

Abstract
Prostate specific membrane antigen (PSMA) is a membrane-bound glutamate carboxypeptidase overexpressed in many forms of prostate cancer. Our laboratory has recently disclosed a class of small molecules, called ARM-Ps (antibody-recruiting molecule targeting prostate cancer) that are capable of enhancing antibody-mediated immune recognition of prostate cancer cells. Interestingly, during the course of these studies, we found ARM-Ps to exhibit extraordinarily high potencies toward PSMA, compared to previously reported inhibitors. Here, we report in-depth biochemical, crystallographic, and computational investigations which elucidate the origin of the observed affinity enhancement. These studies reveal a previously unreported arene-binding site on PSMA, which we believe participates in an aromatic stacking interaction with ARMs. Although this site is composed of only a few amino acid residues, it drastically enhances small molecule binding affinity. These results provide critical insights into the design of PSMA-targeted small molecules for prostate cancer diagnosis and treatment; more broadly, the presence of similar arene-binding sites throughout the proteome could prove widely enabling in the optimization of small-molecule–protein interactions.
Introduction
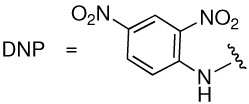
Prostate cancer is a major public health threat against which currently available therapeutic strategies are often ineffective.1,2 Our laboratory has recently disclosed a class of small molecules, called ARM-Ps (antibody-recruiting molecule targeting prostate cancer) that are capable of enhancing antibody-mediated immune recognition of prostate cancer cells.3,4 ARM-Ps accomplish this by binding simultaneously to prostate-specific membrane antigen (PSMA),5 a membrane-bound glycoprotein that is overexpressed on prostate cancer cells, and to anti-dinitrophenyl (anti-DNP) antibodies, which are present endogenously in the human bloodstream (Figure 1A and B).6 PSMA, which is also known as glutamate carboxypeptidase II (GCPII), is a well-studied molecular signature of prostate cancer cells, and has been exploited as a target in both therapeutic and diagnostic strategies for patients with prostate cancer.7–11 PSMA possesses glutamate carboxypeptidase activity, and numerous studies have focused on the identification of small molecules capable of inhibiting this enzyme.12–17 ARM-Ps belong to a class of glutamate urea compounds capable of inhibiting PSMA with high potency.14
Figure 1.

Structure and Function of ARM-Ps (antibody-recruiting molecules targeting prostate cancer). (A) ARM-Ps recruit anti-DNP antibodies to PSMA-expressing prostate cancer cells, and thereby bring about immune-mediated cytotoxicity. (B) ARM-Ps are bifunctional and consist of an antibody-binding terminus (ABT), a linker region, and a cell-binding terminus (CBT).
During the course of developing ARM-Ps, we observed that bifunctional DNP-containing conjugates were strikingly more potent than the parent glutamate urea compounds from which they were derived. Furthermore, we also noted that potency increases were correlated to the length of the linker regions connecting the two poles of the molecule. Here we provide a molecular basis for these findings, which involves the disclosure of a previously unreported arene-binding site on PSMA. These conclusions are supported by extensive biochemical, crystallographic, and computational studies.
Results and discussion
Dependence of linker length on binding affinity
To evaluate in detail the effect of linker length on PSMA binding affinity, we prepared various derivatives of ARM-P (Table 1, 1–12). These compounds consist of glutamate ureas linked to DNP or methoxy groups by oxyethylene moieties of varying lengths. They are named ARM-Px and MeO-Px, respectively, wherein “x” corresponds to the number of oxyethylene units in the linker. Evaluation of these compounds for their ability to inhibit PSMA activity proved quite revealing.3 In all cases, ARM-P derivatives were found to possess Ki values lower in magnitude than their counterparts lacking DNP (compounds 1–7 versus 8–12). In some cases, the affinity difference was up to two orders of magnitude (compound 3 versus 9). This result indicated to us that perhaps the DNP function itself might be playing a role in binding PSMA. Such a hydrophobic interaction involving an aromatic ring and PSMA was not completely unexpected given the proximity of the glutamate-urea binding site to a known hydrophobic pocket in PSMA.18,19 Indeed, inhibitors containing hydrophobic functionality distal to the glutamic acid moiety have exhibited high potency against PSMA.13,20,21
Table 1.
Linker length dependence on PSMA inhibitory potency.
