

Abstract
The Nuclear Factor-kappa B (NF-κB) family of transcription factors regulates the expression of a wide range of genes critical for immune and inflammatory responses, cell survival, immune development, and cell proliferation. Dysregulated NF-κB activity occurs in a number of chronic inflammatory diseases and certain types of cancers making NF-κB signaling an attractive target for the development of anti-inflammatory and anti-cancer drugs. A pivotal regulator of all inducible NF-κB signaling pathways is the IκB kinase (IKK) complex that consists of two kinases (IKKα and IKKβ) and a regulatory subunit named NF-κB essential modulator (NEMO). Genetic analysis of the IKK complex has identified two separate pathways named the classical and non-canonical mechanisms that are dependent on either NEMO and IKKβ (classical) or IKKα alone (non-canonical). To better understand the mechanisms that regulate IKK complex activity and to address the differential functions of IKKα and IKKβ we have molecularly dissected the IKKs. We describe here how these studies have identified a unique inhibitor of pro-inflammatory NF-κB signaling, an unforeseen role for IKKα in the classical NF-κB pathway, and a novel functional domain in IKKβ that is not present in IKKα.
Keywords: I kappa B kinase, Nuclear factor-kappa B, NF-κB essential modulator, NEMO binding domain, Ubiquitin-like domain, Signal transduction
Introduction
Nuclear Factor-kappa B (NF-κB) proteins are a family of transcription factors that regulate the inducible expression of a wide array of genes encoding proteins essential for innate and adaptive immunity, inflammation and cell survival [1-3]. Activation of NF-κB occurs in response to many diverse stimuli including ligation of innate immune receptors (e.g., TLRs), antigen-receptor engagement (BCR and TCR) and pro-inflammatory cytokines (e.g., IL-1 and TNF) [1-3]. NF-κB activation in response to these stimuli is typically rapid and transient leading to the highly regulated expression of target genes. The kinetics of activation are normally controlled by a precise interplay between activating and down regulatory signaling events; however, dysregulated constitutive NF-κB activity occurs in diseases hallmarked by chronic inflammation and has been described in a wide range of solid tumors, leukemias, and lymphomas [4, 5]. In light of this pathophysiological role of dysfunctional NF-κB signaling, understanding the molecular events that regulate its activity is the major goal of many basic researchers and the target of intense scrutiny by the pharmaceutical industry.
The NF-κB family contains five structurally related members named p50 and p52 (the N-terminal fragments of the longer NF-κB1/p105 and NF-κB2/p100 proteins, respectively), p65 (RelA), c-Rel, and RelB [2, 6]. These proteins homo- or heterodimerize to form either transcriptionally active (e.g., p50-p65) or repressive (e.g., p50-p50) NF-κB dimers that are normally sequestered inactive in the cytoplasm of resting cells by members of the inhibitory family of IκB proteins. The IκB family is comprised of IκBα, IκBβ, IκBε, and the C-termini of p105 and p100, and the prototypic NF-κB-IκB complex expressed in the majority of cells types is a heterodimer of p50 and p65 associated with IκBα [1, 2]. Following cell stimulation, IκB proteins are rapidly phosphorylated, ubiquitinated, and then degraded by the 26S proteasome unveiling a nuclear localization sequence on the NF-κB proteins (Fig. 1). NF-κB dimers then migrate to the nucleus where they bind to specific sequences termed κB sites within target gene promoters and together with other transcription factors, regulate gene transcription [1, 2].
Fig. 1.
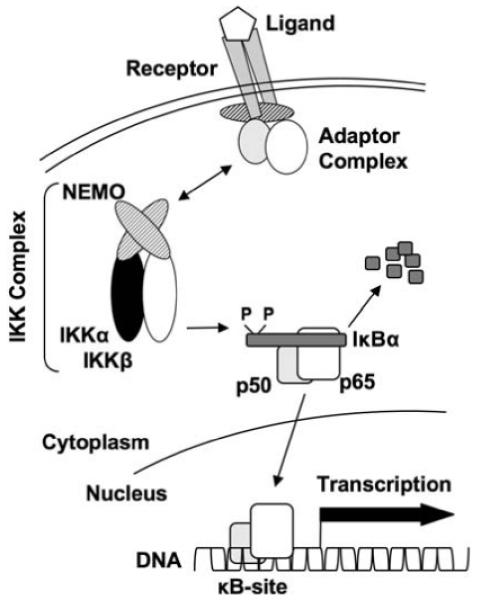
The NF-κB Signaling Pathway: This model depicts the major components of the classical NF-κB pathway. Engagement of a receptor by its ligand (e.g., TNF binding to TNFR) induces the formation of an adaptor protein complex containing adaptor and scaffolding proteins (e.g., TRAFs, MyD88, TIRAP) and kinases (e.g., RIP, IRAK). Assembly of these protein complexes facilitates the recruitment and activation of the IKK complex via NEMO and this in turn leads to the phosphorylation and ultimately ubiquitiniation and proteasomal degradation of IκB proteins (typified by IκBα). IκB degradation reveals a nuclear localization sequence on NF-κB proteins (e.g., a p50-p65 heterodimer) allowing them to migrate to the nucleus, bind to specific κB-sites and regulate target gene transcription. The non-canonical NF-κB pathway follows a similar paradigm; however, the IκB is the C-terminus of NF-κB2/p100 and the resulting nuclear NF-κB species is a p52-RelB heterodimer. The non-canonical pathway does not require NEMO but is dependent upon IKKα and the upstream kinase NIK
The molecular components and signaling events in the many distinct NF-κB activation pathways induced following ligation of separate receptor families (e.g., TNFR, IL-1R, RANK, TCR, BCR, LTβR) are diverse and include the recruitment of wide range of adaptor and scaffolding proteins (e.g., TRAFs, MyD88, TIRAP) and the activation of a number of intermediate kinases (e.g., IRAK, RIP, NIK). Each signaling cascade utilizes unique or overlapping combinations of these upstream intermediates to convey information from the cell surface to the cytoplasmic NF-κB-IκB complex. However, one event that is common to all NF-κB activation pathways is the phosphorylation of the IκB proteins on specific serine residues that tags them for ubiquitination and proteasomal degradation [1, 2, 6, 7]. Given the pivotal role of IκB phosphorylation in all NF-κB signaling cascades, enormous effort from several laboratories in the late 1990s focused on identifying the responsible kinase or kinases. This intense research led to the identification of the high molecular weight IκB kinase (IKK) complex that forms the convergence point for the overwhelming majority of pathways leading to signal-induced NF-κB activation (Figs. 1 and 2) [2, 6-12].
Fig. 2.
