


Abstract
Post-transcriptional regulatory mechanisms are widespread in bacteria. Interestingly, current published data hint that some of these mechanisms may be non-random with respect to their phylogenetic distribution. Although small, trans-acting regulatory RNAs commonly occur in bacterial genomes, they have been better characterized in Gram-negative bacteria, leaving the impression that they may be less important for Firmicutes. It has been presumed that Gram-positive bacteria, in particular the Firmicutes, are likely to utilize cis-acting regulatory RNAs located within the 5′ mRNA leader region more often than trans-acting regulatory RNAs. In this analysis we catalog, by a deep sequencing-based approach, both classes of regulatory RNA candidates for Bacillus subtilis, the model microorganism for Firmicutes. We successfully recover most of the known small RNA regulators while also identifying a greater number of new candidate RNAs. We anticipate these data to be a broadly useful resource for analysis of post-transcriptional regulatory strategies in B. subtilis and other Firmicutes.
INTRODUCTION
A variety of RNA-based regulatory mechanisms have been shown to be important in controlling expression of metabolic pathways, stress responses, developmental processes and pathogenesis. Depending on their structural relationship to the target gene, regulatory RNAs can be divided into two broad categories: those that are co-transcribed (in cis) or transcribed independently (in trans) with respect to the target mRNA. Cis-acting regulatory RNAs are typically located within the 5′ leader region of the target transcript, although they can also be situated within inter-cistronic regions of multi-gene transcripts. Trans-acting regulatory RNAs, which can be encoded either in cis or in trans relative to their target transcript, are transcribed separately from the mRNA target. Both classes of regulatory RNAs are capable of activation or repression of gene expression.
Trans-acting regulatory RNAs in Gram-negative bacteria
In the past several decades, many small (∼70–200 nt) RNAs (‘sRNA’) have been discovered in bacteria (1,2). Overlapping methods have been used for their discovery, including directed cloning and sequencing of sRNA pools, computational approaches, deep sequencing and hybridization of sRNA pools to genomic tiling arrays (3–8). Although a few RNAs serve ‘housekeeping’ functions, such as processing of pre-tRNAs by RNase P, most sRNA candidates identified by these approaches are presumed to function as regulatory agents. For example, one widespread sRNA class regulates gene expression by sequestering an RNA-binding protein (e.g. CsrA) and preventing it from controlling translation of target mRNAs (9). Another widespread class, coined 6S RNAs, regulates expression patterns by structurally mimicking an open promoter and associating with RNA polymerase to prevent transcription initiation of target genes (10). However, most sRNAs are likely to affect gene expression by directly base pairing to one or more target mRNAs (2,11). Some interact with their target mRNA via antisense base paired regions. Typically, these antisense sRNAs associate with the target transcript through formation of long base paired regions of >65 nt in order to regulate mRNA stability, translation or transcription elongation (2). In contrast, many trans-encoded sRNAs associate with their target transcripts through shorter, more imperfect, base pairing interactions (12). Dozens of such sRNAs have been characterized in Escherichia coli and other Gram-negative bacteria. Indeed it has been estimated that 200–300 sRNAs will be present in the average bacterial genome, equivalent in numbers to the total complement of cellular transcription factors (13). Most are individually responsive to different stress conditions; they largely assist the adaptive responses of the microorganism as its local environment undergoes changes. Similar to transcription factors, many sRNA regulators are predicted to regulate multiple targets (14–17); therefore, the full range of their regulatory complexities remains to be determined. Many sRNAs bind near to the start of the coding region in order to affect translation initiation efficiency of the target gene (12,18); however, some reduce translation by interacting further upstream within the 5′ untranslated region (5′ UTR) (15,19,20). Yet others reduce expression by associating with the mRNA further downstream, within the coding region (21).
Cis- and trans-acting regulatory RNAs in B. subtilis and other Gram-positive bacteria
The majority of these studies have occurred with E. coli and other proteobacterial species and it is unclear how meaningful they are for other bacteria. To that end, recent efforts have begun to uncover candidate sRNAs in non-proteobacterial species. Recently, sRNAs have been identified in Gram-positive bacteria, including B. anthracis, Listeria monocytogenes, Staphylococcus aureus, Streptococcus species and Streptomyces coelicolor (22–32). However, the common ‘rules’ for sRNA regulation, as well as their involvement with RNA-binding proteins, are not yet apparent from these analyses (32,33). Historically, B. subtilis has been used as the benchmark model microorganism for Gram-positive bacteria. Yet study of sRNA regulation in this organism is still in an early stage. Therefore, rigorous analysis of the importance and molecular mechanisms for sRNA regulation in B. subtilis is important for elucidation of post-transcriptional regulation in Firmicutes and other Gram-positive bacteria.
Some forms of RNA-mediated regulation have already been demonstrated to be significantly important for B. subtilis. For example, at least 4% of the genome is believed to be subject to control by cis-acting regulatory RNAs alone (34) and is still certain to underestimate the full degree of RNA-mediated regulation. Indeed, little is known about the importance or mechanisms of trans-acting sRNAs, although several individual examples have been identified. A recent publication describing identification of B. subtilis sRNAs via mapping of transcriptionally active regions by high-density oligonucleotide tiling arrays successfully identified most known sRNAs, as well other new candidates (35). Herein, we describe the discovery of sRNAs and other putative regulatory RNA elements using a sister technique to the latter study. Specifically, we utilized a differential RNA-sequencing (dRNA-seq) approach that is selective for transcriptional start sites (TSS) (36). Our approach successfully recovers most known regulatory RNAs as well as a portion of the microarray-predicted sRNAs, also identifying many unique candidates. This catalog of candidate regulatory RNA elements will serve as important reference point for comprehensive analyses of sRNA regulation in B. subtilis and other Bacillus species.
MATERIALS AND METHODS
Bacterial strains and growth condition
Bacillus subtilis strain 168 and NCIB 3610 were used for 454 pyrosequencing and northern blot analyses, respectively. For total RNA extractions, cells were grown overnight until reaching stationary phase (∼20 h) at 37°C in modified glucose minimal medium [(NH4)2SO4 20 g/l, K2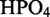 3H2O 183 g/l, KH2PO4 60 g/l,
3H2O 183 g/l, KH2PO4 60 g/l, 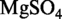 .7H2O 2 g/l, sodium citrate 10 g/l, 0.5% glucose, 0.5 mM CaCl2, 5 µM MnCl2]. For strain 168, tryptophan was added to a final concentration of 50 µg/ml.
.7H2O 2 g/l, sodium citrate 10 g/l, 0.5% glucose, 0.5 mM CaCl2, 5 µM MnCl2]. For strain 168, tryptophan was added to a final concentration of 50 µg/ml.
Total RNA preparation
Total RNA was harvested from B. subtilis strains cultured at 37°C as described (37). Briefly, these cells were pelleted, re-suspended in LETS buffer (0.1M LiCl, 10mM EDTA, 10mM Tris–HCl, 1% SDS), vortexed with acid-washed glass beads (Sigma-Aldrich) for 4 min, and incubated at 55°C for 5 min. The resulting solution was subjected to extraction by TRIzol reagent (Invitrogen) according to the manufacturer instructions. For subsequent pyrosequencing analysis, total RNA preparations were subjected to RNase-free DNase I (Roche) digestion for 30 min at 37°C in the presence of 0.25 mM MgCl2. RNA was then extracted with phenol:isoamyl:chloroform, ethanol precipitated and re-suspended in water. RNA quality was assessed by resolving samples on 1% formaldehyde–agarose gels and quantified via absorbance spectroscopy.
Northern blot analysis
Total RNA samples (15–20 µg) were heated at 65°C for 10 min in 1 × gel loading buffer (45 mM Tris–borate, 4 M urea, 10% sucrose [w/v], 5 mM EDTA, 0.05% SDS, 0.025% xylene cyanol FF, 0.025% bromophenol blue) and resolved by 6% denaturing (8 M urea) polyacrylamide electrophoresis. RNAs were transferred to BrightStar-Plus nylon membranes (Ambion) using a semi-dry electroblotting apparatus (Owl Scientific) according to manufacturer instructions. The blots were UV-crosslinked and hybridized overnight at 42°C in UltraHyb-Oligo buffer (Ambion) with the appropriate 5′-radiolabeled (32P) DNA oligonucleotide (see Supplementary Table S4 for oligonucleotides used in this study). The blots were then washed 2× for 15 min using low stringency wash buffer (1 × SSC, 0.1% SDS, 1 mM EDTA). Radioactive bands were visualized using ImageQuant software and a Typhoon PhosphorImager (Molecular Dynamics).
Four hundred and fifty-four pyrosequencing and data analysis
cDNA cloning and pyrosequencing was performed as described (38) except without size fractionation of RNA. However, the total RNA was split into two samples and for one sample primary transcripts containing 5′-PPP were enriched by treatment of the total RNA samples with Terminator 5′-phosphate-dependent exonuclease (Epicentre). Upon treatment of both samples with tobacco acid pyrophosphatase to generate 5′-monophosphates for linker ligation, RNA samples were poly(A)-tailed using poly(A) polymerase followed by ligation of a RNA adapter to the 5′-phosphate of the small RNAs. First strand cDNA synthesis was then performed using an oligo(dT)-adapter primer and M-MLV H-reverse transcriptase. A detailed protocol for the enrichment, cDNA library generation and subsequent sequencing steps are described (36). cDNA libraries were sequenced on a Roche FLX sequencer at the M.D. Anderson Cancer Center DNA Analysis Core Facility. The 5′-linker and poly-A tail removal were performed using custom-made Python scripts. The resulting cDNA sequences were then mapped onto B. subtilis genome (NC_000964.3) using ‘segemehl’ software (39). Mapped reads were visualized using Integrated Genome Browser (IGB).
