


Abstract
Protein–DNA bindings between transcription factors (TFs) and transcription factor binding sites (TFBSs) play an essential role in transcriptional regulation. Over the past decades, significant efforts have been made to study the principles for protein–DNA bindings. However, it is considered that there are no simple one-to-one rules between amino acids and nucleotides. Many methods impose complicated features beyond sequence patterns. Protein-DNA bindings are formed from associated amino acid and nucleotide sequence pairs, which determine many functional characteristics. Therefore, it is desirable to investigate associated sequence patterns between TFs and TFBSs. With increasing computational power, availability of massive experimental databases on DNA and proteins, and mature data mining techniques, we propose a framework to discover associated TF–TFBS binding sequence patterns in the most explicit and interpretable form from TRANSFAC. The framework is based on association rule mining with Apriori algorithm. The patterns found are evaluated by quantitative measurements at several levels on TRANSFAC. With further independent verifications from literatures, Protein Data Bank and homology modeling, there are strong evidences that the patterns discovered reveal real TF–TFBS bindings across different TFs and TFBSs, which can drive for further knowledge to better understand TF–TFBS bindings.
INTRODUCTION
We first introduce protein–DNA bindings in this section. Existing bioinformatics methods are briefly described, followed by the layout of this article.
Protein–DNA binding
Protein–DNA binding plays a central role in genetic activities such as transcription, packaging, rearrangement, and replication (1,2). Therefor, it is very important to identify and understand the protein–DNA bindings as the basis for further deciphering biological systems. We focus on protein–DNA bindings between transcription factors (TFs) and transcription factor binding sites (TFBSs), which are the primary regulatory activities with abundant data support. TFs bind in a sequence-specific manner to TFBSs to regulate gene transcription (gene expression). The DNA binding domain(s) of a TF can recognize and bind to a collection of similar TFBSs, from which a conserved pattern called motif can be obtained. TFBSs, the nucleotide fragments bound by TFs, are usually short (usually about 5–20 bp) in the cis-regulatory/intergenic regions and can assume very different locations from the transcription start site.
It is expensive and laborious to experimentally identify TF–TFBS binding sequence pairs, for example, using DNA footprinting (3) or gel electrophoresis (4). The technology of chromatin immunoprecipitation (ChIP) (5,6) measures the binding of a particular TF to DNA of co-regulated genes on a genome-wide scale in vivo, but at low resolution. Further processing are needed to extract precise TFBSs (7). TRANSFAC (8) is one of the largest and most representative databases for regulatory elements including TFs, TFBSs, nucleotide distribution matrices of the TFBSs and regulated genes. The data are expertly annotated and manually curated from peer-reviewed and experimentally proved publications. Other annotation databases of TF families and binding domains are also available [e.g. PROSITE (9), Pfam (10)]. It is even more difficult and time-consuming to extract high-resolution 3D TF–TFBS complex structures with X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopic analysis. Nevertheless, the high-quality TF–TFBS binding structures provide valuable insights into verifications of putative principles of binding. The Protein Data Bank (PDB) (11) serves as a representative repository of such experimentally extracted protein–DNA (in particular TF–TFBS) complexes with high resolution at atomic levels. However, the available 3D structures are far from complete. As a result, there is strong motivation to have automatic methods, particularly, computational approaches based on existing abundant data, to provide testable candidates of novel TF domains and/or TFBS motifs with high confidence to guide and accelerate the wet-lab experiments.
Existing methods
The first attempt of computational methods related to TF–TFBS bindings was to discover the motifs of TF domains and TFBSs separately. Moreover, researchers have been trying hard to generalize the one-to-one binding codes from existing 3D structures. Data mining methods have also been proposed with feature transformations and machine learning to decipher complicated binding rules. They are briefly described as follows:
Motif discovery
TF domains and TFBSs sequences are somewhat conserved due to their functional similarity and importance. By exploiting conservation in the sequences, Bioinformatics methods called motif discovery save some of the expensive and laborious laboratory experiments. Motif discovery (6) can be categorized into two types: (i) motif matching and (ii) de novo motif discovery. (i) Motif matching is to identify putative TF domains (9,10) or TFBSs (12) based on motif knowledge obtained from annotated data. (ii) de novo motif discovery predicts conserved patterns without knowledge on their appearances, based on certain motif models and scoring functions (13,14) from a set of protein/DNA promoter sequences with similar regulatory functions. While de novo motif discovery is successful for well-conserved TF functional domain motifs, the counterpart for TFBSs remains very challenging with poor performances on real benchmarks (6,15,16). A significant limitation of motif discovery is the lack of linkage between the binding counterparts for revealing TF–TFBS relationships.
One-to-one binding codes
Numerous studies have been carried out to analyze existing protein–DNA binding 3D structures comprehensively (2,17,18) or with focus on specific families (1) [e.g. zinc fingers (19)]. Various properties have been discovered concerning, e.g. bonding and force types, TF conservation and mutation (1), and bending of the DNA (17). Some are already applicable to predict binding amino acids on the TF side (20). However, annotated data are far from complete. Alternatively, researchers have sought hard for general binding ‘codes' between proteins and DNA, in particular the one-to-one mapping between amino acids from TFs and nucleotides from TFBSs. Despite many proposed one-one binding propensity mappings (1,21,22), it has come to a consensus that there are no simple binding ‘codes' (23).
