


Abstract
Two conserved catalytic arginines, Arg-173 and Arg-292, of the tyrosine site-specific recombinase Cre are essential for the transesterification steps of strand cleavage and joining in native DNA substrates containing scissile phosphate groups. The active site tyrosine (Tyr-324) provides the nucleophile for the cleavage reaction, and forms a covalent 3′-phosphotyrosyl intermediate. The 5′-hydroxyl group formed during cleavage provides the nucleophile for the joining reaction between DNA partners, yielding strand exchange. Previous work showed that substitution of the scissile phosphate (P) by methylphosphonate (MeP) permits strand cleavage by a Cre variant lacking Arg-292. We now demonstrate that MeP activation and cleavage are not blocked by substitution of Arg-173 or even simultaneous substitutions of Arg-173 and Arg-292 by alanine. Furthermore, Cre(R173A) and Cre(R292A) are competent in strand joining, Cre(R173A) being less efficient. No joining activity is detected with Cre(R173A, R292A). Consistent with their ability to cleave and join strands, Cre(R173A) and Cre(R292A) can promote recombination between two MeP-full-site DNA partners. These findings shed light on the overall contribution of active site electrostatics, and tease apart distinctive contributions of the individual arginines, to the chemical steps of recombination. They have general implications in active site mechanisms that promote important phosphoryl transfer reactions in nucleic acids.
INTRODUCTION
Tyrosine family site-specific recombinases and type IB topoisomerases are characterized by a catalytic pentad, Arg-Lys-(His/Lys)-Arg-(His/Trp), that assists the strand cleavage and strand joining steps of DNA recombination and relaxation, respectively (1–6). In Cre recombinase, the pentad residues are Arg-173, Lys-201, His-289, Arg-292 and Trp-315. The nucleophile for strand cleavage, Tyr-324, attacks the scissile phosphodiester bond to form a 3′ covalent adduct with the cleaved strand. The 5′-hydroxyl group formed at the cleavage step carries out nucleophilic attack on the tyrosyl intermediate to promote strand joining. Recombination results when the joining reaction is performed between, rather than, within DNA partners. A recombination event is completed in two steps of concerted single strand exchanges. The first gives rise to a Holliday junction intermediate; the second resolves it into reciprocal recombinant products (3).
Arg-173 and Arg-292 are critical for both strand cleavage and strand joining reactions, presumably to balance the negative charge on the non-bridging phosphate oxygen atoms in the transition state. Consistent with this function, the crystal structures of Cre reveal hydrogen bonding interactions between these two arginines and the scissile phosphate in the ‘disengaged’ (sans-cleavage) and ‘engaged’ (post-cleavage) active sites (2) (Figure 1). A modified DNA substrate in which the negative charge on the scissile phosphate (P) is neutralized by methylphosphonate (MeP) substitution is a poor substrate for strand cleavage by Cre. However, when Arg-292 is replaced by alanine, Cre(R292A) cleaves the MeP–DNA to form the tyrosyl intermediate, which can undergo subsequent hydrolysis (7). This hydrolytic activity promoted by Cre(R292A) has been termed the type I endonuclease activity to distinguish it from a potential type II endonuclease activity that directly targets the MeP bond for hydrolysis (see below). The type II activity has been detected in a mutant of the Cre related-Flp recombinase containing alanine in place of Arg-308 (8), which is the counterpart of Arg-292 in Cre.
Figure 1.

Organization of the catalytic pentad and the tyrosine nucleophile in the Cre active site. The arrangement of the catalytic pentad residues (R-173, K-201, H-289, R-292 and W-315) and the tyrosine nucleophile (Y-324) in the uncleaved (top) and cleaved (bottom) states of the scissile phosphate are shown. Note that Arg-173 and Arg-292 make hydrogen bonding contacts with the phosphate in both states, with slight changes in their relative positioning. The structural representations are based on the Protein Data Bank file 1CRX (2), and is represented using the PyMOL molecular graphics system (DeLano Scientific). Potential hydrogen bond contacts were derived using RasTop v2.6 by imposing a distance constraint of 3.5 Å or less.
The type IB vaccinia topoisomerase (hereafter referred to as just topoisomerase) or its mutant derivative lacking Arg-223 (which corresponds to Arg-292 in Cre) can also perform strand cleavage in MeP–DNA (9,10). However, the tyrosyl intermediate is highly unstable, being rapidly hydrolyzed to the 3′-MeP product. The extreme instability of the MeP–tyrosyl adduct contrasts sharply with the high stability of the P–tyrosyl adduct formed in a wild-type topoisomerase reaction, their respective half-lives at physiological pH being ∼2 min and ∼36 days (9). By contrast, the P–tyrosyl intermediate formed by wild-type Cre and the MeP–tyrosyl intermediate formed by Cre(R292A) are hydrolyzed at comparable rates (7).
In Flp recombinase, lack of Arg-308 elicits a strikingly distinct response to MeP–DNA from that observed in Cre or topoisomerase. Flp(R308A) utilizes water, rather than the catalytic Tyr-343, as the nucleophile to directly hydrolyze the MeP bond (8). This futile type II endonuclease activity is also elicited in wild-type Flp by MeP, but to a much smaller extent (11).
A unifying feature of Cre, Flp and topoisomerase in their reactions with MeP–DNA is the dispensability of a conserved arginine residue in the MeP activation step. However, as alluded to above, the three also differ in the subsequent chemical outcomes in mechanistically interesting ways. Flp, Cre(R292A), topoisomerase and Topo(R223A) mediate strand cleavage in the MeP–substrate using the active site tyrosine nucleophile. However, the MeP–tyrosyl intermediates formed in the recombinase and topoisomerase reactions differ significantly in their relative stabilities with respect to the corresponding P–tyrosyl intermediates (7–10). Flp(R308A) stands apart in the unusual and strong (stronger than Flp) direct hydrolysis reaction that MeP evokes in it. As explained below, these similarities and differences can be rationalized by the organization of the active sites of these enzymes and the DNA–protein complexes within which they perform their respective functions.