RESULTS AND DISCUSSION
A general strategy for high-throughput identification of non-coding and coding RNAs
Bacillus subtilis strain 168 cells were cultured in glucose minimal medium until stationary phase, whereupon total RNA was extracted using standard methods. Most bacterial RNAs, including both mRNAs and sRNAs, contain a triphosphate moiety at their 5′ terminus, whereas processed transcripts, such as rRNAs and tRNAs, contain a 5′ monophosphate. To specifically enrich our samples for primary transcripts, half of the sample was treated with terminator exonuclease that preferentially degrades 5′-monophosphorylated RNAs. cDNA libraries were then prepared from these two pools (‘unenriched’ and ‘enriched’) and analyzed by 454 pyrosequencing. After 5′-linker and poly-A tail removal, a total of 406 531 cDNA reads (>15 nt in length) could be successfully mapped to the B. subtilis genome. Most of these sequences corresponded to ribosomal RNAs and tRNAs, which were excluded from further analysis. At the end, 25 675 and 44 098 cDNA reads were obtained for the unenriched and enriched libraries, respectively, and visualized with IGB (Supplementary Table S1; IGB, Affymetrix). The majority mapped to intergenic regions, due to the enrichment for cDNA reads located proximal to the TSS (36). Based on these data we were able to identify ∼600 potential TSSs in the B. subtilis genome, which appeared to be modestly increased nearer to the origin of replication (Supplementary Figure S1). From this analysis, classes of TSS could be identified for both sense and antisense RNAs (Supplementary Figure S2). Also, signals that were likely to correspond to long 5′ mRNA leader regions could be identified for some genes, whereas other cDNA reads appeared likely to correspond to small regulatory RNAs (sRNAs) (Supplementary Figure S2). We concentrated our efforts on the latter classes, which are most likely to include cis- and trans-acting regulatory RNAs.
Identification of ‘long’ 5′ leader regions
Bacillus subtilis employs a wide variety of cis-acting regulatory RNA elements (34). In contrast to the average length of ∼360 nt (±150 nt) for 5′ leader regions of transcripts including cis-acting regulatory RNAs, the average overall leader length based on the TSS map peaks around 35 nt (Figure 1) (34). This observation suggests that ‘long’ 5′ leader regions are likely to occur only when specialized functions are encrypted within them. One possible function of a long 5′ leader region is to incorporate structural elements that affect the stability of the overall transcript. Alternatively, some long 5′ leader regions include within them sequence and structural components that help guide intracellular mRNA localization. However, the most likely explanation for a long 5′ mRNA leader region is due to inclusion of a cis-acting, signal-responsive regulatory RNA. Therefore, unbiased experimental methods capable of identifying long 5′ leader regions, such as high-throughput sequencing of TSS, offer a potentially powerful approach for discovery of new regulatory RNA elements. Current bioinformatics-based approaches are likely to include bias for phylogenetically widespread and highly conserved regulatory RNAs. In contrast, unbiased mapping of TSSs is expected to uncover 5′ mRNA leader regions without regard for phylogenetic distribution, even when they include poorly conserved, recently evolved, or highly degenerate cis-acting regulatory sequences.
Figure 1.

Length distribution of B. subtilis 5′-leader regions. In total, 600 TSSs were identified based on the dRNA-seq analysis (Supplementary Table S1). The length of putative leader regions was calculated as the distance from the start of the cDNA reads to the start of the downstream coding region. The data is presented as a histogram with bin width = 20. Three individual leader regions longer than 450 nt (‘x1′) are denoted with the dashed line.
To that end, we investigated whether 454 pyrosequencing of B. subtilis stationary phase RNAs was capable of identifying previously established long 5′ leader regions. Previous data have established a minimum of 24 protein-responsive cis-acting regulatory RNAs, 19 tRNA-responsive cis-acting regulatory RNAs, 32 metabolite-responsive cis-acting regulatory RNAs and one metal-sensing regulatory RNA (34,37). Long 5′ leader regions were correctly identified in our data set for 74, 67 and 62% of tRNA-, metabolite- and protein-responsive regulatory RNAs, respectively (Supplementary Table S2A). Moreover, a qualitative assessment of putative start sites determined in our data set matched on average within 1 nt of the previously established start sites (as cataloged by DBTBS; 40). Of the ∼600 putative TSSs identified herein, 93 were located at least 100 nt away from the downstream gene and did not already correspond to a known long leader region (Supplementary Table S1). There are bound to be false positives within this data set, i.e. start sites that do not correspond to synthesis of a long UTR. For example, it is possible that transcription could initiate upstream of a gene for synthesis of a separate, unique sRNA gene, having nothing to do with expression of the downstream gene. Therefore, as a conservative strategy for specifically identifying long leader regions, we assessed most closely those cDNA reads that start within an intergenic region but that cumulatively overlap with the downstream coding sequence (or that end within 10 nt of the downstream gene). We assumed that this arrangement would result in the highest confidence for assigning leader regions. A total of 40 examples fit this description (Table 1; Supplementary Table S3). The fact that the cDNA signals for the remaining 53 stop upstream of the downstream coding region does not automatically eliminate them as corresponding to 5′ leader regions. Indeed, many previously established cis-acting regulatory RNAs resemble the latter pattern, presumably due to the presence of an intrinsic terminator element before the coding region. Also, several transcripts that have been demonstrated to contain long 5′ leader regions of unknown function appear in our data (e.g. srfAA, yxbB), although their cDNA reads do not fully continue into the downstream gene.
Table 1.
Candidates for long 5′ leader regions
| UTR sizea (nt) | cDNA #b | Downstream gene | Referencesc | Distributiond | |
|---|---|---|---|---|---|
| ncr2261 | 264 | 30 | clpX | Gerth et al., 1996e | 2,7,8 |
| ncr102 | 214 | 51 | rpoB | Boor et al., 1995 | 2,7,8 |
| ncr776 | 212 | 30 | pdhA | Gao et al., 2002f | 7 |
| ncr2328 | 199 | 91 | citZ | Jin and Sonenshein, 1994 | 2,7 |
| ncr1471 | 189 | 19 | yybN | This work | (–) |
| ncr2639 | 187 | 8 | cwlO | Bisicchia et al., 2007 | 2 |
| ncr1422 | 183 | 8 | yxjJ | This work | (–) |
| ncr921 | 180 | 748 | ylxS | Shazand et al., 1993 | 2,7,8 |
| ncr1521 | 180 | 8 | ycdA | This work | (–) |
| ncr2264 | 179 | 19 | tig | This work | 2,7,8 |
| ncr1554 | 165 | 48 | ydbN | This work | 2,7 |
| ncr942 | 164 | 13 | ymdA | This work | 2,3,4,7,8,9 |
| ncr551 | 145 | 8 | yhdT | This work | 2,7,8 |
| ncr2017 | 143 | 8 | ypfD | This work | 2,7,8 |
| ncr2103 | 128 | 9 | sodA | This work | 2,7 |
| ncr948 | 124 | 21 | spoVS | Resnekov et al., 1995 | 2,7,8 |
| ncr812 | 123 | 12 | ylbK | This work | 2,12 |
| ncr2692 | 122 | 17 | tagD | Mauel et al., 1995 | (–) |
| ncr2879 | 121 | 65 | yybS | This work | 2,7 |
| ncr95 | 121 | 21 | rplK | This work | Diverse |
| ncr2755 | 111 | 34 | fbaA | This work | 2,7,8 |
| ncr2498 | 110 | 11 | dhbA | Rowland and Taber, 1996 | (–) |
| ncr1278 | 106 | 8 | yuxN | This work | 2 |
| ncr2243 | 104 | 156 | rplU | Barrick et al., 2004g, This work | Diverse |
| ncr2896 | 104 | 14 | rpmH | Ogasawara et al., 1985h | 1,2,3,4,7,8,9,12 |
| ncr665 | 258 | 16 | rex | This work | 2 |
| ncr1323 | 96 | 14 | pelC | This work | (–) |
| ncr1443 | 420 | 617 | yxbB | This work | (–) |
aCandidates for long 5′ leader regions (‘5′ UTR’) are selected based on cDNA signals from intergenic regions (of the enriched sample), which include reads that overlap with or end within 10 nt of the downstream gene. The 5′ UTR size is calculated as the distance from the start of cDNA signals up to the start of the coding region. All 5′ UTR sequences are included in Supplementary Figure S3.
bcDNA # is calculated as the average number of cDNAs corresponding to the first 15 nt from the 5′-end of the overall peak in the enriched sample. Only potential UTRs represented by seven or more cDNA hits are shown in this table. The remaining candidates are listed in Supplementary Table S1.
cThe complete references can be found in Supplementary Materials.
dOrganisms to which blast hits could be detected are denoted as: (–) B. subtilis only (1) Anoxybacillus flavithermus, (2) B. amyloliquefaciens, (3) B. anthracis, (4) B. cereus, (5) B. clausii, (6) B. intermedius, (7) B. licheniformis, (8) B. pumilus, (9) B. thuringiensis, (10) B. weihenstephanensis, (11) Brevibacillus brevis, (12) Geobacillus species, (13) Lysinibacillus sphaericus, (14) Paenibacillus species, (diverse) all of the above Bacillales families with the addition of Staphylococcus, Listeria, Streptococcus and Lactobacillus species.
eThere are two TSS detected for clpX. The TSS described here is 180 nt upstream of the previously characterized start site (Gerth et al., 1996), which was also observed in our data set.
fThe TSS for pdhA detected herein is 139 nt upstream of the one previously characterized by Gao et al. (2002).
gA long 5′ UTR upstream of rplU was predicted previously by Barrick et al. (2004) based on sequence conservation.
hTwo TSS upstream of rpmH are observed in agreement with Ogasawara et al. (1985).
