

Abstract
PHIP was isolated as an insulin receptor substrate 1 (IRS-1) interacting protein. To date, the physiological roles of PHIP remain unknown. Here we show that mice lacking PHIP1, the full-length isoform of PHIP, are born at normal size but suffer a 40% growth deficit by weaning. PHIP1 mutant mice develop hypoglycemia and have an average lifespan of 4 to 5 weeks. PHIP1-deficient mouse embryonic fibroblasts (MEFs) grow markedly slower than wild-type MEFs, but exhibit normal AKT phosphorylation and an increased cell proliferation in response to IGF-1 treatment. Together these results suggest that PHIP1 regulates postnatal growth in an IGF-1/AKT pathway-independent manner.
INTRODUCTION
Insulin and insulin-like growth factor 1 (IGF-1) are key regulators of cell metabolism, proliferation, differentiation and survival [1]. Their potent metabolic and mitogenic effects are initiated by receptor-mediated tyrosine phosphorylation of a family of four insulin receptor substrates (IRS-1 to IRS-4) [2-5]. These proteins, which differ in tissue distribution, subcellular localization and developmental expression, subsequently act as docking sites for the recruitment and activation of a complex network of intracellular Src homology 2-containing signaling molecules [6,7]. Available data from knockout mouse models indicate that IRS-1 and IRS-2, as the principal substrates for insulin and insulin-like growth factor (IGF-1) receptor tyrosine kinases, play a central role in eliciting the pleitropic effects of insulin and IGF-1 [8].
The amino terminus of the IRS proteins contains a highly conserved pleckstrin homology (PH) domain that is critical for insulin/IGF-1 receptor and IRS interactions [9,10]. PHIP (PH-interacting protein) was originally isolated as a 100-kDa (902 amino acids) protein through yeast two-hybrid screening. Biochemical studies revealed that PHIP is a cytoplasmic protein that selectively interacts with the PH domain of IRS-1 [11]. Subsequent functional analysis in human fibroblast cells showed that PHIP is required for insulin receptor-mediated mitogenic and metabolic signal transduction [12]. More recently, a novel 206-kDa (1821 a.a.) isoform of PHIP (PHIP1) containing a WD-40 domain and two bromodomains has been isolated from mouse pancreatic islets [13]. In contrast to PHIP, PHIP1 was shown to be exclusively localized in the nuclear compartment of pancreatic β cells. Based on evidence from overexpression and RNA interference studies in pancreatic β-cell lines, it has been suggested that PHIP1 is involved in control of β-cell proliferation and survival in an IGF-1-dependent and independent manner.
The physiological roles of the PHIP proteins remain unknown. By microinjection of a gene-trapped mouse embryonic stem (ES) cell clone into C57/B6 blastocyts, we have recently generated mice carrying a gene-trapped Phip allele. We report here that mice homozygous for the gene trap mutation in Phip (Phip-/-) suffer from a severe postnatal growth deficit. The average lifespan of viable mutant mice is 4-5 weeks. These mice are slightly glucose intolerant and develop hypoglycemia. Mouse embryonic fibroblasts (MEFs) from Phip-/- mice grow significantly slower than wild-type MEFs, but remain responsive to IGF-1 mitogenic signals. Our data suggest that PHIP1 controls postnatal body growth by functioning independently of the IGF-1/AKT pathway.
MATERIALS AND METHODS
Mice
Phip gene trap mice were generated by micro-injection of mouse embryonic stem cell line RRJ571 (Baygenomics) into C57/BL6 blastocysts. The resulting chimeric male founders were crossed to C57BL/6 females to generate F1 heterozygous (Phip+/−) mice. Phip+/− mice were then intercrossed to generate F2 homozygous Phip-/− mice or embryos. Genotyping of all animals or embryos was done by PCR using primers F4, R1 and R5 (Supplementary Table 1). All animal experiments were performed in accordance with the Cornell Animal Care and Use Guidelines.
Physiological studies
Plasma insulin concentrations were measured using a Rat Insulin RIA Kit (Millipore). Glucose tolerance and insulin tolerance tests (GTT and ITT) were performed essentially as described [14]. Briefly, for glucose tolerance test, mice were fasted for 6-8 hours and then injected intraperitoneally (IP) with 2g per kg body weight of D-glucose. Glucose measurements were taken at 0, 5, 15, 30, 60 and 120 minutes post-injection using an Ascensia Elite XL glucometer (Bayer). Blood was collected from the tail vein at each time point during the glucose tolerance test and plasma insulin levels were determined by ELISA (Crystal Chem). For insulin tolerance test, mice were fasted for 6 h hours and then injected IP with 0.75 U per kg body weight of regular human insulin (Eli Lilly, IN) dissolved in phosphate-buffered saline (PBS). Glucose measurements were taken at 0, 5, 15, 30, 60 and 120 minutes post-injection.
Morphological studies
Hindlimb skeletal muscle, liver and lung were dissected from wild-type and mutant mice at 3 weeks of age, fixed in 4% paraformaldehyde (PFA) in PBS at 4°C overnight and then processed for paraffin embedding. 5-μm sections were mounted on glass slides and stained with hematoxylin and eosin. All images were acquired using an Axiovert 40 microscope (Zeiss) equipped with an AxioCam camera. Hepatocyte and skeletal muscle fiber sizes were measured using the AxioVision software (Version 4.1).