Cre and topoisomerase harbor their active sites entirely within monomers (3,12,13), whereas Flp assembles a shared active site between two neighboring monomers: one donates the pentad, the other the Tyr-343 nucleophile (1,14,15). The coincidence of DNA binding by a monomer and engagement of the scissile phosphate by the tyrosine nucleophile would likely preclude direct hydrolysis of the scissile phosphate by Cre and topoisomerase. The delay between phosphate activation by a DNA-bound Flp monomer and tyrosine delivery by a second such monomer could increase the likelihood of nucleophilic attack by water. The conformational flexibility associated with strand rotation in the DNA relaxation complex might account for the potential susceptibility of the tyrosyl intermediate formed by topoisomerase to hydrolysis. The compact tetrameric organization of Cre and Flp on the synapsed DNA partners and the conformational dynamics of the cleaved strands within the recombination complex might reduce the risk of hydrolysis of the tyrosyl intermediate formed by the recombinases. It has been suggested that Arg-308 positive charge and phosphate negative charge may help exclude water, and prevent abortive hydrolysis, during strand cleavage by Flp and strand joining by topoisomerase, respectively (8–10). Substrate and active site electrostatics may not only contribute to the mechanism of phosphoryl transfer but also help avoid untoward side reactions.
The present work demonstrates that MeP activation by Cre during strand cleavage takes place in the absence of Arg-173, the other conserved arginine of the Cre active site, or simultaneous absence of both Arg-173 and Arg-292. The single Arg-to-Ala mutants are capable of promoting strand joining as well, Cre(R292A) being more efficient than Cre(R173A); the double mutant Cre(R173A, R292A) does not give detectable joining. Furthermore, Cre(R173A) and Cre(R292A) carry out recombination between two MeP–DNA partners. Thus, specific catalytic deficiencies resulting from the loss of one or both conserved arginines of the Cre catalytic pentad can be suppressed to varying extents by neutralizing the negative charge on the scissile phosphate. We consider the mechanistic implications of these findings from the general perspective of phosphoryl transfer reactions in nucleic acids.
MATERIALS AND METHODS
Synthetic oligonucleotides and assembly of half-site substrates
Standard oligonucleotides were purchased from IDT (Coralville, IA). The methylphosphonate containing oligonucleotide (a racemic mixture of the RP and SP forms) was supplied by Trilink Technologies (San Diego, CA). Hybridization of oligonucleotides was performed as described previously (16). A list of the oligonulceotides used in this study is summarized in Table 1.
Table 1.
Synthetic oligonucleotides used in the assembly of half-site and full-site substrates for Cre reactions are listed
| Oligomer | Size (bases) | Sequence |
|---|---|---|
| MeP-half-site (top strand) | 27 | 5′ ACTTGGATCCATAACTTCGTATAA(mp)TGT 3′ |
| P-half-site (top strand) | 27 | 5′ ACTTGGATCCATAACTTCGTATAATGT 3′ |
| P- or MeP-half-site (bottom strand); hydrolysis assays | 30 | 5′ CATACATTATACGAAGTTATGGATCCAAGT 3′ |
| P- or MeP-half-site (bottom strand); strand joining assays | 39 | 5′ TGTATGTTTCATACATTATACGAAGTTATGGATCC AAGT 3′ |
| Template oligo used to synthesize the 54-mer strand (top and bottom) of the short MeP-full-site by Klenow polymerase fill-in reaction | 45 | 5′ ACTTGGATCCATAACTTCGTATAATGTACATTATAC GAAGTTAT(ddC) 3′ |
| Template oligo used to synthesize the 70-mer strand (top and bottom) of the long MeP-full-site by Klenow polymerase fill-in reaction | 61 | 5′ GCATGCATGCATGCATACTTGGATCCATAACTTCGT ATAATGTACATTATACGAAGTTAT(ddC) 3′ |
| 47-mer used to generate a 74-mer strand (top and bottom) of a long MeP-full-site by ligation to the 27-mer shown in row1 | 47 | 5′ ACATTATACGAAGTTATGGATCCAAGTATGCATGCA TGCATGCATGC 3′ |
| splint oligo used in the ligation to generate the 74-mer strand of the long MeP-full-site | 44 | 5′ GATCCATAACTTCGTATAATGTACATTATACGAAGT TATGGATC 3′ |
The string of nucleotides shown in bold represents the Cre binding element. The scissile phosphate or methylphosphonate is indicated by ‘p’ or ‘mp’, respectively. The template oligos employed in the Klenow polymerase reactions contained a 3′-terminal dideoxy-C (ddC).
Construction of P- and MeP-full-sites for recombination reactions
The short P-full-site recombination partner was assembled from two 54-mer synthetic oligonucleotides (7), and the long one by PCR amplification of a DNA segment containing LoxP using appropriate primers.
To construct the MeP-full-site substrates for recombination, the 27-mer oligonucleotide that served as the scissile strand in the MeP half-site was extended by Klenow polymerase (exonuclease minus) using a 45-mer synthetic template strand carrying a 3′ dideoxy-C end. The 54-mer product strand so formed was purified away from the template by urea–polyacrylamide gel electrophoresis (urea–PAGE) and DNA isolation. This DNA strand could hybridize to itself to generate the short MeP–LoxP recombination partner containing a symmetric strand exchange region. To obtain the long MeP–LoxP partner, the MeP containing 27-mer was lengthened at its 3′-end to a 70-mer by the Klenow polymerase chain elongation method using a 61-mer template strand with a 3′ dideoxy-C end. The design of the template strand was such that the polymerized product contained the 54-mer sequence of the short MeP–LoxP plus 16 nt as a 3′ appendage. Self-annealing of the purified 70-mer yielded a variant of the 54-mer palindromic full-site carrying 16-nt long single-stranded 3′ tails on both strands. Note that the blocked 3′-ends (dideoxy-C) of the template strands kept them from being extended during the Klenow polymerase reaction.
A second long MeP-full-site was constructed by a splint-oligo assisted ligation of the 27-mer MeP containing strand to an all phosphate containing 47-mer oligonucleotide, followed by gel purification and self-annealing of the 74-mer. The difference between the two versions of the full-sites was only in the length of the single-stranded tails, 20 versus 16 nt.
Purification of Cre and Cre mutants
Wild-type Cre and its mutant derivatives, harboring a His-6 tag at their N-termini, were purified as described previously (7,17).
