


Abstract
Translation of mRNA into protein is a unidirectional information flow process. Analysing the input (mRNA) and output (protein) of translation, we find that local protein structure information is encoded in the mRNA nucleotide sequence. The Coding Sequence and Structure (CSandS) database developed in this work provides a detailed mapping between over 4000 solved protein structures and their mRNA. CSandS facilitates a comprehensive analysis of codon usage over many organisms. In assigning translation speed, we find that relative codon usage is less informative than tRNA concentration. For all speed measures, no evidence was found that domain boundaries are enriched with slow codons. In fact, genes seemingly avoid slow codons around structurally defined domain boundaries. Translation speed, however, does decrease at the transition into secondary structure. Codons are identified that have structural preferences significantly different from the amino acid they encode. However, each organism has its own set of ‘significant codons’. Our results support the premise that codons encode more information than merely amino acids and give insight into the role of translation in protein folding.
INTRODUCTION
The ribosome is a unidirectional valve that links mRNA, via protein production and protein folding, to protein structure. Accurately predicting the structure of a protein has been referred to as the ‘holy grail’ of structural bioinformatics (1–3). Without a sequence homologue of solved structure, prediction is of limited accuracy (4) but can be improved by using structural restraints to guide the process (5,6). Current prediction algorithms start with the full amino acid sequence; here we consider that synonymous codon usage may encode structural information and so starting with the mRNA sequence could be beneficial. Additional information derived from synonymous codon usage is well characterized in genetics, e.g. splice site recognition and gene expression (7–9). A link between synonymous codon usage, protein production and protein structure has also been proposed previously (10–12), but only recently has the volume of sequence and structure data made a comprehensive study achievable.
Numerous experiments have indicated that the speed and timings of translation may be critical to the formation of a protein's native structure. For example, Komar et al. (13) demonstrated that the removal of rare codons can reduce the specific activity of chloramphenicol acetyltransferase. Képès (14) identified the ‘+70 pause’ in yeast membrane proteins that is thought to aid their correct insertion into the bilayer. Pausing has been identifed during the translation of photosynthetic reaction center protein D1 (15). Pause sites appear to be located after each transmembrane helix and were suggested to aid co-translational binding of chlorophyll and integration of D1 into the thylakoid membrane. This pausing may be caused by local mRNA structures (16). In vitro experiments have shown that synonymous codon mutations can have a subtle but crucial effect on protein structure and/or function (17–21). These translation effects support the theory of co-translational protein folding and the importance of mRNA sequence and/or structure in protein structure formation.
The rate of translation was recently demonstrated to affect the folding efficiency of Escherichia coli protein SufI. Slow translating regions of the gene were defined as segments rich in codons for which the native cognate tRNA concentration is low. By altering the tRNA concentration or inducing synonymous codon mutations in these regions Zhang et al. (17) significantly perturbed folding efficiency. Folding intermediates predicted from these slow translating regions were identified from translationally synchronized ribosomes in vitro. Further, they showed that slow translating regions are often located upstream of domain boundaries (22). Similar results were presented in the earlier work of Makhoul and Trifonov (23) and of Thanaraj and Argos (10) who observed an enrichment of slow codons around domain boundaries. This, though, was disputed by the work of Brunak and Engelbrecht (24). The slow translation of secondary structure termini in globins has also been demonstrated (25).
In general, computational studies have identified a small subset of codons that display structural preferences significantly diverged from those of the residue they encode. Aligning 109 proteins with their mRNA sequences, Adzhubei (26) showed that codons for leucine (Leu), valine (Val), cysteine (Cys), phenylalanine (Phe) and serine (Ser) have different propensities for the amino(N)- and carboxy(C)-termini of secondary structures. Likewise, codon structural preferences for isoleucine (Ile) and arginine (Arg) have been shown to differ between SCOP (27) domain classes, but this was carried out using fewer than 50 proteins per set (28). Tao and Dafu, found 17 human codons that varied in secondary structure propensity from that of their encoded amino acid. However, only two E. coli codons were identified and it was suggested that codon usage bias is organism dependent (29). Gupta et al. (30) also found no universal correlation in synonymous codon usage between organisms, but identified differences in codon usage between α-helices and β-strands. For example, the proline (Pro) codon CCC is over represented in strand while in helix Pro codons CCA and CCT are most abundant (30). More recently, Zhou et al. linked optimal codons, those with near maximal translation speed, to buried residues.
The only large-scale study above (12) utilized the genomes to protein (GTOP) database that links sequenced genomes to Protein Data Bank (PDB) deposited structures (31) via a PSI-BLAST (32) homology search. Proteins of similar sequence usually fold to very similar structures, but the GTOP link is theoretical, uncurated and not accurate enough for a precise study on synonymous codon usage. The second largest study was carried out by Tao and Dafu using 109 human proteins. The Coding Sequence and Structure (CSandS) database provides an accurate, curated mapping between 4406 protein structures and the mRNA that encodes them. This makes a comprehensive analysis of codon-mediated protein structure effects feasible. Here, data is presented at 40% sequence identity for Ecoli (786 proteins), Human (890) and Yeast (301). We find that many synonymous codons vary in their propensity for protein secondary structures when compared to the amino acid that they encode. Furthermore, some codons have a preference to be buried while others exhibit a propensity to be solvent exposed. In accordance with the literature, no universal set of significant codons was found; with structurally significant codons changing between species. Even between species, though, these significant codons are, in general, restricted to the amino acids: glycine (Gly), Ile, Arg, and threonine (Thr). No link was found between the speed of translation and codons assigned here as structurally significant. Translation speed was assessed using the Codon Adaptation Index (CAI) (33), MinMax (34) and tRNA concentration. We found no evidence of an enrichment in slow codons around domain boundaries. In fact, a general deficiency in slow codons both around and directly upstream of domain boundaries is observed. A decrease in translation speed is, however, found to signal the transition into a secondary structure element.