Data mining
In the hope of better understanding for protein–DNA bindings, many data mining approaches have also been proposed (24). Researchers employ and transfer additional detailed information such as base compositions, structures, thermodynamic properties (25,26) as well as expressions (27), into sophisticated features to fit into certain data mining techniques. Although some approaches may provide interpretable rules, most of them have stringent data requirements which cannot be obtained trivially. Existing data beyond sequences are also insufficient and limited for practitioners. These methods usually extract complicated features rather than working on interpretable data directly. Many data mining techniques, such as neural networks, support vector machines (SVM) (28) and regressions (24), may generate rules which are not trivial to interpret. Furthermore, many data mining approaches are based on specific families or particular data sets, where the generality of the results are limited. On the other hand, sequences serve as the most handy primary data that carry important information for protein–DNA bindings (23). It is desirable to make use of the large-scale and comprehensive sequence data to mine explicit and interpretable TF–TFBS binding rules.
Article layout
In this article, we propose a framework based on association rule mining to discover protein–DNA binding sequence patterns from TRANSFAC. The article layout is as follows: the proposed methods are presented in the next section: ‘Materials and Methods' section; experimental results and verifications are reported in sections ‘Results and Analysis' and ‘Verifications' section, respectively; and finally we have the ‘Discussion' section for the approach.
MATERIALS AND METHODS
In this section, we propose a framework for mining, discovering and verifying TF–TFBS bindings on large-scale databases. The framework starts from data cleansing and transformation on TRANSFAC, and then applies association rule mining to discover TF-TFBS binding sequence patterns. Comprehensive 3D verifications and evaluations are carried out on PDB. Detailed bonding analysis is performed to provide strong support to the discovered rules.
In the following subsections, Apriori algorithm for association rule mining is first introduced. We then elaborate how the algorithm is applied to protein–DNA binding pattern discovery. Finally, we present how the data are preprocessed for the task with a running example.
Association rule mining and Apriori Algorithm
Association rule mining (29) aims at discovering frequently co-occurring items, called frequent itemsets, from a large number of data samples above a certain count threshold (minimum support) (30). The support of an itemset is defined as the number of data samples where all the items in the itemset co-occur. In the case of protein–DNA binding, the binding domains of TFs can recognize and form strong bondings with certain sequence-specific patterns of the TFBSs. Therefore, they are likely to co-occur frequently among the combinations between all possible TF and TFBS subsequences, and can be thus identified by association rule mining. In this study, we use the notation of k-mer (a subsequence with k amino acid or nucleotide residues) to represent a candidate item. A frequent TF–TFBS itemset is a TF k-mer and TFBS k-mer (the two k's can be different) pair, or simply a pair, co-occurring with a frequency no less than the minimum support in the TF–TFBS sequence records (TRANSFAC database).
Apriori algorithm proposed by Agrawal et al. (29) is a classical approach to find out frequent itemsets. It is outlined in Algorithm 1 in the Appendix 1. It is a branch and bound algorithm for discovering association rules in a database. With its downward closure property, an optimal performance is guaranteed. The algorithm first obtains frequent 1-itemsets. Iteratively, it uses the frequent n-itemsets (itemsets with n items) to generate all possible candidate (n+1)-itemsets. They are then evaluated for their supports (30). If the support of an (n+1)-itemset is lower than a threshold, the (n+1)-itemset is removed. After the removal, the resultant (n+1)-itemsets are the frequent (n+1)-itemsets. The above procedure is repeated until an empty set is found.
Discovering associated TF–TFBS sequence patterns
To formulate the TF–TFBS sequence pattern discovery problem into association rule mining, we have to transform the protein–DNA binding records into the formats of itemsets (k-mers). An illustrative example for the TF–TFBS binding records from TRANSFAC 2008.3 is shown in Figure 1. The TF (e.g. T01333 RXR-γ) can bind to several TFBS DNA sequences. The DNA sequences may be different in lengths due to experimental methods and noises. Both the TF and TFBS sequences are chopped into overlapping short k-mers, as illustrated in Figure 2 (first part). They together with the corresponding reverse complements (e.g. GACCT and reverse complement: AGGTC) form one data sample. To generate the itemsets, all the k-mers are recorded in a binary array where appearing k-mers are marked 1; and 0 otherwise. Thus, the length of the array depends on the number of all possible TF k-mers and TFBS k-mers (Figure 2, second part). Since k is usually short (4–6), all the possible 4k combinations of TFBS DNA k-mers can be adopted. However, it is computationally infeasible to obtain all the possible 20k combinations of TF k-mers. Thus a data-driven approach is employed by scanning the whole TRANSFAC to obtain frequent TF amino acid k-mers.
Figure 1.

TFBS sequences of a TF (TRANSFAC 2008.3 ID: T01333).
Figure 2.

Flowchart of the proposed framework to discover association rules from TRANSFAC.
Since there are multiple TFBSs for each TF (e.g. Figure 1), a question arises: how to define the ‘commonly found' TFBS k-mers of a TF? Without loss of generality, the majority rule (31) is applied. If the majority of a TF's TFBS sequences contains a certain DNA residue k-mer, then the k-mer is considered ‘commonly found'. We set the majority to be 50% for TFBS k-mers. We only count the number of TFBS sequences in which a certain k-mer appears, in order not to be biased by multiple occurrences of the k-mer appearing in only a few TFBS sequences. Figure 1 illustrates an example where there are five TFBS sequences. The TFBS DNA k-mer AGGTC (or its reverse complement: GACCT) can be found in three of the TFBS sequences. The k-mer appears in 60% (3/5) of the TFBS sequences of the TF, and thus is considered ‘commonly found'. On the other hand, GTTCA is not considered ‘commonly found' because it only appears in 2 (40%) out of the 5 TFBS sequences of the TF.