RNA isolation and quantitative RT-PCR
RNA was isolated using the TRIZOL RNA Isolation Kit (Invitrogen). For quantitative RT-PCR analysis, total RNA was treated with DNase I for 10 minutes and purified using the RNAqueous-Micro Kit (Ambion). cDNA was synthesized using SuperScript III Reverse Transcriptase (Invitrogen). Quantitative PCR (qPCR) was performed using Power SYBR Green PCR Master Mix on an ABI Prism 7000 Sequence Detection System (Applied Biosystems). All qPCR primers (Supplementary Table 1) were designed using the PrimerSelect program of Lasergene 7.1 Sequence Analysis Software (DNAStar). Quantification of expression and normalization were done essentially as described [15].
Cell proliferation assays
Wild-type and Phip-/- MEF cells were isolated and cultured as described [16]. For IGF-1 treatment, MEF cells were plated at a density of 2.4 × 104 cells per 60-mm dish in complete medium. After 12 hours of culture, the cells were serum-starved overnight and then 100 ng/ml of human IGF-1 (National Hormone and Peptide Program, UCLA) was added into the serum-free medium. Mock treatment was carried out by adding an equal volume of PBS into the culture medium. Cells were counted daily for 6 consecutive days.
[3H] Thymidine incorporation assay was performed essentially as described [17]. Briefly, wild-type and Phip-/- MEF cells were plated in triplicates in 48-well plates at a density of 7.5 × 103 cell/well and grown in complete medium overnight. MEFs were serum-starved for 24 hour and subsequently cultured in basal medium with 25, 50, 100 and 200 ng/ml of recombinant human IGF-1 for 18 hours in the presence of [Methyl-3H] thymidine at a final concentration of 1 μCi/well (MP Biochemicals, CA). MEFs were then washed 3 times with cold PBS. DNA was precipitated with 500 μl of cold 10% trichloroacetic acid and solubilized by the addition of 100 μl of 0.3 N NaOH. The amount of [3H] thymidine incorporated into DNA was measured by liquid scintillation counting and normalized to the amount of total cellular protein.
AKT phosphorylation and Western blot analysis
Phip+/+ and Phip-/− MEFs were seeded at 5 × 106 cells per 100-cm dish and cultured for 16 hours in complete medium. The MEFs were serum-starved overnight and treated with 100 ng/ml IGF-1 for 0, 5, 10 and 30 minutes. Total proteins were extracted using cell extraction buffer containing 0.05 M Tris-HCl at pH8.0, 0.15 M NaCl, 5.0mM EDTA, 1% NP-40 and a protease inhibitor cocktail (MBL) at 4°C. After centrifugation, soluble protein in the extract was quantified using bicinchoninic acid (BCA) assay (Pierce, Rockford, IL). Proteins were fractionated by SDS-10% PAGE, transferred to nitrocellulose membranes and probed with primary antibodies recognizing total AKT and AKT phosphorylated on serine 473 (Cell Signaling). Primary antibody binding was detected by using horseradish peroxidase coupled to goat anti-rabbit immunoglobulin G (IgG) or donkey anti-goat IgG (1:10,000). The antibody complex was visualized by incubation with the Lumi-Light Western blotting substrate (Roche-Mannhein, Mannhein, Germany).
Statistical Analysis
Differences between compared groups were evaluated by performing two-tailed Student’s t-test and p < 0.05 is considered significant.
RESULTS
Generation and molecular characterization of Phip gene-trap mutant (Phip-/-) mice
The Phip gene-trap mice were generated by microinjection of a commercially available mouse embryonic stem (ES) cell clone, RRJ571, into C57/B6 blastocysts. This clone contains an exon-trap cassette in intron 4 of the Phip gene. To identify the insertion site for the gene trap, we carried out PCR-based chromosome walking using a series of forward and reverse primers specific to intron 4. The trap is located approximately 1516-bp downstream of exon 4 (Fig. 1A).
Figure 1. Generation and characterization of Phip gene-trap mice.