Interestingly, several transcripts with newly identified long leader regions can be grouped relative to their expected functional roles. For example, five such transcripts contained moderately ‘long’ 5′ leader regions (104, 104, 119, 121 and 143 nt) upstream of genes encoding ribosomal protein homologues. Post-initiation regulation of ribosomal protein genes is common in bacteria. Oftentimes, ribosomal proteins (r-proteins) bind to structural motifs located within the leader region to control expression of the downstream genes, in order to coordinate their overall stoichiometry with other r-proteins (41–44). It is possible that the moderately long 5′ leader regions identified herein are required for similar regulatory mechanisms. Certain RNase enzymes have also been demonstrated to post-transcriptionally autoregulate their expression by interacting within their 5′ leader region (45). Therefore, the fact that the recently identified RNase Y (ymdA) gene appears to be preceded by a 164 nt leader region could be suggestive of a similar mechanism. Also, transcripts encoding for certain core transcription elongation subunits (rpoB, greA, nusA—located in an operon with ylxS) also appear from our data to contain a long 5′ leader region, suggesting they also may be subjected to post-initiation control.
Another functionally related group of transcripts within this list encode for central metabolism genes. For example, the 5′ leader regions for certain tricarboxylic acid cycle transcripts, including pyruvate dehyrogenase (pdhA), citrate synthase (citZ) and succinate dehydrogenase (odhA), appear to be 212, 199 and 100 nt in length. Similarly, the 5′ leader region for an oxidative phosphorylation gene, menaquinol oxidase (qoxA), is 248 nt in length. A single glycolysis-related transcript, which encodes for fructose 1,6 bisphosphate aldolase (fbaA), also contains a long 5′ leader region (111 nt). It remains to be determined whether there are any common sequence or structural features between the 5′ leader portions of these central metabolism transcripts. Further experimentation will be required to assess whether the new 5′ leader regions identified in this study contain within them elements that are important for post-transcriptional regulation of their associated genes.
Identification and detection of small RNAs
One of our primary motivations for performing the experimentation described herein was to validate previously established sRNAs and, more preferably, to discover new examples. There have been 14 non-housekeeping sRNAs identified previously in B. subtilis, although only a few have been studied in detail. Two have been identified as 6S RNAs (6S-1 and 6S-2; 46,47). Other studies have predicted a small suite of candidates, some of which may be under control of sporulation-specific sigma factors (35,48,49). Of these candidates, mRNA targets have been experimentally identified for only a few. For example, two antisense RNAs have been demonstrated to regulate a toxin gene (txpA) and an unknown gene (yabE), respectively (50,51). One sRNA, SR1, controls expression of a transcriptional activator of arginine catabolism, AhrC (52), while another, FsrA, controls iron-responsive genes (sdhCAB, citB, yvfW, leuCD) (53). Recent discoveries in S. aureus revealed a sRNA (RsaE) that is widely conserved amongst Gram-positive species, including B. subtilis. It appears to target central metabolism genes and cstA, which encodes for a ‘carbon starvation’ gene (27). A more recent investigation, which examined the global transcriptional profile of B. subtilis by high-density oligonucleotide tiling arrays, resulted in identification of 54 new sRNA candidates (35). This raised the total number of sRNAs proposed for B. subtilis to ∼70. To find novel sRNA candidates using 454 pyrosequencing of stationary phase RNAs, we searched for cDNA peaks that occurred specifically and entirely within intergenic regions, and which oftentimes included an identifiable intrinsic transcription terminator at the 3′ terminus. Of the 14 previously identified sRNAs, our analysis recovered seven: 6S-1, 6S-2, fsrA, bsrE, bsrF (SR2), bsrG and bsrH (Tables 2 and 3, Supplementary Table S2B; 46,47,49,53). Additionally, pRNA, a small RNA oligonucleotide that is synthesized by RNA polymerase using 6S as a template, could be detected for 6S-1 but not 6S-2 (Figure 2). The complicated relationship between the 6S and pRNA expression profiles will be addressed more fully in a different, future publication (R. Hartmann, personal communication). SurA, another previously identified sRNA, appeared from our analysis to be an antisense RNA since it appeared to overlap with the adjacent yndL gene. A putative sRNA was also previously identified within the polC-ylxS locus. This particular candidate was not specifically found within our analysis; instead, our data exhibited cDNA reads at the same locus but that appeared to correspond to a long 5′ leader region for the ylxS gene. The remaining previously identified sRNA candidates (bsrC, bsrD, bsrI, SR1, SurC) couldn’t be detected in our data set, potentially due to limited expression during our growth conditions. SurC, for example, is exclusively expressed during sporulation (48). Also, from the 54 putative sRNAs identified by Rasmussen et al. (35), we detected 11 (20%) total. Finally, our analysis detected 50 new unique sRNA candidates (Tables 2 and 3). We did not specifically investigate whether these RNAs exhibited putative open reading frames; therefore, we cannot exclude that a subset might encode for small peptides.
Table 2.
Predicted sRNAs
| Peaka (nt) | Start | End | cDNA # | Prev gene | Next gene | Gene direction | Name | Distributionb | |
|---|---|---|---|---|---|---|---|---|---|
| ncr1160 | 70 | 2697037 | 2697106 | 920 | yqaJ | yqaI | /−/+/−/ | – | (–) |
| ncr1159 | 50 | 2692882 | 2692931 | 352 | yqaO | yqaN | /−/+/−/ | – | (–) |
| ncr982 | 80 | 1917501 | 1917580 | 324 | yndN | lexA | /+/+/−/ | – | 8 |
| ncr1058 | 297 | 2273533 | 2273829 | 260 | yolA | yokL | /−/+/−/ | ncr46/bsrG | 2,7 |
| ncr1562 | 60 | 532583 | 532642 | 197 | ydcO | ydcP | /+/−/+/ | – | 8 |
| ncr1932 | 184 | 2273701 | 2273884 | 112 | yolA | yokL | /−/−/−/ | – | 2 |
| ncr1175 | 107 | 2773780 | 2773886 | 90 | yrhK | cypB | /+/+/−/ | – | 2 |
| ncr1937 | 69 | 2283685 | 2283753 | 89 | yokC | yokB | /−/−/−/ | – | (–) |
| ncr2768 | 56 | 3852061 | 3852116 | 54 | pbpG | ywhD | /+/−/−/ | – | 2,6,8 |
| ncr724 | 56 | 1451260 | 1451315 | 46 | stoA | zosA | /+/+/+/ | – | (–) |
| ncr1857 | 260 | 2069869 | 2070128 | 44 | yobI | yobJ | /−/−/−/ | bsrE | 2,3,4,7,8 |
| ncr1019 | 171 | 2069821 | 2069991 | 35 | yobI | yobJ | /−/+/−/ | ncr39 | 2,7,8 |
| ncr1575 | 199 | 606407 | 606605 | 34 | vmlR | ydgF | /−/−/−/ | ncr10 | 2,3,4,9,10,13 |
| ncr2184 | 232 | 2779137 | 2779368 | 32 | yrzI | yrhG | /−/−/−/ | ncr60 | 2,7,8 |
| ncr471 | 173 | 820666 | 820838 | 24 | yfmI | yfmG | /−/+/+/ | – | (–) |
| ncr264 | 248 | 376678 | 376925 | 23 | hxlR | srfAA | /+/+/+/ | – | (–) |
| ncr952 | 151 | 1780404 | 1780554 | 20 | mutL | ymzD | /+/+/−/ | – | 2 |
| ncr2424 | 58 | 3146126 | 3146183 | 20 | mntA | menC | /−/−/−/ | – | 7 |
| ncr1915 | 59 | 2208755 | 2208813 | 18 | yopM | yopL | /−/−/−/ | – | (–) |
| ncr629 | 117 | 1233429 | 1233545 | 16 | yizD | yjbH | /−/+/−/ | ncr22/rsaE | Diverse |
| ncr1015 | 120 | 2053989 | 2054108 | 14 | pps | xynA | /−/+/−/ | – | 2,7,11,14 |
| ncr1155 | 233 | 2678729 | 2678961 | 13 | yqdB | yqbM | /+/+/−/ | ncr58/bsrH | 2,5,7,8,11 |
| ncr1241 | 128 | 3225697 | 3225824 | 13 | yugI | alaT | /−/+/−/ | – | 2,7 |
| ncr2299 | 99 | 2913485 | 2913583 | 13 | trxA | xsa | /−/−/−/ | – | (–) |
| ncr738 | 58 | 1467704 | 1467761 | 13 | ykwD | pbpH | /−/+/+/ | – | 2 |
| ncr969 | 58 | 1868404 | 1868461 | 12 | ymzA | ymaA | /+/+/+/ | – | 2 |
| ncr2637 | 74 | 3573045 | 3573118 | 11 | yvcI | trxB | /−/−/−/ | – | 1,2,7,8 |
| ncr178 | 124 | 199857 | 199980 | 11 | glmM | glmS | /+/+/+/ | – | 2 |
| ncr560 | 230 | 1056390 | 1056619 | 11 | yhaZ | yhaX | /−/+/+/ | ncr18 | 2 |
| ncr992 | 72 | 1925548 | 1925619 | 7 | yneK | cotM | /−/+/−/ | – | 2 |
| ncr620 | 100 | 1219702 | 1219801 | 7 | trpS | oppA | /−/+/+/ | – | 2,7 |
| ncr585 | 201 | 1150478 | 1150678 | 7 | gerPA | yisI | /−/+/−/ | ncr20 | (–) |
| ncr1957 | 60 | 2316348 | 2316407 | 6 | ypbR | ypbQ | /−/−/−/ | – | 2 |
aCandidates for small RNA are selected based on cDNA signals from the intergenic regions (of the enriched sample), which do not correspond to a known gene, with at least 25 nt distance from both the upstream and downstream genes. The length is measured from the start to the end of the cDNA signals.