MATERIALS AND METHODS
Coding sequence and structure database
The CSandS database was compiled through the cross-linking and evaluation of many existing databases. Our starting point was work on a PDB to UniProt (35,36), mapping carried out by the Martin and coworkers (37) and PDBTOSP (www.expasy.ch/cgi-bin/lists?pdbtosp.txt). The extracted protein identifiers were run through the protein identification cross-reference (PICR) (38) service to locate correlated entries in the PDB, TREMBL (39), EMBL (40) and UniProt. Results were ranked on the number of times the match occurred in our data sets and the highest ranked matches were maintained for each protein chain. At this stage, we had 49 941 PDB files, comprising of 152 310 protein chains, linked to mRNA identifiers. The secondary structure assignment and solvent accessibility of each residue in a protein chain was extracted using JOY (41). At this processing stage, 496 chains (<1% of our data set) were lost; these consisted of 316 protein models, 148 with no side chain data and 32 with formatting errors.
All protein coding, mRNA, sequences were downloaded from EMBL on 9 June 2009. Each sequence was run against our protein chain database using BLASTx (32) with an E-value cut-off of e−20 to ensure only high-accuracy matches. In analysing the results, the coding sequence and protein chain were only paired if they came from the same organism (‘matched’). If the PICR web server had also identified this PDB to mRNA link then it was classed as ‘confirmed’. Protein chain to mRNA matches were only maintained if the mRNA sequences had no alignment gaps. Gaps in the protein chain were allowed given that it is not always possible to experimentally resolve every amino acid; however, in all cases the protein chain must cover at least 90% of the mRNA sequence.
CSandS is a 1:1 mapping; however, during development it was redundant. For example, a PDB file containing two identical protein chains would create two ‘confirmed’ hits in our BLASTx analysis. In cases such as this only one hit is maintained.
CSandS contains 4406 protein chains. As CSandS is to investigate the effect of nucleotide sequence on protein structure, the database is made non-redundant at the nucleotide level. That is, we accept similar protein sequences/structures if the mRNA encoding them is significantly different. CD-HIT (42) is used to create a non-redundant database: there remain 4021 and 3151 protein chains at 90 and 40% sequence identity, respectively.
Domain data is added to the database via SCOP (27) (release 1.75) parsable files. In all cases, the mRNA sequence is as provided by EMBL and uses thymine T rather than uracil U. These are protein coding sequences, processed mRNA, and do not relate directly to open reading frames within the genome. The CSands database is freely available at www.stats.ox.ac.uk/bioinfo/resources/.
Relating mRNA to protein structure
Codon secondary structure preference
This measure relates codon usage to protein structure at the residue level without reference to translation speed. We have defined nine secondary structure classes. The three major secondary structures: helix (H), strand (E) and coil (C) are assigned by JOY and each is further classified into three sections (Table 1). These are the start, centre and end of a secondary structure element.
Table 1.
Secondary structure classifications
| Class | Definition | Fragments |
|---|---|---|
| H1 | Start of a helix | CHH or EHH |
| H2 | Within a helix | HHH |
| H3 | End of a helix | HHC or HHE |
| E1 | Start of a strand | CEE or HEE |
| E2 | Within a strand | EEE |
| E3 | End of a strand | EEC or EEH |
| C1 | Start of a coil | HCC or ECC |
| C2 | Within a coil | CCC |
| C3 | End of a coil | CCH or CCE |
The class code is given in column 1 with a longer description in column 2. The secondary structures that qualify for this class are given in column 3, where H is helix, C is coil and E is strand. The central residue/codon of the fragment is assigned to that class.
Through CSandS each codon is mapped to a particular residue and hence a secondary structure assignment. For classification into our secondary structure sets, each codon is considered along with its neighbouring codons; e.g. to assign a secondary structure to codon X the codons X−1, X and X + 1 are used. For example, if the secondary structure pattern HHHCC is encoded by TGCATGTTGCAGAAA then the central codon (TTG which is an H) is classified as H3 as it is in the trio HHC. For each organism, the number of observations of codon Cdn in secondary structure SS is stratified by gene and summed over all genes. Subsequently, two statistical tests of significance are undertaken with the null hypothesis: ‘within a particular family of synonymous codons for amino acid A, the counts in SS are independent of the codon used to encode A’.
Mantel–Haenszel test: we carry out the Mantel–Haenszel (MH) test in an analogous fashion to that described by Zhou et al. (12). For each codon and secondary structure classification, a 2 × 2 contingency table is constructed with the data stratified by gene and synonymous codon family. Thus, for amino acid A that is encoded by codons Cdn1…Cdn3 we create three (the number of codons of A) 2 × 2 tables for each secondary structure classification (e.g. H1) for each gene. Then, using the MH test we can see how, over all genes, the observations of codon Cdn1 diverge from that of Cdn2 and Cdn3.
Chi-squared test: the MH test is stratified by gene and only contingency tables that contain more than two counts are considered. This results in 97 875 counts being ignored (an average of 1659 per codon as Met, Trp and stop codons are excluded). For this reason, we also examine the data as a whole using the chi-squared test. For each codon, the expected number of observations in each secondary structure (CdnexpSS) is calculated and is compared to the observed counts (CdnobsSS) in the following way: (CdnobsSS} − CdnexpSS)2 / CdnexpSS. For each SS the result is summed over all synonymous codons and compared to the chi-distribution using NCdn − 1 degrees of freedom, where NCdn is the number of synonymous codons in the family.
Significance: for both tests, we take significance to be at the 5% level. That is, if the P-value 0.05 or less the null hypothesis is rejected.
Propensity: the MH and chi-squared tests indicate whether a codon is structurally significant within its family of synonymous codons. They do not directly indicate the nature of this significance, e.g. whether the codon is over- or under-represented in the particular secondary structure classification. This can be achieved using the propensity [Equation (1)].
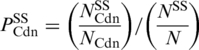 |
(1) |
Codon, Cdn, has propensity, PCdnSS, for a secondary structure, SS. PCdnSS is given by the number of times Cdn is observed in secondary structure SS, NCdnSS divided by the total occurrences of Cdn and NCdn. The result of which is divided by the background distribution: all observations of the secondary structure SS, NSS, over the total number of observations N. Likewise, PASS is the propensity of amino acid A for secondary structure SS. A propensity >1 means that the codon is over-represented in the secondary structure and a propensity <1 indicates that the codon is under-represented.