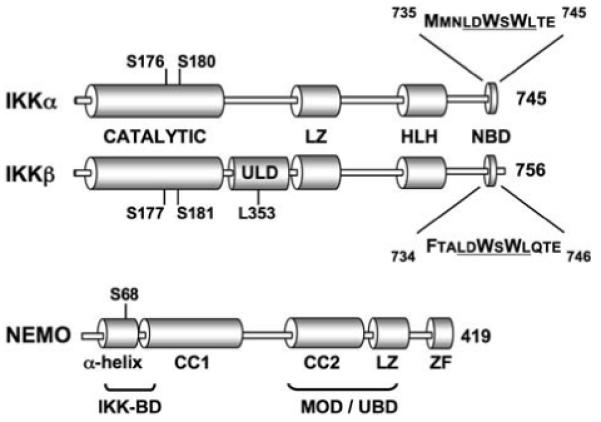
The IκB Kinase complex: The positions of the catalytic, leucine zipper (LZ), helix-loop-helix (HLH), and NEMO binding domains (NBD) of IKKα and IKKβ are shown. IKKβ also contains a novel UbL-Like domain (ULD) that is not present in IKKα [92]. The T loop serine residues within the catalytic domains of both kinases that are required for kinase activation are shown. The NBDs of both kinases are expanded showing the shared hexapeptide sequence (underlined) and the residues that form hydrophobic bonds with NEMO are in larger font [29]. The domain structure of NEMO is depicted (CC, Coiled coil; ZF, zinc finger) and the positions of the N-terminal IKK binding domain (IKK-BD) and the minimal oligomerization domain (MOD) are shown. The MOD also overlaps with a ubiquitin-binding domain (UBD) that is required for IKK activation. Phosphorylation of the serine at position 68 within the IKK-BD negatively regulates IKK complex activity [17]
The IKK complex has been extensively studied at the molecular, biochemical, and genetic levels and remarkably, this has led to the identification and characterization of two distinct NF-κB signaling mechanisms (discussed below). We have also learned a great deal about the mechanisms regulating IKK complex activity; however, a full understanding of the precise biochemical events and molecular interactions that control signal-induced activation and down regulation of IKK remains distant [2, 6, 7]. These gaps in understanding drive our principal research goal to determine the precise mechanisms through which the IKK complex responds to diverse upstream signals. Our overarching hypothesis is that understanding the molecular and biochemical events regulating IKK complex activity will reveal novel and highly selective targets for the development of specific therapeutic strategies targeting pathophysiological NF-κB activation.
The IKK complex
The IKK complex contains two catalytic subunits named IKKα (IKK1) and IKKβ (IKK2), and a non-catalytic regulatory subunit named NEMO (NF-κB essential modulator) or IKKγ (Fig. 2) [2, 6, 7]. IKKα and IKKβ share significant structural identity and each contains an N-terminal catalytic domain, a central leucine zipper motif through which they heterodimerize and a C-terminal helix-loop-helix domain [2, 6, 7]. NEMO contains several separate domains through which it dimerizes, oligomerizes, interacts with the IKKs and associates with upstream signaling intermediates (Fig. 2) [10, 13-21]. It has been proposed that multiple combinations of the IKK complex subunits exist including homodimers of IKKα or IKKβ either associated or distinct from NEMO [22-24], however most biochemical evidence indicates that the majority of cellular IKK complexes contain a core of IKKα-IKKβ-NEMO in the ratio of 1:1:2 [2, 6, 7]. In vitro studies of IKK complex assembly strongly suggest that this combination of subunits can form higher order complexes though multimerization thereby accounting for the high molecular weight observed when the IKK complex is subjected to gel filtration chromatography [25].
Activation of IKK activity requires phosphorylation of either IKKα or IKKβ on two specific serine residues within the T loop of the catalytic domain in each kinase (Fig. 2) [2, 6, 7]. Substitution of these serine residues with alanine renders IKKα or IKKβ catalytically inactive, whereas phosphomimic mutations in which the residues are substituted with glutamic acid generate constitutively active IKKs [2, 6, 7]. The kinase or kinases responsible for T loop phosphorylation remain unclear, however two possible mechanisms have been proposed. The first of these is that the IKKs phosphorylate each other by transautophosphorylation facilitated by either conformational changes within the IKK complex or induced proximity through the oligomerization of multiple core IKK complexes [26]. The second potential mechanism for T loop phosphorylation could be the existence of separate IKK-kinases that target these residues and it is again possible that conformational changes within the complex expose the T loop for phosphorylation by such an IKK-K [2]. Alternatively, recruitment of the IKK complex to upstream receptor/adaptor protein platforms may bring the IKKs into contact with pathway-specific IKK-Ks. It is also feasible that both mechanisms (i.e., transautophosphorylation and an IKK-K) can occur independently or together in a manner that is dependent upon the nature of the specific upstream signaling pathway.
While the specific mechanisms regulating T loop phosphorylation remain to be determined, the critical role of NEMO for induced IKK activation has been definitively established. In this regard, cells lacking NEMO fail to activate NF-κB in response to most pro-inflammatory, immune-regulatory, and pro-survival stimuli [21, 27, 28]. Furthermore NEMO-deficient mice die during embryogenesis through massive hepatocyte apoptosis due to the inability to activate pro-survival NF-κB signaling in response to TNF in the developing liver [27, 28]. Exactly how NEMO functions to activate the IKK complex is the focus of ongoing research by a number of laboratories and a model for this function is beginning to emerge. NEMO associates as a dimer with both IKKα and IKKβ [16, 29] and it can also oligomerize via a minimal oligomerization domain (MOD) (Fig. 2) [13, 20]. Induced oligomerization of NEMO following stimulation appears to be essential for IKK activation [13, 14, 16-20, 25] and mutations within the MOD block IKK activation [13, 20]. The precise role of oligomerization is not yet known, but it may facilitate IKK transautophosphorylation or T loop phosphorylation by an IKK-K through induced proximity or conformational changes in the complex [19]. Much work remains to be done to define the specific molecular and biochemical details of IKK complex activation, however signal-induced NEMO oligomerization clearly plays a major role in this process.
In addition to functioning in the activation of the IKK complex, NEMO also appears to control the downregulation phase of transient IKK activity. NEMO serves as the docking platform for several negative regulatory proteins including the deubiquitinases A20 and CYLD [30-32] and the protein phosphatases PP2A and PP2C [33-35] that may inhibit IKK activity by removing phosphates from the T loop serines [8]. It is not clear how NEMO switches roles from a regulator of IKK activation to a mediator of repression, but several lines of evidence point to a potential mechanism. First of these was our demonstration that an IKKβ mutant unable to bind to NEMO exhibited enhanced basal catalytic activity suggesting that it cannot be “turned off” [36] and implying that the interaction of IKKβ with NEMO is required for the kinase to be downregulated. So why do cells lacking NEMO not display constitutive IKK activity? The answer to this question remains to be determined, but likely hinges on the crucial role of NEMO in providing the initial activation signal for IKK activity. Additional evidence for a mutual negative regulatory interaction between IKKβ and NEMO was obtained from studies showing that IKKβ mutants harboring phosphomimic mutations at serine 740 (Fig. 2) within the C-terminal region required for NEMO interaction (Fig. 2; see below) were inactive [19, 36, 37]. Furthermore Palkowitsch and co-workers recently demonstrated that specific phosphorylation of NEMO on serine 68 within the IKK-binding region (see below) leads to downregulation of IKK activity [17]. These accumulated findings support a model in which IKK C-terminal autophosphorylation and phosophorylation of NEMO within the IKK-binding region leads to conformational changes that possibly recruit negative regulatory proteins to inhibit IKK function [17, 19, 36, 37]. This negative feedback model represents a working hypothesis [2] and studies are underway to determine whether this and/or other as yet unknown mechanisms are responsible for IKK downregulation following NEMO-dependent activation.