After all valid TF data samples are transformed into itemsets, Apriori algorithm is applied to generate frequent TF–TFBS k-mer sequence patterns (the links in Figure 2, second part). The special feature in this study is that the co-occurring pairs should contain both TF and TFBS k-mer items, as illustrated in the third part of Figure 2. In the current study, we only consider one TF k-mer with one TFBS k-mer in the frequent itemsets, but it is straightforward to generalize it to be multiple TF and TFBS k-mers in principle. The huge computational intensity for the generalization, when applied on the large TRANSFAC database, prevents us from doing so at this time. Finally, the association rules are computed based on the confidence measurements for the frequent itemsets, which are defined as follows:
where conf(k-merDNA ⇒ k-merAA) is called forward confidence, conf(k-merDNA ⇐ k-merAA) is called backward confidence and support(X) is the support of itemset X. For each association rule, its forward confidence measures the posterior probability that the corresponding amino acid k-mer can be found in a TF's sequence if the DNA k-mer is commonly found in the TF's TFBS sequences. Its backward confidence measures the posterior probability that the corresponding DNA k-mer can be commonly found in a TF's TFBS sequences if the amino acid k-mer is found in the TF's sequence. The minimum of them is taken as confidence in this article. The higher the confidence, the better the association rule is (Figure 2, fourth part). The whole proposed approach is summarized in Figure 2.
Data preparation
To apply the methodology on TRANSFAC, TF and TFBS data were downloaded and extracted from the flat files of TRANSFAC 2008.3 [a free public (older) version is also available (http://www.gene-regulation.com/pub/database.html)]. The entries without sequence data were discarded. Since a TF can bind to one or more TFBSs, TFBS data were grouped by TF. TFBS sequences were extracted for each TF to form a TF data set—a TF sequence and the corresponding TFBS sequences—and finally to be transformed into itemsets. To avoid sampling error, TF data sets with less than five TFBS sequences were discarded. Furthermore, the redundancy of TF sequences was removed by BLASTClust using 90% TF sequence identity (32). Only one TF data set was selected for each cluster. Note that we only used sequence data in TRANSFAC. None of the prior information (e.g. the binding domains of TFs) other than sequences was used. Importantly, it turns out that the results of the proposed approach can be verified by annotations, 3D structures from PDB and even homology modeling as described in the subsequent sections.
After data preparation, the 631 TF data sets (listed in Table 5 in the Appendix 1) were selected. The minimum support (30) was set to seven TF data sets to avoid sampling error. For the values of k, we try 4–6 for both TF k-mers and TFBS k-mers, resulting in 9 (3 × 3) different combinations. In particular, 256 DNA 4-mers, 1024 DNA 5-mers and 4096 DNA 6-mers were adopted for TFBS, whereas 99 621 amino acid 4-mers, 82 561 amino acid 5-mers, and 39 320 amino acid 6-mers were adopted for TF, as the frequent 1-itemsets.
Table 5.
631 TRANSFAC 2008.3 IDs and factor names used in this article
| ID | Factor name | ID | Factor name | ID | Factor name | ID | Factor name | ID | Factor name | ID | Factor name |
|---|---|---|---|---|---|---|---|---|---|---|---|
| T00003 | AS-CT3 | T00842 | Tra-1(long form) | T01950 | HNF-1α-B | T04378 | Mad | T08676 | STAT6 | T09986 | NF-AT4 |
| T00008 | Adf-1 | T00843 | Ttk69K | T01951 | HNF-1α-C | T04446 | Nkx5-1 | T08787 | ARF1isoform-1 | T09990 | CDP-isoform1 |