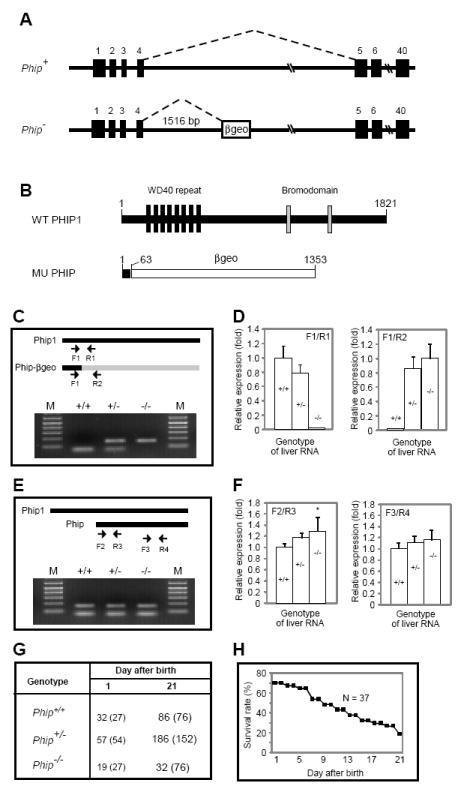
(A) Schematic representation of wild-type (Phip+) and mutant (Phip-) Phip alleles. Filled boxes represent exons; the open box in Phip- denotes the gene trap cassette. Dashed lines indicate RNA splicing events. (B) Schematic representation of wild-type and mutant PHIP peptides generated from Phip+ and Phip-, respectively. The number of amino acids for each peptide is indicated. The mutant peptide is a fusion protein containing the N-terminal 63 amino acids of PHIP1 and full-length βgeo. (C and E) RT-PCR analysis of liver RNAs from Phip+/+, Phip+/- and Phip-/- mice. PCR primers (arrows) and their locations in corresponding cDNAs are indicated on the top. For each genotype, PCR products from the indicated two primer pairs were pooled and resolved using a 2% agarose gel. In C, upper and lower bands represent products amplified by F1/R2 and F1/R1, respectively. In E, upper and lower bands represent products amplified by F2/R3 and F3/R4, respectively. (D and F) Quantification of transcripts in C and E by quantitative RT-PCR. Genotypes of the RNA are indicated as +/+, +/- and -/-, respectively. qPCR primers are shown on top of the bar graph. N = 3 mice per genotype, *, p < 0.05 Phip-/- versus Phip+/+ mice. (G) Distribution of Phip+/+, Phip+/- and Phip-/- mice. Numbers in parentheses indicate expected distribution. (H) Survival rate of Phip-/- mice during postnatal period. A total of 37 newborn Phip-/- pups were monitored in a 3-week window.
The gene-trap cassette in Phip contains a strong splicing acceptor site and a β-galactosidase-neomycin (βgeo) fusion gene. This cassette is expected to block splicing between exon 4 and 5, resulting in a fusion protein containing the 63 amino acids of the N-terminus of PHIP1 and (βgeo (Fig. 1B). To assess this, we performed semi- and quantitative RT-PCR analyses of liver RNAs from wild-type (Phip+/+), heterozygous (Phip+/-) and homozygous (Phip-/-) mice using Phip and βgeo specific primers (Fig. 1C, arrows). As expected, a wild-type amplicon was detected in Phip+/+and Phip+/- mice, but not in Phip-/-mice; in contrast, a Phip-βgeo fusion amplicon was detected in Phip+/- and Phip-/- mice, but not in Phip+/+ mice (Fig. 1C and 1D). Available information in the NCBI Genbank database predicts multiple alternatively spliced transcripts for the mouse Phip gene. To assess this, we carried out additional RT-PCR analysis using isoform-specific primers (Fig. 1E). Indeed, at least one smaller transcript, encoding the 902 a.a. isoform of PHIP, was detected and found to be up-regulated in Phip-/- mice (Fig. 1E and 1F). Together, these results indicate that Phip-/- mice are void of the full-length isoform of PHIP (PHIP1), but retain the 902-a.a. and possibly other isoforms of PHIP peptides. Thus, the Phip-/- mice reported here are not PHIP- null mutants.
Heterozygous mice (Phip+/-) appeared morphologically normal and were fertile. Of the 272 weaned offspring from intercrossing Phip+/- mice, only 32 Phip-/- mice (11.7%) were found (Fig. 1G). No Phip-/- mice was found 6 weeks after birth. To determine whether homozygous mutants were under-represented at birth, we determined the distribution ratio of wild-type, heterozygous and homozygous animals at birth. Of 108 new born mice, only 19 Phip-/- mice (17.6%) were found (Fig. 1G), suggesting that some Phip-/- embryos died during development. Phip-/- mice had an average life-span of about 4 weeks, with the longest living mouse so far at 5 weeks of age (Fig. 1H). Overall, these results indicate that PHIP1 plays a non-essential role during embryogenesis, but is critically required for postnatal growth and survival of mice.
PHIP1 regulates postnatal tissue growth and functioning
All Phip-/- mice that survived to weaning appeared to be markedly smaller than their wild-type littermate control mice (Fig. 2A). To determine whether the growth deficiency of Phip-/- mice was the result of fetal or postnatal growth deficiency, we monitored the growth rate of Phip-/- mice from birth to weaning (Fig. 2B). New born Phip-/- pups had comparable weights to their wild-type and Phip+/- littermates. However, by 1 week of age, Phip-/- mice appeared to be smaller than their wild-type and Phip+/- mice littermates. During the next 2-3 weeks, the body weight difference between Phip-/- and wild-type mice increased progressively. To further assess the growth phenotype of Phip-/- mice, we compared the tissue/body weight ratio for various tissues between Phip-/- and wild-type mice (Table 1). No significant difference was found between the two genotypes in the relative weight of heart, liver, lung, hindlimb muscle, pancreas and spleen. However, Phip-/- mice had a significantly lower adipose/body weight ratio than wild-type mice. Next we sought to determine whether cell size plays a role for the observed growth phenotype in Phip-/- mice. Phip-/- myocytes were significantly reduced in size as compared to wild-type myocytes (Fig. 2C-E). Phip-/- hepatocytes, although morphologically normal, also appeared to be significantly smaller than wild-type hepatocytes (Fig. 2F-H). These results are in line with the notion that a major contributor to the postnatal growth phenotype of Phip-/- mice is hypotrophy of somatic cells.