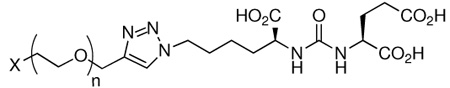 | ||||
|---|---|---|---|---|
| Compound | X | n | IC50 (nM)b | Ki (nM)c |
| 1, ARM-P0 | DNPa | 0 | 1.76 ± 0.41 | 0.078 ± 0.018 |
| 2, ARM-P1 | DNP | 1 | 1.05 ± 0.11 | 0.047 ± 0.005 |
| 3, ARM-P2 | DNP | 2 | 0.54 ± 0.18 | 0.024 ± 0.008 |
| 4, ARM-P4 | DNP | 4 | 0.46 ± 0.18 | 0.020 ± 0.008 |
| 5, ARM-P6 | DNP | 6 | 2.29 ± 0.60 | 0.101 ± 0.027 |
| 6, ARM-P8 | DNP | 8 | 3.29 ± 1.14 | 0.145 ± 0.050 |
| 7, ARM-P12 | DNP | 12 | 37.3 ± 16.2 | 1.65 ± 0.72 |
| 8, MeO-P0 | OMe | 0 | 30.5 ± 12.1 | 1.35 ± 0.54 |
| 9, MeO-P2 | OMe | 2 | 40.8 ± 9.4 | 1.81 ± 0.42 |
| 10, MeO-P4 | OMe | 4 | 30.2 ± 14.4 | 1.34 ± 0.64 |
| 11, MeO-P8 | OMe | 8 | 131.0 ± 57.6 | 5.79 ± 2.54 |
| 12, MeO-P12 | OMe | 12 | 165.2 ± 58.9 | 7.30 ± 2.60 |
IC50 values represent the mean of triplicate experiments.
Ki values were calculated from IC50 and Km values using the Cheng-Prusoff equation as described in the supporting information.
A model involving binding of the DNP moiety to the hydrophobic pocket adjacent to the S1 site did not explain the decrease in affinity between ARM-P2 and derivatives with shorter oxyethylene linkers (i.e., ARM-P0 and ARM-P1). Indeed, one would expect ARM-P0 and ARM-P1 to exhibit enhanced potency versus ARM-P2 because of the close proximity of the accessory hydrophobic pocket to the P1’ glutamate binding cavity. The opposite trend suggested perhaps the presence of an alternative hydrophobic binding site, situated at a substantial distance away from this cavity. This hypothesis is supported by observations in related systems in which bifunctional ligands bind proteins at two remote sites. In such systems, an ideal linker length between binding poles is required for maximum affinity; shorter linkers are unable to form optimal interactions in both binding sites, while longer linkers pay a relatively high entropic penalty upon bivalent binding without any compensatory enthalpic benefit.22–25
Notably, compounds within the MeO-P series (8–12) containing relatively short linkers all bind PSMA with comparable affinity. The presence of linkers consisting of 8 oxyethylene groups or longer appears to inhibit compound binding, perhaps because bulky PEG chains prevent access of the glutamate urea moiety to its binding site on PSMA.26 The increased sensitivity of ARM-P derivatives to changes in linker length as compared to MeO-P compounds suggests that factors other than simple steric bulk are operating for the ARM-Ps.
Structure–activity studies: effect of varying aromatic groups on binding affinity
To test further our model for bidentate binding, we set out to probe the impact of the phenyl ring substituent on inhibitor potency. We therefore synthesized analogues of ARM-P2 replacing the DNP moiety with a range of electronically distinct aromatic species (3, 13–17, Table 2). As shown, these analogues included nitrophenyl (ortho and para to the linker), p-methoxyphenyl, phenyl, and cyclohexyl derivatives. Consistent with the hypothesis that the DNP moiety occupies a novel binding site, profound changes in affinity were observed in this series. Interestingly, the parent dinitrophenyl-containing compound (ARM-P2) possessed the highest potency of all the analogues tested (Ki = 24 pM), and removal of nitro groups led to three-fold decreases in affinity in the p-nitrophenyl (13) and o-nitrophenyl (14) analogues (Ki = 60 and 78 pM, respectively). The similarities between these analogues suggests that inhibitor potency is dictated by electronic rather than steric effects. Phenyl (15) and methoxyphenyl (16) analogues proved an additional order of magnitude less potent than mononitrated derivatives (Ki = 730 and 970 pM, respectively), and the cyclohexyl-substituted derivative (17) proved yet another order of magnitude worse than the least potent aryl compounds (Ki = 15.1 nM). This may result from the enhanced steric bulk of the cyclohexyl substituent versus planar arenes.
Table 2.
Dependence of Ki on substituents and electronics of aromatic ring.
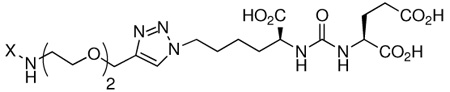 | ||||
|---|---|---|---|---|
| Compound | X | ELUMO (eV)a | IC50 (nM)b | Ki (nM)c |
| 3, ARM-P2 | DNP | −1.44 | 0.54 ± 0.18 | 0.024 ± 0.008 |
| 13 | o-NO2-Ph | −0.92 | 1.78 ± 0.15 | 0.078 ± 0.007 |
| 14 | p-NO2-Ph | −0.91 | 1.36 ± 0.18 | 0.060 ± 0.008 |
| 15 | Ph | 0.50 | 16.6 ± 6.3 | 0.73 ± 0.28 |
| 16 | p-MeO-Ph | 0.43 | 21.9 ± 9.9 | 0.97 ± 0.44 |
| 17 | Cyclohexyl | N/A | 342.3 ± 170.5 | 15.1 ± 7.5 |
ELUMO energies of arene rings (XŠNHMe) were calculated using the PDDG/PM3 method implemented in the BOSS software package, and reported in electron-volts as detailed in the Supporting Information.