bOrganisms to which blast hits could be detected are denoted as: (–) B. subtilis only (1) Anoxybacillus flavithermus, (2) B. amyloliquefaciens, (3) B. anthracis, (4) B. cereus, (5) B. clausii, (6) B. intermedius, (7) B. licheniformis, (8) B. pumilus, (9) B. thuringiensis, (10) B. weihenstephanensis, (11) Brevibacillus brevis, (12) Geobacillus species, (13) Lysinibacillus sphaericus, (14) Paenibacillus species, (diverse) all of the above Bacillales families with the addition of Staphylococcus species.
Table 3.
Predicted sRNAs exhibiting lowered abundance
| Peaka (nt) | Start | End | cDNA # | Prev gene | Next gene | Gene direction | Name | Distributionb | |
|---|---|---|---|---|---|---|---|---|---|
| ncr1855 | 94 | 2069075 | 2069168 | 5 | yobI | yobJ | /−/−/−/ | – | 2,7,8 |
| ncr2507 | 85 | 3302792 | 3302876 | 5 | yuzG | guaC | /−/−/+/ | – | (–) |
| ncr1670 | 68 | 1077246 | 1077313 | 5 | hinT | ecsA | /−/−/+/ | – | 2,7 |
| ncr214 | 108 | 275609 | 275716 | 5 | garD | ycbJ | /+/+/+/ | – | (–) |
| ncr2360 | 188 | 3036340 | 3036527 | 5 | rpsD | tyrS | /+/−/−/ | – | 2 |
| ncr2173 | 97 | 2734262 | 2734358 | 5 | yrdB | yrdA | /−/−/−/ | – | (–) |
| ncr2185 | 159 | 2780319 | 2780477 | 4 | yrhG | yrhF | /−/−/−/ | – | 2,7 |
| ncr2166 | 83 | 2678994 | 2679076 | 4 | yqdB | yqbM | /+/−/−/ | – | 2,7 |
| ncr1876 | 140 | 2099817 | 2099956 | 4 | yozO | yozC | /−/−/−/ | – | 2 |
| ncr1566 | 79 | 559532 | 559610 | 4 | cspC | ydeB | /+/−/−/ | – | 2,7,8 |
| ncr976 | 52 | 1900528 | 1900579 | 4 | yncF | cotU | /+/+/−/ | – | (–) |
| ncr2160 | 259 | 2647405 | 2647663 | 3 | yqeG | yqeF | /−/−/−/ | – | 2,7,12 |
| ncr1421 | 223 | 3996388 | 3996610 | 3 | pepT | yxjJ | /+/+/+/ | – | (–) |
| ncr1755 | 187 | 1453368 | 1453554 | 3 | zosA | ykvY | /+/−/+/ | ncr35 | (–) |
| ncr1733 | 107 | 1357727 | 1357833 | 3 | ykcC | htrA | /+/−/−/ | ncr26 | (–) |
| ncr1118 | 86 | 2540930 | 2541015 | 3 | yqhR | yqhQ | /−/+/+/ | – | 2,7 |
| ncr2339 | 57 | 2991183 | 2991239 | 3 | ytsJ | dnaE | /−/−/−/ | – | (–) |
| ncr2665 | 77 | 3631679 | 3631755 | 3 | yvyD | yvzG | /−/−/−/ | – | (–) |
| ncr1935 | 104 | 2282621 | 2282724 | 2 | yokD | yokC | /+/−/−/ | – | (–) |
| ncr1052 | 183 | 2221800 | 2221982 | 2 | yonR | yonP | /−/+/+/ | ncr44 | (–) |
| ncr721 | 112 | 1446806 | 1446917 | 2 | ykzR | ykvR | /+/+/+/ | ncr34 | (–) |
| ncr181 | 158 | 204991 | 205148 | 2 | adaB | ndhF | /+/+/+/ | ncr4 | (–) |
| ncr465 | 51 | 796025 | 796075 | 2 | yetO | ltaSA | /+/+/+/ | – | (–) |
| ncr2897 | 118 | 157 | 274 | 2 | startc | dnaA | /+/+/+ | – | (–) |
| ncr2857 | 103 | 4122960 | 4123062 | 2 | yyzE | yydK | /−/−/+/ | – | (–) |
| ncr977 | 106 | 1901991 | 1902096 | 2 | cotU | thyA | /−/+/+/ | – | (–) |
| ncr2752 | 256 | 3804713 | 3804968 | 2 | rho | ywjI | /−/−/−/ | ncr75 | 2,7,8 |
| ncr1565 | 61 | 554497 | 554557 | 2 | yddR | yddS | /+/−/+/ | – | (–) |
| ncr2658 | 60 | 3625573 | 3625632 | 2 | ftsE | cccB | /−/−/−/ | – | 2 |
| ncr826 | 72 | 1596338 | 1596409 | 2 | sbp | ftsA | /+/+/+/ | – | 2,8 |
| ncr1221 | 73 | 3072289 | 3072361 | 2 | ythP | ytzE | /−/+/+/ | – | 2 |
| ncr2179 | 111 | 2752023 | 2752133 | 2 | yraI | yraH | /−/−/−/ | – | 2,7 |
aCandidates for small RNA are selected based on cDNA signals from the intergenic regions (of the enriched sample), which do not correspond to a known gene, with at least 25 nt distance from both the upstream and downstream genes. The length is measured from the start to the end of cDNA signals.
bOrganisms to which blast hits could be detected are denoted as: (–) B. subtilis only (1) Anoxybacillus flavithermus, (2) B. amyloliquefaciens, (3) B. anthracis, (4) B. cereus, (5) B. clausii, (6) B. intermedius, (7) B. licheniformis, (8) B. pumilus, (9) B. thuringiensis, (10) B. weihenstephanensis, (11) Brevibacillus brevis, (12) Geobacillus species, (13) Lysinibacillus sphaericus and (14) Paenibacillus species.
cStart indicates the start of the genomic replication (0o).
Figure 2.

Visualization of enriched cDNA reads for B. subtilis 6S RNAs. The upper panel shows the sequence and predicted secondary structure for 6S-1. Denoted in red is the sequence, which is transcribed by RNAP from 6S as an RNA template into the short product RNA (pRNA). The bottom panel shows the genomic context and the distribution of cDNA reads mapped to both 6S-1 and 6S-2 loci. Arrows denote the direction of transcription. The cDNA reads for 6S-1 and 6S-2 are shown in the same relative scale. In contrast, the cDNA reads corresponding to the pRNA are approximately 10-fold less abundant compared to the 6S and, thus, are shown as a close-up for visualization purposes.
An interesting feature of sRNAs in Gram-negative bacteria is their phylogenetic distribution. For example, it is not uncommon to find sRNAs that are well conserved among the γ-proteobacterial species. It is not yet clear why these sRNAs have not evolved more rapidly among these organisms but is generally assumed that the primary sequence and secondary structure conservation for certain sRNAs has been retained to maintain intermolecular interactions with a common mRNA target. However, it is also possible that certain sRNAs exhibit phylogenetic conservation because they are constructed from exceptionally successful structural scaffolds, which are optimized for both interactions with target mRNAs and protection against RNases. Of the sRNA candidates identified in this study, most can be identified only in B. subtilis or the most closely related Bacillus species that have been sequenced. However, a few B. subtilis sRNA candidates also appeared to be present in genome sequences of other Bacilluls species (Tables 2 and 3). Overall, this suggests that the B. subtilis sRNAs are likely to be more limited in their phylogenetic distribution than their proteobacterial counterparts.
Most striking in its phylogenetic distribution is RsaE, which has been identified in two prior studies (27,35) and can be found in diverse Gram-positive bacteria, including Staphylococcus, Lysinibacillus, Geobacillus, Listeria and Bacillus species. In B. subtilis, the top mRNA candidate for interaction with RsaE is cstA, which encodes for an uncharacterized carbon homeostasis protein (27,35). However, this gene does not appear to be a target in Staphylococcus species. Therefore, it is still unclear why this particular sRNA exhibits such high, albeit lineage-sporadic, distribution.