Codon translation speed
Measures of codon usage
Codon usage is often used to infer the speed of translation (33,34). In this study, we calculate codon usage in two ways. Firstly, via the relative occurrence of synonymous codons within a set of coding sequences (e.g. a genome) and secondly, via the abundance of tRNA with a complementary anti-codon. For the former, we utilize two measures of relative synonymous codon usage: the CAI (33) and MinMax (34). Both measures are organism specific and provide a speed score per codon that relates to the relative abundance of that codon to all other codons encoding the same amino acid (f(Cdn)). Methionine (Met) and tryptophan (Trp) are each encoded by only one codon and so are allocated maximal translation speed. Two sets of coding sequences are used for calculating f(Cdn): all EMBL coding sequences and coding sequences present in the top 5% of expressed genes. For E. coli and Yeast, we also examine the CAI ‘relative adaptiveness’ scores originally calculated by Sharp and Li (33). Their Relative Synonymous Codon Usage (RCSU) values were also tested but did not alter the results.
The tRNA abundance data was only assessed in Ecoli with data taken from Ref (43). In this case, Met and Trp are not necessarily translated at maximal speed .
In all cases, our scores provide an ‘estimate’ of the translation speed and probably do not relate to the actual speed of translation. In many cases, they are measures of codon optimality in anology to the definition provide by Zhou et al. (12). However, for ease of reading they are from here on referred to as the speed of translation.
Codon speed
For all our measures, the translation speed assigned to a codon is the arithmetic mean of codon speeds within a sliding window centred on the codon of interest. We tested windows of size 1, 3, 5, 7,…,19; here data is presented for windows of size 3 (one codon either side considered), 9 and 19.
Gene expression levels
Microarray gene expression data was downloaded from Gene Expression Omnibus (44) on 4 June 2009. Data ‘soft-files’ held gene identifiers that could be linked to EMBL. Gene names were provided for E. coli and these were converted to EMBL codes (via SWISSPROT) using a cross-reference database from cytoscape (accessed on 7 July 2009). For each organism, the top 5% of expressed genes were identified and subsequently used to calculate the relative codon usage. Further details are available in the Supplementary Data.
Measures involving codon translation speed
Codon speed around domain boundaries
We test whether slow codons are more frequent than expected around domain boundaries. Only proteins assigned, by SCOP, as having two domains are considered. The number of qualifying proteins in CSandS is 121, 120 and 51 for Ecoli, Human and Yeast, respectively. The frequency of slow codons within a section of codons centred on the domain boundary is compared to the mean frequency of slow codons in all codon sections of size S. Any section starting within S/2 residues either side of the domain boundary is not considered as it would overlap significantly with the section assigned to the domain boundary. S is tested at 8, 12, 16, 20, 24, 28, 32, 36, 40 and 44 residues. In each case, the domain boundary as assigned in SCOP is at codon position S/2. Slow codons are those with one of the slowest 20 speeds in the mRNA sequence. The translational speed of two codons can be equal and in this way it is common that more than 20 codons throughout the mRNA sequence are assigned as slow. The equivalent test was carried out for fast codons.
Mean translation speed
When comparing the translation speed of different sets of amino acids, the mean translation speed of each set is used. In the examination of secondary structure transitions, the speed of each codon is divided by the mean translation speed of the fragment. Taking the log provides a result centred around zero, where a slower than average codon is negative and a faster than average codon is positive.
Other measures
In the course of this study, other tests were carried out, and a brief description of these and their results can be found in the Supplementary Data.
RESULTS
Throughout the article, our data sets are referred to as Ecoli, Human and Yeast. Where the organisms itself is mentioned we use italics. The CSandS database contains over 4000 protein sequences; their JOY-assigned protein secondary structures; and their corresponding mRNA coding sequence. The database is not limited to particular species but is dominated by three species: E. coli, Human and Yeast. Table 2 provides a breakdown of the database by numbers. The extent of the database has made viable a comprehensive analysis of RNA-coding sequences and their relationship to the corresponding protein structures. With over 4000 detailed mappings, the CSandS database is much larger than other comparable resources. For example, the Integrated Sequence and Structure Database (45) held 105 non-homologous mammalian proteins. It was updated to 279 proteins (46) but has since gone off-line. More recently, a mRNA-mapping to ASTRAL-SCOP was made, with 648 domains linked to their mRNA-coding sequence (47). Only the GTOP database (48) can challenge CSandS in its sequence and structure coverage. GTOP is based on a theoretical, uncurated mapping between assigned gene sequences and PDB deposited structures. The mapping is based on PSI-BLAST-assessed homology, given the understanding that similar sequences share a common 3D conformation (49). However, such a mapping cannot be accurate enough to assign specific start and end points to secondary structures or recognize subtle changes in orientation or solvent accessibility. Additionally, given its reliance on amino acid mappings the GTOP database cannot effectively compare codon usage. Thus, CSandS is currently a unique tool in the mapping of mRNA at the codon level to protein structure at the residue level.
Table 2.
The number of protein chains present for the top six represented organisms in the CSandS database
| Sequence identity cut-off |
|||
|---|---|---|---|
| Organism | 40% | 60% | 90% |
| Human | 890 | 1017 | 1164 |
| E.coli | 786 | 808 | 871 |
| Yeast | 301 | 305 | 322 |
| Mouse | 198 | 219 | 252 |
| Bovine (Bovin) | 137 | 140 | 154 |
| Bacillus subtilis (Bacsu) | 145 | 146 | 148 |
Sequence identity is calculated at the nucleotide level using CD-HIT (42). Organism abbreviations come from UniProtKB.
As mentioned, we use ‘translation speed’ to mean our estimated value of translation speed from CAI, MinMax or tRNA concentration.
Synonymous codons have different secondary structure preferences
‘Significant’ codons are those that have a secondary structure preference that differ significantly from that expected within their synonymous codon family. There is no universal set of significant codons across organisms. It is unusual for the same structural trait to be exhibited even where the same codon shows up as significant in two organisms. For example, on only seven occasions the same structural trait was observed within Ecoli and Human. No codon-specific structural trait-is observed over all three species.
The MH test identifies more structural significances (84 in Ecoli) than the chi-squared test (60 in Ecoli). In general, the tests are in agreement (see Supplementary Data) but in one instance the Chi-squared test identifies a structurally significant codon family (Cys) over-looked by the MH test. This is probably due to the low counts of Cys in individual genes resulting in a large amount of data loss under the MH test, where only tables with greater than two counts are considered. Over the three organisms, only the amino acids Gly, Ile, Arg and Thr all have codons that show significant structural traits under MH. No amino acid's synonymous codon family is significant in all organisms when the chi-squared test is considered. Here, we present some of the structural traits as examples and highlight agreement and contradictions with previous work. Throughout, P-values (P) are derived from the MH test.