Figure 2. Inactivation of PHIP1 impairs postnatal whole body growth.
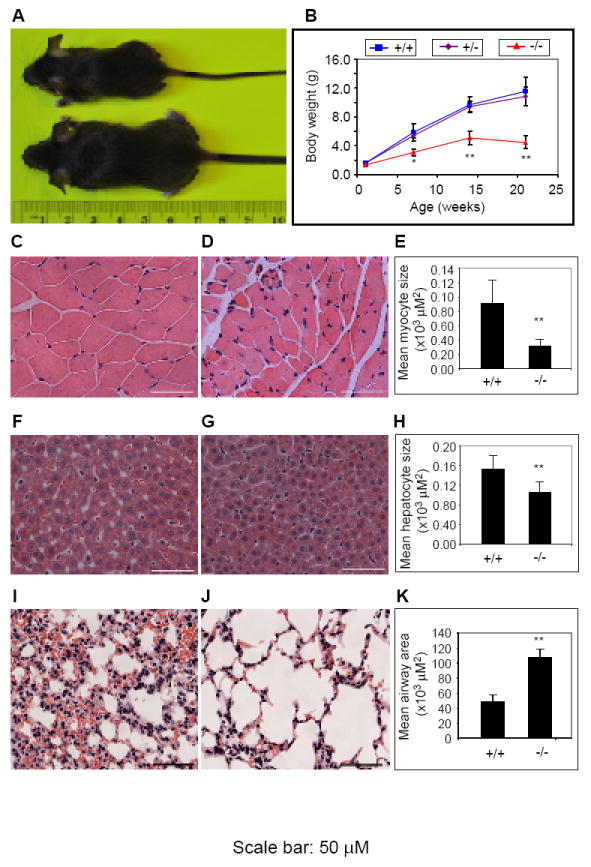
(A) Representative images of Phip+/+ (bottom) and Phip-/- (top) mice at 4 weeks of age. (B) Postnatal body weight gain profiles of Phip+/+, Phip+/- and Phip-/- mice. N = 10 per genotype, *, p < 0.05, **, p < 0.01 Phip-/- versus Phip+/+ mice. (C-D) H&E staining of hindlimb skeletal muscle cross sections from Phip+/+ (C) and Phip-/- (D) mice. (F-G) H&E staining of liver sections from Phip+/+ (F) and Phip-/- (G) mice. (I-J) H&E staining of lung sections from Phip+/+ (I) and Phip-/- (J) mice. (E, H and K) Quantification of sizes of myocytes (E), hepatocytes (H) and lung airways from Phip+/+ and Phip-/- mice. N = 3 per genotype, **, p < 0.01 Phip-/- versus Phip+/+ mice. All data are expressed as mean ± standard deviation. Scale bar: 50 μM.
Table 1.
Organ/body weight ratios of Phip+/+ and Phip-/- mice
| Body weight (g) | Adipose (%) | Heart (%) | Liver (%) | Lung (%) | Hindlimb muscle (%) | Pancreas (%) | Spleen (%) | |
|---|---|---|---|---|---|---|---|---|
| Phip+/+ | 14.8 ± 0.73 | 1.79 ± 0.26 | 0.52 ± 0.05 | 3.72 ± 0.39 | 1.94 ± 0.19 | 1.32 ± 0.30 | 1.02 ± 0.07 | 0.40 ± 0.05 |
| Phip -/- | 8.9 ± 1.63 | **0.25 ± 0.14 | 0.51 ± 0.19 | 4.18 ± 0.32 | 2.38 ± 0.42 | 0.81 ± 0.19 | 1.34 ± 0.29 | 0.32 ± 0.10 |
Phip+/+ (n = 4) and Phip-/- (n = 3) mice were sacrificed at three weeks of age. The major visceral organs, abdominal fat and hindlimb skeletal muscle were weighed and averaged according genotype. The averaged organ weight was used to calculate an organ to body weight ratio. The data are expressed as mean ± standard deviation. Note that mutant mice had a significantly lower abdominal fat content than wild-type mice.
Phip-/- mice appeared to have a healthy heart and had no symptom of tumor or infectious disease development at the time of their death. We speculated that their death may relate to the improper development or failure of some internal organs. Thus, we carried out histological analysis of the lung in Phip-/- mice. The lung epithelium in Phip-/- mice was poorly developed, had severely dilated airways and appeared to contain significantly fewer red blood cells (Fig. 2I-K). Based on this observation, it is possible Phip-/- mice die due to a progressively failing lung function. Further studies are needed to determine whether functional abnormalities are present in other organs or tissues of Phip-/- mice.