| T00011 | ADR1 | T00851 | T3R-β1 | T01973 | REST-form2 | T04539 | RPN4 | T08797 | MCB1 | T10028 | SREBP-1c |
| T00019 | AhR | T00863 | Ubx | T01992 | Abd-A | T04610 | SXR | T08805 | WRKY1 | T10030 | POU3F2 |
| T00026 | Antp | T00886 | v-ErbA | T02003 | Cdx-3 | T04651 | ER-β | T08823 | E2F | T10059 | GCMa |
| T00028 | YAP1 | T00891 | HNF-1β-A | T02008 | Ems | T04665 | Xvent-1 | T08853 | myogenin | T10068 | COUP-TF2 |
| T00033 | AP-2α | T00893 | v-Jun | T02030 | Sd | T04674 | IRF-7A | T08858 | REVERB-α | T10083 | HNF-3α |
| T00063 | Bcd | T00894 | Vmw65 | T02033 | HsfA1 | T04675 | MRF-2- isoform1 | T08863 | S8 | T10144 | Gfi1b |
| T00077 | CACCC- binding factor | T00895 | v-Myb | T02039 | HAC1 | T04679 | dri | T08868 | CTCF | T10187 | NF-E2p45 |
| T00079 | Cad | T00899 | WT1 | T02050 | Nkx6-2 | T04728 | CDC5L | T08878 | Opaque-2 | T10207 | GATA-6 |
| T00080 | CBF1 | T00910 | YB-1 | T02054 | HOX11 | T04733 | Alfin1 | T08972 | EAR2 | T10209 | Nkx2-1 |
| T00104 | C/EBPα | T00915 | YY1 | T02063 | KNOX3 | T04734 | Topors- isoform1 | T08978 | Dl-A | T10211 | Evi-1 |
| T00106 | C/EBP | T00917 | Zen-1 | T02068 | PU.1 | T04783 | mtTFA | T08985 | Pti4 | T10265 | LRH-1 |
| T00109 | C/EBPδ | T00918 | Zeste | T02099 | Zen-2 | T04784 | PF1 | T08989 | Fra-1 | T10276 | Erm |
| T00112 | c-Ets-1 | T00923 | Zta | T02100 | Zeste | T04811 | FOXP1a | T08994 | HIF-1α- isoform1 | T10282 | Otx2 |
| T00113 | c-Ets-2 | T00925 | AMT1 | T02128 | SAP-1b | T04817 | LIM1 | T09001 | BPC1 | T10317 | IA-1 |
| T00115 | c-Ets-168 | T00937 | HBP-1a | T02142 | OCA-B | T04819 | EmBP-1a | T09018 | N-Myc | T10331 | NRF-1 |
| T00117 | CF1 | T00938 | HBP-1b | T02216 | TFIIA-α/β precursor (major) | T04886 | Tel-2b | T09033 | TEF-1 | T10392 | GATA-3 |
| T00120 | CF2-II | T00969 | POU3F1 | T02217 | TFIIA-α/β precursor (minor) | T04931 | p73α | T09051 | AhR | T10393 | GATA-2 |
| T00128 | HOXA4 | T01005 | MEF2A- isoform1 | T02235 | PEBP2αB1 | T04957 | EKLF | T09059 | SEF2-1B | T10429 | PU.1 |
| T00140 | c-Myc | T01017 | CRE-BP2 | T02248 | StuAp | T04961 | GLI2α | T09071 | AG | T10459 | Alx-3 |
| T00151 | CP2a | T01019 | Elf-1 | T02256 | AML1a | T04996 | ZBP89 | T09089 | PIF3 | T10462 | Prop-1 |
| T00163 | CREB | T01027 | BAS1 | T02288 | HFH-1 | T04998 | Tel-2a | T09093 | IPF1 | T10473 | TEF-5 |
| T00167 | ATF-2-xbb4 | T01035 | Isl-1α | T02290 | FOXD3 | T04999 | Tel-2c | T09097 | SRY | T10482 | AP-2γ |
| T00176 | CTF-1 | T01051 | FOXA4a | T02291 | Croc | T05021 | NERF-1a | T09098 | SREBP-2 | T10484 | TEF-3 |
| T00177 | CTF-2 | T01053 | HNF-3β | T02294 | FOXI1a | T05051 | BTEB3 | T09102 | FOXO4 | T10543 | Sox5 |
| T00179 | CUP2 | T01059 | MNB1a | T02302 | GCM | T05137 | CIZ6-1 | T09106 | RelA-p65 | T10573 | DREB1A |
| T00183 | DBP | T01072 | TEF | T02313 | MIBP1 | T05181 | DSF | T09117 | E2F-1 | T10588 | Snai3 |
| T00193 | Dfd | T01074 | Ap | T02330 | G/HBF-1 | T05553 | MYBAS1 | T09129 | BCL-6 | T10638 | HY5 |
| T00204 | E12 | T01078 | GBF1 | T02361 | CREBβ | T05587 | BZI-1 | T09156 | TGIF-isoform2 | T10644 | MTF-1 |
| T00208 | E74A | T01083 | NF-μNR | T02378 | USF1 | T05682 | ERRα1 | T09158 | BZR1 | T10664 | Gfi1 |
| T00217 | EcR | T01085 | abaA | T02419 | Sp3 | T05705 | GATA-1 | T09159 | PITX2A | T10666 | SRY |
| T00253 | En | T01109 | TCF-1(P) | T02420 | Sox13 | T05706 | GATA-2 | T09162 | Pax-3 | T10674 | MafK |
| T00262 | ER-α | T01112 | EBF1-L | T02422 | HNF-4α2 | T05707 | GATA-3 | T09177 | MyoD | T10712 | DMRT1 |
| T00264 | ER-α | T01147 | SF-1isoform2 | T02429 | HNF-4α1 | T05708 | GATA-4 | T09178 | C/EBPα | T10720 | GCR1 |
| T00272 | Eve | T01152 | T3R-α1 | T02463 | GBF1 | T05737 | PCF3 | T09182 | Pax-5 | T10721 | DMRT2 |
| T00295 | Ftz | T01154 | c-Rel | T02469 | AP-2β | T05743 | ABI4 | T09183 | WRKY53 | T10723 | DMRT3 |
| T00296 | FTZ-F1 | T01258 | MSN4 | T02529 | PPARγ1 | T05770 | DREB1A | T09184 | Pax-8 | T10725 | DMRT7 |