In Ecoli, the codon GAA (Glu) is over-represented at the start of helices (H1, P = 0.001; Figure 1), its synonymous codon GAG is under-represented (P = 0.001). CTC (Leu) is also over-represented in H1 (P = ;0.004) but no Leu codon is under-represented in this Ecoli structural category. Human has fewer structurally significant codons (MH = 41, χ = 16). These include, GTT (Val) that is over-represented at the start of helices (H1, P = 0.029) and end of coils (C3, P = 0.0001). TCA (Ser) is over-represented at the start of helices (H1, P = 0.011; Figure 1) and the Ile codon ATT is under-represented at the start of strand (E1, P = 0.0003). Yeast has still fewer codons of structural significance (MH = 12, χ = 8) and this may be due to the smaller number of protein/gene sequences found in CSandS. In the centre of coils (C2), the codon GGA (Gly) is over-represented (P = 0.0007) while GGT (Gly) is under-represented (P = 0.0002). The codons ACA (Thr) and ACG (Thr) are both over-represented at the end of helices (H3) with P-values of 0.009 and 0.03, respectively.
Figure 1.

Examples of codons that are over-represented at the start of helices compared to other codons in their synonymous codon family. In each case the codon of interest is displayed in red and all synonymous codons are shown in blue. Significant codon positioning is highlighted using ball and stick representation. The Glu codon GAA is over-represented at the start of helices in Ecoli (A) and in Human the Ser codon TCA is over-represented at H1 (B). PDB structures 2GFF (A) and 1L9L (B) are used to illustrate the examples. Image created using Chimera (50).
Most of the significant codons we identify result in a propensity change within a secondary structure type rather than a change in the favoured secondary structure. We, unlike Gu et al. (28), found no evidence that CGA (Arg) is over-represented at the termini of helices. Furthermore, there is no evidence in CSandS that CCC (Pro) is over-represented in strand, nor that CCA (Pro) and CCT (Pro) are most abundant in helices as found by Gupta et al. (30). In fact, in Ecoli the Pro codon CCC is found to be over-represented at the start of helices. Adzhubei (26) found that certain codons had a pronounced preference for the N- or C-terminus of secondary structures. Likewise, in CSandS a number of codons are found to be significantly over- or under-represented at secondary structure termini. In Ecoli, 40 codons exhibit opposite propensities for the N- and C-termini of α-helices (Supplementary Data).
The results described above clearly link codons with local secondary structure. It is often hypothesized that these codon effects are manifestations of changes in codon translation speed (10,23,34,51–53). This is supported by the protein structure changes invoked by synonymous codon mutation (17,18,54); however, there is little computational evidence to support this hypothesis. Similarly, we find no link between significant codons and their speed of translation. However, codons that differ most in propensity from their encoded amino acid come primarily from those codons translated most slowly (Supplementary Data).
Codon frequency and tRNA abundance are not correlated
Previous studies investigating a link between codon translational speed and protein structure have used the CAI to assign codon speed, e.g. (10,55). A more recent study used cellular tRNA concentrations (22). We calculate CAI and MinMax from all EMBL coding sequences as well as from highly expressed genes. Relative speeds assigned by CAI and MinMax are correlated (R2 = 0.61–0.93) but do not correlate well with those assigned by tRNA concentration (R2 = 0.03) or the values originally published by Sharp and Li (33) (R2 = 0.13). For details, see Supplementary Data.
Codon usage measures are dependent on the coding sequences used to compile them. The correlation between speeds assigned from highly expressed genes and all coding sequences is thus unexpected. Recent experiments have indicated that codon usage is linked to the cell cycle (56,57). It may be that our highly expressed genes are a representative set covering all stages of the cell cycle. The lack of correlation with the original CAI data of Sharp and Li (33) who used 18 mostly ribosomal proteins supports this conclusion.
The tRNA concentration can respond to changes in cellular conditions (43,58). Changes in tRNA concentration have also been linked to changes in gene expression (57,59) and viral virulence (60,61). The lack of correlation between speeds assigned from tRNA concentration and all other measures is not surprising; this further supports the idea that our measures of codon usage are averaged over all phases of the cell cycle. Results obtained using tRNA concentration, here only available for Ecoli, produced the most informative and robust picture of the effect translational speed has on protein structure. In the rest of this article we focus on these results.
Domain boundaries are not enriched in slow codons
Domains are commonly thought of as structurally stable, individual folding units within the protein. The placement of domain boundaries is based on knowledge of protein structure and folding. However, the particular amino acid assigned to define the domain boundary is in essence an arbitrary selection and varies even between well-regarded databases (62) such as SCOP, CATH (63) and Pfam (64). Domain boundary definitions are also updated and change over time. Here, structurally defined domain boundaries as given by SCOP (release 1.75) are used.
Like Brunak and Engelbrecht (24), we found no evidence that slow codons are clustered around domain boundaries in any of the three organisms in the study. This is true for all different length sections centred on the domain boundary. However, we observe some evidence that domain boundaries avoid slow codons and that they are enriched in fast codons (Figure 2). Equivalent results are produced if CATH defined domain boundaries are used; Pfam definitions produce a slight variance in results for ‘fast’ codons (Supplementary Data). The results may be due to domains in SCOP and CATH being structurally defined while in Pfam they are assigned via sequence consideration.
Figure 2.

Domain boundaries are deficient in slow codons and enriched in fast codons. Data is shown using translation speed calculated from tRNA concentration (A) and the original CAI (B). Solid lines represent fast codons and dotted lines slow codons. Enrichment (Y-axis), the percentage increase over that found in the protein as a whole, is shown for different length sections (X-axis) centred on the domain boundary. For tRNA concentration (A) only Ecoli data is available, with data displayed for all three codon speed windows considered in this study (3, 9 and 19). In (B) data is displayed for Ecoli (black) and Yeast (grey) using a codon speed window of 19.
Recently, taking a small set of proteins, Zhang and coworkers (17,22) demonstrated that slow translating regions are found around domain boundaries. We are able to reproduce figures found in these publications and suggest that translational pausing at domain boundaries is not a general trait; rather that pauses may be incorporated in the nucleotide sequence where required for high-fidelity folding.
