acknowledgments
This project was supported by grant OH 03561 from the National Institute for Occupational Safety and Health of the Center for Disease Control and Prevention and in part by the National Institute of Environmental Health Sciences Center grant ES00260.
Abbreviations
- AP-1
Activator protein-1
- BAI
bioavailable iron
- CWP
coal workers’ pneumoconiosis
- DFO
deferoxamine
- ERK
extracellular signal-regulated kinases
- FBS
fetal bovine serum
- GM-CSF
granulocyte macrophage-colony stimulating factor
- IL
interleukin
- JNK
c-Jun-NH2-terminal kinase
- MAPK
mitogen-activated protein kinase
- MEM
minimal essential medium
- NFAT
nuclear factor of activated T cells
References
- 1.Attfield M, Wagner G. Respiratory Disease in Coal Miners. In: Rom WN, editor. Environmental and Occupational Medicine. 2. Little, Brown, and Company; Boston: 1993. pp. 325–344. [Google Scholar]
- 2.Attfield MD, Castellan RM. Epidemiological data on US coal miners’ pneumoconiosis, 1960 to 1988. Am J Public Health. 1992;82:964–970. doi: 10.2105/ajph.82.7.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hurley JF, Alexander WP, Hazledine DJ, Jacobsen M, Maclaren WM. Exposure to respirable coalmine dust and incidence of progressive massive fibrosis. Br J Ind Med. 1987;44:661–672. doi: 10.1136/oem.44.10.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgan WKC, Burgess DB, Jacobson G, O’Brien RJ, Pendergrass E, Reger RB, Shoub EP. The prevalence of coal workers’ pneumoconiosis in US coal miners. Arch Environ Health. 1973;27:221–230. doi: 10.1080/00039896.1973.10666356. [DOI] [PubMed] [Google Scholar]
- 5.Attfield MD, Seixas NS. Prevalence of pneumoconiosis and its relationship to dust exposure in a cohort of US bituminous coal miners and exminers. Am J Ind Med. 1995;27:137–151. doi: 10.1002/ajim.4700270113. [DOI] [PubMed] [Google Scholar]
- 6.Walton WH, Dodgson J, Hadden GG, Jacobsen M. The effect of quartz and other non-coal dusts in coal workers’ pneumoconiosis. In: Walton WH, editor. Inhaled particles IV. Pergamon Press; Oxford: 1977. pp. 669–689. [PubMed] [Google Scholar]
- 7.Zhang Q, Dai JS, Ali AM, Chen LC, Huang X. Roles of bioavailable iron and calcium in coal dust-induced oxidative stress: possible implications in coal workers’ lung disease. Free Radic Res. 2002;36:285–294. doi: 10.1080/10715760290019309. [DOI] [PubMed] [Google Scholar]
- 8.Huang X, Fournier J, Koenig K, Chen LC. Buffering capacity of coal and its acid-soluble Fe2+ content: possible role in coal workers’ pneumoconiosis. Chem Res Toxicol. 1998;11:722–729. doi: 10.1021/tx970151o. [DOI] [PubMed] [Google Scholar]
- 9.Castranova V, Vallyathan V. Silicosis and coal workers’ pneumoconiosis. Environ Health Perspect. 2000;108(Suppl 4):675–684. doi: 10.1289/ehp.00108s4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smart DE, Vincent KJ, Arthur MJ, Eickelberg O, Castellazzi M, Mann J, Mann DA. JunD regulates transcription of the tissue inhibitor of metalloproteinases-1 and interleukin-6 genes in activated hepatic stellate cells. J Biol Chem. 2001;276:24414–24421. doi: 10.1074/jbc.M101840200. [DOI] [PubMed] [Google Scholar]
- 11.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 12.Kelly K, Chu Y. The regulation of MAP kinase pathways by MAP kinase phosphatases. In: Gutkind JS, editor. Signaling networks and cell cycle control. Human Press; Totowa, NJ: 2000. pp. 165–182. [Google Scholar]
- 13.Ding M, Shi X, Dong Z, Chen F, Lu Y, Castranova V, Vallyathan V. Freshly fractured crystalline silica induces activator protein-1 activation through ERKs and p38 MAPK. J Biol Chem. 1999;274:30611–30616. doi: 10.1074/jbc.274.43.30611. [DOI] [PubMed] [Google Scholar]
- 14.Huang C, Ma WY, Li J, Dong Z. Arsenic induces apoptosis through a c-Jun NH2-terminal kinase-dependent, p53-independent pathway. Cancer Res. 1999;59:3053–3058. [PubMed] [Google Scholar]
- 15.Huang C, Ding M, Li J, Leonard SS, Rojanasakul Y, Castranova V, Vallyathan V, Ju G, Shi X. Vanadium-induced nuclear factor of activated T cells activation through hydrogen peroxide. J Biol Chem. 2001;276:22397–22403. doi: 10.1074/jbc.M010828200. [DOI] [PubMed] [Google Scholar]
- 16.Huang C, Chen N, Ma WY, Dong Z. Vanadium induces AP-1-and NFkappB-dependent transcription activity. Int J Oncol. 1998;13:711–715. doi: 10.3892/ijo.13.4.711. [DOI] [PubMed] [Google Scholar]
- 17.Huang C, Bode AM, Chen NY, Ma WY, Li J, Nomura M, Dong Z. Transactivation of AP-1 in AP-1-luciferase reporter transgenic mice by arsenite and arsenate. Anticancer Res. 2001;21:261–267. [PubMed] [Google Scholar]
- 18.Rincon M, Flavell RA. Transcription mediated by NFAT is highly inducible in effector CD4+ T helper 2 (Th2) cells but not in Th1 cells. Mol Cell Biol. 1997;17:1522–1534. doi: 10.1128/mcb.17.3.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang C, Ma W, Bowden GT, Dong Z. Ultraviolet B-induced activated protein-1 activation does not require epidermal growth factor receptor but is blocked by a dominant negative PKClambda/iota. J Biol Chem. 1996;271:31262–31268. doi: 10.1074/jbc.271.49.31262. [DOI] [PubMed] [Google Scholar]