The IKK-NEMO interaction
The crucial role of NEMO in regulating inducible IKK activity led us, and others to determine the mechanism through which NEMO associates with the IKK complex. Interaction analysis of truncation mutants identified the N-terminal region of NEMO (residues 47–111) encompassing an α-helical domain as necessary and sufficient for association with IKKα and IKKβ (Fig. 2) [16, 29, 38]. This portion of NEMO forms an SDS-resistant dimer and Marienfeld et al. demonstrated that distinct α-helical subdomains within this region are required for dimerization and NEMO binding, respectively [16]. Recent elegant crystallographic studies have confirmed these accumulated biochemical data and revealed that this dimeric conformation forms two 800Å α-helical IKK-binding pockets [29].
A similar truncation mutation approach identified a small region within the extreme C-termini of IKKα and IKKβ that is necessary for their interaction with NEMO (Fig. 2) [36, 38]. We named this region the NEMO binding domain (NBD) and initially described this as the six residues (LDWSWL) shared between 738–743 in IKKα and 737–742 in IKKβ [36, 38]. Subsequent biochemical and structural analysis extended the minimal NBD to include the 11 amino acids surrounding this hexapeptide in IKKβ [39], and the residues F734 in IKKβ and M735 in IKKα were recently shown to be required for binding to NEMO [29]. Remarkably, the NBD is evolutionarily conserved and its existence in oyster and zebrafish IKK demonstrates the importance of this specific molecular interaction for NF-κB activation [40-42].
Detailed mutational analysis of the IKKβ NBD demonstrated that D738, W739, and W741 are critical for the interaction with NEMO [36, 38]. The equivalent aspartate and tryptophans in the IKKα NBD are also necessary for its binding to NEMO; however, we found that the absolute requirement for these residues in IKKα is less stringent [36]. This difference possibly accounts for the higher affinity of IKKβ for NEMO compared with IKKα [36] although Rushe and colleagues recently confirmed the importance of these residues in both kinases for the overall architecture of the binding site [29]. Consequently the precise reasons for the different affinities of IKKα and IKKβ for NEMO are not known but as we will discuss later, determining this may help to understand the distinct functions of the IKKs.
The recently solved crystal structure of the NEMO-IKK interaction demonstrates that the two NBD trpytophan residues together with F734 in IKKβ or M735 in IKKα form hydrophobic interactions with residues in the α-helical binding cleft in NEMO [29]. The leucine residues flanking the shared hexapeptide sequence most likely form a seal enclosing the tryptophans that excludes water from the hydrophobic pocket. In addition, D738 and S740 form an intramolecular bond via a salt bridge that kinks the backbone of the NBD and D738 also forms a hydrogen bond with R101 in NEMO [29]. Disruption of the D738-S740 interaction by phosphorylation of S740 (see above) [17, 19, 36] may therefore alter the way that the tryptophans insert into the NEMO pocket and generate a conformational change that affects the overall structure of the interaction [29].
Our original biochemical analysis led us to predict that the NBD was an interaction “hotspot” that is defined as a region at the center of a binding interface rich in tryptophan, tyrosine, and arginine residues that frequently also contains aspartate [36, 43]. Another characteristic required to maintain high-energy hydrophobic interactions in binding hotspots is the occlusion of solvent [43] and as discussed above, the structure of the NBD strongly suggests that this occurs within this region. Overall therefore the combined biochemical and structural analysis of the NBD predicts that the NEMO-IKK interaction is an ideal target for the development of small molecule inhibitors designed to disrupt the IKK complex [29, 36, 38, 39, 44]. In the following section we will briefly discuss an approach using cell-penetrating peptides (CPPs) that we successfully employed to test this hypothesis.
The NEMO binding domain peptide
To determine whether the NEMO-IKK interaction could be blocked in vitro we generated a peptide encompassing residues 735–745 of IKKβ and tested this in binding assays using recombinant IKKα, IKKβ, and NEMO [36, 38]. We found that the wild type, but not a mutant version of the NBD containing alanines at the critical tryptophan positions (W739A and W741A), inhibited the interaction of NEMO with both kinases [36, 38]. These assays revealed the distinct differences in affinity of the IKKs for NEMO as blocking the IKKβ interaction required higher concentrations of NBD peptide than that which inhibited IKKα [36]. Furthermore the NBD peptide could disrupt preformed IKK complexes suggesting that it readily competes with and displaces the IKKs from the hydrophobic binding pocket in NEMO [36]. Subsequent workers used similar approaches to define the precise residues constituting the minimal NBD required for maximal inhibition in vitro [29, 39] and it was suggested recently that a significantly longer peptide (i.e., 705–745 of IKKβ) binds with the highest affinity to NEMO [44]. This finding would indicate that as yet unknown N-terminal residues play a role in binding to NEMO, however this remains to be resolved as it was not apparent from the crystal structure of the NEMO-IKK interaction [29].
The ability to block the interaction in vitro led us to question whether the NBD peptide could be delivered to cells to disrupt NF-κB signaling. To accomplish this we utilized the ability of an expanding list of small naturally occurring or synthetic peptides to traverse cell membranes [45-48]. These peptides are named CPPs and coupling of bioactive cargo such as an inhibitory peptide to a CPP allows cellular uptake of the otherwise refractory cargo [45-48]. We therefore generated a fusion consisting of the IKKβ NBD (735–745) with a CPP derived from the drosophila antennapedia protein named AntP that is one of the most widely used and best-characterized CPPs [49]. Remarkably this AntP-NBD fusion peptide rapidly entered HeLa cells and blocked both TNF- and IL-1-induced NF-κB activation, and consistent with our in vitro interaction studies, a mutant version of peptide containing W→A mutations did not block NF-κB signaling [38].
Cellular delivery of the NBD peptide using separate CPPs has been demonstrated by a number of groups [46, 50-55]. These CPPs include a peptide derived from the HIV TAT protein and a synthetic CPP modeled on TAT named PTD-5 [46]. In each case, however, the resulting NBD peptide has been shown to block NF-κB signaling in a range of cell types and to inhibit responses in cellular models of immune activation, inflammation, infection, cell survival, and cancer [46, 50-55]. For example, the wild type NBD but not the mutant control peptide inhibited TNF-induced leukocyte adhesion molecule expression by vascular endothelial cells and NO release by macrophages, both of which are NF-κB-dependent responses [38]. The NBD also blocked osteoclast differentiation in vitro [52, 56] and has been extensively reported to inhibit the activation of intestinal epithelial cells, macrophages, neutrophils, dendritic cells, T cells, and B cells [57]. Furthermore, the wild type NBD but not the mutant peptide effectively blocks dysregulated constitutive NF-κB activity and promotes cell death in cancer cell lines including those derived from pancreatic and breast tumors and multiple myeloma [57].
The most exciting feature of the NBD peptide is its ability to function in vivo. This was first demonstrated in mouse models of peritonitis and edema where intraperitoneal (i.p.) injection or topical administration of the peptide, respectively blocked inflammation [38]. The NBD peptide has now been shown to function in an array of in vivo models of both acute and chronic inflammatory disease including blockade of osteoclastogenesis, bone erosion and joint destruction in collagen-induced arthritis [52, 56], and inhibition of pro-inflammatory cytokine expression and bowel injury in separate models of colitis [58-60]. A remarkable characteristic of CPPs is their ability to transduce cargo across the blood–brain barrier and consistent with this, i.p. injection of the NBD effectively alleviated pathology in mouse and rat models of inflammation-induced brain injury [61-64] and a murine model of Parkinson’s disease [65]. In each of these studies the WT NBD was tested alongside the mutant peptide that was uniformly ineffective. Furthermore, the many separate routes of administration used to deliver the NBD (including i.p., i.v., topical, intranasal, intracranial) demonstrate that it can function systemically and reach distant tissue sites although the precise pharmacokinetics remain to be determined [57]. Finally, several reports have demonstrated that the NBD peptide blocks inflammation in large animal models of disease including a piglet model of acute respiratory distress syndrome [66-68]. This therefore supports the exciting promise that this approach holds for possible future translational studies in the clinic.