When we consider the region immediately downstream from (N-terminal to) the domain boundary our measures of codon translation speed provide different results. Using CAI, an increase in slow codons is observed in Yeast, but not Ecoli, proteins. Using tRNA, no such trend is observed (Supplementary Data). In general, the domain boundary is thought to be less structurally conserved than intra-domain loops and as such less codon selection is perhaps expected.
Translation speed of domains
The translation speed of a set of amino acids is examined by calculating their mean codon translation speed. In general, the first 20 residues of a protein are translated more slowly than the last 20 residues. For Ecoli, this is true for 69% of proteins using a window of size 19. The same trend is observed for MinMax and CAI.
For 121 Ecoli two-domain proteins, the mean translation speed of the first domains (4.07) is similar to the mean translation speed of the second domains (4.17) and the distribution of speeds are similar. However, if the speeds of the two domains that constitute a single protein are compared, 68% of the time the second domain is translated faster. Examining this effect within SCOP classes it is clear that the behaviour varies by SCOP class. The second domain is translated faster in 60, 100, 74 and 50% of cases for two-domain α, β, α/β and α+β Ecoli proteins, respectively. As expected the first 20 residues of domain one are found to be translated more slowly than the first 20 residues in domain two. Thus, when considered with the finding that domain boundaries are enriched in fast codons, a consistent picture is built up of the first domain being translated more slowly than the second domain. Previous research has shown that codon usage is non-random near the point of translation initiation, with an enrichment of non-preferred codons observed (65,66). This may, at least partially, explain our results.
Translation speed of secondary structure transitions
Here the translation speed of secondary structure transitions is explored. Taking the transition from coil into helix as an example, all structure fragments of the form XCCCHHHX, where X is any secondary structure type, are considered. The translation speed of each codon, s, is compared to the mean translation speed of the fragment 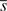 . Taking the logarithm of the relative speed (
. Taking the logarithm of the relative speed (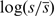 ) produces results centred on zero. The mean translation speed may be sensitive to the number of codons taken to assign the fragment. For this reason, this test is carried out using fragments of length 8 (XCCCHHHX) to 16 (XXXXXCCCHHHXXXXX). No significant differences are observed for the different fragment lengths and in all cases a clear decrease in translation speed on transition from coil to helix is observed (Figure 3A). Error bars based on SD or standard error are not appropriate given the non-normal distribution of speeds over each codon position. Thus, we perform both the Wilcoxon and Kolmogorov–Smirnov tests to assess significance (Supplementary Data). These tests indicate that the distribution of speeds for the transition codon is significantly different from those not adjacent to it. Further, there is no evidence of a difference between codons −5 and −1 or −4 and −3 for example. Given the small fragment sizes, a window of three codons is used to assign translation speed. Data is presented for tRNA concentration; other measures of codon translation speed (CAI and MinMax) do not show a clear signal.
) produces results centred on zero. The mean translation speed may be sensitive to the number of codons taken to assign the fragment. For this reason, this test is carried out using fragments of length 8 (XCCCHHHX) to 16 (XXXXXCCCHHHXXXXX). No significant differences are observed for the different fragment lengths and in all cases a clear decrease in translation speed on transition from coil to helix is observed (Figure 3A). Error bars based on SD or standard error are not appropriate given the non-normal distribution of speeds over each codon position. Thus, we perform both the Wilcoxon and Kolmogorov–Smirnov tests to assess significance (Supplementary Data). These tests indicate that the distribution of speeds for the transition codon is significantly different from those not adjacent to it. Further, there is no evidence of a difference between codons −5 and −1 or −4 and −3 for example. Given the small fragment sizes, a window of three codons is used to assign translation speed. Data is presented for tRNA concentration; other measures of codon translation speed (CAI and MinMax) do not show a clear signal.
Figure 3.

Change in relative translation speed (Y-axis) on the transition between secondary structures (X-axis). In moving from coil into helix (A) or coil into strand (B) a clear decrease in translation speed is observed. The transition from helix into coil (C) or strand into coil (D) is also characterized by a decrease in translation speed. In this instance, around three residues downstream of (N-terminal to) the transition site. This is followed by an increase in translation speed as the coil region is produced. Data shown for Ecoli using a speed window of three codons.
Equivalent tests were carried out for the transition from helix to coil (XHHHCCCX), coil to strand (XCCCEEEX) and strand to coil (XEEECCCX). They all show clear patterns in relative translation speed at the transition point (Figure 3). In general, when starting production of a helix or strand there is a decrease in the translation speed that begins about three codons before the start of the helix. Similarly, the translation speed decreases just before the helix or strand terminates (Figure 3C and D). Notably, on the transition from coil to strand, the translation speed increases immediately before the sharp decrease as strand production begins (Figure 3B). If we assume strand is translated more slowly than coil, this trend may be due to a large number of turns between consecutive β-strands.
The data presented only includes transitions to and from coil, thus transitions directly from strand into helix are excluded. If we consider all transitions into helix, the same general pattern is observed, although the magnitude of the speed distributions is reduced slightly. Generally, the results oppose those of Thanaraj and Argos (55) who found that slow codons have a higher propensity to encode strand and coils. Here, fast codons are a signature of starting production of a coil and slow codons a signal that a transition into helix or strand is imminent. Current secondary structure prediction algorithms are highly accurate; however, predicting the actual termini of secondary structures is still relatively imprecise (67,68). It may be that consideration of codon translation speed could improve secondary structure prediction, particularly in the region of secondary structure transitions.
DISCUSSION
Our newly compiled CSandS database has allowed us to carry out a comprehensive study of mRNA coding sequence data and its relationship to protein structure. In a number of cases, the mRNA is shown to be more informative about the protein structure than the amino acid sequence alone. For example, in Ecoli GAA (Glu) is over-represented at the start of helices (H1, P = 0.001) whereas its synonymous codon GAG is under-represented (P = 0.001).