In vitro Cre reactions
In vitro Cre reactions were carried out under conditions described earlier (7). Assays were initially optimized using a range of Cre and Cre variant amounts per Cre binding element of the DNA substrate employed. Subsequent experiments were performed with the optimized protein amounts. Reactions were terminated by addition of sodium dodecyl sulfate (SDS) to a final concentration of 0.1%. For assaying the Cre–DNA covalent adduct, samples were analyzed directly by SDS polyacrylamide gel electrophoresis (SDS–PAGE). For assaying hydrolysis, DNA was isolated by phenol–chloroform extraction followed by ethanol precipitation at −20°C. The DNA samples were washed twice with 80% ethanol, and analyzed by 12% urea–PAGE. In this assay, the proteinase K treatment that normally precedes DNA extraction was omitted. By doing so, following electrophoresis, the hydrolysis product could be visualized without interference from the strand cleavage product carrying short Cre peptides at the 3′ end. DNA samples isolated from recombination reactions between two full-LoxP-sites were also analyzed by 12% urea–PAGE.
Kinetic analysis of the type I hydrolysis reaction by Cre(R173A) and Cre(R173A, R292A)
Kinetic parameters were derived according to a two-step reaction scheme, strand cleavage by the tyrosine nucleophile of Cre followed by hydrolysis of the DNA–tyrosyl intermediate.
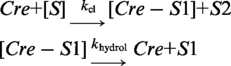 |
S is the substrate, and kcl and khydrol are the first order rate constants for cleavage and hydrolysis, respectively. Cre-S1 refers to the covalent adduct, S2 to the trinucleotide released during strand cleavage and S1 to the hydrolysis product. Each of the two reaction steps is assumed to be irreversible. Curve fitting by non-linear regression was performed by using KinTek Global Kinetic Explorer simulation/dynamic visualization software (18,19). For a given starting concentration of S, each rate constant and the output scaling factor were adjusted to obtain the optimal global fit to the experimental data.
Kinetic analysis of strand joining by Cre(R173A) and Cre(292A)
The first order rate constants for the strand joining reaction were estimated according to the scheme:
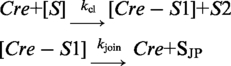 |
SJP denotes the product of strand joining. Curve fitting was performed as described above for the strand cleavage and hydrolysis reactions.
Quantification of reaction products
Gels in which reactions were analyzed were dried and exposed to a storage phosphor screen (Bio-Rad) or X-ray film to detect radioactive bands. Exposure times were optimized to obtain reliable signals while avoiding saturation of the screen. Phosphor screens were scanned by a phosphor-imager (GE-Healthcare). Image analysis was performed by using the software Quantity One (version 4.5.1; Bio-Rad).
RESULTS
Experimental layout
Assays were carried out primarily using MeP substrates (7), in the form of half-LoxP or full-LoxP sites, supplied as a racemic mixture of the RP and SP stereoisomers. A subset of reactions were performed on P substrates as controls. A half-site contains only one Cre-binding element and one scissile strand. When the Tyr-324 nucleophile performs strand cleavage in a half-site, the short trinucleotide product of the reaction (with a 5′-hydroxyl group) will not remain stably hydrogen bonded to the opposite strand. The diffusion of the trinucleotide away from the reaction center makes the cleavage reaction effectively irreversible. If the non-scissile strand of the half-site harbors a 5′-hydroxyl end, a pseudo-joining reaction may occur by the attack of the hydroxyl on the cleaved tyrosyl intermediate. In principle, a scissile P- or MeP bond in the DNA substrate activated by a bound Cre or the corresponding tyrosyl intermediate formed by strand cleavage could be hydrolyzed as a result of nucleophilic attack by water. In the experiments described below, strand cleavage, strand joining and potential hydrolytic reactions were assayed. Even though the Cre active site is contained within a monomer, its full chemical competence may depend on allosteric activation by a second Cre monomer. Thus, the half-site reactions may occur in the context of dimeric, trimeric or tetrameric [Cre-half-site] complexes (20,21).
In reactions that monitored hydrolysis promoted by a Cre mutant, the 5′-hydroxyl of the strand complementary to the scissile one was blocked by phosphorylation. This hydroxyl was left unmodified when assaying strand joining. The covalent tyrosyl adduct formed by strand cleavage was revealed by SDS–PAGE. The products of hydrolysis, strand joining and recombination were visualized by electrophoresis under strand denaturing conditions (urea–PAGE). The substrates used in the MeP-full-site reaction contained a symmetrized 6-bp strand exchange region. Schematic representations of specific reactions are included in figures for clarity.
Strand cleavage in the MeP-half-site by Cre(R173A): hydrolysis of the MeP–tyrosyl intermediate
In previous work, we found that unlike Flp(R308A), the equivalent Cre(R292A) does not support direct hydrolysis of the MeP bond (type II endonuclease activity) (7). Instead, it promotes formation of the MeP–tyrosyl intermediate, which may then be targeted for hydrolysis by the type I endonuclease activity. The suggested explanation is that the cis-acting active site of a Cre monomer fully engages the scissile P or MeP upon DNA binding, thus excluding water from the reaction center. An alternative possibility is that it is Arg-173 of Cre (corresponding to Arg-191 of Flp), which also contacts the scissile phosphate (2) (Figure 1), that is responsible for repelling water during strand cleavage by Cre. We wished to know whether the R173A mutation in Cre would induce it to exhibit the type II endonuclease activity.
Reactions with Cre(R173A) revealed the early formation of the MeP–tyrosyl intermediate (Figure 2A), followed by its subsequent decay. The 24-mer hydrolysis product was also formed in the reaction but with a time delay compared to the tyrosyl adduct (Figure 2B). The correspondence between the diminution of this adduct and the build-up of the 24-mer was consistent with a precursor–product relationship between the two. Furthermore, in agreement with this notion, blocking strand cleavage by eliminating the Tyr-324 nucleophile also blocked 24-mer formation. Neither the covalent cleaved intermediate (Figure 3A) nor the 24-mer (Figure 3B) was detected in a reaction with Cre(R173A, Y324G). The mobility of the major 24-mer product corresponded to the presence of MeP at its 3′ end, while that of the faint band just above indicated a 3′-hydroxyl end (7). Cre(R173A) was inactive on a P-half-site (Figure 4).
Figure 2.