Several sRNA candidates that were identified herein but that were also discovered by prior studies (bsrE, bsrH, ncr39, ncr10 and ncr60) can also be found within the genomes of other Firmicutes, most often for Bacillus species (Tables 2 and 3). Comparative sequence alignments of these sRNA genes reveals several instances of covarying residues within putative helices, which together predict the occurrence of secondary structure features common for each sRNA class (data not shown). Additionally, a few novel sRNAs discovered by our current analysis appear to be conserved amongst genomes of a few other Bacillus species. For example, ncr1015 can be identified in the genomes of B. subtilis, B. amyloliquefaciens, B. licheniformis, Brevibacillus species and Paenibacillus species. Similarly, ncr2637 can be found in Anoxybacillus flavithermus, B. subtilis, B. amyloliquefaciens, B. licheniformis and B. pumilus. It is not yet obvious why these particular sRNA candidates are conserved in these other organisms, although a common mRNA target would be the primary assumption.
These data together help create an inventory of sRNA candidates in B. subtilis. However, demonstrating they are functionally required for genetic regulation is a challenging endeavor. Three experimental methods are traditionally used to add more confidence in individual sRNA candidates: (i) independent detection by alternative experimentation (e.g. by northern blot analysis), (ii) demonstration of a reliance upon Hfq for stability and (iii) prediction and validation of mRNA targets. The role(s) of Hfq in Gram-positive bacteria is still poorly defined; therefore, this was not taken into account in the current study. Instead, we chose 11 of the longest and most highly expressed sRNA candidates for validation by northern blot analyses. All of the sRNA candidates that were chosen for northern analysis could be successfully detected (Figures 3, 4 and 6). Also, several appeared to be subjected to intracellular processing, given that they corresponded to lengths shorter than their predicted size (Figures 3 and 4). Other sRNA candidates were not assessed thusly as they appeared to exhibit lowered expression levels that are likely to be within the range of detection by deep sequencing methodology but not by northern blot analyses. Therefore, although much more experimentation is still yet required, preliminary experimentation on a subset of the candidate sRNAs appeared to validate their intracellular presence.
Figure 3.

The expression, predicted secondary structure, and genomic context of B. subtilis sRNA candidates: ncr1175 (A), ncr982 (B), ncr1241 (C) and ncr1015 (D). For each of these RNAs, the expression level was assessed by northern blotting using total samples obtained from stationary phase cells cultured in minimal media. ‘Asterisk’ indicates that the size of sRNA detected by northern blotting is in agreement with the size of the putative sRNA as predicted by sequencing data. ‘Filled triangle’ denotes a sRNA with a different predicted size (e.g. due to processing or termination events). The genomic locus of each sRNA is shown with its enriched cDNA hits and the two flanking genes. The transcriptional unit is indicated by an arrow. An open circle denotes a potential intrinsic terminator. Candidate mRNA targets, as predicted by TargetRNA software, for are also included in the figure. The region of the sRNA predicted to associate with the target mRNA is highlighted in gray. Secondary structures were predicted using RNAfold and RNAz software.
Figure 6.

Novel TA systems predicted by deep sequencing analysis. (A) Genomic locus of three new TA systems in B. subtilis. The toxic protein (gray arrow) and the RNA antitoxin (black arrow) are all arranged in tail-to-tail configuration. Note that the txpA and ratA system had been previously characterized (46). (B) Northern blotting of the toxin and antitoxin RNAs. ‘Asterisk’ indicates that the size of the sRNA as predicted by northern blot is in agreement with sequencing data. ‘Filled triangle’ denotes sRNA with different predicted size (e.g. due to processing or termination events). The expression level for bsrH and as-bsrH were too low to be detected by northern blotting in our analysis. (C) Putative sequences for as-bsrE, bsrG and bsrH toxins. Predicted membrane spanning regions are highlighted in gray. (D) Sequence alignment of the bsrE, as-bsrG and as-bsrH RNA antitoxins. Regions with base-pairing potentials are shown with different colors and labeled as P1–P4.
Figure 4.

The expression, predicted secondary structure, and genomic context of B. subtilis sRNA candidates: ncr1575 (A), ncr952 (B) and RsaE/ncr629 (C). For each of these RNAs, the expression level was assessed by northern blotting using total samples obtained from stationary phase cells cultured in minimal media. ‘Asterisk’ indicates that the size of sRNA detected by northern blotting is in agreement with the size of the putative sRNA as predicted by the sequencing data. ‘Filled triangle’ denotes a sRNA with a different predicted size (e.g. due to processing or termination events). The genomic locus of each sRNA is shown with its enriched cDNA hits and the two flanking genes. The transcriptional unit is indicated by an arrow. An open circle denotes a potential intrinsic terminator. Candidate mRNA targets, as predicted by TargetRNA software, for are also included in the figure. The region of the sRNA predicted to associate with the target mRNA is highlighted in gray.
In order to begin assessing putative mRNA targets for these sRNA motifs we subjected the sRNA candidates to analysis by TargetRNA, a program designed to search for interrupted base pairing interactions within intergenic regions (54). As it is difficult to differentiate false positives from actual mRNA targets using this software alone, we interpret these predictions with caution. Only a subset of the target predictions, which exhibited particularly low estimated P-values, is highlighted in Figures 3 and 4. One possible explanation for the lack of mRNA targets for certain sRNA candidates is that the latter may target portions of protein coding sequences to affect mRNA stability or translation (21), an interaction that is not addressed by current prediction software. Additional experimentation will be required in order to determine whether these and other mRNAs represent actual targets for the sRNA candidates newly identified herein.
Prophage regions contain sRNAs and multiple RNA-based toxin–antitoxin systems
The genome of B. subtilis contains several prophages (SPβ, skin, PBSX) and prophage-like (pro1-pro7) regions, which are typically characterized by higher-than-background A + T nucleotide composition (55–58). Prophages, like plasmids, conjugative transposons and introns, are mobile elements that can be transferred horizontally, occasionally causing genomic rearrangements in bacteria. These elements often carry beneficial traits, such as antibiotic resistance cassettes or virulence factors that could help the host adapt to their environment.
From our analysis, we detected 16 putative sRNAs originating mainly from the SPβ, skin, pro6 and pro7 loci (Figure 5). Some of these sRNA candidates, in fact, were the highest expressed sRNAs in our data set (data not shown). None of the putative sRNAs described herein (Tables 2 and 3) were identified within the PBSX or pro1-pro5 regions. It is generally assumed that a subset of phage genes expressed during lysogenic phase either confer a particular selective advantage for the host or are important for maintaining the phage-host equilibrium. We predict that some of the sRNAs identified herein are likely to perform similar functions. However, it remains to be determined whether these sRNAs target genes within the phage loci or specific host genes, although there is precedence for both scenarios in other prokaryotes as well as eukaryotes (59–63).
Figure 5.

Putative sRNAs encoded within prophage regions. 16 sRNA candidates (denoted by arrow) originated from prophage or prophage-like regions (SPβ, skin, P6, P7) and are shown relative to their genomic location. Genes immediately upstream and downstream of the sRNA are also listed.
Interestingly, six of the sRNA candidates within phage-like regions are predicted to interact in pairs through antisense interactions. These three sRNA pairs are co-organized within intergenic regions in distinct tail-to-tail arrangements; their 3′ terminal ∼100 nt overlap and therefore are predicted to interact via antisense pairings (Figure 6). Several of the sRNAs that we predict to be organized in this manner have been identified previously, although their corresponding antisense partners were not (49). Specifically, our data suggest that sRNA candidates bsrE, bsrG and bsrH, which were identified previously, pair through intermolecular antisense interactions with newly identified ncr1019, ncr1058 and ncr1155, respectively. For convenience, we refer to these various sRNAs as bsrE, bsrG, bsrH, as-bsrE, as-bsrG and as-bsrH in order to denote their antisense pairings. The three pairs of these RNA molecules are located within different prophage or prophage-like regions: bsrE/as-bsrE in pro6, bsrG/as-bsrG in SPβ and bsrH/as-bsrH in skin. In addition, we also noticed that one of the pairs (bsrH/as-bsrH) is situated adjacent to a previously established toxin–antitoxin (TA) system, txpA/ratA (Figure 6; 50). Of particular note is that the txpA/ratA TA system shares a similar overall arrangement with the newly identified antisense-based RNA pairs (Figure 6). Most TA modules consist of two components: a stable toxin and a labile antitoxin. The txpA/ratA system represents a typical type I TA system that includes an mRNA encoding for a short, toxic peptide (TxpA) and an antitoxin that is comprised of an antisense RNA (RatA). In contrast, type II TA systems rely on a protein factor as the antitoxin (64–66).
Based on these observations we examined each of the six sRNA candidates for small open reading frames. Interestingly, only one sRNA from each pairings exhibited the potential to encode for a peptide of ∼30 amino acids and included an appropriately spaced ribosome binding site (as-bsrE, bsrG, bsrH; Figure 6). All three peptides are predicted to contain a single α-helical transmembrane domain of ∼20 amino acids with several additional charged residues at the C-terminus. This arrangement is consistent with type I toxins (Figure 6; 66). TxpA encodes a similar peptide but with a modestly longer C-terminus. The remaining sRNAs (bsrE, as-bsrG, as-bsrH) did not exhibit any similar peptide-encoding potential, consistent with a potential role as an antitoxin. However these latter RNAs shared some primary sequence features and a common overall secondary structure arrangement, which consists of four stem-loop regions (Figure 6). Approximately 100 nt, located between two of the helical elements (P2 and P4) exhibited base pairing potential to the 3′-end of the respective peptide-encoding mRNAs. RatA appears to have a similar secondary structure arrangement but with a longer 5′-portion (data not shown). The molecular mechanisms for how these antisense RNAs control toxin expression are still unclear. However, it has been proposed previously (50) that extensive 3′-end base pairing could promote simultaneous degradation of both RNAs.