Results from CSandS can be split into two groups, those that are independent of codon translation speed and those that are not. Most previous studies have identified particular significant codons in a limited set of organisms (28–30,69). Our speed independent measures indicate a more general trend that there is protein structural information contained in the mRNA nucleotide sequence that is not found in the protein primary sequence. Structurally significant codons come predominantly from those amino acids encoded by four or more codons (70% in Ecoli). It is in these sets of synonymous codons that greater differences in translation speed are found. However, no direct link between significant codons and translation speed is elucidated. From analysis of CSandS it is evident that the set of significant codons is not universal. For example, within Ecoli and Human the Gln codon CAA is over-represented in the centre of strands (E2); this is not the case in Yeast. Further, GGT (Gly) is over-represented in E2 in Ecoli but under-represented in E2 in Yeast.
It is hypothesized that the protein structural effects ascribed to mRNA result from changes in translation speed (10,23,34,51–53). Previously, linking these nucleotide-mediated features to translation speed has been difficult. CAI and MinMax scores are related (R2 = 0.6); but neither CAI nor MinMax is correlated to experimental tRNA concentrations (R2 = 0.03). Using tRNA concentration data produces more consistent results and the importance of tRNA concentration has also recently been demonstrated by the work of Romano et al. (70). They showed that folding phase transitions can be successfully modelled using tRNA concentrations.
Opposed to many studies (10,17,22) but in agreement with Brunak and Engelbrecht (24), we find that domain boundaries are not enriched with slow codons. Our study indicates that domain boundaries are deficient in slow codons and show a small enrichment in fast codons. The sequence that connects two domains and contains the domain boundary is often thought to be less structurally constrained than intra-domain loops and this lack of constraint may be linked to its faster translation. An increase in translation speed is also observed when terminating a secondary structure and starting coil production. In fact, secondary structure transitions are, in general, ‘signed’ within the mRNA. For example, slow codons have a higher information content at the start of helices (Supplementary Data) and a relative decrease in translation speed is observed at the point of transition from coil to helix and coil to strand.
This work is one of the largest to date; however, there are still cases where the volume of data is not large enough to draw definite conclusions, e.g domain boundaries. Still, there is no contradiction between our results for domain boundaries and secondary structure transitions; i.e. that domain boundaries are enriched in ‘fast’ codons and the transition into secondary structure is relatively slow. Domain boundaries occur at a secondary structure transition only 24% of the time in our Ecoli two-domain data set. Most domain boundaries occur in coil regions that our results suggest are translated quickly. Moreover, we observe a relative decrease in translation speed rather than an increase in rare codons occurs at the transition into secondary structure.
Zhou et al. demonstrated that buried core residues are likely to be encoded by fast translating codons. Using Ooi number (71) as a measure of residue burial, we found that highly buried residues were on average translated more quickly (Supplementary Data). Codon optimality is not the only possible mechanism by which codon usage could affect the rate of translation. It was recently demonstrated that local mRNA structures near the 5′-end can alter protein expression (65) and in 1985, it was found that the mRNA for highly expressed E. coli proteins contains many more hairpins than a random sequence encoding the same amino acid sequence (51). It was proposed that mRNA hairpins slowed down translation to increase the accuracy of folding. Further, a 2006 study found that three haplotypes of Human ‘Catechol-O-methyltransferase’ (COMT) had different levels of enzyme activity and that COMT activity was correlated to the number of mRNA stem–loops (72). Examining codons observed to significantly increase the likelihood of amino acid burial in Ecoli proteins, we found that these codons destabilized local mRNA structures (Supplementary Data). Though preliminary work, it suggests that the structure of mRNA may be important in the formation of protein structure and is in keeping with the understanding that mRNA secondary structure must be unfolded to enter the ribosome (73).
CONCLUSION
The coverage and detail of the CSandS makes it a unique tool in the examination of links between mRNA and protein. Analysing the data contained in the CSandS database using tRNA concentration as a measure of codon translation speed has produced a consistent picture of how translation can affect local protein structures in Ecoli. Information about protein structure beyond that of the amino acid sequence is contained in the mRNA-coding sequence. We demonstrate that this structural information is species specific and maybe linked to translation speed. N-terminal regions are generally translated slower than C-terminal regions and this could be related to co-translational folding. There is a clear decrease in translation speed at the start of secondary structures in Ecoli, a relationship that could be exploited in the accurate prediction of secondary structure termini.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Oxford University Doctoral Training Centres (Industrial and Systems Biology; in part to C.M.D.). Funding for open access charge: Oxford University.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank both Dr Simon Meyers and Professor Graham Wood for their help and advice on statistical testing. Also, Seb Kelm for multiple readings of this manuscript.
REFERENCES
- 1.Jones DT. Progress in protein structure prediction. Curr. Opin. Struct. Biol. 1997;7:377–387. doi: 10.1016/s0959-440x(97)80055-3. [DOI] [PubMed] [Google Scholar]
- 2.Koehl P, Levitt M. A brighter future for protein structure prediction. Nat. Struct. Mol. Biol. 1999;6:108–111. doi: 10.1038/5794. [DOI] [PubMed] [Google Scholar]
- 3.Moult J. Predicting protein three-dimensional structure. Curr. Opin. Biotechnol. 1999;10:583–588. doi: 10.1016/s0958-1669(99)00037-3. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008;18:342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skolnick J, Kolinski A, Ortiz AR. MONSSTER: a method for folding globular proteins with a small number of distance restraints. J. Mol. Biol. 1997;265:217–241. doi: 10.1006/jmbi.1996.0720. [DOI] [PubMed] [Google Scholar]