These accumulated in vitro and in vivo studies of the NBD confirm our hypothesis that dissecting specific molecular interactions within the IKK complex identifies targets for the development of potentially effective therapeutic strategies. Nevertheless, while the NBD peptide serves as a convincing “proof of principle” for this approach, much remains to be accomplished before reagents targeting this domain can be brought to clinical trials. We need to obtain a full understanding of the pharmacokinetics and potential toxicity of the NBD and we must establish the effects of long-term treatment. However it is possible that the NBD peptide will serve as a model for the future development of new generations of inhibitors such as small molecules or peptidomimetics designed to insert into the IKK binding groove of NEMO [29]. One further question that remains to be addressed is the potential differential effects that the NBD peptide may have on the activity of IKKα and IKKβ. This is particularly important to determine as we will describe in the following section the surprising finding that these kinases regulate completely separate NF-κB signaling pathways.
Two pathways to NF-κB
Despite their significant similarities, a series of elegant genetic studies revealed that IKKα and IKKβ in fact play distinct roles within the overall paradigm of NF-κB signaling [2, 6, 7, 69]. In this regard, rapid and transient TNF-induced IκBα degradation occurs though a pathway that depends upon IKKβ and NEMO. Like NEMO-deficient animals, mice lacking IKKβ died during development from massive TNF-induced hepatocyte apoptosis due to the inability to mount an NF-κB-dependent anti-apoptotic response [70, 71]. This phenotype was also observed in mice lacking the p65 NF-κB subunit [72] thereby genetically linking NEMO, IKKβ, and p65 in a common signaling cascade. This NF-κB signaling pathway is now named the “classical” mechanism and it is defined as NEMO- and IKKβ-dependent IκB phosphorylation and degradation liberating NF-κB complexes typified by the ubiquitous p50-p65 heterodimer (model depicted in Fig. 1). The classical pathway regulates the vast majority of genes activated by NF-κB including those encoding pro-inflammatory and immunomodulatory cytokines (e.g., IL-1, IL-2, IL-6, TNF), chemokines (e.g., CXCL8, CCL2, CCL3), leukocyte adhesion molecules (e.g., E-selectin, ICAM-1, and VCAM-1), and various pro-survival and anti-apoptotic genes (e.g., Bcl2, Bcl-XL, XIAP) [2, 69]. In the current model of classical NF-κB activation, all stimuli that induce IκBα degradation including TNF, IL-1, LPS, and antigen-receptor engagement, are considered to be activators of the NEMO- and IKKβ-dependent classical NF-κB pathway [2, 69]. However, as we will discuss later, our recent findings question the rigid requirements for these specific IKK complex subunits for classical signaling to occur.
Analysis of knock-in mice harboring inactive mutant IKKα revealed an unpredicted role for this kinase in processing of the IκB-like C-terminus of NF-κB2/p100 to generate p52 [73]. The NF-κB2/p100 targeted by IKKα is complexed with RelB and its processing results in the generation of p52-RelB NF-κB dimers that migrate to the nucleus. This mechanism is named the non-canonical or alternative pathway and it functions in cells lacking both IKKβ and NEMO [73-76]. The non-canonical pathway is absolutely dependent upon an upstream kinase named NIK (NF-κB-inducing kinase) that phosphorylates and activates IKKα, and p100 processing is absent in alymphoplasia mice that have a mutated NIK gene (NIKaly/aly) [77]. NIK is rapidly turned over in resting cells through the ubiquitin ligase activity of the TRAF3 adaptor protein that ubiquitinates NIK triggering its proteasomal degradation [78]. Ligation of non-canonical pathway-inducing receptors sequesters TRAF3 leading to the accumulation of newly synthesized NIK and activation of IKKα [78]. This requirement for new protein synthesis and the longer kinetics of activation compared with classical signaling are hallmarks of the non-canonical pathway [73-76].
Non-canonical NF-κB signaling is induced by ligation of a subset of TNF receptor family members including the lymphotoxin-β receptor (LTβR), CD40, RANK, and BAFF-R (B cell-activating factor receptor). Most stimuli that activate the classical pathway such as TNF, IL-1, and antigen-receptor ligation do not activate the non-canonical pathway; however, the non-canonical stimuli can activate both NF-κB pathways [69]. The major functions of non-canonical NF-κB (i.e., p52-RelB) are in regulating lymphoid organogenesis and B cell survival and maturation and mice lacking components of the non-canonical pathway (i.e., NIK, IKKα, p100, or RelB) exhibit defects in these developmental processes [2, 69]. Consistent with the phenotype of these animals, the limited panel of non-canonical NF-κB-dependent genes include BAFF (the ligand for BAFF-R) and the chemokines CXCL12, CXCL13, CCL19, and CCL21, that function during lymphoid organogenesis.
These accumulated findings demonstrate that signaling via the IKK complex regulates two distinct pathways that utilize separate subunits of the complex. Many questions remain concerning the molecular mechanisms underlying the distinct roles of the IKKs and whether both pathways indeed use the tripartite IKK holocomplex containing IKKα, IKKβ, and NEMO. In this regard, the non-canonical pathway is commonly considered to utilize an IKKα homodimer; however, little biochemical evidence supports the existence of this separate complex [22-24]. We have focused our attention on two specific questions regarding the distinct roles of IKKα and IKKβ within the core complex. The first of these concerns the ability of IKKα to bind via its NBD to NEMO. The current models of classical and non-canonical signaling suggest that the interaction of NEMO with IKKα is redundant however, as we will discuss in the next section, we have identified a novel role for NEMO and IKKα in the classical NF-κB pathway. Our second area of interest lies in determining the molecular basis of the distinct functions of the IKKs. IKKα and IKKβ share extensive sequence and structural identity (Fig. 2), however as we will describe, we have identified a novel domain present only in IKKβ and we are currently seeking to determine the precise role of this domain in NF-κB signaling.
Why does IKKα associate with NEMO?
The role of IKKα in regulating non-canonical NF-κB signaling does not require its ability to bind to NEMO as the pathway is activated in NEMO-deficient cells [69, 73-76]. Furthermore, classical NF-κB signaling in response to TNF occurs in the absence of IKKα suggesting that the interaction of IKKα with NEMO plays no role in regulating this pathway [71, 79]. Despite this however, several lines of evidence indicate that IKKα may indeed play a role in classical pathway activation. A number of groups have reported that NF-κB-dependent gene expression in response to some classical pathway stimuli occurs in IKKβ-deficient murine embryonic fibroblasts (MEFs) [70, 80, 81] and Cao et al. demonstrated that IKKα is the key kinase responsible for Receptor Activator of NF-κB (RANK)-induced classical NF-κB activation in mammary epithelial cells [82]. In light of these discrepancies we sought to determine whether the interaction of NEMO with IKKα plays a role in regulating classical NF-κB signaling.