IC50 values represent the mean of triplicate experiments.
Ki values were calculated using IC50 and Km values via the Cheng-Prusoff equation as outlined in the supporting information. A Km value of 925 nM was used in these calculations.
To examine the relationship between arene electronics effects and PSMA inhibitory potency, we performed semiempirical calculations of arene LUMO energies using the PDDG/PM3 method implemented in the BOSS software package.27,28 An excellent correlation was observed between the LUMO energies of aromatic substituents and experimentally determined Ki values (Figure 2). Electron poor aromatic rings are expected to experience strong π-stacking interactions with electron-rich arenes,29,30 suggesting that such interactions may be dominant in dictating binding affinity in this system. These results are strongly indicative of multisite binding in the ARM-P series, and led us to test this hypothesis further using X-ray crystallography.
Figure 2.

Correlation between Ki values and ELUMO of aromatic ring component of ARM-P analogue. Measured Ki values (mean of triplicate experiments ± standard deviation) are plotted versus PDDG/PM3 LUMO energies calculated using the BOSS software package.
Crystallographic Studies
Initial refinement and analysis
Crystal structures were determined for PSMA in complex with ARM-P ligands containing 2, 4, and 8 oxyethylene units in the linker region (3, 4, and 6) and with MeO-P4 (10), which lacks the DNP moiety, and were refined at the resolution of 1.69 Ǻ, 1.59 Ǻ, 1.59 Ǻ, and 1.78 Ǻ, respectively. Individual compounds were fit into the positive peaks on the difference Fo-Fc electron density map in the final stages of refinement. For all four inhibitors, clear interpretable densities were observed for the C-terminal part encompassing the P1’ glutarate, the urea linkage, the lysine linker and the triazole ring. Although density corresponding to the DNP phenyl ring is defined in all ARM-P complexes, density corresponding to the nitro groups is absent suggesting that the DNP moiety is present in at least two different conformations. Also, electron density peaks corresponding to the poly-oxyethylene linker were absent from all complexes, consistent with a lack of intermolecular contacts between this flexible element and the protein.
Models of ARM-P2, ARM-P4, and ARM-P8 in complex with PSMA, built from the electron density map, are depicted in Figure 3. Despite the attachment of large oxyethylene linkers, the glutamate urea portions of all inhibitors interact with the protein active site in a fashion reminiscent of previously reported complexes with urea13,14,18 and phosphonate31 inhibitors, and the substrate N-acetyl-aspartyl-glutamate (NAAG).32 In all structures, positioning of the P1’ glutarate is enforced by H-bonds (indicated as dashed lines) with Arg210, Asn257, Tyr552, Lys699, Tyr700, and active-site water molecules, and hydrophobic interactions with the side chains of Phe209 and Leu428. The ureido nitrogen atoms serve as H-bond donors in interactions with Glu424 and the Gly518 main chain carbonyl, and the carbonyl oxygen makes contacts with both the catalytic zinc atom and Tyr552, and His553. The P1 α-carboxylate in all inhibitors structurally overlaps with the equivalent groups of previously reported complexes, and is held in place by interactions with an arginine-rich patch (Arg463, Arg534, Arg536) along with H-bonding contacts to Asn519, the Ser517 main-chain carbonyl, and water molecules.18,19
Figure 3.

Close-up of PSMA active site bound to bifunctional glutamate urea inhibitors ARM-P2 (gold), ARM-P4 (grey), and ARM-P8 (blue). Structures were superimposed on with corresponding (or equivalent) Cα atoms. Inhibitors are shown in stick representation and protein residues are shown as lines. Hydrogen bonding interactions are indicated by dashed lines. The zinc ions and chloride ion in the active site are labeled as grey and green dotted spheres, respectively, and water molecules are depicted as red spheres. In both protein and inhibitor structures, carbon atoms are colored as indicated above, and other atoms are colored red (oxygen), and blue (nitrogen).
Discovery of an arene-binding cleft
A key site of interaction between PSMA and all ARM-P derivatives is the triazole ring, which was observed to pack against the side chains of Tyr552 and Tyr700 in all complexes (Figure 3). The steric hindrance caused by the oxyethylene linker emanating from the triazole ring prevents closure of the enzyme’s entrance lid (amino acids Trp541 – Gly548), as observed for PSMA complexes with smaller ligands.19 A key consequence of the entrance lid’s open conformation is the revelation of a previously unreported binding cleft for the DNP ring (Figure 4).19
Figure 4.

The PSMA/ARM-P2 complex reveals a previously unreported arene-binding cleft. (A) Global view of PSMA with a close-up of arene-binding site. Residues making up the arene-binding cleft are labeled in cyan. The entrance lid (residues 542–548), which resides in an open conformation in the ARM-P2 complex, is indicated as a red loop. Overlaid on this complex is the entrance lid in its closed conformation (colored blue), which would come into steric conflict with the linker region of the inhibitor. (B and C) Close-up images of the urea binding sites in structures containing both open and closed entrance loops (colored in blue). Residues forming the DNP binding site (W541, R511, and R463) are colored red. In all panels, structural data for PSMA with a the closed entrance lid comes from the complex with the small urea-based inhibitor DCIBzL (PDB ID – 3IWW).33 The zinc ions in the active site are labeled as orange spheres and the ARM-P2 carbons are colored gold. The DCIBzL carbons in B and C are colored purple.