- 6.Kolinski A, Skolnick J. Assembly of protein structure from sparse experimental data: an efficient monte carlo model. Proteins. 1998;32:475–494. [PubMed] [Google Scholar]
- 7.Duret L, Mouchiroud D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA. 1999;96:4482–4487. doi: 10.1073/pnas.96.8.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parmley JL, Hurst LD. Exonic splicing regulatory elements skew synonymous codon usage near intron-exon boundaries in mammals. Mol. Biol. Evol. 2007;24:1600–1603. doi: 10.1093/molbev/msm104. [DOI] [PubMed] [Google Scholar]
- 9.Chamary J-V, Hurst LD. Biased codon usage near intronexon junctions: selection on splicing enhancers, splice-site recognition or something else? Trends Genet. 2005;21:256–259. doi: 10.1016/j.tig.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Thanaraj TA, Argos P. Ribosome-mediated translational pause and protein domain organization. Protein Sci. 1996;5:1594–1612. doi: 10.1002/pro.5560050814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biro JC. Indications that “codon boundaries” are physicochemically defined and that protein-folding information is contained in the redundant exon bases. Theor. Biol. Med. Model. 2006;3:28. doi: 10.1186/1742-4682-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou T, Weems M, Wilke CO. Translationally optimal codons associate with structurally sensitive sites in proteins. Mol. Biol. Evol. 2009;26:1571–1580. doi: 10.1093/molbev/msp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komar AA, Lesnik T, Reiss C. Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett. 1999;462:387–391. doi: 10.1016/s0014-5793(99)01566-5. [DOI] [PubMed] [Google Scholar]
- 14.Kepes F. The “+70 pause”: hypothesis of a translational control of membrane protein assembly. J. Mol. Biol. 1996;262:77–86. doi: 10.1006/jmbi.1996.0500. [DOI] [PubMed] [Google Scholar]
- 15.Kim J, Klein PG, Mullet JE. Ribosomes pause at specific sites during synthesis of membrane-bound chloroplast reaction center protein D1. J. Biol. Chem. 1991;266:14931–14938. [PubMed] [Google Scholar]
- 16.Zama M. Discontinuous translation and mRNA secondary structure. Nucleic Acids Symp. Ser. 1995;35:97–98. [PubMed] [Google Scholar]
- 17.Zhang G, Hubalewska M, Ignatova Z. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol. 2009;16:274–280. doi: 10.1038/nsmb.1554. [DOI] [PubMed] [Google Scholar]
- 18.Hamano T, Matsuo K, Hibi Y, Victoriano AF, Takahashi N, Mabuchi Y, Soji T, Irie S, Sawanpanyalert P, Yanai H, et al. A single-nucleotide synonymous mutation in the gag gene controlling human immunodeficiency virus type 1 virion production. J. Virol. 2007;81:1528–1533. doi: 10.1128/JVI.01596-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 20.Komar AA. Genetics. SNPS, silent but not invisible. Science. 2007;315:466–467. doi: 10.1126/science.1138239. [DOI] [PubMed] [Google Scholar]
- 21.Cortazzo P, Cervenansky C, Marin M, Reiss C, Ehrlich R, Deana A. Silent mutations affect in vivo protein folding in Escherichia coli. Biochem. Biophys. Res. Commun. 2002;293:537–541. doi: 10.1016/S0006-291X(02)00226-7. [DOI] [PubMed] [Google Scholar]
- 22.Zhang G, Ignatova Z. Generic algorithm to predict the speed of translational elongation: implications for protein biogenesis. PLoS ONE. 2009;4:e5036. doi: 10.1371/journal.pone.0005036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makhoul CH, Trifonov EN. Distribution of rare triplets along mRNA and their relation to protein folding. J. Biomol. Struct. Dyn. 2002;20:413–420. doi: 10.1080/07391102.2002.10506859. [DOI] [PubMed] [Google Scholar]
- 24.Brunak S, Engelbrecht J. Protein structure and the sequential structure of mRNA: alpha-helix and beta-sheet signals at the nucleotide level. Proteins. 1996;25:237–252. doi: 10.1002/(SICI)1097-0134(199606)25:2<237::AID-PROT9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 25.Krasheninnikov IA, Komar AA, Adzhubei IA. Nonuniform size distribution of nascent globin peptides, evidence for pause localization sites, and a contranslational protein-folding model. J. Protein Chem. 1991;10:445–453. doi: 10.1007/BF01025472. [DOI] [PubMed] [Google Scholar]
- 26.Adzhubei AA. Non-random usage of degenerate codons is related to protein three-dimensional structure. FEBS Lett. 1996;399:78–82. doi: 10.1016/s0014-5793(96)01287-2. [DOI] [PubMed] [Google Scholar]
- 27.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 28.Gu W, Sun X, Lu Z. Folding type specific secondary structure propensities of synonymous codons folding type specific secondary structure propensities of synonymous codons. IEEE Transactions Nanobioscience. 2003;2:150–157. doi: 10.1109/tnb.2003.817024. [DOI] [PubMed] [Google Scholar]
- 29.Xie T, Ding D, Tao X, Dafu D. The relationship between synonymous codon usage and protein structure. FEBS Lett. 1998;434:93–96. doi: 10.1016/s0014-5793(98)00955-7. [DOI] [PubMed] [Google Scholar]
- 30.Gupta SK, Majumdar S, Bhattacharya TK, Ghosh TC. Studies on the relationships between the synonymous codon usage and protein secondary structural units. Biochem. Biophys. Res. Commun. 2000;269:692–696. doi: 10.1006/bbrc.2000.2351. [DOI] [PubMed] [Google Scholar]
- 31.Bernstein FC, Koetzle TF, Williams GJ, Jr, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 32.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharp PM, Li WH. The codon adaptation index.a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clarke TF, Clark PL. Rare codons cluster. PLoS ONE. 2008;3:e3412. doi: 10.1371/journal.pone.0003412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.UniProt Consortium. The universal protein resource (UniProt) Nucleic Acids Res. 2007;35:D193–D197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.UniProt Consortium. The universal protein resource (uniprot) 2009. Nucleic Acids Res. 2009;37:D169–D174. doi: 10.1093/nar/gkn664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin AC. Mapping PDB chains to UniProtKB entries. Bioinformatics. 2005;21:4297–4301. doi: 10.1093/bioinformatics/bti694. [DOI] [PubMed] [Google Scholar]
- 38.Côté RG, Jones P, Martens L, Kerrien S, Reisinger F, Lin Q, Leinonen R, Apweiler R, Hermjakob H. The protein identifier cross-referencing (PICR) service: reconciling protein identifiers across multiple source databases. BMC bioinformatics. 2007;8:401. doi: 10.1186/1471-2105-8-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bairoch A, Apweiler R. The SWISS-PROT protein sequence data bank and its supplement TREMBL. Nucleic Acids Res. 1997;25:31–36. doi: 10.1093/nar/25.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamm GH, Cameron GN. The EMBL data library. Nucleic Acids Res. 1986;14:5–9. doi: 10.1093/nar/14.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizuguchi K, Deane CM, Blundell TL, Johnson MS, Overington JP. JOY: protein sequence-structure representation and analysis. Bioinformatics. 1998;14:617–623. doi: 10.1093/bioinformatics/14.7.617. [DOI] [PubMed] [Google Scholar]