PHIP1-deficient mice develop hypoglycemia
PHIP1 is highly expressed in the pancreatic β cells and was shown to be required for pancreatic β-cell proliferation [13]. In addition, PHIP, an alternatively spliced isoform of PHIP1, was isolated as an IRS-1 interacting protein and was previously showed to be involved in insulin signal transduction [12,18]. Thus, we next assessed whether PHIP1-deficient mice were defective in glucose metabolism. Basal blood glucose levels of Phip+/+ and Phip-/- mice were monitored from birth to 3 weeks of age. No significant difference was observed in basal blood glucose levels among the three genotypes at birth. However, 5 days after birth Phip-/- mice showed a lower blood glucose level than Phip+/+ mice; and they progressively developed severe hypoglycemia during the next two weeks (Fig. 3A). We monitored the daily activity and feeding behavior of these mice and found that Phip-/- mice were physically active and appeared to have normal feeding behavior. We also determined the plasma insulin levels of Phip+/+ and Phip-/- mice and found that Phip-/- mice had a comparable fasting serum insulin level as Phip+/+ mice (Fig. 3B). These results suggest that neither under-nutrition nor hyperinsulinemia contributed to the hypoglycemia in Phip-/- mice. Given the fact the transcript encoding PHIP is up-regulated in Phip-/- mice, we tested the hypothesis that Phip-/- mice may be more sensitive to insulin. Indeed, insulin tolerance tests (ITT) indicated that the same dosage of insulin had stronger glucose-lowering effect in Phip-/- mice than in wild-type control mice (Fig. 3C). Despite the proposed role of PHIP1 in promoting β-cell proliferation, Phip-/- mice appeared to have adequate pancreatic function, as revealed by glucose intolerance tests (Fig. 3C). Together, these data indicate that PHIP1 deficiency sensitizes mice to insulin action, but does not affect insulin production in the pancreas.
Figure 3. Phip-/- mice progressively develop hypoglycemia.
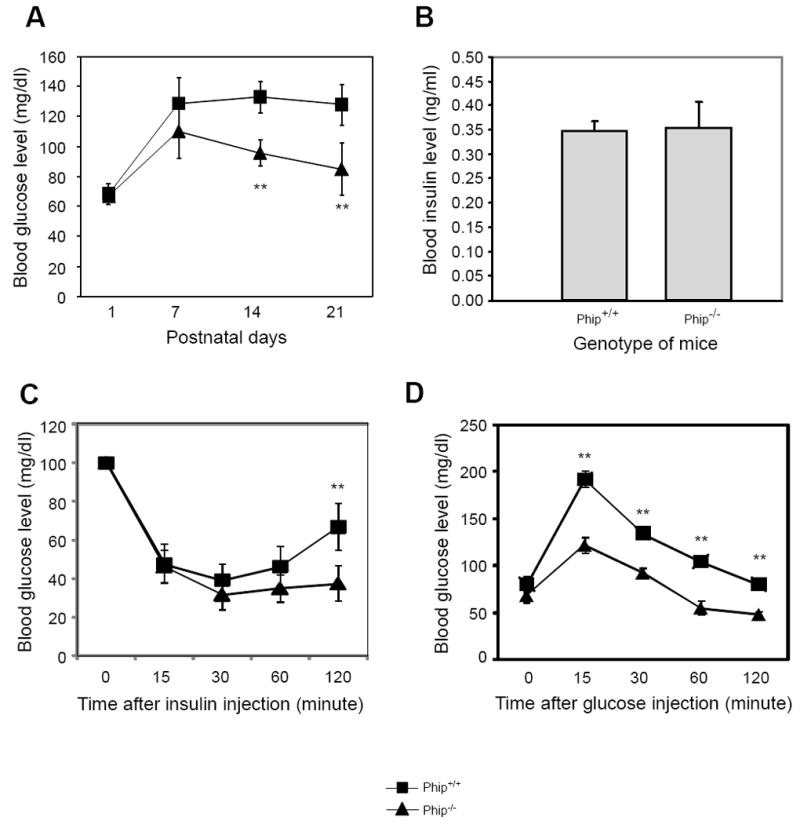
(A) Fasting blood glucose levels of Phip+/+ and Phip-/- mice during postnatal period. Mice were fasted for 8 hours and blood glucose concentrations were measured at postnatal day 1, 7, 14 and 21. N = ? per genotype; *, p < 0.05, **, p < 0.01 Phip-/- versus Phip+/+ mice. (B) Fasting serum insulin concentrations in Phip+/+ and Phip-/- mice. Mice were fasted for 8 hours and plasma insulin concentrations were determined by ELISA. N = 3 per genotype. (C) Insulin tolerance test. Phip+/+ and Phip-/- mice at 3 weeks of age were IP injected with 0.75 U per kg body weight of human insulin. Glucose measurements were taken at 0, 15, 30, 60 and 120 minutes post insulin injection. (D) Glucose tolerance test. 3-week old Phip+/+ and Phip-/- mice fasted for 8 hours were IP injected with 2g per kg body weight of D-glucose. Glucose measurements were taken at 0, 15, 30, 60 and 120 minutes post glucose injection. *, p<0.05, **, p < 0.01 Phip-/- vs Phip+/+ mice.