The arene-binding region, formed from the indole group of Trp541 and the guanidinium group of Arg511 holds the DNP ring in close contact with these groups at distances of 3.1 Å and 3.9 Å, respectively. The bottom of the cleft is lined by the Arg463 side chain. Positioning of the phenyl ring creates a plane virtually parallel to both indole and guanidinium functionalities, suggesting that simultaneous π-cation (DNP-Arg511) and π-stacking (DNP-Trp541) interactions may both contribute to inhibitor binding.34,35 Critically, the arene-binding region is only revealed upon opening of the entrance lid (Figure 4B); closure of the entrance lid, as in the overlaid complex between PSMA and the small urea-based inhibitor DCIBzL,33 would lead to significant steric overlap with the triazole moiety as well as closure of the arene-binding site (Figure 4C). Thus, the protein is capable of adopting two separate conformations, each suited to accommodate high-affinity binding interactions with distinct classes of glutamate-urea inhibitors.
A key structural feature was observed in the MeO-P4 complex (Figure 5). Here, unlike in the ARM-P complexes, Trp541 exists in two distinct conformations. The non-stacking conformation is rotated approximately 4 Å from what is seen in ARM-P complexes, and blocks the arene-binding groove. The conformational flexibility exhibited by Trp541 in the PSMA/MeO-P4 complex suggests that when present, the dinitroarene stabilizes the side chain indole moiety via π-stacking, as implied by the ARM-P structures depicted above. Taken together, these data provide strong support for a model in which ARM-Ps bind PSMA through interactions at both the enzyme active site and at a newly reported arene-binding cleft.
Figure 5.

Close-up view of the active site of PSMA bound to MeO-P4. Hydrogen bonding interactions are indicated by dashed lines. The zinc ions in the active site and adjacent chloride ion are labeled as grey and green dotted spheres, respectively, and water molecules are depicted as red spheres. In both protein and inhibitor structures, carbon atoms are colored in olive, and other atoms are colored red (oxygen), and blue (nitrogen).
Notably, the complex between PSMA and MPE,36 a methotrexate-derived phosphonate, was also shown to possess an open entrance lid like the complexes disclosed herein.19 It was concluded from the PSMA/MPE complex that the protein’s ability to adopt an open conformation serves to enable its binding to relatively large substrates, such as folyl-poly-γ-glutamates. One might imagine that the revelation of an arene-binding site upon opening of the entrance lid might serve to enhance affinity for these arene-containing enzyme substrates. Interestingly, however, the pendant pteroyl ring in the MTE complex was not observed to interact with the PSMA arene-binding cleft, perhaps due to its relatively short linkage to the zinc-binding phosphonate region. The observations reported herein suggest that perhaps larger natural poly-γ-glutamate substrates are able to make use of the arene-binding site, however further studies are necessary to test this possibility.
Molecular dynamics (MD) simulations
To clarify the nature of the protein-ligand interactions in the ARM-P complexes, explicit solvent molecular dynamics (MD) simulations were carried out using crystallographic data for PSMA complexes with ARM-P0, ARMP-P2, ARM-P4, ARM-P8 and MeO-P0. Each protein-ligand complex was modeled with the OPLS-AA force field,37 embedded in a triclinic box of TIP3P water molecules.38 Dynamics were simulated for 50 ns using the Desmond software package.39
These simulations revealed a number of noteworthy features (see Supporting Information for video files for all simulations). Although the PSMA active site and glutamate urea moieties are fairly rigid throughout the timescale of MD simulations, distal protein-ligand interactions exhibit highly dynamic behavior. For example, the simulation of the MeO-P0–PSMA complex revealed that the arene-binding site is unstable in the absence of DNP; Trp541 tends to rotate toward Arg511, thus obscuring the arene-binding site (Figure 6, panels a–c). This observation directly correlates with the disorder in Trp541 observed in the MeO-P4–PSMA crystal structure (Figure 3). Furthermore, the PEG moieties in all complexes are highly dynamic and do not seem to form specific interactions with PSMA, suggesting that these make minimal enthalpic contributions to binding affinity. These observations also explain the absence of electron density corresponding to linker regions in all crystal structures.
Figure 6.