- 42.Li W, Godzik A. CD-HIT: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 43.Dong H. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996;260:649–663. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- 44.Barrett T, Edgar R. Gene expression omnibus: microarray data storage, submission, retrieval, and analysis. Meth. Enzymo. 2006;411:352–369. doi: 10.1016/S0076-6879(06)11019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adzhubei IA, Adzhubei AA, Neidle S. An integrated sequence-structure database incorporating matching mRNA sequence, amino acid sequence and protein three-dimensional structure data. Nucleic Acids Res. 1998;26:327–331. doi: 10.1093/nar/26.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adzhubei IA, Adzhubei AA. ISSD version 2.0: taxonomic range extended. Nucleic Acids Res. 1999;27:268–271. doi: 10.1093/nar/27.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jia M, Luo L, Liu C. Statistical correlation between protein secondary structure and messenger RNA stem-loop structure. Biopolymers. 2004;73:16–26. doi: 10.1002/bip.10496. [DOI] [PubMed] [Google Scholar]
- 48.Kawabata T, Fukuchi S, Homma K, Ota M, Araki J, Ito T, Ichiyoshi N, Nishikawa K. GTOP: a database of protein structures predicted from genome sequences. Nucleic Acids Res. 2002;30:294–298. doi: 10.1093/nar/30.1.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- 50.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 51.Shpaer EG. The secondary structure of mRNAs from Escherichia coli: its possible role in increasing the accuracy of translation. Nucleic Acids Res. 1985;13:275–288. doi: 10.1093/nar/13.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Komar AAA. A pause for thought along the co-translational folding pathway. Trends Biochem. Sci. 2008;34:16–24. doi: 10.1016/j.tibs.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 53.Marin M. Folding at the rhythm of the rare codon beat. Biotechnol. J. 2008;3:1047–1057. doi: 10.1002/biot.200800089. [DOI] [PubMed] [Google Scholar]
- 54.Crombie T, Boyle JP, Coggins JR, Brown AJ. The folding of the bifunctional TRP3 protein in yeast is influenced by a translational pause which lies in a region of structural divergence with Escherichia coli indoleglycerol-phosphate synthase. Eur. J. Biochem. / FEBS. 1994;226:657–664. doi: 10.1111/j.1432-1033.1994.tb20093.x. [DOI] [PubMed] [Google Scholar]
- 55.Thanaraj TA, Argos P. Protein secondary structural types are differentially coded on messenger RNA. Protein Sci. 1996;5:1973–1983. doi: 10.1002/pro.5560051003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Najafabadi HSS, Goodarzi H, Salavati R. Universal function-specificity of codon usage. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkp792. doi:10.1093/nar/gkp792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MRR, Pan T. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009;37:7268–7280. doi: 10.1093/nar/gkp787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kanduc D. Changes of tRNA population during compensatory cell proliferation: differential expression of methionine-tRNA species. Arch. Biochem. Bioph. 1997;342:1–5. doi: 10.1006/abbi.1996.9869. [DOI] [PubMed] [Google Scholar]
- 59.García-Contreras R, Zhang X-SS, Kim Y, Wood TK. Protein translation and cell death: the role of rare tRNAs in biofilm formation and in activating dormant phage killer genes. PLoS ONE. 2008;3:e2394. doi: 10.1371/journal.pone.0002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gu W, Li M, Zhao WMM, Fang NXX, Bu S, Frazer IH, Zhao K-NN. tRNASer(CGA) differentially regulates expression of wild-type and codon-modified papillomavirus l1 genes. Nucleic Acids Res. 2004;32:4448–4461. doi: 10.1093/nar/gkh748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bailly-Bechet M, Vergassola M, Rocha E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007;17:1486–1495. doi: 10.1101/gr.6649807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tai C-HH, Lee W-JJ, Vincent JJ, Lee B. Evaluation of domain prediction in CASP6. Proteins. 2005;61(Suppl. 7):183–192. doi: 10.1002/prot.20736. [DOI] [PubMed] [Google Scholar]
- 63.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH–a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 64.Sonnhammer EL, Eddy SR, Durbin R. Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins. 1997;28:405–420. doi: 10.1002/(sici)1097-0134(199707)28:3<405::aid-prot10>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 65.Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gu W, Zhou T, Wilke CO. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput. Biol. 2010;6:e1000664. doi: 10.1371/journal.pcbi.1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilson CL, Hubbard SJ, Doig AJ. A critical assessment of the secondary structure alpha-helices and their termini in proteins. Protein Eng. 2002;15:545–554. doi: 10.1093/protein/15.7.545. [DOI] [PubMed] [Google Scholar]
- 68.Wilson CL, Boardman PE, Doig AJ, Hubbard SJ. Improved prediction for N-termini of alpha-helices using empirical information. Proteins. 2004;57:322–330. doi: 10.1002/prot.20218. [DOI] [PubMed] [Google Scholar]
- 69.Oresic M, Dehn M, Korenblum D, Shalloway D. Tracing specific synonymous codon-secondary structure correlations through evolution. J. Mol. Evol. 2003;56:473–484. doi: 10.1007/s00239-002-2418-x. [DOI] [PubMed] [Google Scholar]
- 70.Romano MC, Thiel M, Stansfield I, Grebogi C. Queueing phase transition: theory of translation. Phys. Rev. Lett. 2009;102:198104. doi: 10.1103/PhysRevLett.102.198104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nishikawa K, Ooi T. Prediction of the surface-interior diagram of globular proteins by an empirical method. Int. J. Pept. Protein Res. 1980;16:19–32. doi: 10.1111/j.1399-3011.1980.tb02931.x. [DOI] [PubMed] [Google Scholar]
- 72.Nackley AG, Shabalina SA, Tchivileva IE, Satterfield K, Korchynskyi O, Makarov SS, Maixner W, Diatchenko L. Human catechol-o-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science. 2006;314:1930–1933. doi: 10.1126/science.1131262. [DOI] [PubMed] [Google Scholar]
- 73.Marzi S, Myasnikov AG, Serganov A, Ehresmann C, Romby P, Yusupov M, Klaholz BP. Structured mRNAs regulate translation initiation by binding to the platform of the ribosome. Cell. 2007;130:1019–1031. doi: 10.1016/j.cell.2007.07.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.