To address this question, we examined classical NF-κB activation in separate lines of MEFs lacking each of the IKK complex subunits. We stimulated these cells with TNF and IL-1 and consistent with previous reports both cytokines-induced IκBα degradation in WT and IKKα−/− MEFs, but not in NEMO-deficient cells [83]. Surprisingly however, although TNF did not induce IκBα degradation in IKKβ-deficient MEFs, IL-1-induced IκBα degradation and classical NF-κB (p50-p65) migration to the nucleus was intact in these cells [83]. This suggested that IL-1 but not TNF could activate the classical pathway in a NEMO- and IKKα-dependent manner and to explore this hypothesis, we treated IKKβ−/− MEFs with the NBD peptide. We found that the NBD peptide completely inhibited IL-1-induced NF-κB activation in these cells demonstrating that IL-1 induced NF-κB requires the interaction of IKKα with NEMO and occurs in the absence of IKKβ.
Our findings led us to surmise that the current model of classical NF-κB signaling is incomplete and that the interaction of IKKα with NEMO plays a role in its activation in response to at least a subset of stimuli. While it is clear that NEMO is critical for all classical NF-κB pathways, IL-1 and TNF have different absolute requirements for the IKK subunits necessary for phosphorylating IκBα (Fig. 3). We speculate that the separate upstream signaling cascades differentially interface with NEMO resulting in distinct downstream IKK activation profiles. This notion is supported by studies showing that separate domains of NEMO are required for IL-1- and TNF-induced signaling [15, 84]. It is therefore possible that association of the terminal signaling components of each pathway with these discrete NEMO domains directs the activation of either IKKβ alone in response to TNF or in the case of IL-1, either IKKβ or IKKα. Further studies exploring the function of the separate regions of NEMO and the downstream kinases activated by diverse signaling pathways are clearly required and these may identify novel targets for selectively disrupting pro-inflammatory signaling via the IKK complex. In addition, it will be necessary to determine on a signal by signal basis which stimuli activate the classical pathway in a strictly NEMO–IKKβ-dependent manner and which can utilize both IKKβ and IKKα. One critical future experiment that will help to elucidate these issues will be the generation of a knock-in mouse strain lacking only the IKKα NBD. Such a model will provide valuable insight into both the significance of this interaction in vivo and the potential outcome of targeting the IKKα NBD using selective pharmacological approaches.
Fig. 3.

Distinct modes of IKK complex activity transduce TNF and IL-1 signaling to NF-κB: Our findings verify that TNF-induced IκBα degradation and classical NF-κB activation is critically dependent upon NEMO and IKKβ (left). Similarly IL-1 signaling absolutely requires intact NEMO (right). However, IL-1-induced IκBα degradation, NF-κB nuclear translocation and NF-κB transcriptional activity occurs in the absence of IKKβ demonstrating that NEMO and IKKα can form a signaling complex capable of activating the classical NF-κB pathway in response to certain pro-inflammatory stimuli [83]
It will also be intriguing to determine whether the interaction of NEMO with IKKα plays a role in regulating any of the non-signaling functions of the kinase. In this regard IKKα has been shown to regulate keratinocyte development in a manner that is unrelated to its catalytic activity [79, 85] and it is not known whether this function requires the association of NEMO with IKKα. A genetic model of mice lacking the IKKα NBD would allow this to be investigated in detail. Furthermore, IKKα has been shown to translocate to the nucleus where it regulates NF-κB-dependent gene expression by either phosphorylating histones [86, 87] or NF-κB proteins themselves [88, 89]. Again it is not clear whether these nuclear functions of IKKα require its association with NEMO. Establishing whether these non-signaling functions of IKKα are regulated via its interaction with NEMO will be critical for fully determining the potential benefits or pitfalls of therapeutically targeting the NBD.
Why are IKKα and IKKβ functionally distinct?
Despite existing in the same holocomplex, IKKα and IKKβ clearly exhibit distinct functions in the regulation of classical and non-canonical NF-κB signaling. As we described above, IKKα also regulates keratinocyte development and controls nuclear NF-κB activity although these functions may represent the activity of an IKKα pool separate from the tripartite complex with IKKβ and NEMO. Furthermore IKKβ phosphorylates non-IκB substrates including the adaptor proteins Bcl10 and CARMA that are required for antigen receptor-induced NF-κB activation [2, 3, 6, 7] and we have shown that IKKβ but not IKKα regulates caspase-independent cell death although the precise molecular target of IKKβ in this response is not known [90]. IKKα and IKKβ share over 50% sequence identity, their domain structures are similar (Fig. 2) and both IKKs associate with NEMO [2, 6, 7]. In light of these sequence and structural similarities, the precise molecular basis for the distinct functions of IKKα and IKKβ is unclear.
One possibility is that the different affinity of the kinases for NEMO contributes to their functional differences [36]. Evidence supporting this concept came from experiments by Kwak and colleagues who swapped the C-termini containing the NBDs of IKKα and IKKβ and found that IKKα with the IKKβ C-terminus functioned similarly to IKKβ in the classical pathway [91]. Based on these findings Hayden and Ghosh have suggested that the lower affinity of IKKα for NEMO may allow NIK access to the T loop of IKKα but not IKKβ; however, this hypothesis remains to be formally tested [2]. Another difference that exists between the kinases that has to date been overlooked is the fact that IKKβ is 12 amino acids longer than IKKα. This extension occurs at the C-terminus immediately after the NBD and contains five negatively charged glutamic acid residues [37, 38]. It is possible that this highly charged region of IKKβ contributes to the functional differences between the kinase although this remains to be addressed.
In seeking to identify possible differences that may exist between the IKKs, we performed a series of proteomic database searches using web-based proWle, motif and protein family analysis programs. Surprisingly, analysis of the complete amino acid sequence of human IKKβ using the ExPASy (Expert Protein Analysis System) molecular biology server and the PROSITE database identified a region of 77 amino acids from L307 to M384 corresponding to the Ubiquitin_2 (Type 2 ubiquitin-like) profile (PROSITE document PDOC00271; PROSITE accession number PS50053). A similar domain was not detected in IKKα demonstrating that this region is unique for IKKβ [92]. This domain is similar to the integral ubiquitin-like (UbL) domains that are present in a family of functionally divergent proteins [93] and we therefore named this region the UbL-like domain (ULD) of IKKβ (Fig. 2) [92].
To determine whether the IKKβ ULD was required for activity of the kinase, we generated a deletion mutant lacking the region and found that this failed to activate NF-κB in response to IL-1 or TNF [92]. The mutant was incorporated into the core IKK complex showing that the ULD plays no role in the intermolecular interactions between the IKKs and NEMO. More directed site-specific mutations within the ULD identified the leucine at position 353 (Fig. 2) as necessary for IKKβ to activate NF-κB-dependent gene expression [92]. Remarkably, however, the L353 mutant was able to phosphorylate IκBα suggesting that the defect in NF-κB activation was not at the level of inhibition of IκB degradation. L353 aligns with the conserved hydrophobic patch spanning isoleucine 44 in ubiquitin that allows ubiquitin to associate with its multiple interaction partners [93]. Several UbL domain-containing proteins also use this hydrophobic patch to interact with ubiquitin-binding domains (UBDs) in target proteins and the IKK-related kinase TBK1 was shown recently to contain a ULD through which it interacts with its substrate interferon-regulatory factor (IRF)-3 [94]. However, the IKKβ ULD does not appear to play a role in IκB protein interaction suggesting that these domains have evolved distinct roles to regulate the signaling functions of IKKβ and TBK1 [92]. Instead we found that the IKKβ ULD mutants strongly associated with the NF-κB p65 subunit in the absence or presence of stimuli, whereas wild-type IKKβ could not be detected in a complex with p65 [92]. These findings led us to propose a model in which the ULD plays a role in release of NF-κB following IκBα phosphorylation and we are addressing this hypothesis in our ongoing studies.