Selected snapshots from the MD simulations of PSMA/ARM-P complexes. The ligands are represented in colored sticks, Arg463, Arg511 and Trp541 are represented in orange sticks. Figure created with the program VMD.40
By far the most stable intermolecular contact in the arene-binding site in ARM-P–PSMA complexes is the stacking interaction between DNP and Trp541. For all ARMs, the DNP moieties participate in face-to-face interactions with Trp541 side chain indole moieties for significant time periods throughout MD simulations. Simulations of the ARM-P0–PSMA complex revealed a remarkable level of flexibility in the triazole-alkyl region, which enables π-stacking contacts in the arene-binding site to remain intact even in the absence of an oxyethylene linker (Figure 6, panels d–f). When stacked with the Trp541 side chain indole, the DNP ring is observed to rotate in-plane, supporting the hypothesis that the lack of well defined electron density corresponding to nitro groups in crystal structures is due to the presence of multiple arene conformations. However, in all ARM-P complexes, the nitro groups in the DNP ring are frequently observed pointing toward the Arg463 side chain guanidinium group, suggesting possible hydrogen bonding or electrostatic interactions between these groups. Furthermore, although crystallographic data support a role for π-cation interactions with Arg511 in the arene-binding site, this residue is highly disordered in MD simulations, and does not form long-lived contacts with the ligand. This observation is consistent with the data presented in Table 2 and Figure 2, which suggest that cation-π interactions play a relatively minor role versus π-stacking interactions in stabilizing these systems.
Notably, during the course of MD simulations for both ARM-P2 (panels g-i) and ARM-P8 (panels m-o), the DNP ring dissociates from the arene-binding cleft, whereas this interaction remains intact in the ARM-P4 simulation (panels j–l). Taken together, these data suggest that the DNP–Trp541 interactions are relatively weak. Interestingly, the contact with Trp541 reforms rapidly during the simulations of ARM-P2, but not ARM-P8; this may either reflect a higher entropic penalty associated with bivalent binding because of the longer linker group, or the tendency of the linker to occupy the arene-binding site, thus preventing the DNP group’s return to Trp541.22–25 Additional studies to clarify the contribution of the linker group to the binding thermodynamics of the ARM-P ligands would be desirable. From a functional standpoint, the propensity of ARM-P8 to disengage from the PSMA arene-binding site enables it to form ternary complexes with prostate cancer cells and antibodies, which is critical to its cytotoxic activity.3 However, this functionality comes at the expense of PSMA binding affinity. Furthermore, the model reported herein suggests the possibility of ultra high-affinity ARM-P analogues capable of interacting simultaneously with the PSMA arene-binding site and anti-DNP antibodies.
Conclusion
Here we detail the discovery of an arene-binding site on prostate-specific membrane antigen (PSMA), which gives rise to unusually high affinity binding interactions with designed bifunctional antibody-recruiting small molecules (ARMs). Our conclusions are supported by extensive crystallographic, biochemical, and computational data, which, taken together, strongly suggests a model in which bidentate binding of ARM-Ps to PSMA leads to substantial increases in inhibitor potency. The serendipitous nature of the discovery reported herein along with the relative simplicity of the PSMA arene-binding site – which consists merely of three amino acids only one of which (Trp541) is responsible for affinity enhancement – suggest that low-affinity binding sites for arenes could be quite prevalent among proteins. Along these lines, it is well-documented that a large proportion of circulating immunoglobulin possess high-affinity binding activity against nitroarene ligands,41 and between 1 and 10% of myeloma proteins bind nitrophenyl ligands.42 The possibility that such binding sites arise from conserved folds within immunoglobulin domains has been suggested,43 however, this trend may also result from the unique immunogenicity of nitroarenes,44,45 a property that has also been attributed to their propensity to form hydrophobic contacts with proteins.44 In either case, although structural data exists demonstrating the unique propensity of nitroarenes to engage in π-stacking interactions with aromatic amino acid side chains,46,47 the proteomic prevalence of nitroarene-binding motifs has not been systematically explored. The widespread existence of such binding sites could enable facile optimization of small molecule ligands for proteins identified through high-throughput screening, and could find ready utility in fragment-based approaches to inhibitor design.48
Although underexplored, strategies that utilize small molecules to enhance recognition of pathogens by the human immune system promise to leverage the strengths of both antibody- and small-molecule-based therapeutic approaches. The results reported herein suggest the possibility for improving such technologies for treating prostate cancer. For example, ultra-high-affinity ARM-Ps could be constructed by exploiting the presence of the arene-binding site in PSMA and converting the highly flexible first-generation ARM-Ps into more rigid scaffolds. More broadly, the high-level expression of PSMA (GCPII) on prostate cancer cell surfaces and on tumor neovasculature,49 as well as its putative role in the pathophysiology of schizophrenia,50 have rendered it an extremely useful and popular target for inhibitor design. The results presented herein therefore could substantially impact the development of effective diagnostic and therapeutic approaches for patients suffering from cancer and other diseases.
Supplementary Material
Acknowledgements
This work was funded by the National Institutes of Health through the NIH Director’s New Innovator Award Program (DP22OD002913 to D.A.S.), and through NIH Grant GM32136 (to W.L.J.). C.B. acknowledges financial support from the MEYS (project ME10031) and the Institutional Research Support of the IBT (CEZ:AV0Z50520701). The use of the Advanced Photon Source was supported by the U.S. Department of Energy (W-31-109-Eng38). J.L. acknowledges financial support from the National Cancer Institute, Center for Cancer Research. This research was partially supported by the National Science Foundation through Teragrid resources provided by the Texas Advanced Computing Center under grant number TG-CHE090106 (to J.M.), and by a Marie Curie International Outgoing Fellowship (J.M.) within the 7th European Community Framework Programme (FP7-PEOPLE-2008-16704-1-IOF, 234796-PPIdesign).