PHIP1 regulates somatic cell growth independent of the IGF-1/AKT signaling pathway
To investigate the molecular mechanisms underpinning the growth deficiency of Phip-/- mice, we isolated mouse embryonic fibroblast cells from E13.5 Phip-/- embryos and characterized their growth. Phip-/- MEFs, while morphologically indistinguishable from Phip+/+ MEFs, grew significantly more slowly than wild-type MEFs (Fig. 4A). This result, which is consistent with the previous finding that PHIP1 is involved in regulating pancreatic β-cell proliferation [13], suggests that hypoplasia likely contributed to the growth deficiency of Phip-/- mice. Since PHIP1 was shown to promote cell proliferation in both IGF-1-dependent and independent manners [13], and the growth phenotype of Phip-/- mice resemble that of IGF-1 receptor (Igf1r) knockout mice [19], we tested whether PHIP1 functionally interacts with the IGF-1/AKT signaling pathway. Mouse embryonic fibroblast (MEF) cells from E13.5 Phip+/+ and Phip-/- embryos were treated with 100 ng/ml human IGF-1 for various times and AKT phosphorylation was assessed by Western blot analysis. Phip-/- MEFs showed normal levels of AKT phosphorylation at the 473 serine residue upon IGF-1 treatment (Fig. 4B). This result suggests that inactivation of PHIP1 does not impair IGF-1/AKT signal transduction. We next assessed whether Phip-/- MEFs increase DNA synthesis in response to IGF-1 treatment using [3H]-thymidine incorporation assay. Phip-/- MEFs, upon treatment with various concentrations of IGF-1, exhibited a dose-dependent increase of DNA synthesis in response to IGF-1 (Fig. 4C). Taken together, these data are consistent with the notion that PHIP1 is required for postnatal growth and functions independently of the IGF-1/AKT pathway.
Figure 4. PHIP1 regulates MEF cell proliferation in an IGF-1 independent manner.
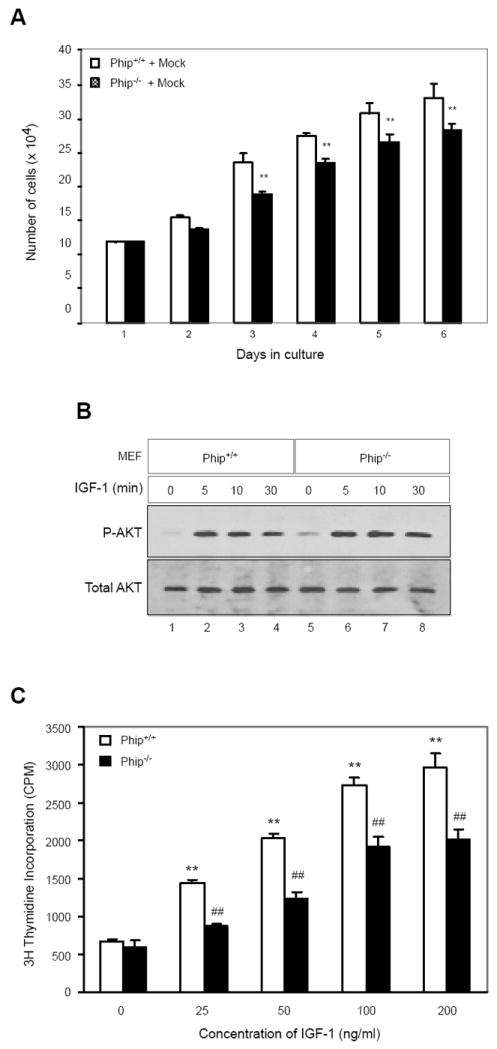
(A) Rate of Phip+/+ and Phip-/- MEF cell proliferation. MEFs from E13.5 Phip+/+ and Phip-/- embryos were plated at a density of 2.4 × 104 per 60-mm plate and cultured for a week. Cells were counted every 24 hours. *, p < 0.05, **, p < 0.01 Phip-/- versus Phip+/+ MEFs. (B) Western blot analysis of AKT phosphorylation in Phip+/+ and Phip-/- MEFs. MEFs of defined genotypes were plated at a density of 5.6 × 106 per 100-mm dish. After serum-starving for 24 hours, the MEFs were treated with IGF-1 (100 ng/ml) for 0, 5, 10 and 30 minutes. MEF cell lysates were prepared in RIPA lysis buffer and resolved on 7% SDS PAGE. Total and phosphorylated (serine 473) AKT were detected using anti-AKT and anti-pAKT antibodies. (C) [3H] Thymidine incorporation assay of DNA synthesis of Phip+/+ and Phip-/- MEFs in response to IGF-1. Phip+/+ and Phip-/- MEFs were plated in triplicates in 48-well plates (7.5 × 103 cells/well), grown and serum-starved as described in (B). MEFs were treated with 25, 50, 100 and 200 ng/ml of human IGF-1 for 18 hours in the presence of 1 μCi of [3H] thymidine per well. *, p < 0.05, **, p < 0.01 non-treated versus treated Phip+/+ MEFs. #, p < 0.05, ##, p < 0.01 non-treated versus treated Phip-/- MEFs.