It remains to be determined whether the ULD accounts for the differences in function between the IKKs; however, deletion of the corresponding region in IKKα did not affect its ability to regulate the non-canonical pathway [92]. In-depth structure-function analysis of the ULD is clearly required to define its role in IKKβ regulation but it is tempting to speculate that the ULD provides a promising target for drugs aimed at specifically blocking the activity of IKKβ and the classical NF-κB pathway while maintaining IKKα function and non-canonical signaling.
Conclusion
We have learned an enormous amount about the IKK complex since its discovery 11 years ago, yet many important questions concerning the regulation of its activity remain to be answered [2, 6, 7]. Finding solutions to these questions will be critical if we are to fully exploit the IKK complex as a target for the development of drugs aimed at blocking NF-κB signaling. The studies we have described here open up new avenues of research in this area and highlight the importance of understanding the molecular and biochemical events controlling the diverse functions of the IKK complex. Given the multiple roles of NF-κB signaling in immune, inflammatory, developmental, and cell survival responses, a major goal in therapeutically targeting NF-κB will be the development of pathway-specific drugs that block aberrant signaling and maintain normal function. Understanding the precise biochemical events and molecular interactions that regulate the IKK complex will help us to attain this crucial goal, and finding these answers will keep researchers busy for a long time to come.
Acknowledgments
Work in the May laboratory is supported by the National Institutes of Health (NIH/NHLBI: 1RO1HL080612) and the WW Smith Charitable Trust (H0703). Laura Solt is a graduate student in the Immunology Graduate Group and is supported by NIH/NIAID 5T32AI055428.
Abbreviations
- CPP
Cell penetrating peptide
- IKK
IκB kinase
- IκB
Inhibitor of NF-κB
- NBD
NEMO binding domain
- NEMO
NF-κB essential modulator
- NF-κB
Nuclear factor-kappa B
- NIK
NF-κB-inducing kinase
- ULD
Ubiquitin like domain
Contributor Information
Laura A. Solt, Department of Animal Biology, University of Pennsylvania School of Veterinary Medicine, 3800 Spruce Street (OVH 200E), Philadelphia, PA 19104-6045, USA
Michael J. May, Department of Animal Biology, University of Pennsylvania School of Veterinary Medicine, 3800 Spruce Street (OVH 200E), Philadelphia, PA 19104-6045, USA; Mari Lowe Center for Comparative Oncology, University of Pennsylvania School of Veterinary Medicine, 3800 Spruce Street, Philadelphia, PA 19104-6045, USA
References
- 1.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–4. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 2.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–62. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 3.Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–80. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 4.Courtois G, Gilmore TD. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–43. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 5.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 6.Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25:6685–705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- 7.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;357:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 8.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–54. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 9.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–6. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 10.Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 11.Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV. IkappaB kinase-beta: NF-kappaB activation and complex formation with IkappaB kinase-alpha and NIK. Science. 1997;278:866–9. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 12.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–52. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 13.Agou F, Courtois G, Chiaravalli J, Baleux F, Coic YM, Traincard F, et al. Inhibition of NF-kappa B activation by peptides targeting NF-kappa B essential modulator (nemo) oligomerization. J Biol Chem. 2004;279:54248–57. doi: 10.1074/jbc.M406423200. [DOI] [PubMed] [Google Scholar]
- 14.Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, et al. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–31. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- 15.Makris C, Roberts JL, Karin M. The carboxyl-terminal region of IkappaB kinase gamma (IKKgamma) is required for full IKK activation. Mol Cell Biol. 2002;22:6573–81. doi: 10.1128/MCB.22.18.6573-6581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marienfeld RB, Palkowitsch L, Ghosh S. Dimerization of the I kappa B kinase-binding domain of NEMO is required for tumor necrosis factor alpha-induced NF-kappa B activity. Mol Cell Biol. 2006;26:9209–19. doi: 10.1128/MCB.00478-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palkowitsch L, Leidner J, Ghosh S, Marienfeld RB. Phosphorylation of serine 68 in the IkappaB kinase (IKK)-binding domain of NEMO interferes with the structure of the IKK complex and tumor necrosis factor-alpha-induced NF-kappaB activity. J Biol Chem. 2008;283:76–86. doi: 10.1074/jbc.M708856200. [DOI] [PubMed] [Google Scholar]
- 18.Poyet JL, Srinivasula SM, Lin JH, Fernandes-Alnemri T, Yamaoka S, Tsichlis PN, et al. Activation of the Ikappa B kinases by RIP via IKKgamma/NEMO-mediated oligomerization. J Biol Chem. 2000;275:37966–77. doi: 10.1074/jbc.M006643200. [DOI] [PubMed] [Google Scholar]
- 19.Schomer-Miller B, Higashimoto T, Lee YK, Zandi E. Regulation of IkappaB kinase (IKK) complex by IKKgamma-dependent phosphorylation of the T-loop and C terminus of IKKbeta. J Biol Chem. 2006;281:15268–76. doi: 10.1074/jbc.M513793200. [DOI] [PubMed] [Google Scholar]
- 20.TegethoV S, Behlke J, Scheidereit C. Tetrameric oligomerization of IkappaB kinase gamma (IKKgamma) is obligatory for IKK complex activity and NF-kappaB activation. Mol Cell Biol. 2003;23:2029–41. doi: 10.1128/MCB.23.6.2029-2041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, et al. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;93:1231–40. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- 22.Fontan E, Traincard F, Levy SG, Yamaoka S, Veron M, Agou F. NEMO oligomerization in the dynamic assembly of the IkappaB kinase core complex. FEBS J. 2007;274:2540–51. doi: 10.1111/j.1742-4658.2007.05788.x. [DOI] [PubMed] [Google Scholar]
- 23.Khoshnan A, Kempiak SJ, Bennett BL, Bae D, Xu W, Manning AM, et al. Primary human CD4 + T cells contain heterogeneous I kappa B kinase complexes: role in activation of the IL-2 promoter. J Immunol. 1999;163:5444–52. [PubMed] [Google Scholar]
- 24.Mercurio F, Murray BW, Shevchenko A, Bennett BL, Young DB, Li JW, et al. IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol. 1999;19:1526–38. doi: 10.1128/mcb.19.2.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drew D, Shimada E, Huynh K, Bergqvist S, Talwar R, Karin M, et al. Inhibitor kappaB kinase beta binding by inhibitor kappaB kinase gamma. Biochemistry. 2007;46:12482–90. doi: 10.1021/bi701137a. [DOI] [PubMed] [Google Scholar]