Type I TA systems were originally discovered as an important component of plasmid maintenance mechanisms in E. coli (67). More recently they have also been discovered in many bacterial chromosomes (66,68–70). It has been theorized that coupling TA systems to control of plasmid replication would ensure that plasmid-free cells are killed by toxin accumulation, a phenomenon termed ‘post-segregational killing’ (71). Similarly, the txpA/ratA antisense module was proposed to be important for ensuring propagation of an accompanying phage genome in host cells (50). Our analysis therefore uncovers three more potential TA systems that are distributed in different prophage regions, suggesting that RNA-based TA mechanisms could be more common than previously recognized. Interestingly, we also note that two of the toxin-encoding mRNAs (as-bsrE and bsrG) are predicted to contain a ResD binding site within their putative promoter regions, suggesting a potential linkage between toxin expression and oxygen limitation (data not shown; 72).
Identification of antisense RNAs
In addition to the newly identified TA systems described above, our analysis also revealed other putative antisense RNAs (asRNAs). These different asRNAs could be assigned to several different categories based upon the arrangement with their target mRNA. A subset of the asRNAs exhibited the potential to fully pair with the entire target mRNA, while others pair with only a portion of the target mRNA through head-to-head (5′-overlap) or tail-to-tail (3′-overlap) interactions. In total, 29 candidate asRNAs were identified in our analysis (Table 4; Supplementary Table S2B). Two of them (ratA and as-yabE) were previously shown to regulate expression of target mRNAs (the toxin-encoding txpA mRNA and yabE, an mRNA encoding for a cell-wall binding protein, respectively) (Supplementary Table S2B; 50,51). SurA, which overlaps a σK-regulated transcript, yndL, has been shown to accumulate during sporulation but its regulatory capabilities remain to be demonstrated (48). Ten of the asRNA candidates discovered herein were also identified via the high-density tiling array analysis (35). These include asRNAs for the major vegetative growth phase sigma factor sigA, the teichoic acid biosynthesic enzyme, ggaA, a leucine biosynthetic enzyme, leuA, a choline transporter, opuBD and a cryptic SpoIIIJ-associated protein, jag. The remaining asRNAs identified herein are novel and are predicted to pair with a variety of characterized and uncharacterized genes (Table 4).
Table 4.
Predicted novel antisense RNA (asRNA) candidates
| Peaka (nt) | Start | End | cDNA # | Opposite gene | Overlapb | asRNA direction | Name | |
|---|---|---|---|---|---|---|---|---|
| ncr2706 | 47 | 3738263 | 3738309 | 114 | ywqA | N | − | |
| ncr1430 | 70 | 4035606 | 4035675 | 58 | bglP | 5′ | + | |
| ncr1687 | 24 | 1154737 | 1154760 | 26 | wprA | G | − | |
| ncr1296 | 236 | 3460215 | 3460450 | 26 | opuBD | G | + | shd102 |
| ncr1207 | 231 | 2997369 | 2997599 | 15 | ytoI | G | + | shd84 |
| ncr1334 | 103 | 3669968 | 3670070 | 12 | ggaA | 3′ | + | shd112 |
| ncr1812 | 239 | 1915034 | 1915272 | 11 | yndL | 5′ | − | SurA |
| ncr1265 | 218 | 3307602 | 3307819 | 10 | yutK | G | + | |
| ncr1193 | 130 | 2892567 | 2892696 | 10 | leuA | G | + | shd83 |
| ncr2153 | 101 | 2641931 | 2642031 | 9 | comER | G | − | |
| ncr1383 | 204 | 3862340 | 3862543 | 8 | ywfM | 3′ | + | shd115 |
| ncr1135 | 160 | 2600156 | 2600315 | 7 | sigA | 3′ | + | shd77 |
| ncr1186 | 17 | 2849374 | 2849390 | 7 | nadB | G | + | |
| ncr1006 | 219 | 2002176 | 2002394 | 6 | yoeA | 5′ | + | |
| ncr1799 | 25 | 1775732 | 1775756 | 6 | mutS | 5′ | − | |
| ncr1479 | 232 | 4213213 | 4213444 | 6 | jag | G | + | shd127 |
| ncr1046 | 71 | 2190676 | 2190746 | 5 | yoqZ | G | + | shd60 |
| ncr1557 | 159 | 519216 | 519374 | 5 | ndoA | 3′ | − | shd23 |
| ncr2058 | 110 | 2483671 | 2483780 | 5 | yqzJ | G | − | |
| ncr394 | 76 | 646225 | 646300 | 5 | ydiF | G | + | shd26 |
| ncr2160 | 259 | 2647405 | 2647663 | 3 | sda | G | − | |
| ncr1351 | 227 | 3747160 | 3747386 | 2 | mbl | 3′ | + | |
| ncr1565 | 61 | 554497 | 554557 | 2 | yddR | 3′ | − | |
| ncr2885 | 106 | 4186388 | 4186493 | 2 | yyaQ | 3′ | − | |
| ncr1546 | 50 | 452734 | 452783 | 2 | mtlD | 3′ | − | |
| ncr507 | 30 | 924181 | 924210 | 2 | yfhD | 3′ | + | |
| ncr2410 | 249 | 3123766 | 3124014 | 2 | ytoA | 3′ | − |
aCandidates for antisense RNA are selected based on cDNA signals (of the enriched sample) that either start within genes with the opposite orientation or end within 50 nt away from such genes. The length is measured from the start to the end of cDNA signals.
bOverlapping region is classified as follows: G = asRNA is fully complementary to the opposite gene. 5′ = asRNA 5′ end is complementary to the 5′ region of the opposite gene. 3′ = asRNA 3′ end is complementary to the 3′ region of the opposite gene. N = no overlap; the distance between 3′-end of the asRNA and the opposite gene is less than 50 nt and there is no predicted intrinsic terminator.
One potentially interesting asRNA candidate is ncr1430, which overlaps with the 5′leader region of bglP (Table 4; Figure 7). bglP encodes for a sugar phosphotransferase system (PTS) component that is involved in the utilization of β-glucosides, such as arbutin and salicin. It is transcribed as an operon with bglH, which encodes for the enzyme to metabolize the sugars (73). The full synthesis of the bglPH operon had been shown to be under regulatory control by CcpA-mediated catabolite repression and a transcription attenuation mechanism mediated by the RNA antiterminator (RAT) element located within the 5′ leader region (74). Under inducing conditions and in the absence of glucose, an antiterminator protein, LicT, binds to the RAT element to stabilize formation of an antiterminator structure, thereby allowing transcription to proceed to the downstream coding region (75). Based upon its location, ncr1430 is predicted to base pair with the bglPH mRNA within the region between the RAT element and the downstream bglP open reading frame, perhaps to repress translation. We hypothesize that this arrangement could provide yet a second layer of post-transcriptional regulation to allow finer control over the level of proteins needed to import and process the appropriate sugar. If true, these data highlight a unique example of regulation of a single gene by a transcriptional mechanism as well as by both cis- and trans-acting regulatory RNAs.
Figure 7.

Novel arrangement of an antisense RNA predicted to base pair with the bglP 5′ leader region, which includes a cis-acting regulatory RNA. IGB representation of enriched cDNA reads corresponding to ncr1430 (top) and bglP UTR (bottom). Each transcriptional unit is represented by an arrow and the potential intrinsic terminator region is shown by a circle. ncr1430 RNA is predicted to base-paired with the ribosomal binding site of bglP (denoted by gray box).
CONCLUSION
Bacillus species, which are typically motile, aerobic endospore-forming microorganisms, have been isolated from diverse locales including soil, water sources and plant root systems (76). Contributing to its adaptive abilities, B. subtilis is capable of differentiating from dividing cells to metabolically inactive spores, which are resistant to many chemicals, irradiation and desiccation. As mutually exclusive alternative lifestyles, B. subtilis can also initiate developmental pathways that culminate with cells that are competent (capable of DNA uptake and primed for homologous recombination), or function as dedicated producers of biofilm extracellular matrix constituents (77,78). Indeed, B. subtilis can form a multicellular community, consisting of spatially and temporally located cellular subtypes (77–79). Responsible in part for these biological properties are many transcription factors and a suite of alternative sigma factors that regulate transcription of the developmental pathway genes. However, given their general importance in Gram-negative bacteria, it stands to reason that RNA-based regulatory strategies are also likely to be important for coordination of multicellular behaviors and developmental pathways.
Collectively, our results argued for broader roles for small RNA regulators in B. subtilis. In combination with other data, we increase the number of potential small RNAs in B. subtilis to upwards of 100 candidates. The next step will be to identify the biological functions of these RNAs. We hypothesize that due to the differences in several key proteins involved in RNA metabolism between Gram-positive and Gram-negative bacteria (e.g. ribonucleases, Hfq and Rho), future studies of these regulatory RNAs will reveal novel mechanisms and add to the repertoire of bacterial RNA-based genetic regulatory strategies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The University of Texas Southwestern Medical Center Endowed Scholars Program; The Searle Scholars Program; National Institutes of Health (GM081882); The Welch Foundation (I-1643). Sara and Frank McKnight fellowship (to I.I.). Funding for open access charge: Searle Scholars Program UT Southwestern Medical Center Endowed Scholars Program Welch Foundation (I-1643) National Institutes of Health (GM081882).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENT
We are grateful to the members of the Winkler lab for helpful discussions.