Strand cleavage and type I endonuclease activities of Cre(R173A) on an MeP-half-site substrate. In the schematic representation of the half-site substrate, the asterisk indicates 32P-label at the 5′-end, and ‘mp’ the scissile methylphosphonate. The parallel arrows denote the Cre binding element. Short stretches of additional nucleotides extending from the binding element to its left on both strands are not shown. The type I endonuclease activity proceeds via formation of the MeP–tyrosyl intermediate, followed by its hydrolysis. Reactions were split into halves to analyze the covalent Cre–DNA adduct (indicated by the circular knob linked to the labeled DNA chain) by SDS–PAGE (A) and the potential hydrolysis product(s) by denaturing urea–PAGE (B). The end-labeled half-site (A) or its labeled strand (B) is denoted by ‘S’, and the hydrolysis product by ‘HP’. The labeled strand in the MeP-half-site is 27-nt long, and HP is a 24-mer. In the control reaction, the MeP-half-site was incubated in the recombination buffer for 48 h in the absence of Cre(R173A). The plots represent mean values from three separate experiments with Cre(R173A). The global fit to the kinetic data was performed using KinTek Global Kinetic Explorer, as explained in ‘Materials and Methods’ section. For a given starting concentration of the MeP-half-site, each rate constant and the output scaling factor were adjusted to obtain the optimal global fit.
Figure 3.

Lack of endonucleolytic activity in MeP-half-site reactions of Cre(R173A, Y324G). Reactions and product analysis were performed as described under Figure 2. The results of SDS–PAGE and denaturing PAGE analyses are shown in A and B, respectively. The predicted positions of migration of the covalent intermediate and the hydrolysis product (HP) are indicated. The incubation time for the control reaction without addition of Cre(R173A, Y324G) was 24 h.
Figure 4.

Lack of strand cleavage or hydrolysis in a P-half-site by Cre(R173A). The half-site substrate was identical in sequence to that employed in the reactions shown in Figures 2 and 3, except that it contained a scissile phosphate instead of methylphosphonate. Reactions were analyzed as described in the legends for Figures 2 and 3. (A) SDS-PAGE. (B) Urea-PAGE.
Thus, the absence of Arg-173 or that of Arg-292 in Cre does not render the scissile MeP bond vulnerable to direct hydrolysis. However these individual mutations do not prevent MeP activation and formation of the MeP–tyrosyl intermediate. The strand cleavage step during Cre recombination is presumably protected by the monomeric cis-organization, rather than the electrostatic properties, of its active site.
The first order rate constants for the strand cleavage and type I endonuclease activities of Cre(R173A) were estimated as kcl = 4.4 × 10−4 s−1 and khydrol = 2.7 × 10−4 s−1, respectively (Table 2). Unlike the situation with topoisomerase, hydrolysis of the MeP–tyrosyl intermediate is not dramatically accelerated by Cre(R173A) or Cre(R292A) relative to that of the P–tyrosyl intermediate by wild-type Cre (khydrol = 2.8 × 10−5 s−1) (7). The corresponding rate constants for topoisomerase differ by a factor of >104 between the MeP- and P–tyrosyl intermediates (9).
Table 2.
Activities of Cre mutants containing alanine replacements at Arg-173 or Arg-292 or both positions deduced from this study are summarized
| Cre mutant | Activities on MeP DNA substrates |
||||
|---|---|---|---|---|---|
| Strand cleavage | Type I endonuclease (hydrolysis of MeP–tyrosyl intermediate) | Type II endonuclease (direct hydrolysis of MeP bond in DNA) | Strand joining | Recombination | |
| Cre(R173A) | Active (kcl = 4.4 × 10−4s−1) | Active (khydrol = 2.7 × 10−4s−1) | Inactive | Active (kjoin = 8.6 × 10−6s−1) | Active |
| Cre(R292A) | Active (kcl = 3.2 × 10−4s−1) | Active (kcl = 8.8 × 10−4s−1) | Inactive | Active (kjoin = 2.8 × 10−5s−1) | Active |
| Cre(R173A, R292A) | Active 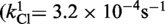 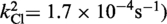
|
Active 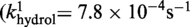 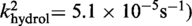
|
Inactive | Inactive | Inactive |
The kinetic data for the strand cleavage and endonuclease activities of Cre(R173A, R292A) yielded better global fit by assuming two contributing first-order rate constants for each activity. The distinct reactivities of the RP and SP forms of the MeP–substrate (provided as an achiral mixture of the two forms) could potentially account for the binary rate constants.
Strand cleavage and type I endonuclease activities of Cre(R173A, R292A)
In addition to the conserved twin arginines, the Cre catalytic pentad contains additional positive charges in the form of Lys-201 and His-289. Thus, there is a cradle of positive charge within the active site that presumably adds a significant electrophilic character to the chemical steps of recombination (2,3). In the crystal structure, His-289 forms hydrogen bonding contact with the scissile phosphate after it has been cleaved but not prior to cleavage (2) (Figure 1). Trp-315 of the pentad is within hydrogen bonding distance of the scissile phosphate in both its uncleaved and cleaved states (Figure 1). We wondered whether, even in the combined absence of Arg-173 and Arg-292, MeP activation might be feasible through the residual positive charge in the active site and/or the hydrogen bonding interactions through His-289 and Trp-315. A related question, assuming that Cre(R173A, R292A) promotes MeP activation, is whether the double mutation might cause susceptibility of MeP to direct hydrolysis (type II endonuclease activity).
Cre(R173A, R292A) not only produced the covalent intermediate in the MeP-half-site but, rather surprisingly, the initial rate of its formation was similar to that observed in the Cre(R173A) reaction (Figure 5A). The MeP–tyrosyl intermediate was longer lived in the double mutant reaction, peaking at ∼4 h. Its disappearance with concomitant formation of the 24-mer hydrolysis product (via the type I endonuclease mechanism) did not become significant until 6–8 h from start (Figure 5B). A potential type II endonuclease activity of Cre(R173A, R292A) was ruled out by incorporating into it the Y324G mutation as well. The triple mutant was unable to generate the covalent intermediate as expected, or to promote direct MeP hydrolysis (Figure 6A and B).
Figure 5.