| T00301 | GAGA factor | T01265 | MAC1 | T02636 | CBF1 | T05834 | CBF2 | T09190 | AGL15 | T10727 | DMRT4 |
| T00302 | GAL4 | T01274 | ABF2 | T02639 | ANT | T05835 | DRF1.1 | T09194 | NF-AT1C | T10731 | DMRT5 |
| T00303 | GAL80 | T01275 | mat1-Mc | T02654 | ERF2 | T05837 | DRF1.3 | T09195 | SPL14 | T10739 | MRP1 |
| T00315 | GBF | T01286 | ROX1 | T02669 | EmBP-1a | T05929 | SUSIBA2 | T09196 | HSF2A | T10745 | HSFA2 |
| T00329 | Glass | T01313 | ATF3 | T02672 | GBF1 | T05943 | FOXP1d | T09199 | STAT5A | T10747 | MTF-1 |
| T00330 | GLI1 | T01333 | RXR-γ | T02690 | Dof2 | T05975 | E2F1 | T09218 | Msx-1 | T10754 | ABF1 |
| T00331 | GLI3 | T01346 | Arnt | T02691 | Dof3 | T05977 | PEND | T09225 | En-1 | T10760 | HAP1 |
| T00337 | GR-α | T01350 | T3R-β2 | T02772 | GCNF | T05982 | POTH1 | T09226 | Lhx2 | T10795 | C/EBPγ |
| T00349 | HAP2 | T01352 | PPARα | T02786 | RITA-1 | T06004 | DeltaNp63α | T09230 | Prep1 | T10849 | STB5 |
| T00350 | HAP3 | T01388 | C/EBP | T02789 | bZIP910 | T06029 | Sox17 | T09243 | MafG | T10854 | GCN4 |
| T00368 | HNF-1α-A | T01400 | Ets-1deltaVII | T02790 | bZIP911 | T06043 | AGP1 | T09287 | MITF-A2 | T10881 | TRAB1 |
| T00377 | HOXA5 | T01422 | ste11 | T02807 | OSBZ8 | T06137 | p73β | T09304 | Smad4 | T10928 | TGA2 |
| T00383 | HSF | T01427 | p300 | T02809 | ROM1 | T06168 | p63α | T09319 | IRF-1 | T10958 | ATHB-2 |
| T00385 | HSF1 | T01431 | c-Maf (long form) | T02810 | ROM2 | T06341 | BEL5 | T09323 | IRF-1 | T10959 | PCF1 |
| T00386 | HSTF | T01470 | Ik-2 | T02818 | GLN3 | T06356 | Rim101p | T09343 | SRF | T10960 | PCF2 |
| T00395 | Hb | T01471 | Ik-3 | T02825 | gaf2 | T06404 | WRKY38 | T09355 | Alx-4 | T11115 | ZIC1 |
| T00401 | ICP4 | T01476 | Abd-B | T02841 | FACB | T06429 | HIC-1- isoform2 | T09356 | HOXA3 | T11136 | DEC2 |
| T00445 | KNIRPS | T01477 | BR-CZ1 | T02846 | UAY | T06532 | NAC69-1 | T09383 | GABP-α | T11158 | HELIOS-B |
| T00456 | Kr | T01478 | BR-CZ2 | T02878 | TCF-4E | T06533 | MYB80 | T09424 | WRKY2 | T11164 | FOXJ1 |
| T00458 | LAC9 | T01479 | BR-CZ3 | T02897 | Sox6-Isoform1 | T06537 | Ci | T09426 | Sp3-isoform1 | T11166 | FOXF1 |
| T00459 | C/EBPβ(LAP) | T01480 | BR-CZ4 | T02905 | LEF-1 | T08158 | ABZ1 | T09427 | RAP-1-xbb1 | T11180 | Gli1 |
| T00480 | MAL63 | T01481 | Pbx1a | T02907 | MYB305 | T08251 | FBI-1 | T09431 | Sp1 | T11200 | DEC1 |
| T00487 | MATα2 | T01482 | Exd | T02929 | MYB340 | T08252 | NF-AT3 | T09441 | RBP-Jκ | T11217 | Gzf1 |
| T00488 | MATa1 | T01484 | Cdx-1 | T02936 | FOXO1 | T08279 | USF1 | T09444 | CPRF-3 | T11246 | ZIC2 |
| T00489 | Max-isoform2 | T01492 | STAT1α | T02983 | Pax-4a | T08291 | GATA-1 | T09449 | CPRF-2 | T11250 | Brachyury |
| T00490 | MAZ | T01517 | Twi | T02999 | OCSBF-1 | T08292 | GATA- 1isoform1 | T09450 | CPRF-1 | T11256 | GCMb |
| T00497 | MBP-1(1) | T01527 | RORα1 | T03031 | Pax-2.1 | T08293 | GATA-1 | T09462 | Egr-1 | T11258 | GCMa |
| T00500 | MCM1 | T01528 | RORα2 | T03178 | SQUA | T08298 | Kaiso | T09478 | TGA1a | T11310 | MafA |
| T00509 | MIG1 | T01556 | SREBP-1a | T03227 | CAT8 | T08300 | ER-α-L | T09507 | Sox-xbb1 | T11372 | HOXB8 |
| T00529 | MZF1B-C | T01590 | P (long form) | T03256 | HNF-3β | T08313 | USF2a | T09514 | HTF4γ | T11383 | HOXD13 |
| T00535 | NF-1 | T01592 | C1 (long form) | T03258 | HNF-6β | T08318 | Elf-1 | T09531 | ATF-4 | T11390 | Cart-1 |
| T00594 | RelA-p65 | T01599 | LCR-F1 | T03388 | Meis-1a | T08319 | Zec | T09540 | c-Krox | T11394 | PR-β |
| T00625 | ZEB(1124AA) | T01615 | Su | T03389 | Meis-1b | T08321 | p53-isoform1 | T09548 | IRF-3 | T11402 | Crx |
| T00627 | NIT2 | T01649 | HES-1 | T03447 | LHX3b | T08323 | p53 | T09561 | Roaz | T11425 | Chx10 |
| T00642 | POU2F1 | T01660 | PR-α | T03481 | SKN7 | T08340 | Egr-2 | T09569 | Hlf | T11440 | FAC1-xbb1 |