DISCUSSION
We report here that mice homozygous for a gene-trap mutation in the Phip gene (Phip-/-) exhibit severe whole body growth deficiency during postnatal period. Mutant hepatocytes and myocytes appear to be significantly smaller than those of wild-type mice. Mutant MEFs are smaller in size and grow significantly slower than wild-type control MEFs. Taken together, these findings suggest that Phip is an important regulator of somatic cell growth and cell sizes in mice. To the best of our knowledge, this represents the first in vivo functional study for the mammalian Phip gene.
Available sequence information in the NCBI Genbank database indicates that the mouse Phip gene has multiple alternative spliced transcripts. These transcripts are predicted to encode at least 4 PHIP proteins with sizes ranging from 902 to 1821 amino acids [13]. The cDNAs encoding PHIP and PHIP1, the 902 and 1821 a.a. variant, respectively, were previously cloned. It has been shown that PHIP, a cytosolic protein, selectively interacts with the PH domain in IRS-1 and is involved in insulin and IGF-1 signaling [11,12]. In contrast, PHIP1, the 1821 a.a. isoform, was found to be exclusively localized in the nucleus of mouse pancreatic β cells, an indication that PHIP1 may function as a transcription factor [13]. Indeed, overexpression of PHIP1 promotes cyclin D2 gene expression, whereas RNAi-mediated knocking-down of PHIP1 suppresses cyclin D2 expression [13]. We show by semi- and quantitative RT-PCR that the full length Phip transcript, encoding PHIP1, is absent in Phip-/- mice (Fig. 1C-D). However, the transcript that encodes the 902 a.a isoform of PHIP is detectable and, interestingly, up-regulated in the homozygous mutants (Fig. 1E-F). These results strongly suggest that Phip- is a hypermorphic allele and that it is the inactivation of PHIP1, the 1821 a.a. isoform, that is responsible for the observed postnatal growth phenotype in Phip-/- mice.
Phip-/- mice gradually develop hypoglycemia during postnatal period, albeit that they have a comparable serum insulin level to their wild-type littermate control mice. The molecular mechanisms underlining this glucose metabolic phenotype remains unclear. Given the fact that the Phip transcripts encoding the 902 a.a. isoform of PHIP is up-regulated in Phip-/- mice, and that this isoform of PHIP was previously implicated in insulin signal transduction [11,12], it is possible that Phip-/- mice may be more sensitive to insulin due to an enhanced insulin signaling pathway. Further biochemical studies are needed to determine the molecular defects of the insulin signaling pathway.
Phip-/- mice exhibit a growth deficiency that is similar in severity to that of IGF-1 null mice (60% of the body weight of wild-type mice) [19,20]. Additionally, a previous study in vitro showed that PHIP1 promotes pancreatic β cell proliferation in an IGF-1/IRS-2 dependent and independent manner [13]. Given the previous finding that PHIP1 is exclusively localized in the nucleus [13], we tested whether PHIP1 functions as a downstream target of the IGF-1/AKT signaling pathway. We show that PHIP1-deficient MEFs exhibit an intrinsically slower growth than wild-type MEFs (Fig. 4A). Surprisingly, in response to exogenous IGF-1 PHIP1-deficient MEFs increase DNA synthesis in an IGF-1 dose-dependent manner (Fig. 4C). Further, we show by Western blot analysis that Phip-/- MEFs have normal AKT phosphorylation following IGF-1 treatment. Together, these observations suggest that PHIP1 regulates postnatal growth in an IGF-1/AKT pathway independent manner. Several functional pathways, namely the IGF-1/PI3/AKT, IGF-1/RAS/MAPK and IGF-1/PDK1/p70S6K pathways, have been implicated in the transduction of IGF-1 signals [1,21-23]. It remains to be determined whether PHIP1 functions as a downstream target of the IGF-1/RAS/MAPK and IGF-1/PDK1/p70S6k pathways.
In conclusion, we report that mice deficient for the full-length isoform of PHIP, PHIP1, are severely retarded during postnatal body growth. PHIP1 appear to act in an IGF-1/AKT-independent manner. The mouse genome contains two Phip-related genes: Brwd1 and Brwd3 (Bromodomain and WD-repeat containing genes). BRWD1, PHIP1 and BRWD3 are structurally conserved and seem to have overlapping expression patterns [24]. Mice deficient for BRWD1, however, were largely normal except that both males and females are infertile [24], suggesting that members of the BRWD family may have redundant functions. Further proof of this requires the generation of BRWD1 and PHIP1 double mutant mice.