Strand cleavage and endonuclease activities of Cre(R173A, R292A) on an MeP-half-site. Reactions and analyses were carried out as described under Figure 2. In the control reaction, the MeP-half-site was incubated without Cre(R173A, R292A) in the reaction buffer for 24 h. The values from three separate experiments are plotted in the graph. Assumption of two contributing first order rate constants, rather than a single rate constant, for the strand cleavage and type I endonuclease reactions gave better global fit to the experimental data. (A) SDS-PAGE. (B) Urea-PAGE.
Figure 6.

Lack of endonucleolytic activity in MeP-half-site reactions of Cre(R173A, R292A, Y324G). Reactions and product analysis were performed as described under Figure 2. The predicted positions of migration of the covalent intermediate and the hydrolysis product (HP) are indicated. The incubation time for the control reaction without addition of Cre(R173A, R192A, Y324G) was 24 h. (A) SDS-PAGE. (B) Urea-PAGE.
The kinetic data in Figure 5 could not be easily accommodated by an irreversible two-step reaction model, with a single rate constant for each step. A better fit was obtained by introducing two contributing rate constants for strand cleavage 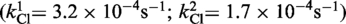 and hydrolysis
and hydrolysis 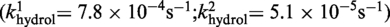 . One possibility is that the RP and SP forms present in the achiral mixture of the MeP-half-site are kinetically distinct in their reactions with Cre(R173A, R292A). A previous study (7) showed that Cre(R292A) has a strong bias favoring SP over RP. The stereochemical preference of Cre(R173A) or Cre(R173A, R292A) has not yet been determined.
. One possibility is that the RP and SP forms present in the achiral mixture of the MeP-half-site are kinetically distinct in their reactions with Cre(R173A, R292A). A previous study (7) showed that Cre(R292A) has a strong bias favoring SP over RP. The stereochemical preference of Cre(R173A) or Cre(R173A, R292A) has not yet been determined.
Strand joining by Cre(R173A), Cre(R292A) and Cre(R173A, R292A)
The strand cleavage potential of the single and double alanine substitution mutants of Arg-173 and Arg-292 raise questions of their potential catalytic competence in strand joining as well as their effects on the competition between the joining reaction and the type I endonuclease activity, both of which target the tyrosyl intermediate. To test joining, Cre(R173A), Cre(R292A) and Cre(R173A, R292A) were reacted with an MeP-half-site containing a longer 5′ single-stranded extension of the bottom strand (with a hydroxyl end) than the standard MeP-half-site used in strand cleavage reactions. This tail could loop back and pair with itself to position the 5′-hydroxyl in the joining orientation (Figure 7).
Figure 7.

Strand joining reactions of Cre(R173A) (A), Cre(R292A) (B) and Cre(R173A, R292A) (C) in an MeP- half-site and Cre (D) in a P-half-site. In the schematic diagram, the scissile MeP or P position is indicated by the filled circle. The bottom strand of the half-site designed to follow strand joining was longer than that of the standard MeP-half-site used in the strand cleavage/hydrolysis assays, and contained a free 5′-hydroxyl group. Looping back of the single-stranded region would place the 5′-hydroxyl in position to attack the tyrosyl intermediate formed by strand cleavage. This intra-half-site reaction would produce a hairpin (63-mer). Strand joining could potentially occur between two half-sites to yield a ‘pseudo-full-site’. The individual strands of the pseudo-full-site and the hairpin would be identical in sequence, and display the same mobility during denaturing PAGE. The lower panels show urea–PAGE analysis to assay type I endonuclease activity. ‘JP’ refers to this joined product, ‘HP’ to the hydrolysis product and ‘S’ to the labeled strand of the half-site. ‘C’ is a control reaction incubated for 24 h in the absence of Cre or a Cre variant.
Cre(R173A) yielded ∼1.5% of the joined product (Figure 7A), whereas Cre(R292A) was more efficient, the output leveling off at ∼6% (Figure 7B). Cre(R173A, R292A) did not give detectable joining reaction (Figure 7C). For comparison, in a P-half-site reaction with Cre, ∼50% of the input substrate was converted to the joined product in about 6 h (Figure 7D).
For Cre(R173A) and Cre(R292A) reactions, the joined product was formed in significantly higher yields than the 24-mer hydrolysis product, which was reduced to near background levels (Figure 7A and B). This prominence of strand joining relative to hydrolysis was also true of the wild-type Cre reaction with the P-half-site (Figure 7D). Thus, the 5′-hydroxyl group effectively out-competes water in nucleophilic attack on the MeP–tyrosyl or P–tyrosyl intermediate.
The yields of strand joining by Cre(R173A) (Figure 7A) and Cre(R292A) (Figure 7B) were considerably diminished in comparison to those of hydrolysis by these mutants from the MeP-half-site with the shorter bottom strand lacking the 5′-hydroxyl nucleophile (7) (Figure 2A). The reduction was from 30–40% hydrolysis to ∼6% or less joining (and practically no hydrolysis) in the relevant substrates. It is possible that the extended bottom strand tail of the half-site, with its ability to fold back, and/or the 5′-hydroxyl group could interfere with Tyr-324 mediated cleavage, lowering the levels of the tyroysl intermediate available for either hydrolysis or joining. Alternatively, lack of Arg-173 or Arg-292 might reduce the efficiency of strand joining, with the 5′-hydroxyl group positioned within the active site additionally blocking nucleophilic attack by water due to steric interference. To test these possibilities, we assayed the extent of covalent adduct formation by each of the Cre mutants in the joining substrate.
In Cre(R173A), Cre(R292A) and Cre(R173A, R292A) reactions with the joining substrate, we did not detect an accumulation of the MeP–tyrosyl intermediate that is predicted for its inefficient conversion to the hydrolysis or joining products. In all three cases, the cleaved intermediate reached a maximum between 4 and 6% (Figure 8A). Its depletion in the Cre(R173A) and Cre(R292A) reactions was in step with the increase in the joined product. Furthermore, neither strand cleavage nor hydrolysis was improved appreciably when the bottom strand in the joining substrate was phosphorylated at the 5′-end (Figure 8B; data not shown).
Figure 8.

Strand cleavage and strand joining by Cre(R173A), Cre(R292A) and Cre(R173A, R292A). The MeP-half-sites contained either a 5′-hydroxyl (A) or a 5′-phosphate group (B) on the bottom strand. The plots in A were obtained by using the data presented in Figure 7. In the reactions shown in (B), no strand joining was possible because of the blocked 5′-end of the bottom strand. Global fits to the kinetic data were generated using KinTek Global Kinetic Explorer. Points that fall below the flat portions of the simulated curves in (B) may be due to slow breakdown of the tyrosyl intermediate by a weak type I endonuclease activity, which was not assayed in this analysis.