| T00644 | POU2F1a | T01661 | PRA | T03491 | MED8 | T08348 | RXR-α | T09571 | MYB1 | T11453 | TAF-1 |
| T00651 | POU5F1 | T01664 | TR2-11 | T03500 | MOT3 | T08358 | GATA-4 | T09588 | E4BP4 | T13753 | HsfB1 |
| T00653 | POU5F1(Oct-5) | T01667 | RFX2 | T03524 | PDR1 | T08409 | GAMYB | T09608 | Kid3 | T13760 | ABF1 |
| T00669 | Ovo-B | T01669 | RFX2 | T03525 | PDR3 | T08410 | PBF | T09623 | ATF6 | T13794 | TGA1 |
| T00677 | Pax-1 | T01670 | RFX3 | T03538 | RCS1 | T08411 | SED | T09629 | MYBJS1 | T13809 | AGL2 |
| T00689 | PHO2 | T01671 | RFX3 | T03541 | RFX1 | T08415 | CBT | T09635 | AP1 | T13810 | Dof4 |
| T00690 | PHO4 | T01673 | RFX1 | T03556 | RGT1 | T08431 | PPARα | T09649 | cel-let-7 | T13811 | AGL3 |
| T00691 | Pit-1A | T01675 | Nkx2-5 | T03593 | Pax-9a | T08441 | Sox10 | T09701 | cel-miR-84 | T14002 | GKLF |
| T00696 | PRB | T01679 | PacC | T03594 | Pax-9b | T08445 | Elk-1-isoform1 | T09706 | hsa-let-7a | T14118 | ASR-1 |
| T00697 | PRB | T01692 | T3R-β1 | T03600 | SIP4 | T08466 | c-Jun | T09707 | hsa-let-7b | T14187 | AIRE-isoform1 |
| T00699 | Prd | T01705 | HOXA7 | T03612 | NK-4 | T08475 | GR-α | T09718 | hsa-miR-23a | T14230 | WRKY40 |
| T00709 | qa-1F | T01710 | HoxA-9 | T03707 | XBP1 | T08482 | VDR | T09727 | hsa-miR-103 | T14231 | RP58 |
| T00710 | R | T01735 | HOXB7 | T03717 | ZAP1 | T08487 | AR | T09729 | hsa-miR-107 | T14234 | WRKY18 |
| T00715 | RAP1 | T01737 | HOXB8 | T03718 | WRKY1 | T08492 | LRH-1-xbb1 | T09731 | dme-miR-2a | T14258 | Nkx3-2 |
| T00719 | RAR-α1 | T01755 | HOXD9 | T03722 | ZAP1 | T08493 | c-Fos | T09732 | dme-miR-2b | T14268 | MIZF |
| T00725 | REB1 | T01757 | HOXD10 | T03975 | SPF1 | T08505 | COUP-TF1 | T09737 | dme-miR-7 | T14302 | C1-Myb |
| T00731 | RME1 | T01784 | MEF-2A | T03994 | ID1 | T08520 | TBP | T09741 | dme-miR-13a | T14317 | Myb-15 |
| T00737 | SAP-1a | T01786 | E12 | T04001 | ATHB-9 | T08528 | AR | T09742 | dme-miR-13b | T14381 | ATHB-1 |
| T00746 | SGF-3 | T01799 | Tal-1 | T04096 | Smad3 | T08544 | MOVO-B | T09793 | dme-let-7 | T14382 | ATHB-5 |
| T00751 | Sn | T01814 | Pax-6/Pd-5a | T04146 | HLTF | T08546 | Ovo1a | T09806 | hsa-miR-1 | T14442 | STF1 |
| T00761 | SRF | T01823 | Pax-2 | T04166 | FOXD3 | T08571 | GATA-2 | T09810 | hsa-miR-124a | T14444 | TGA1 |
| T00763 | SRF | T01838 | Sox4 | T04169 | FOXJ2 (long isoform) | T08577 | ZBRK1 | T09812 | hsa-miR-130a | T14447 | PBF |
| T00767 | Sry-δ | T01841 | WT1-del2 | T04176 | FOXO4 | T08580 | STAT3 | T09819 | hsa-miR-125a | T14485 | XBP-1 |
| T00769 | Sry-β | T01851 | HMGI | T04255 | Nkx3-1 | T08583 | CCA1 | T09824 | hsa-miR-206 | T14491 | CBNAC |
| T00776 | SWI5 | T01865 | Oct-2.3 | T04280 | FOXP3 | T08584 | LHY | T09840 | hsa-miR-130b | T14517 | Zic3 |
| T00788 | T-Ag | T01866 | Oct-2.4 | T04297 | Nkx6-1 | T08613 | ZNF219 | T09880 | dre-miR-430a | T14521 | ZF5 |
| T00789 | Tll | T01867 | Oct-2.6 | T04312 | NURR1- isoform1 | T08615 | PLZFB | T09892 | c-Myb-isoform1 | T14543 | CBF1 |
| T00798 | TBP | T01882 | unc-86 | T04323 | Nkx2-5 | T08619 | WEREWOLF | T09914 | SF-1 | T14573 | FUS3 |
| T00810 | TFE3-L | T01888 | POU6F1(c2) | T04324 | DREF | T08621 | HAHB-4 | T09923 | RREB-1 | T14681 | Spz1 |
| T00812 | TFEB-isoform1 | T01897 | Cf1a | T04336 | Nkx2-8 | T08624 | Sox9 | T09942 | HNF-3β | T14827 | DEAF-1 |
| T00814 | TFE3-S | T01900 | PDM-1 | T04337 | Nkx2-2 | T08630 | CAR | T09949 | FOXC1 | T14951 | Ncx |
| T00830 | TGA1b | T01944 | NF-AT1 | T04345 | TBX5-L | T08667 | SZF1-1 | T09960 | TR4 | T14954 | OG-2 |
| T14992 | Pitx3 |
Apriori algorithm was then applied to discover frequently co-occurring TF–TFBS k-mer pairs (2-itemsets). Finally, the resultant pairs were rescanned in TRANSFAC to measure their forward and backward confidences (33).