REFERENCES
- 1.Wassarman KM, Zhang A, Storz G. Small RNAs in Escherichia coli. Trends Microbiol. 1999;10:37–45. doi: 10.1016/s0966-842x(98)01379-1. [DOI] [PubMed] [Google Scholar]
- 2.Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altuvia S. Identification of bacterial small non-coding RNAs: experimental approaches. Curr. Opin. Microbiol. 2007;10:257–261. doi: 10.1016/j.mib.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Livny J, Waldor MK. Identification of small RNAs in diverse bacterial species. Curr. Opin. Microbiol. 2007;10:96–101. doi: 10.1016/j.mib.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Landt SG, Abeliuk E, McGrath PT, Lesley JA, McAdams HH, Shapiro L. Small non-coding RNAs in Caulobacter crescentus. Mol. Microbiol. 2008;68:600–614. doi: 10.1111/j.1365-2958.2008.06172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Livny J, Teonadi H, Livny M, Waldor MK. High-throughput, kingdom-wide prediction and annotation of bacterial non-coding RNAs. PLoS ONE. 2008;3:e3197. doi: 10.1371/journal.pone.0003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JC, Vogel J. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 2008;4:e1000163. doi: 10.1371/journal.pgen.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma CM, Vogel J. Experimental approaches for the discovery and characterization of regulatory small RNA. Curr. Opin. Microbiol. 2009;12:536–546. doi: 10.1016/j.mib.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Babitzke P, Baker CS, Romeo T. Regulation of translation initiation by RNA-binding proteins. Annu. Rev. Microbiol. 2009;63:27–44. doi: 10.1146/annurev.micro.091208.073514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wassarman KM. 6S RNA: a regulator of transcription. Mol. Microbiol. 2007;65:1425–1431. doi: 10.1111/j.1365-2958.2007.05894.x. [DOI] [PubMed] [Google Scholar]
- 11.Gottesman S, McCullen CA, Guillier M, Vanderpool CK, Majdalani N, Benhammou J, Thompson KM, FitzGerald PC, Sowa NA, FitzGerald DJ. Small RNA regulators and the bacterial response to stress. Cold Spring Harb. Symp. Quant. Biol. 2006;71:1–11. doi: 10.1101/sqb.2006.71.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottesman S. Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet. 2005;21:399–404. doi: 10.1016/j.tig.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 13.Hershberg R, Altuvia S, Margalit H. A survey of small RNA-encoding genes in Escherichia coli. Nucleic Acids Res. 2003;31:1813–1820. doi: 10.1093/nar/gkg297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bejerano-Sagie M, Xavier KB. The role of small RNAs in quorum sensing. Curr. Opin. Microbiol. 2007;10:189–198. doi: 10.1016/j.mib.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 15.Sharma CM, Darfeuille F, Plantinga TH, Vogel J. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev. 2007;21:2804–2817. doi: 10.1101/gad.447207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valentin-Hansen P, Johansen J, Rasmussen AA. Small RNAs controlling outer membrane porins. Curr. Opin. Microbiol. 2007;10:152–155. doi: 10.1016/j.mib.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Papenfort K, Vogel J. Multiple target regulation by small noncoding RNAs rewires gene expression at the post-transcriptional level. Res. Microbiol. 2009;160:278–287. doi: 10.1016/j.resmic.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Bouvier M, Sharma CM, Mika F, Nierhaus KH, Vogel J. Small RNA binding to 5′ mRNA coding region inhibits translational initiation. Mol. Cell. 2008;32:827–837. doi: 10.1016/j.molcel.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 19.Darfeuille F, Unoson C, Vogel J, Wagner EG. An antisense RNA inhibits translation by competing with standby ribosomes. Mol. Cell. 2007;26:381–92. doi: 10.1016/j.molcel.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Vecerek B, Moll I, Blasi U. Control of Fur synthesis by the noncoding RNA RyhB and iron-responsive decoding. EMBO J. 2007;26:965–975. doi: 10.1038/sj.emboj.7601553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfeiffer V, Papenfort K, Lucchini S, Hinton JC, Vogel J. Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat. Struct. Mol. Biol. 2009;16:840–846. doi: 10.1038/nsmb.1631. [DOI] [PubMed] [Google Scholar]
- 22.Christiansen JK, Nielsen JS, Ebersbach T, Valentin-Hansen P, Søgaard-Andersen L, Kallipolitis BH. Identification of small Hfq-binding RNAs in Listeria monocytogenes. RNA. 2006;12:1383–1396. doi: 10.1261/rna.49706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halfmann A, Kovács M, Hakenbeck R, Brückner R. Identification of the genes directly controlled by the response regulator CiaR in Streptococcus pneumoniae: five out of 15 promoters drive expression of small non-coding RNAs. Mol. Microbiol. 2007;66:110–126. doi: 10.1111/j.1365-2958.2007.05900.x. [DOI] [PubMed] [Google Scholar]
- 24.Mandin P, Repoila F, Vergassola M, Geissmann T, Cossart P. Identification of new noncoding RNAs in Listeria monocytogenes and prediction of mRNA targets. Nucleic Acids Res. 2007;35:962–974. doi: 10.1093/nar/gkl1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, et al. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulatory Rot by an antisense mechanism. Genes Dev. 2007;21:1353–66. doi: 10.1101/gad.423507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swiercz JP, Hindra, Bobek J, Bobek J, Haiser HJ, Di Berardo C, Tjaden B, Elliot MA. Small non-coding RNAs in Streptomyces coelicolor. Nucleic Acids Res. 2008;36:7240–7251. doi: 10.1093/nar/gkn898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geissmann T, Chevalier C, Cros MJ, Boisset S, Fechter P, Noirot C, Schrenzel J, Francois P, Vandenesch F, Gaspin C, et al. A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res. 2009;37:7239–7257. doi: 10.1093/nar/gkp668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nielsen JS, Lei LK, Ebersbach T, Olsen AS, Klitgaard JK, Valentin-Hansen P, Kallipolitis BH. Defining a role for Hfq in Gram-positive bacteria: evidence for Hfq-dependent antisense regulation in Listeria monocytogenes. Nucleic Acid Res. 2009;38:907–919. doi: 10.1093/nar/gkp1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Passalacqua KD, Varadarajan A, Ondov BD, Okou DT, Zwick ME, Bergman NH. Structure and complexity of a bacterial transcriptome. J. Bacteriol. 2009;191:3203–3211. doi: 10.1128/JB.00122-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perez N, Trevino J, Liu Z, Ho SC, Babitzke P, Sumby P. A genome-wide analysis of small regulatory RNAs in the human pathogen group A Streptococcus. PLoS One. 2009;4:e7668. doi: 10.1371/journal.pone.0007668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsui HC, Mukherjee D, Ray VA, Sham LT, Feig AL, Winkler ME. Identification and characterization of noncoding small RNAs in Streptococcus pneumoniae serotype 2 strain D39. J. Bacteriol. 2010;192:264–279. doi: 10.1128/JB.01204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romby P, Charpentier E. An overview of RNAs with regulatory functions in gram-positive bacteria. Cell Mol. Life Sci. 2009;67:217–237. doi: 10.1007/s00018-009-0162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jousselin A, Metzinger L, Felden B. On the facultative requirement of the bacterial RNA chaperone, Hfq. Trends Microbiol. 2009;17:399–405. doi: 10.1016/j.tim.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Irnov, Kertsburg A, Winkler WC. Genetic control by cis-acting regulatory RNAs in Bacillus subtilis: general principles and prospects for discovery. Cold Spring Harb. Symp. Quant. Biol. 2006;71:239–249. doi: 10.1101/sqb.2006.71.021. [DOI] [PubMed] [Google Scholar]
- 35.Rasmussen S, Nielsen HB, Jarmer H. The transcriptionally active regions in the genome of Bacillus subtilis. Mol. Microbiol. 2009;73:1043–1057. doi: 10.1111/j.1365-2958.2009.06830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiβ S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 37.Dann CE, Wakeman CA, Sieling CL, Baker SC, Irnov I, Winkler WC. Structure and mechanism of a metal-sensing regulatory RNA. Cell. 2007;130:878–892. doi: 10.1016/j.cell.2007.06.051. [DOI] [PubMed] [Google Scholar]
- 38.Berezikov E, Thuemmler F, van Laake LW, Kondova I, Bontrop R, Cuppen E, Plasterk RH. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet. 2006;38:1375–1377. doi: 10.1038/ng1914. [DOI] [PubMed] [Google Scholar]