It is the long tail (perhaps in its folded form), rather than the free 5′-hydroxyl group, that likely contributes to the inhibition of strand cleavage. This non-standard configuration of the half-site could impede its self-association into dimeric, trimeric and tetrameric assemblies. The consequent adverse effect on the allosteric interaction between two neighboring Cre monomers is expected to be detrimental to strand cleavage. Strand cleavage in full-sites has been shown to be stimulated strongly by synapsis between two partner sites (20). However, despite the unfavorable effect of the special half-site design on strand cleavage, the data in Figure 7 demonstrate the distinct joining efficiencies of wild-type Cre in the P-half-site and Cre(R173A), Cre(R292A) and Cre(R173A, R292A) in the corresponding MeP-half-site.
The more or less equivalent yields of the MeP–tyrosyl intermediate from the strand joining substrate by the single and double mutants (Figure 8A), contrast with the distinct outputs of the joined product by them (Figure 7). The 4-fold better joining by Cre(R292A) than Cre(R173A), with no detectable joining by Cre(R173A, R292A), suggests that Arg-173 is more critical than Arg-292 for strand joining in the MeP-half-site.
Recombination between MeP-full-site partners by Cre(R292A)
The ability of Cre(R292A) to promote strand cleavage and strand joining (although with reduced efficiency) in MeP-half-sites suggests that this mutant may promote recombination between two full-site partners containing MeP at the scissile positions. A successful reaction would indicate that neither MeP substitution nor the lack of this catalytic arginine poses a strong barrier to the conformational dynamics of strand exchange between DNA partners. Cre(R173A) may also perform recombination in MeP-full-sites, perhaps with lower efficiency because of its weaker joining capacity.
In the MeP-full-site substrates, because of the nature of their construction (see ‘Materials and Methods’ section), the recombination target sites were rendered palindromic in sequence. The symmetrized strand exchange region (5′ATGT-ACAT3′) would not affect recombination (22), although the reaction would lose the directionality associated with asymmetric strand exchange regions, as dictated by the requirement of homology between them. One of the reaction partners was lengthened at either end by polymerizing an additional oligonucleotide tail. DNA exchange between the radioactively labeled short substrate and the unlabeled long one would result in a readily identifiable recombinant strand (Figure 9).
Figure 9.

Recombination mediated by Cre(R173A) and Cre(R292A) between LoxP sites containing MeP at the scissile positions. Strand exchange reactions between two MeP-full-sites or two P-full-sites are diagrammed schematically at the top. In the MeP-full-sites, because of their assembly protocol, the strand exchange regions as well as the flanking sequences are fully palindromic, that is, the top and bottom strands are identical in sequence. The single-stranded tails in the long substrate (S2) permit distinction of the recombinant product formed by strand exchange with the end-labeled short substrate (S1) when analyzed by urea–PAGE. The scissile MeP positions are indicated by filled circles. In (A), S2 to S1 molar ratios were 0.25, 0.5 and 1.0 in lanes 3, 4 and 5, respectively, with a constant reaction time of 3 h; in B and C, S2 to S1 ratio was held at 4:1, and reaction times were varied. (B and C) Depict distinct sections of the same gel, with the control lance (1 in B and 1′ in C) being identical. The incubation for the mock reaction without Cre was 4 h. The reaction shown in D utilized P-full-sites (S′ and S″) with the native asymmetric strand exchange regions. In lanes 2–4, the molar ratio of S″ to S′ was maintained as 1:1, with serial 2- and 4-fold increases in the amount of Cre. The reaction time was 1 h. The recombinant products from the MeP- and P-full-site reactions are represented as R and R′, respectively.
In the MeP-full-site reaction, Cre(R292A) generated the predicted 74-mer (Figure 9A) or the 70-mer (Figure 9B) product of strand exchange. The size difference was due to the 4-nt shorter tail length of the unlableled substrate (S2) in the reaction shown in Figure 9B. The 70-mer yield of ∼5% in 4–6 h is in line with the ∼3% yield of the joined product obtained in the same time period during the joining reaction in the MeP-half-site (Figure 7B). Cre(R173A) also carried out recombination in the MeP-full-site, its activity being weaker than that of Cre(R292A) (Figure 9C). This result is consistent with the poor joining efficiency of Cre(R173A). For comparison, recombination between two P-full-sites by Cre is shown in Figure 9D.
Thus, Cre(R173A) and Cre(R292A) are weak but functional tyrosine recombinases for DNA substrates in which the negative charge at the scissile positions is neutralized by MeP modification. The data also indicate that neither the lack of Arg-173 or Arg-292 in the Cre active site nor the MeP substitution in DNA precludes the conformational dynamics associated with the strand exchange process.
DISCUSSION
In this study, we have addressed the role of electrostatic complementarity between the Cre recombinase active site and the scissile phosphate in the strand cleavage and joining steps of recombination. Substrate and active site electrostatics were modulated by modifying the scissile phosphate to methylphosphonate and replacing specific arginine residues with alanine, respectively. As deduced from crystal structures, interactions between the phosphate and each of the two conserved arginines, Arg-173 and Arg-292, of the catalytic pentad provide the major electrostatic contribution towards stabilizing the transition state (2,3). The catalytic properties of active sites lacking either one or both of these positively charged side chains towards the charge-neutralized scissile MeP reveal mechanistic features of recombination that could not have been gleaned from reactions with DNA substrates containing native phosphate groups at the scissile positions. The cumulative results from this study are summarized in Table 2, and provide the framework for active site mechanisms considered below.