RESULTS AND ANALYSIS
In this section, the discovered rules are reported, followed by analysis with different measurements.
Rules discovered
Varying k from 4 to 6 for both TF k-mers and TFBS k-mers, we have obtained nine sets of associated pairs. For each set of pairs, the forward and backward confidences of each pair were calculated. Then, the pairs in the same set were sorted by the minima of their forward and backward confidences in descending order. The nine sets of rules (pairs) exhibit a similar trend that the number of rules decreases as the association criterion becomes more stringent (with higher confidence levels). The TFBS 5-mers settings in general show the most available rules when the confidence level is high (≥0.5), indicating more conserved and significant results. Therefore, we focus on them and use TFBS 5-mer–TF 5-mer as the representative example throughout the article. The results for all other settings are available in the Supplementary Data.
The number of rules (pairs) discovered is summarized in Table 1. For instance, there are 70 TF 5-mer–TFBS 5-mer pairs without any further removal (in the N column) with both forward and backward confidences ≥0.5. Considering direct and reverse complement TFBS DNA k-mers as equivalent, we further removed the duplicated pairs (e.g. leaving AGGTC–CEGCK and removing GACCT–CEGCK because AGGTC and GACCT are reverse complements). The results are shown in the N′ column in Table 1. For instance, the 70 TF 5-mer–TFBS 5-mer pairs were reduced to 35 at a confidence level of 0.5. Furthermore, we found that most pairs could be merged together to form a longer pair. For instance, GGTCA–SGYHY and GGTCA–GYHYG could be merged to form a pair GGTCA–SGYHYG. Thus the pairs have been merged and the rule numbers are shown in the Nm column in Table 1. For instance, 35 TF 5-mer–TFBS 5-mer pairs are merged to form 11 merged pairs when the confidence level is equal to 0.5.
Table 1.
Number of the TFBS 5-mer–TF 5-mer pairs across different confidence levels
| Confidence | 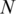 |
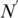 |
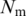 |
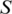 |
|---|---|---|---|---|
| 0.0 | 262 | 131 | 29 | 9.88 ± 3.68 |
| 0.1 | 262 | 131 | 29 | 9.88 ± 3.68 |
| 0.2 | 240 | 120 | 24 | 10.14 ± 3.73 |
| 0.3 | 180 | 90 | 23 | 10.63 ± 4.11 |
| 0.4 | 126 | 63 | 21 | 11.40 ± 4.59 |
| 0.5 | 70 | 35 | 11 | 13.63 ± 5.05 |
| 0.6 | 24 | 12 | 8 | 15.08 ± 5.28 |
| 0.7 | 6 | 3 | 2 | 10.33 ± 2.36 |
| 0.8 | 0 | 0 | 0 | N/A |
| 0.9 | 0 | 0 | 0 | N/A |
| 1.0 | 0 | 0 | 0 | N/A |
N, number of pairs, N, number of pairs (duplicated pairs removed); 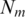 , number of merged pairs; S, mean and SD of the support of the pairs in
, number of merged pairs; S, mean and SD of the support of the pairs in 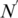 .)
.)
Quantitative analysis
To evaluate the number of TF data sets supporting each pair (support), the support for each pair was counted. In general, more supports are found when the confidence level is increased. For instance, the average support of the TFBS 5-mer–TF 5-mer pairs is generally increased when the confidence level is increased in the S column of Table 1. The overall results are summarized in Supplementary Table S4.
Support is considered the degree of co-occurrence between a TF amino acid k-mer and a TFBS DNA k-mer. Forward and backward confidences consider the cases when either one of them is absent. Some may have questions about the remaining case. How about the case when both of them are absent? To take the case into account, ϕ-coefficients (35) were measured for each pair, as shown in the ϕ column in Table 2. The overall results are summarized in Supplementary Table S5. Most values are >0.4, indicating that positive correlations exist among pairs.
Table 2.