- 26.Tang ED, Inohara N, Wang CY, Nunez G, Guan KL. Roles for homotypic interactions and trans-autophosphorylation in IkappaB kinase beta IKKbeta) activation [corrected] J Biol Chem. 2003;278:38566–70. doi: 10.1074/jbc.M304374200. [DOI] [PubMed] [Google Scholar]
- 27.Makris C, Godfrey VL, Krahn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, et al. Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol Cell. 2000;5:969–79. doi: 10.1016/s1097-2765(00)80262-2. [DOI] [PubMed] [Google Scholar]
- 28.Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, et al. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 2000;14:854–62. [PMC free article] [PubMed] [Google Scholar]
- 29.Rushe M, Silvian L, Bixler S, Chen LL, Cheung A, Bowes S, et al. Structure of a NEMO/IKK-associating domain reveals architecture of the interaction site. Structure. 2008;16:798–808. doi: 10.1016/j.str.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 30.Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 31.Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424:801–5. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 32.Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12:301–11. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
- 33.Fu DX, Kuo YL, Liu BY, Jeang KT, Giam CZ. Human T-lymphotropic virus type I tax activates I-kappa B kinase by inhibiting I-kappa B kinase-associated serine/threonine protein phosphatase 2A. J Biol Chem. 2003;278:1487–93. doi: 10.1074/jbc.M210631200. [DOI] [PubMed] [Google Scholar]
- 34.Hong S, Wang LC, Gao X, Kuo YL, Liu B, Merling R, et al. Heptad repeats regulate protein phosphatase 2a recruitment to I-kappaB kinase gamma/NF-kappaB essential modulator and are targeted by human T-lymphotropic virus type 1 tax. J Biol Chem. 2007;282:12119–26. doi: 10.1074/jbc.M610392200. [DOI] [PubMed] [Google Scholar]
- 35.Prajapati S, Verma U, Yamamoto Y, Kwak YT, Gaynor RB. Protein phosphatase 2Cbeta association with the IkappaB kinase complex is involved in regulating NF-kappaB activity. J Biol Chem. 2004;279:1739–46. doi: 10.1074/jbc.M306273200. [DOI] [PubMed] [Google Scholar]
- 36.May MJ, Marienfeld RB, Ghosh S. Characterization of the Ikappa B-kinase NEMO binding domain. J Biol Chem. 2002;277:45992–6000. doi: 10.1074/jbc.M206494200. [DOI] [PubMed] [Google Scholar]
- 37.Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284:309–13. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- 38.May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000;289:1550–4. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 39.Strnad J, McDonnell PA, Riexinger DJ, Mapelli C, Cheng L, Gray H, et al. NEMO binding domain of IKK-2 encompasses amino acids 735–745. J Mol Recognit. 2006;19:227–33. doi: 10.1002/jmr.766. [DOI] [PubMed] [Google Scholar]
- 40.Correa RG, Matsui T, Tergaonkar V, Rodriguez-Esteban C, Izpisua-Belmonte JC, Verma IM. Zebrafish IkappaB kinase 1 negatively regulates NF-kappaB activity. Curr Biol. 2005;15:1291–5. doi: 10.1016/j.cub.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 41.Escoubas JM, Briant L, Montagnani C, Hez S, Devaux C, Roch P. Oyster IKK-like protein shares structural and functional properties with its mammalian homologues. FEBS Lett. 1999;453:293–8. doi: 10.1016/s0014-5793(99)00737-1. [DOI] [PubMed] [Google Scholar]
- 42.Xiong X, Feng Q, Chen L, Xie L, Zhang R. Cloning and characterization of an IKK homologue from pearl oyster, Pinctada fucata. Dev Comp Immunol. 2008;32:15–25. doi: 10.1016/j.dci.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 43.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 44.Lo YC, Maddineni U, Chung JY, Rich RL, Myszka DG, Wu H. High-affinity interaction between IKKbeta and NEMO. Biochemistry. 2008;47:3109–16. doi: 10.1021/bi702312c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deshayes S, Morris MC, Divita G, Heitz F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell Mol Life Sci. 2005;62:1839–49. doi: 10.1007/s00018-005-5109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tilstra J, Rehman KK, Hennon T, Plevy SE, Clemens P, Robbins PD. Protein transduction: identification, characterization and optimization. Biochem Soc Trans. 2007;35:811–5. doi: 10.1042/BST0350811. [DOI] [PubMed] [Google Scholar]
- 47.Wadia JS, Dowdy SF. Protein transduction technology. Curr Opin Biotechnol. 2002;13:52–6. doi: 10.1016/s0958-1669(02)00284-7. [DOI] [PubMed] [Google Scholar]
- 48.Zorko M, Langel U. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv Drug Deliv Rev. 2005;57:529–45. doi: 10.1016/j.addr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 49.Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, Prochiantz A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J Biol Chem. 1996;271:18188–93. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- 50.Choi M, Rolle S, Wellner M, Cardoso MC, Scheidereit C, Luft FC, et al. Inhibition of NF-kappaB by a TAT-NEMO-binding domain peptide accelerates constitutive apoptosis and abrogates LPS-delayed neutrophil apoptosis. Blood. 2003;102:2259–67. doi: 10.1182/blood-2002-09-2960. [DOI] [PubMed] [Google Scholar]
- 51.Clohisy JC, Yamanaka Y, Faccio R, Abu-Amer Y. Inhibition of IKK activation, through sequestering NEMO, blocks PMMA-induced osteoclastogenesis and calvarial inflammatory osteolysis. J Orthop Res. 2006;24:1358–65. doi: 10.1002/jor.20184. [DOI] [PubMed] [Google Scholar]
- 52.Dai S, Hirayama T, Abbas S, Abu-Amer Y. The I{kappa}B Kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks osteoclastogenesis and bone erosion in in Xammatory arthritis. J Biol Chem. 2004;279:37219–22. doi: 10.1074/jbc.C400258200. [DOI] [PubMed] [Google Scholar]
- 53.Jones SW, Christison R, Bundell K, Voyce CJ, Brockbank SM, Newham P, et al. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br J Pharmacol. 2005;145:1093–102. doi: 10.1038/sj.bjp.0706279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rehman KK, Bertera S, Bottino R, Balamurugan AN, Mai JC, Mi Z, et al. Protection of islets by in situ peptide-mediated transduction of the Ikappa B kinase inhibitor Nemo-binding domain peptide. J Biol Chem. 2003;278:9862–8. doi: 10.1074/jbc.M207700200. [DOI] [PubMed] [Google Scholar]
- 55.Salanova B, Choi M, Rolle S, Wellner M, Scheidereit C, Luft FC. The effect of fever-like temperatures on neutrophil signaling. FASEB J. 2005;19:816–8. doi: 10.1096/fj.04-2983fje. [DOI] [PubMed] [Google Scholar]
- 56.Jimi E, Aoki K, Saito H, D’Acquisto F, May MJ, Nakamura I, et al. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med. 2004;10:617–24. doi: 10.1038/nm1054. [DOI] [PubMed] [Google Scholar]
- 57.Orange JS, May MJ. Cell penetrating peptide inhibitors of nuclear factor-kappa B. Cell Mol Life Sci. 2008 doi: 10.1007/s00018-008-8222-z. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dave SH, Tilstra JS, Matsuoka K, Li F, Karrasch T, Uno JK, et al. Amelioration of chronic murine colitis by peptide-mediated transduction of the I{kappa}B Kinase inhibitor NEMO binding domain peptide. J Immunol. 2007;179:7852–9. doi: 10.4049/jimmunol.179.11.7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Plaen IG, Liu SX, Tian R, Neequaye I, May MJ, Han XB, et al. Inhibition of nuclear factor-kappaB ameliorates bowel injury and prolongs survival in a neonatal rat model of necrotizing enterocolitis. Pediatr Res. 2007;61:716–21. doi: 10.1203/pdr.0b013e3180534219. [DOI] [PubMed] [Google Scholar]