Dispensability of Arg-173 and Arg-292 in strand cleavage by Tyr-324 in MeP–DNA
The present work, in conjunction with previous findings (7), demonstrates that Cre variants lacking either or both of the conserved active site arginines (Arg-173 and Arg-292) are capable of MeP activation and cleavage using Tyr-324 nucleophile (Figures 2A and 5A). Cleavage competence in the combined absence of the conserved catalytic arginines was not anticipated from structural data or previous biochemical results with Cre mutants on substrates containing scissile phosphates. When the nominal negative charge in the transition state is reduced from −2 to −1 by P to MeP substitution, an active site with severe electrostatic impairment is still able to promote the first step of recombination with reasonable efficiency. The MeP transition state with its lone negative charge could be stabilized through electrophilic or other types of interactions from the intact pentad residues (Lys-201, His-289 and Trp-315), perhaps with additional assistance from non-pentad residues. According to the crystal structure, both His-289 and Trp-315 are within hydrogen bonding distance in the active site containing the P–tyrosyl intermediate, and Trp-315 is also in contact with the scissile phosphate when it is not cleaved (2). Furthermore, the active site cavity resulting from the absence of Arg-173 and/or Arg-292 may permit residues that are normally non-catalytic to relocate themselves. For example, if Gln-133 or Arg-326 could swing into favorable positions, they might serve a surrogate role in transition state stabilization by the mutant active sites.
The single arginine mutants and the double mutant are also capable of hydrolyzing the cleaved intermediate by the type I endonuclease activity (Figures 2B and 5B) (7). These Cre mutants are strikingly different from a Flp mutant lacking Arg-308 (equivalent to Arg-292 in Cre), which causes direct hydrolysis of the MeP bond by the type II endonuclease activity (8). This distinction between Cre and Flp appears to follow from their cis-acting monomeric and trans-acting shared active sites, respectively. The Cre mutants are akin to topoisomerase and the topoisomerase mutant lacking Arg-223 (equivalent to Arg-292 in Cre and Arg-308 in Flp), both of which exhibit the type I endonuclease activity towards the MeP–tyrosyl intermediate (9,10). However, the dramatic difference in stabilities between the P–tyrosyl intermediate and MeP–tyrosyl intermediate seen in topoisomerase or topoisomerase mutant reactions is not observed in Cre or Cre mutant reactions. Unlike the toposiomerase situation, the role of phosphate electrostatics per se in protecting the cleaved intermediate against hydrolysis during strand joining by Cre appears not to be prominent. This protection is likely derived from the compact organization of the Cre tetramer in the recombination synapse plus the alignment of the 5′-hydroxyl groups in their joining-competent orientation concomitant with cleavage.
Arg-173 is more important than Arg-292 in strand joining of the MeP–tyrosyl intermediate: recombination in MeP–DNA by Cre(R292A)
The sum of the strand cleavage and joining results from the MeP-half-site signify the non-equivalent roles of Arg-173 and Arg-292 in recombination chemistry. Whereas strand cleavage proceeds in the absence of both arginines (7) (Figures 2A and 5A); detectable strand joining requires at least one of the two to be present (Figure 7). Relative cleavage and joining efficiencies of Cre(R173A) and Cre(R292A) suggest that Arg-173 is more important in joining than Arg-292. In Flp, the situation is quite different. As shown previously (8), replacement of Arg-308 in Flp (counterpart of Arg-292 in Cre) blocks strand cleavage by the catalytic tyrosine, causing abortive hydrolysis of MeP. Since little or no tyrosyl intermediate is formed by Flp(R308A), strand joining by this mutant has to be correspondingly inept or absent. By contrast, we find that Flp(R191A), lacking the equivalent of Arg-173 in Cre, can mediate strand cleavage as well as strand joining (11). Consistent with their abilities to promote both these reactions, Cre(R173A), Cre(R292A) and Flp(R191A) are functional recombinases towards MeP DNA substrates (Figure 9) (11).
Stereochemistry of strand cleavage and strand joining in the MeP-half-site reactions
In the crystal structure (Figure 1), Arg-173 and Arg-292 are favorably located to make potential contacts with the pro-RP and pro-SP oxygen atoms, respectively, of the scissile phosphate in the intact DNA backbone. When DNA is cleaved by Tyr-324, Arg-173 position is modulated to be able to contact either of these two oxygen atoms. Consistent with the structural data, our previous work (7) revealed a strong preference by Cre(R292A) for the SP form of the MeP-half-site (in which the pro-RP oxygen is replaced by the methyl group and the pro-SP oxygen is retained) over the RP form. The simple expectation is that the preference would be switched to the RP form for Cre(R173A), at least during the cleavage reaction. Because of the modulation of phosphate contacts by Arg-173 following cleavage, potential stereochemical bias of Cre(R173A) in the type I hydrolysis reaction or the joining reaction is difficult to anticipate. A more incisive stereochemical analysis of the Arg-173/Arg-292 single and double mutants of Cre using pure RP and SP forms of the MeP substrate is being performed.
Substrate and active site electrostatics in DNA phosphoryl transfer: more than just mechanistic?
The findings from this study, along with those previously published (7–10), highlight the significance of electrostatic complementarity between active sites and substrates during the phosphoryl transfer steps of site-specific recombination by tyrosine recombinases and DNA relaxation by a type IB topoisomerase. Adverse catalytic effects of mutations that eliminate critical positive charges within the active site can be ameliorated by removing a negative charge from the substrate by P to MeP substitution. The extent of suppression is variable depending on the particular active site mutation as well as the particular chemical step of the reaction, stand cleavage or joining. Cre and Flp recombinase mutants that are rescued for both chemical steps (albeit to different extents) are also able to perform recombination reaction in MeP–DNA substrates (this study) (11). Chemical modification of the phosphate by methyl replacement of non-bridging oxygen and mutational alterations of active site residues are not seriously detrimental to the conformational dynamics required for strand exchange between two DNA partners.
Phosphate and active site electrostatics, critical as they are in phosphoryl transfer chemistry, may transcend purely catalytic roles. In toposiomerase reaction, the MeP–tyrosyl intermediate is prone to rapid hydrolysis whereas its P–tyrosyl counterpart is extremely resistant to this futile reaction (9,10). In Flp reaction, the absence of the active site Arg-308 makes the MeP bond the target of direct hydrolysis, thus aborting recombination at the cleavage step. This activity, in a much weaker form, has also been observed in a wild-type Flp/MeP-half-site reaction (11) whereas direct hydrolysis of the phosphodiester bond by Flp has been undetectable or insignificant under normal reaction conditions. Manifestation of such aberrant reactions as a result of phosphate modification with or without catalytic mutations suggests that substrate and active site electrostatics may be utilized for protecting desired reactions against wanton side reactions that damage genome integrity.