Footnotes
Supporting Information Available: Detailed experimental procedures and compound characterizations are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.American Cancer Society. [accessed July 7, 2010];"Cancer Facts and Figures 2010,". 2010 Available at http://www.cancer.org/Research/CancerFactsFigures/CancerFactsFigures/cancer-facts-and-figures-2010.
- 2.Olson WC, Heston WDW, Rajasekaran AK. Rev. Recent Clin. Trials. 2007;2:182–190. doi: 10.2174/157488707781662724. [DOI] [PubMed] [Google Scholar]
- 3.Murelli RP, Zhang AX, Michel J, Jorgensen WL, Spiegel DA. J. Am. Chem. Soc. 2009:17090–17092. doi: 10.1021/ja906844e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For lead examples of other bifunctional materials that redirect antibodies to target cells, see: Parker CG, Domaoal RA, Anderson KS, Spiegel DA. J. Am. Chem. Soc. 2009;131:16393. doi: 10.1021/ja9057647. Shokat KM, Schultz PG. J. Am. Chem. Soc. 1991;113:1861–1862. Naicker KP, Li H, Heredia A, Song H, Wang L. Org. and Biomolec. Chem. 2004;2:660–664. doi: 10.1039/b313844e. Li J, Zacharek S, Chen X, Wang JQ, Zhang W, Janczuk A, Wang PG. Bioorg. Med. Chem. 1999;7:1549–1558. doi: 10.1016/s0968-0896(99)00099-1. Perdomo MF, Levi M, llberg MS, Vahlne A.Proc. Natl. Acad. Sci. U.S.A 2008619118201 Bertozzi CR, Bednarski MD. J. Am. Chem. Soc. 1992;114:5543–5546. Bertozzi CR, Bednarski MD. J. Am. Chem. Soc. 1992;114:2242–2245. Krishnamurthy V, Quinton L, Estroff L, Metallo S, Isaacs J, Mizgerd J, Whitesides G. Biomaterials. 2006:12. doi: 10.1016/j.biomaterials.2006.02.006. Metallo SJ, Kane RS, Holmlin RE, Whitesides GM. J. Am. Chem. Soc. 2003;125:4534–4540. doi: 10.1021/ja030045a. Carlson C, Mowery P, Owen R, Dykhuizen EC, Kiessling L. ACS Chem. Biol. 2007;2:119–127. doi: 10.1021/cb6003788. Owen RM, Carlson CB, Xu J, Mowery P, Fasella E, Kiessling LL. ChemBioChem. 2007;8:68–82. doi: 10.1002/cbic.200600339. Lu Y, You F, Vlahov I, Westrick E, Fan M, Low PS, Leamon CP. Mol. Pharm. 2007;4:695–706. doi: 10.1021/mp070050b. Lu Y, Low PS. Cancer Immunol. Immunother. 51:153–163. doi: 10.1007/s00262-002-0266-6. Popkov M, Gonzalez B, Sinha S, Barbas CF. Proc. Natl. Acad. Sci. U.S.A. 2009;106:4378–4383. doi: 10.1073/pnas.0900147106. Rader C, Sinha SC, Popkov M, Lerner RA, Barbas CF., III Proc. Natl. Acad. Sci. U.S.A. 2003;100:5396–5400. doi: 10.1073/pnas.0931308100. Guo F, Das S, Mueller BM, Barbas CF, III, Lerner RA, Sinha SC. Proc. Natl. Acad. Sci. U.S.A. 2006;103:11009–11014. doi: 10.1073/pnas.0603822103. Wuellner U, Gavrilyuk JI, Barbas CF. Angew. Chem. Int. Ed. [Online early access]. DOI : 10.1002/anie.201001736 Published Online: July 19, 2010. O'Reilly MK, Collins BE, Han S, Liao L, Rillahan C, Kitov PI, Bundle DR, Paulson JC. J. Amer. Chem. Soc. 2008;130:7736–7745. doi: 10.1021/ja802008q.