- 60.Shibata W, Maeda S, Hikiba Y, Yanai A, Ohmae T, Sakamoto K, et al. Cutting edge: the I{kappa}B Kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks in Xammatory injury in murine colitis. J Immunol. 2007;179:2681–5. doi: 10.4049/jimmunol.179.5.2681. [DOI] [PubMed] [Google Scholar]
- 61.Nadjar A, Bluthe RM, May MJ, Dantzer R, Parnet P. Inactivation of the cerebral NFkappaB pathway inhibits interleukin-1beta-induced sickness behavior and c-Fos expression in various brain nuclei. Neuropsychopharmacology. 2005;30:1492–9. doi: 10.1038/sj.npp.1300755. [DOI] [PubMed] [Google Scholar]
- 62.Nadjar A, Tridon V, May MJ, Ghosh S, Dantzer R, Amedee T, et al. NFkappaB activates in vivo the synthesis of inducible Cox-2 in the brain. J Cereb Blood Flow Metab. 2005;25:1047–59. doi: 10.1038/sj.jcbfm.9600106. [DOI] [PubMed] [Google Scholar]
- 63.Nijboer CH, Heijnen CJ, Groenendaal F, May MJ, van Bel F, Kavelaars A. Strong neuroprotection by inhibition of NF-{kappa}B after neonatal hypoxia-ischemia involves apoptotic mechanisms but is independent of cytokines. Stroke. 2008;39:2127–37. doi: 10.1161/STROKEAHA.107.504175. [DOI] [PubMed] [Google Scholar]
- 64.Nijboer CH, Heijnen CJ, Groenendaal F, May MJ, van Bel F, Kavelaars A. A dual role of the NF-{kappa}B pathway in neonatal hypoxic-ischemic brain damage. Stroke. 2008 doi: 10.1161/STROKEAHA.108.516401. in press. [DOI] [PubMed] [Google Scholar]
- 65.Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, et al. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci USA. 2007;104:18754–9. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ankermann T, Reisner A, Wiemann T, Krams M, Kohler H, Krause MF. Topical inhibition of nuclear factor-kappaB enhances reduction in lung edema by surfactant in a piglet model of airway lavage. Crit Care Med. 2005;33:1384–91. doi: 10.1097/01.ccm.0000166371.18066.5e. [DOI] [PubMed] [Google Scholar]
- 67.Mora AL, Lavoy J, McKean M, Stecenko A, Brigham KL, Parker R, et al. Prevention of NF-{kappa}B activation in vivo by a cell permeable NF-{kappa}B inhibitor peptide. Am J Physiol Lung Cell Mol Physiol. 2005;289:L536–44. doi: 10.1152/ajplung.00164.2005. [DOI] [PubMed] [Google Scholar]
- 68.von Bismarck P, Klemm K, Wistadt CF, Winoto-Morbach S, Uhlig U, Schutze S, et al. Surfactant “fortification” by topical inhibition of nuclear factor-kappaB activity in a newborn piglet lavage model. Crit Care Med. 2007;35:2309–18. doi: 10.1097/01.ccm.0000281472.47067.45. [DOI] [PubMed] [Google Scholar]
- 69.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–8. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 70.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284:321–5. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 71.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, et al. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–45. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–70. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 73.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–9. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 74.Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. 2002;3:958–65. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- 75.Coope HJ, Atkinson PG, Huhse B, Belich M, Janzen J, Holman MJ, et al. CD40 regulates the processing of NF-kappaB2 p100 to p52. EMBO J. 2002;21:5375–85. doi: 10.1093/emboj/cdf542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17:525–35. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 77.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–9. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 78.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kB inducing kinase by TRAF3-induced degradation. J Biol Chem. 2004;279:26243. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 79.Hu Y, Baud V, Oga T, Kim KI, Yoshida K, Karin M. IKKalpha controls formation of the epidermis independently of NF-kappaB. Nature. 2001;410:710–4. doi: 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- 80.Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, et al. IKKalpha, IKKbeta, and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program. J Biol Chem. 2002;277:45129–40. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Massa PE, Li X, Hanidu A, Siamas J, Pariali M, Pareja J, et al. Gene expression profiling in conjunction with physiological rescues of IKKalpha-null cells with wild type or mutant IKKalpha reveals distinct classes of IKKalpha/NF-kappaB-dependent genes. J Biol Chem. 2005;280:14057–69. doi: 10.1074/jbc.M414401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, et al. IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. 2001;107:763–75. doi: 10.1016/s0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- 83.Solt LA, Madge LA, Orange JS, May MJ. Interleukin-1-induced NF-kappaB activation is NEMO-dependent but does not require IKKbeta. J Biol Chem. 2007;282:8724–33. doi: 10.1074/jbc.M609613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang TT, Feinberg SL, Suryanarayanan S, Miyamoto S. The zinc finger domain of NEMO is selectively required for NF-kappa B activation by UV radiation and topoisomerase inhibitors. Mol Cell Biol. 2002;22:5813–25. doi: 10.1128/MCB.22.16.5813-5825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sil AK, Maeda S, Sano Y, Roop DR, Karin M. IkappaB kinase-alpha acts in the epidermis to control skeletal and craniofacial morphogenesis. Nature. 2004;428:660–4. doi: 10.1038/nature02421. [DOI] [PubMed] [Google Scholar]
- 86.Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–63. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- 87.Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature. 2003;423:655–9. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- 88.Hoberg JE, Popko AE, Ramsey CS, Mayo MW. IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol. 2006;26:457–71. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–43. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 90.May MJ, Madge LA. Caspase inhibition sensitizes inhibitor of NF-kappaB kinase beta-deficient fibro-blasts to caspase-independent cell death via the generation of reactive oxygen species. J Biol Chem. 2007;282:16105–16. doi: 10.1074/jbc.M611115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kwak YT, Guo J, Shen J, Gaynor RB. Analysis of domains in the IKKalpha and IKKbeta proteins that regulate their kinase activity. J Biol Chem. 2000;275:14752–9. doi: 10.1074/jbc.m001039200. [DOI] [PubMed] [Google Scholar]
- 92.May MJ, Larsen SE, Shim JH, Madge LA, Ghosh S. A novel ubiquitin-like domain in IkappaB kinase beta is required for functional activity of the kinase. J Biol Chem. 2004;279:45528–39. doi: 10.1074/jbc.M408579200. [DOI] [PubMed] [Google Scholar]
- 93.Hartmann-Petersen R, Gordon C. Integral UBL. Domain proteins: a family of proteasome interacting proteins. Semin Cell Dev Biol. 2004;15:247–59. doi: 10.1016/j.semcdb.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 94.Ikeda F, Hecker CM, Rozenknop A, Nordmeier RD, Rogov V, Hofmann K, et al. Involvement of the ubiquitin-like domain of TBK1/IKK-i kinases in regulation of IFN-inducible genes. EMBO J. 2007;26:3451–62. doi: 10.1038/sj.emboj.7601773. [DOI] [PMC free article] [PubMed] [Google Scholar]