Marked instability by hydrolysis of either the DNA backbone or that of the tyrosyl intermediate by MeP substitution in conjunction with removal of catalytic arginines has not been observed in Cre (this study) (7). Features other than electrostatics such as active site organization, tightness of the DNA–protein reaction complex and the conformational dynamics of DNA cleavage and joining may also serve to exclude water. Given that a wide variety of biologically critical transesterification reactions (DNA recombination, DNA transposition and RNA splicing, to name a few) must occur in the context of an overwhelming excess of water (∼56M), strategies to minimize hydrolysis must be an inherent part of the evolutionary designs of active sites performing these reactions.
Methylphosphonate modification, combined with appropriate active site mutations, provides a useful and convenient way of probing electrostatic attributes of phosphoryl transfer mechanisms. An added bonus is that the analysis may also speak to the potential significance of substrate and active site electrostatics as a water repelling strategy.
FUNDING
National Institutes of Health award (GM035654 to M.J.); Robert F Welch Foundation grant (F-1274); UT Austin Faculty research Award. Funding for open access charge: National Institutes of Health award (GM035654 to M.J.).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank K. A. Johnson for his generous help with the use of the KinTek Global Kinetic Explorer software
REFERENCES
- 1.Chen Y, Narendra U, Iype LE, Cox MM, Rice PA. Crystal structure of a Flp recombinase-Holliday junction complex: assembly of an active oligomer by helix swapping. Mol. Cell. 2000;6:885–897. [PubMed] [Google Scholar]
- 2.Guo F, Gopaul DN, Van Duyne GD. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997;389:40–46. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- 3.Van Duyne GD. A structural view of tyrosine recombinase site-specific recombination. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington DC: ASM Press; 2002. pp. 93–117. [Google Scholar]
- 4.Krogh BO, Shuman S. Catalytic mechanism of DNA topoisomerase IB. Mol. Cell. 2000;5:1035–1041. doi: 10.1016/s1097-2765(00)80268-3. [DOI] [PubMed] [Google Scholar]
- 5.Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WG. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Rice PA. New insight into site-specific recombination from Flp recombinase-DNA structures. Annu. Rev. Biophys. Biomol. Struct. 2003;32:135–159. doi: 10.1146/annurev.biophys.32.110601.141732. [DOI] [PubMed] [Google Scholar]
- 7.Ma CH, Kachroo AH, Maciaszek A, Chen TY, Guga P, Jayaram M. Reactions of Cre with methylpzhosphonate DNA: similarities and contrasts with Flp and vaccinia topoisomerase. PLoS ONE. 2009;4:e7248. doi: 10.1371/journal.pone.0007248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma CH, Rowley PA, Maciaszek A, Guga P, Jayaram M. Active site electrostatics protect genome integrity by blocking abortive hydrolysis during DNA recombination. EMBO J. 2009;28:1745–1756. doi: 10.1038/emboj.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tian L, Claeboe CD, Hecht SM, Shuman S. Guarding the genome: electrostatic repulsion of water by DNA suppresses a potent nuclease activity of topoisomerase IB. Mol. Cell. 2003;12:199–208. doi: 10.1016/s1097-2765(03)00263-6. [DOI] [PubMed] [Google Scholar]
- 10.Tian L, Claeboe CD, Hecht SM, Shuman S. Mechanistic plasticity of DNA topoisomerase IB: phosphate electrostatics dictate the need for a catalytic arginine. Structure. 2005;13:513–520. doi: 10.1016/j.str.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Rowley PA, Kachroo AH, Ma CH, Maciaszek AD, Guga P, Jayaram M. Electrostatic suppression allows tyrosine site-specific recombination in the absence of a conserved catalytic arginine. J. Biol. Chem. doi: 10.1074/jbc.M110.112292. doi:10.1074/jbc.M110.112292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng C, Kussie P, Pavletich N, Shuman S. Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell. 1998;92:841–850. doi: 10.1016/s0092-8674(00)81411-7. [DOI] [PubMed] [Google Scholar]
- 13.Perry K, Hwang Y, Bushman FD, Van Duyne GD. Structural basis for specificity in the poxvirus topoisomerase. Mol. Cell. 2006;23:343–354. doi: 10.1016/j.molcel.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 14.Chen JW, Lee J, Jayaram M. DNA cleavage in trans by the active site tyrosine during Flp recombination: switching protein partners before exchanging strands. Cell. 1992;69:647–658. doi: 10.1016/0092-8674(92)90228-5. [DOI] [PubMed] [Google Scholar]
- 15.Lee J, Jayaram M, Grainge I. Wild-type Flp recombinase cleaves DNA in trans. EMBO J. 1999;18:784–791. doi: 10.1093/emboj/18.3.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J, Whang I, Jayaram M. Assembly and orientation of Flp recombinase active sites on two-, three- and four-armed DNA substrates: implications for a recombination mechanism. J. Mol. Biol. 1996;257:532–549. doi: 10.1006/jmbi.1996.0183. [DOI] [PubMed] [Google Scholar]
- 17.Shaikh AC, Sadowski PD. The Cre recombinase cleaves the lox site in trans. J. Biol. Chem. 1997;272:5695–5702. doi: 10.1074/jbc.272.9.5695. [DOI] [PubMed] [Google Scholar]
- 18.Johnson KA, Simpson ZB, Blom T. Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 19.Johnson KA, Simpson ZB, Blom T. FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 2009;387:30–41. doi: 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh K, Lau CK, Gupta K, Van Duyne GD. Preferential synapsis of loxP sites drives ordered strand exchange in Cre-loxP site-specific recombination. Nat. Chem. Biol. 2005;1:275–282. doi: 10.1038/nchembio733. [DOI] [PubMed] [Google Scholar]
- 21.Woods KC, Martin SS, Chu VC, Baldwin EP. Quasi-equivalence in site-specific recombinase structure and function: crystal structure and activity of trimeric Cre recombinase bound to a three-way Lox DNA junction. J. Mol. Biol. 2001;313:49–69. doi: 10.1006/jmbi.2001.5012. [DOI] [PubMed] [Google Scholar]
- 22.Hoess RH, Wierzbicki A, Abremski K. The role of the loxP spacer region in P1 site-specific recombination. Nucleic Acids Res. 1986;14:2287–2300. doi: 10.1093/nar/14.5.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]