- 5.Holmes EH, Greene TG, Tino WT, Boynton AL, Aldape HC, Misrock SL, Murphy GP. Prostate Suppl. 1996;7:25–29. [PubMed] [Google Scholar]
- 6.Ortega E, Kostovetzky M, Larralde C. Mol. Immunol. 1984;21:883–888. doi: 10.1016/0161-5890(84)90143-3. [DOI] [PubMed] [Google Scholar]
- 7.Humblet V, Misra P, Bhushan KR, Nasr K, Ko Y-S, Tsukamoto T, Pannier N, Frangioni JV, Maison W. J. Med. Chem. 2009;52:544–550. doi: 10.1021/jm801033c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jayaprakash S, Wang X, Heston WD, Kozikowski AP. ChemMedChem. 2006;1:299–302. doi: 10.1002/cmdc.200500044. [DOI] [PubMed] [Google Scholar]
- 9.Kularatne SA, Wang K, Santhapuram H-KR, Low PS. Mol. Pharm. 2009;6:780–789. doi: 10.1021/mp900069d. [DOI] [PubMed] [Google Scholar]
- 10.Kularatne SA, Zhou Z, Yang J, Post CB, Low PS. Mol. Pharm. 2009;6:790–800. doi: 10.1021/mp9000712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pomper MG, Musachio JL, Zhang J, Scheffel U, Zhou Y, Hilton J, Maini A, Dannals RF, Wong DF, Kozikowski AP. Mol. Imaging. 2002;1:96–101. doi: 10.1162/15353500200202109. [DOI] [PubMed] [Google Scholar]
- 12.Jackson PF, Tays KL, Maclin KM, Ko Y-S, Li W, Vitharana D, Tsukamoto T, Stoermer D, Lu X-CM, Wozniak K, Slusher BS. J. Med. Chem. 2001;44:4170–4175. doi: 10.1021/jm0001774. [DOI] [PubMed] [Google Scholar]
- 13.Kozikowski A, Zhang J, Nan F, Petukhov P, Grajkowska E, Wroblewski J, Yamamoto T, Bzdega T, Wroblewska B, Neale J. J. Med. Chem. 2004;47:1729–1738. doi: 10.1021/jm0306226. [DOI] [PubMed] [Google Scholar]
- 14.Kozikowski AP, Nan F, Conti P, Zhang J, Ramadan E, Bzdega T, Wroblewska B, Neale JH, Pshenichkin S, Wroblewski JT. J. Med. Chem. 2001;44:298–301. doi: 10.1021/jm000406m. [DOI] [PubMed] [Google Scholar]
- 15.Majer P, et al. J. Med. Chem. 2003;46:1989–1996. doi: 10.1021/jm020515w. [DOI] [PubMed] [Google Scholar]
- 16.Slusher BS, Vornov J, Thomas AG, Hurn PD, Harukuni I, Bhardwaj A, Traystman RJ, Robinson MB, Britton P, Lu XCM, Tortella FC, Wozniak SKM, Yudkoff M, Potier BM, Jackson PF. Nat. Med. 1999;5:1396–1402. doi: 10.1038/70971. [DOI] [PubMed] [Google Scholar]
- 17.Tsukamoto T, Wozniak K, Slusher B. Drug Discovery Today. 2007;12:767–776. doi: 10.1016/j.drudis.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Barinka C, Byun Y, Dusich CL, Banerjee SR, Chen Y, Castanares M, Kozikowski AP, Mease RC, Pomper MG, Lubkowski J. J. Med. Chem. 2008;51:7737–7743. doi: 10.1021/jm800765e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barinka C, Hlouchova K, Rovenska M, Majer P, Dauter M, Hin N, Ko Y-S, Tsukamoto T, Slusher BS, Konvalinka J, Lubkowski J. J. of Mol. Biol. 2008;376:1438–1450. doi: 10.1016/j.jmb.2007.12.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maung J, Mallari JP, Girtsman TA, Wu LY, Rowley JA, Santiago NM, Brunelle AN, Berkman CE. Bioorg. Med. Chem. 2004;12:4969–4979. doi: 10.1016/j.bmc.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Foss CA, Byun Y, Nimmagadda S, Pullambhatla M, Fox JJ, Castanares M, Lupold SE, Babich JW, Mease RC, Pomper MG. J. Med. Chem. 2008;51:7933–7943. doi: 10.1021/jm801055h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Contakes SM, Juda GA, Langley DB, Halpern-Manners NW, Duff AP, Dunn AR, Gray HB, Dooley DM, Guss JM, Freeman HC. Proc. Natl. Acad. Sci. 2005;102:13451–13456. doi: 10.1073/pnas.0506336102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.May JF, Splain RA, Brotschi C, Kiessling LL. Proc. Natl. Acad. Sci. 2009;106:11851–11856. doi: 10.1073/pnas.0901407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, Whitesides GM. J. Am. Chem. Soc. 2007;129:1312–1320. doi: 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kane RS. Langmuir. 2010;26:8636–8640. doi: 10.1021/la9047193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ke S, Wright JC, Kwon GS. Bioconjug. Chem. 2007;18:2109–2114. doi: 10.1021/bc700204k. [DOI] [PubMed] [Google Scholar]
- 27.Repasky MP, Chandrasekhar J, Jorgensen WL. J. Comput. Chem. 2002;23:1601–1622. doi: 10.1002/jcc.10162. [DOI] [PubMed] [Google Scholar]
- 28.Jorgensen WL, Tirado-Rives J. J. Comput. Chem. 2005;26:1689–1700. doi: 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]
- 29.Cockroft SL, Hunter CA, Lawson KR, Perkins J, Urch CJ. J. Am. Chem. Soc. 2005;127:8594–8595. doi: 10.1021/ja050880n. [DOI] [PubMed] [Google Scholar]
- 30.Cockroft SL, Perkins J, Zonta C, Adams H, Spey SE, Low CMR, Vinter JG, Lawson KR, Urch CJ, Hunter CA. Org. Biomol. Chem. 2007;5:1062–1080. doi: 10.1039/b617576g. [DOI] [PubMed] [Google Scholar]
- 31.Barinka C, Rovenska M, Mlcochova P, Hlouchova K, Plechanovova A, Majer P, Tsukamoto T, Slusher BS, Konvalinka J, Lubkowski J. J. Med. Chem. 2007;50:3267–3273. doi: 10.1021/jm070133w. [DOI] [PubMed] [Google Scholar]
- 32.Klusák Vc, Bařinka C, Plechanovová A, Mlčochová P, Konvalinka J, Rulšíek Lr, Lubkowski J. Biochemistry. 2009;48:4126–4138. doi: 10.1021/bi900220s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Byun Y, Barinka C, Pullambhatla M, Bhang H-eC, Fox JJ, Lubkowski J, Mease RC, Pomper MG. Bioorg. Med. Chem. Lett. 2010;20:392–397. doi: 10.1016/j.bmcl.2009.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duffy EM, Kowalczyk PJ, Jorgensen WL. J. Am. Chem. Soc. 1993;115:9271–9275. [Google Scholar]
- 35.Gallivan JP, Dougherty DA. Proc. Natl. Acad. Sci. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The abbreviation MPE is used to stand for 2-[(3-{4-[(2-amino-4-hydroxy-pteridin-6-ylmethyl)-amino]-benzoylamino}-3-carboxy-propyl)-hydroxy-phosphinoylmethyl]-pentanedioic acid.