Supplementary Material
Acknowledgments
We thank Dr. Bruce Currie (Cornell University) for stimulating discussions and critical comments during preparation of the manuscript. The financial support for this research project was provided by the College of Agricultural and Life science, Cornell University to QML and NIH to JCS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klammt J, Pfaffle R, Werner H, Kiess W. IGF signaling defects as causes of growth failure and IUGR. Trends Endocrinol Metab. 2008;19:197–205. doi: 10.1016/j.tem.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Lavan BE, Fantin VR, Chang ET, Lane WS, Keller SR, Lienhard GE. A novel 160-kDa phosphotyrosine protein in insulin-treated embryonic kidney cells is a new member of the insulin receptor substrate family. J Biol Chem. 1997;272:21403–7. doi: 10.1074/jbc.272.34.21403. [DOI] [PubMed] [Google Scholar]
- 3.Lavan BE, Lane WS, Lienhard GE. The 60-kDa phosphotyrosine protein in insulin-treated adipocytes is a new member of the insulin receptor substrate family. J Biol Chem. 1997;272:11439–43. doi: 10.1074/jbc.272.17.11439. [DOI] [PubMed] [Google Scholar]
- 4.Sun XJ, et al. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature. 1991;352:73–7. doi: 10.1038/352073a0. [DOI] [PubMed] [Google Scholar]
- 5.Sun XJ, et al. Role of IRS-2 in insulin and cytokine signalling. Nature. 1995;377:173–7. doi: 10.1038/377173a0. [DOI] [PubMed] [Google Scholar]
- 6.White MF. The IRS-signalling system: a network of docking proteins that mediate insulin action. Mol Cell Biochem. 1998;182:3–11. [PubMed] [Google Scholar]
- 7.White MF. Regulating insulin signaling and beta-cell function through IRS proteins. Can J Physiol Pharmacol. 2006;84:725–37. doi: 10.1139/y06-008. [DOI] [PubMed] [Google Scholar]
- 8.Sesti G, Federici M, Hribal ML, Lauro D, Sbraccia P, Lauro R. Defects of the insulin receptor substrate (IRS) system in human metabolic disorders. Faseb J. 2001;15:2099–111. doi: 10.1096/fj.01-0009rev. [DOI] [PubMed] [Google Scholar]
- 9.Myers MG, Jr, et al. The pleckstrin homology domain in insulin receptor substrate-1 sensitizes insulin signaling. J Biol Chem. 1995;270:11715–8. doi: 10.1074/jbc.270.20.11715. [DOI] [PubMed] [Google Scholar]
- 10.Yenush L, Makati KJ, Smith-Hall J, Ishibashi O, Myers MG, Jr, White MF. The pleckstrin homology domain is the principal link between the insulin receptor and IRS-1. J Biol Chem. 1996;271:24300–6. doi: 10.1074/jbc.271.39.24300. [DOI] [PubMed] [Google Scholar]
- 11.Farhang-Fallah J, Yin X, Trentin G, Cheng AM, Rozakis-Adcock M. Cloning and characterization of PHIP, a novel insulin receptor substrate-1 pleckstrin homology domain interacting protein. J Biol Chem. 2000;275:40492–7. doi: 10.1074/jbc.C000611200. [DOI] [PubMed] [Google Scholar]
- 12.Farhang-Fallah J, Randhawa VK, Nimnual A, Klip A, Bar-Sagi D, Rozakis-Adcock M. The pleckstrin homology (PH) domain-interacting protein couples the insulin receptor substrate 1 PH domain to insulin signaling pathways leading to mitogenesis and GLUT4 translocation. Mol Cell Biol. 2002;22:7325–36. doi: 10.1128/MCB.22.20.7325-7336.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Podcheko A, Northcott P, Bikopoulos G, Lee A, Bommareddi SR, Kushner JA, Farhang-Fallah J, Rozakis-Adcock M. Identification of a WD40 repeat-containing isoform of PHIP as a novel regulator of beta-cell growth and survival. Mol Cell Biol. 2007;27:6484–96. doi: 10.1128/MCB.02409-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sachdeva MM, Claiborn KC, Khoo C, Yang J, Groff DN, Mirmira RG, Stoffers DA. Pdx1 (MODY4) regulates pancreatic beta cell susceptibility to ER stress. Proc Natl Acad Sci U S A. 2009;106:19090–5. doi: 10.1073/pnas.0904849106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Francisco AB, et al. Deficiency of suppressor enhancer Lin12 1 like (SEL1L) in mice leads to systemic endoplasmic reticulum stress and embryonic lethality. J Biol Chem. 2010;285:13694–703. doi: 10.1074/jbc.M109.085340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorn SR, Purup S, Cohick WS, Vestergaard M, Sejrsen K, Boisclair YR. Leptin does not act directly on mammary epithelial cells in prepubertal dairy heifers. J Dairy Sci. 2006;89:1467–77. doi: 10.3168/jds.S0022-0302(06)72214-7. [DOI] [PubMed] [Google Scholar]
- 18.Kaburagi Y, et al. Role of IRS and PHIP on insulin-induced tyrosine phosphorylation and distribution of IRS proteins. Cell Struct Funct. 2007;32:69–78. doi: 10.1247/csf.07003. [DOI] [PubMed] [Google Scholar]
- 19.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 20.Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- 21.Glass DJ. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol. 2003;5:87–90. doi: 10.1038/ncb0203-87. [DOI] [PubMed] [Google Scholar]
- 22.Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279:707–10. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- 23.Song YH, Godard M, Li Y, Richmond SR, Rosenthal N, Delafontaine P. Insulin-like growth factor I-mediated skeletal muscle hypertrophy is characterized by increased mTOR-p70S6K signaling without increased Akt phosphorylation. J Investig Med. 2005;53:135–42. doi: 10.2310/6650.2005.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Philipps DL, et al. The dual bromodomain and WD repeat-containing mouse protein BRWD1 is required for normal spermiogenesis and the oocyte-embryo transition. Dev Biol. 2008;317:72–82. doi: 10.1016/j.ydbio.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.