- 20.Huang C, Ma WY, Young MR, Colburn N, Dong Z. Shortage of mitogen-activated protein kinase is responsible for resistance to AP-1 transactivation and transformation in mouse JB6 cells. Proc Natl Acad Sci USA. 1998;95:156–161. doi: 10.1073/pnas.95.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang C, Ma WY, Dong Z. Requirement for phosphatidylinositol 3-kinase in epidermal growth factor-induced AP-1 transactivation and transformation in JB6 P+ cells. Mol Cell Biol. 1996;16:6427–6435. doi: 10.1128/mcb.16.11.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang C, Ma WY, Dawson MI, Rincon M, Flavell RA, Dong Z. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc Natl Acad Sci USA. 1997;94:5826–5830. doi: 10.1073/pnas.94.11.5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang X, Dai JS, Fournier J, Ali AM, Zhang Q, Frenkel K. Ferrous ion autoxidation and its chelation in iron-loaded human liver HepG2 cells. Free Radic Biol Med. 2002;32:84–92. doi: 10.1016/s0891-5849(01)00770-5. [DOI] [PubMed] [Google Scholar]
- 24.Huang X, Zalma R, Pezerat H. Factors that influence the formation and stability of hydrated ferrous sulfate in coal dusts. Possible relation to the emphysema of coal miners. Chem Res Toxicol. 1994;7:451–457. doi: 10.1021/tx00039a025. [DOI] [PubMed] [Google Scholar]
- 25.Vallyathan V, Goins M, Lapp LN, Pack D, Leonard S, Shi X, Castranova V. Changes in bronchoalveolar lavage indices associated with radiographic classification in coal miners. Am J Respir Crit Care Med. 2000;162:958–965. doi: 10.1164/ajrccm.162.3.9909074. [DOI] [PubMed] [Google Scholar]
- 26.Wallaert B, Lassalle P, Fortin F, Aerts C, Bart F, Fournier E, Voisin C. Superoxide anion generation by alveolar inflammatory cells in simple pneumoconiosis and in progressive massive fibrosis of nonsmoking coal workers. Am Rev Respir Dis. 1990;141:129–133. doi: 10.1164/ajrccm/141.1.129. [DOI] [PubMed] [Google Scholar]
- 27.Rom WN. Relationship of inflammatory cell cytokines to disease severity in individuals with occupational inorganic dust exposure. Am J Ind Med. 1991;19:15–27. doi: 10.1002/ajim.4700190104. [DOI] [PubMed] [Google Scholar]
- 28.Perrin-Nadif R, Auburtin G, Dusch M, Porcher JM, Mur JM. Blood antioxidant enzymes as markers of exposure or effect in coal miners. Occup Environ Med. 1996;53:41–45. doi: 10.1136/oem.53.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borm PJ, Meijers JM, Swaen GM. Molecular epidemiology of coal worker’s pneumoconiosis: application to risk assessment of oxidant and monokine generation by mineral dusts. Exp Lung Res. 1990;16:57–71. doi: 10.3109/01902149009064699. [DOI] [PubMed] [Google Scholar]
- 30.Huang X, Zalma R, Pezerat H. Chemical reactivity of the carbon-centered free radicals and ferrous iron in coals: role of bioavailable Fe2+ in coal workers pneumoconiosis. Free Radic Res. 1999;30:439–451. doi: 10.1080/10715769900300481. [DOI] [PubMed] [Google Scholar]
- 31.Hsu TC, Young MR, Cmarik J, Colburn NH. Activator protein 1 (AP-1)- and nuclear factor kappaB (NF-kappaB)-dependent transcriptional events in carcinogenesis. Free Radic Biol Med. 2000;28:1338–1348. doi: 10.1016/s0891-5849(00)00220-3. [DOI] [PubMed] [Google Scholar]
- 32.Vallyathan V, Shi X. The role of oxygen free radicals in occupational and environmental lung diseases. Environ Health Perspect. 1997;105:165–177. doi: 10.1289/ehp.97105s1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476–2489. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]
- 34.Macian F, Garcia-Rodriguez C, Rao A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J. 2000;19:4783–4795. doi: 10.1093/emboj/19.17.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chow CW, Rincon M, Davis RJ. Requirement for transcription factor NFAT in interleukin-2 expression. Mol Cell Biol. 1999;19:2300–2307. doi: 10.1128/mcb.19.3.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cockerill PN, Bert AG, Jenkins F, Ryan GR, Shannon MF, Vadas MA. Human granulocyte-macrophage colony-stimulating factor enhancer function is associated with cooperative interactions between AP-1 and NFATp/c. Mol Cell Biol. 1995;15:2071–2079. doi: 10.1128/mcb.15.4.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11:653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schenk H, Klein M, Erdbrugger W, Droge W, Schulze-Osthoff K. Distinct effects of thioredoxin and antioxidants on the activation of transcription factors NF-kappa B and AP-1. Proc Natl Acad Sci USA. 1994;91:1672–1676. doi: 10.1073/pnas.91.5.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abate C, Patel L, Rauscher FJ, 3rd, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249:1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]