- 37.Jorgensen WL, Maxwell DS, Tirado-Rives J. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 38.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 39.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Proceedings of the 2006 ACM/IEEE SC|06 Conference; 2006. [Google Scholar]
- 40.Humphrey W, Dalke A, Schulten K. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 41.Antibodies recognizing the 2,4-dinitrophenyl (DNP) epitope have been estimated to constitute 1% of circulating IgM (approx. 10 µg/mL in human serum) and 0.8% of circulating IgG (approx. 40–120 µg/mL in human serum). See Karjalainen K, Makela O. Eur. J. Immunol. 1976;6:88–93. doi: 10.1002/eji.1830060204. Farah FS. Immunology. 1973;25:217–226. Rowe DS, Anderson SG, Skegg J. In: Immunoglobulins. Merler E, editor. National Academy of Sciences Press; 1970. p. 361. The prevalence of anti-DNP antibodies has been estimated at between 18–90% of humans (see Ortega E, Kostovetzky M, Larralde C. Mol. Immunol. 1984;21:883–888. doi: 10.1016/0161-5890(84)90143-3. Jormalainen S, Makela O. Eur. J. Immunol. 1971;1:471–478. doi: 10.1002/eji.1830010613.
- 42.Michaelides MC, Eisen HN. J. Exp. Med. 1974;140:687–702. doi: 10.1084/jem.140.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parker CW, Osterland CK. Biochemistry. 1970;9:1074–1082. doi: 10.1021/bi00807a004. [DOI] [PubMed] [Google Scholar]
- 44.Grünewald J, Hunt GS, Dong L, Niessen F, Wen BG, Tsao M-L, Perera R, Kang M, Laffitte BA, Azarian S, Ruf W, Nasoff M, Lerner RA, Schultz PG, Smider VV. Proc. Natl. Acad. Sci. 2009;106:4337–4342. doi: 10.1073/pnas.0900507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grünewald J, Tsao M-L, Perera R, Dong L, Niessen F, Wen BG, Kubitz DM, Smider VV, Ruf W, Nasoff M, Lerner RA, Schultz PG. Proc. Natl. Acad. Sci. 2008;105:11276–11280. doi: 10.1073/pnas.0804157105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brunger AT, Leahy DJ, Hynes TR, Fox RO. J. Mol. Biol. 1991;221:239–256. doi: 10.1016/0022-2836(91)80217-i. [DOI] [PubMed] [Google Scholar]
- 47.Nitroarene-protein complexes were downloaded from the RCSB protein data bank, and include the following PDB IDs: 1VID, 3BWM, 3BWY, 3GE5, 1RSM, 1W4O, 1NEN, 2ROY, 2B14, 2B15, 2B16, 2RAZ, 1D1A, 1D1B, 1D1C, 2BMQ, 1BAF, 2BFG, 1H2J, 4A3H, 1QI2, 1OAU, 1GW1, 1GVY, 1IDT, 1OO6, 1GVO, 1GVS, 1VYP, 1VYR, 1VYS, and 1GVR. Out of a total of 32 available complexes, 16 indicated π-stacking interactions between the nitroarene and an aromatic amino acid sidechain.
- 48.Erlanson DA, Wells JA, Braisted AC. Annu. Rev. Biophys. Biomol. Struct. 2004;33:199–223. doi: 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- 49.Anilkumar G, Barwe SP, Christiansen JJ, Rajasekaran SA, Kohn DB, Rajasekaran AK. Microvasc. Res. 2006;72:54–61. doi: 10.1016/j.mvr.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 50.Zhou J, Neale JH, Pomper MG, Kozikowski AP. Nat. Rev. Drug Discovery. 2005;4:1015–1026. doi: 10.1038/nrd1903. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.