- 39.Hoffmann S, Otto C, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermüller J. Fast mapping of short sequences with mismatches, insertions, and deletions using index structures. PLoS Comput. Biol. 2009;5:e1000502. doi: 10.1371/journal.pcbi.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sierro N, Makita Y, de Hoon M, Nakai K. DBTBS: a database of transcriptional regulation in Bacillus subtilis containing upstream intergenic conservation information. Nucleic Acids Res. 2008;36:D93–96. doi: 10.1093/nar/gkm910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nomura M, Yates JL, Dead D, Post LE. Feedback regulation of ribosomal protein gene expression in Escherichia coli: structural homology of ribosomal RNA and ribosomal protein mRNA. Proc. Natl Acad. Sci. USA. 1980;77:7084–7088. doi: 10.1073/pnas.77.12.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grundy F, Henkin TM. The rpsD gene, encoding ribosomal protein S4, is autogenously regulated in Bacillus subtilis. J. Bacteriol. 1991;173:4595–4602. doi: 10.1128/jb.173.15.4595-4602.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choone N, Even S, Zig L, Putzer H. Ribosomal protein L20 controls expression of the Bacillus subtilis infC operon via a transcription attenuation mechanism. Nucleic Acids Res. 2007;35:1578–1588. doi: 10.1093/nar/gkm011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X, Lindahl L, Sha Y, Zengel JM. Analysis of the Bacillus subtilis S10 ribosomal protein gene cluster identifies two promoters that may be responsible for transcription of the entire 15-kilobase S10-spc-alpha cluster. J. Bacteriol. 1997;179:7046–7054. doi: 10.1128/jb.179.22.7046-7054.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuck A, Diwa A, Belasco JG. RNase E autoregulates its synthesis in Escherichia coli by binding directly to a stem-loop in the rne 5′ untranslated region. Mol. Microbiol. 2009;72:470–478. doi: 10.1111/j.1365-2958.2009.06662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barrick JE, Sudarsan N, Weinberg Z, Ruzzo WL, Breaker RR. 6S RNA is a widespread regulator of eubacterial RNA polymerase that resembles an open promoter. RNA. 2005;11:774–784. doi: 10.1261/rna.7286705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trotochaud AE, Wassarman KM. A highly conserved 6S RNA structure is required for regulation of transcription. Nat. Struct. Mol. Biol. 2005;12:313–319. doi: 10.1038/nsmb917. [DOI] [PubMed] [Google Scholar]
- 48.Silvaggi JM, Perkins JB, Losick R. Genes for small, noncoding RNAs under sporulation control in Bacillus subtilis. J. Bacteriol. 2006;188:532–541. doi: 10.1128/JB.188.2.532-541.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saito S, Kakeshita H, Nakamura K. Novel small RNA-encoding genes in the intergenic regions of Bacillus subtilis. Gene. 2009;428:2–8. doi: 10.1016/j.gene.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 50.Silvaggi JM, Perkins JB, Losick R. Small untranslated RNA antitoxin in Bacillus subtilis. J. Bacteriol. 2005;187:6641–6650. doi: 10.1128/JB.187.19.6641-6650.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eiamphungporn W, Helmann JD. Extracytoplasmic function sigma factors regulate expression of the Bacillus subtilis yabE gene via a cis-acting antisense RNA. J. Bacteriol. 2009;191:1101–1105. doi: 10.1128/JB.01530-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heidrich N, Chinali A, Gerth U, Brantl S. The small untranslated RNA SR1 from the Bacillus subtilis genome is involved in the regulation of arginine catabolism. Mol. Microbiol. 2006;62:520–36. doi: 10.1111/j.1365-2958.2006.05384.x. [DOI] [PubMed] [Google Scholar]
- 53.Gaballa A, Antelmann H, Aguilar C, Khakh SK, Song KB, Smaldone GT, Helmann JD. The Bacillus subtilis iron-sparing response is mediated by a Fur-regulated small RNA and three small, basic protein. Proc. Natl Acad. Sci. USA. 2008;105:11927–11932. doi: 10.1073/pnas.0711752105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tjaden B, Goodwin SS, Opdyke JA, Guillier M, Fu DX, Gottesman S, Storz G. Target prediction for small, noncoding RNAs in bacteria. Nucleic Acids Res. 2006;34:2791–2802. doi: 10.1093/nar/gkl356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zahler SA, Korman RZ, Rosenthal R, Hemphill HE. Bacillus subtilis bacteriophage SPβ: localization of the prophage attachment site and specialized transduction. J. Bacteriol. 1977;190:6983–6995. doi: 10.1128/jb.129.1.556-558.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wood HE, Dawson MT, Devine KM, McConnell DJ. Characterization of PBSX, a defective prophage of Bacillus subtilis. J. Bacteriol. 1990;172:2667–2674. doi: 10.1128/jb.172.5.2667-2674.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takemaru K, Mizuno M, Sato T, Takeuchi M, Kobayashi Y. Complete nucleotide sequence of a skin element excised by DNA rearrangement during sporulation in Bacillus subtilis. Microbiology. 1995;141:323–327. doi: 10.1099/13500872-141-2-323. [DOI] [PubMed] [Google Scholar]
- 58.Nicolas P, Bize L, Muri F, Hoebeke M, Rodolphe F, Ehrlich SD, Prum B, Bessières P. Mining Bacillus subtilis chromosome heterogeneities using hidden Markov models. Nucleic Acids Res. 2002;30:1418–1426. doi: 10.1093/nar/30.6.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pfeiffer S, Zavolan M, Grässer FA, Chien M, Russo JJ, Jingyue J, John B, Enright AJ, Marks D, Sander C, et al. Identification of virus-encoded microRNAs. Science. 2004;304:734–736. doi: 10.1126/science.1096781. [DOI] [PubMed] [Google Scholar]
- 60.Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature. 2005;435:682–686. doi: 10.1038/nature03576. [DOI] [PubMed] [Google Scholar]
- 61.Castillo-Keller M, Vuong P, Misra R. Novel mechanism of Escherichia coli porin regulation. J. Bacteriol. 2006;188:576–586. doi: 10.1128/JB.188.2.576-586.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, Braich R, Manoharan M, Soutchek, Ohler U, et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450:1096–1099. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grey F, Meyers H, White EA, Spector DH, Nelson J. A human cytomegalovirus-encoded microRNA regulates expression of multiple viral genes involved in replication. PLoS Pathogen. 2007;3:1593–1602. doi: 10.1371/journal.ppat.0030163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gerdes K, Christensen SK, Lobner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005;3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 65.Gerdes K, Wagner EGH. RNA antitoxins. Curr. Opin. Microbiol. 2007;10:117–124. doi: 10.1016/j.mib.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 66.Fozo EM, Hemm MR, Storz G. Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 2008;72:579–589. doi: 10.1128/MMBR.00025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gerdes K, Larsen JE, Molin S. Stable inheritance of plasmid R1 requires two different loci. J. Bacteriol. 1985;161:292–298. doi: 10.1128/jb.161.1.292-298.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pedersen K, Gerdes K. Multiple hok genes on the chromosome of Escherichia coli. Mol. Microbiol. 1999;32:1090–1102. doi: 10.1046/j.1365-2958.1999.01431.x. [DOI] [PubMed] [Google Scholar]
- 69.Fozo EM, Makarova KS, Shabaline SA, Yutin N, Koonin EV, Storz G. Abundance of type I toxin-antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq054. doi:10.1093/nar/gkq045 [Epub ahead of print 19 February 2010] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kwong SM, Jensen SO, Firth N. Prevalence of Fst-like toxin-antitoxin systems. Microbiology. 2010;156:975–977. doi: 10.1099/mic.0.038323-0. [DOI] [PubMed] [Google Scholar]
- 71.Hayes F. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science. 2003;301:1496–1499. doi: 10.1126/science.1088157. [DOI] [PubMed] [Google Scholar]
- 72.Hartig E, Geng H, Hartmann A, Hubacek A, Munch R, Ye RW, Jahn D, Nakano MM. Bacillus subtilis ResD induces expression of the potential regulatory genes yclJK upon oxygen limitation. J. Bacteriol. 2004;186:6477–6484. doi: 10.1128/JB.186.19.6477-6484.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Le Coq D, Lindner C, Kruger S, Steinmetz M, Stulke J. New beta-glucoside (bgl) genes in Bacillus subtilis: the bglP gene product has both transport and regulatory functions similar to those of BglF, its Escherichia coli homolog. J. Bacteriol. 1995;177:1527–1535. doi: 10.1128/jb.177.6.1527-1535.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aymerich S, Steinmetz M. Specificity determinants and structural features in the RNA target of the bacterial antiterminator proteins of the BglG/SacY family. Proc. Natl Acad. Sci. USA. 1992;89:10410–10414. doi: 10.1073/pnas.89.21.10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kruger S, Hecker M. Regulation of the putative bglpH operon for aryl-beta-glucoside utilization in Bacillus subtilis. J. Bacteriol. 1995;177:5590–5597. doi: 10.1128/jb.177.19.5590-5597.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Earl AM, Losick R, Kolter R. Ecology and genomics of Bacillus subtilis. Trends in Microbiol. 2008;16:269–275. doi: 10.1016/j.tim.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aguilar C, Vlamakis H, Losick R, Kolter R. Thinking about Bacillus subtilis as a multicellular organism. Curr. Opin. Microbiol. 2007;10:638–643. doi: 10.1016/j.mib.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.López D, Vlamakis H, Kolter R. Generation of multiple cell types in Bacillus subtilis. FEMS Microbiol. Rev. 2008;33:152–163. doi: 10.1111/j.1574-6976.2008.00148.x. [DOI] [PubMed] [Google Scholar]
- 79.López D, Kolter R. Extracellular signals that define distinct and coexisting cell fates in Bacillus subtilis. FEMS Microbiol. Rev. 2009;34:134–149. doi: 10.1111/j.1574-6976.2009.00199.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.