Quantitative measurements for the TFBS 5-mer–TF 5-mer pairs across different confidence levels
| Confidence | 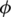 |
L | FC | BC |
|---|---|---|---|---|
| 0.0 | 0.49 ± 0.11 | 17.92 ± 7.34 | 1.89 ± 0.67 | 3.50 ± 2.29 |
| 0.1 | 0.49 ± 0.11 | 17.92 ± 7.34 | 1.89 ± 0.67 | 3.50 ± 2.29 |
| 0.2 | 0.51 ± 0.11 | 18.32 ± 7.46 | 1.94 ± 0.68 | 3.51 ± 2.30 |
| 0.3 | 0.54 ± 0.10 | 19.81 ± 7.79 | 2.02 ± 0.64 | 3.46 ± 2.31 |
| 0.4 | 0.58 ± 0.09 | 21.41 ± 8.53 | 2.23 ± 0.66 | 3.61 ± 2.40 |
| 0.5 | 0.64 ± 0.07 | 22.57 ± 10.46 | 2.49 ± 0.70 | 4.35 ± 2.65 |
| 0.6 | 0.71 ± 0.06 | 25.80 ± 13.76 | 3.33 ± 0.57 | 4.21 ± 2.55 |
| 0.7 | 0.79 ± 0.03 | 42.07 ± 14.87 | 3.70 ± 0.29 | 4.87 ± 0.00 |
| 0.8 | N/A | N/A | N/A | N/A |
| 0.9 | N/A | N/A | N/A | N/A |
| 1.0 | N/A | N/A | N/A | N/A |
ϕ, mean and SD of ϕ-coefficient; L, mean and SD of lift; FC, mean and SD of forward conviction; BC, mean and SD of backward conviction.
Consider the following scenario: if a TFBS DNA k-mer and a TF amino acid k-mer are both frequently found in the data sets, it will be very likely that they co-occur frequently merely by chance. To tackle such scenario, forward and backward confidences do play their important roles in pruning them. But for clarity, lift (36) that estimates the ratio of the actual support to the expected support was measured for each pair, where the expected support was calculated from the random model that the TFBS DNA k-mer is independent of the TF amino acid k-mer for each pair. For instance, the average lift for the TFBS 5-mer–TF 5-mer pairs is shown in the L column in Table 2. The overall results are summarized in Supplementary Table S6. Most values of the lift are >5. Thus the DNA residue k-mer and the amino acid residue k-mer of most pairs co-occur at least five times more frequently than the prediction based on the independent assumption made by the lift measurement.
To estimate the validity of the pairs, both forward and backward convictions (the same directions as the forward and backward confidences, respectively) (36) were measured for each pair. The measurements were averaged for each set of pairs. For instance, the average forward and backward convictions for the TFBS 5-mer–TF 5-mer pairs is shown in the FC and BC columns in Table 2. The overall results are summarized in Supplementary Tables S7 and S8. Most values are >1. The pairs commit fewer errors than the prediction based on the statistically independent assumption made by the measurements: forward and backward convictions. In other words, the pairs would have committed more errors if the association between its TFBS k-mer and TF k-mer had happened purely by chance.
Annotation analysis
If the pairs in our results are the actual binding cores between TFs and TFBSs, most of their TF amino acid k-mers should be inside DNA binding domains. Thus, the TF amino acid k-mers were scanned in TRANSFAC to check whether they were within the annotated DNA binding domains. As stated in the previous section, the set of TFBS 4-mer–TF 4-mer pairs constitutes all the pairs in the other sets by the downward closure property. Thus only the TF amino acid 4-mers of the set of TFBS 4-mer–TF 4-mer pairs were needed for the checking: of the 792 TF amino acid 4-mers, 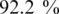 of them were found within the DNA binding domains listed in the ‘PFAM 18’ list downloaded from DBD (37) on 25 January 2010.
of them were found within the DNA binding domains listed in the ‘PFAM 18’ list downloaded from DBD (37) on 25 January 2010.
Empirical analysis
Since the numbers of results are quite large, they are tabulated in a statistical perspective in the previous sections. This section provides readers with empirical insights into the results obtained. Comparing with the other sets, the set of TFBS 5-mer–TF 5-mer pairs shows its relative invariability to confidence level pruning. Thus, it motivates us to have an in-depth empirical analysis on them. They are listed in Table 3.
Table 3.
The set of TFBS 5-mer–TF 5-mer pairs (duplicated pairs removed and sorted in alphabetical order)
| Confidence | Forward confidence | Backward confidence | Pairs | Confidence | Forward confidence | Backward confidence | Pairs | Confidence | Forward confidence | Backward confidence | Pairs |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.7 | 0.7 | 0.8 | AAACA–HNLSL | 0.4 | 0.4 | 0.7 | AGGTC–CQYCR | 0.3 | 0.5 | 0.3 | GCCAC–ARRSR |
| 0.5 | 0.5 | 0.7 | AAACA–IRHNL | 0.2 | 0.2 | 0.6 | AGGTC–CVVCG | 0.4 | 0.5 | 0.4 | GCCAC–ESARR |
| 0.5 | 0.5 | 0.6 | AAACA–KPPYS | 0.6 | 0.6 | 0.7 | AGGTC–EGCKG | 0.4 | 0.4 | 0.6 | GCCAC–KQSNR |
| 0.4 | 0.4 | 0.7 | AAACA–NLSLN | 0.2 | 0.2 | 0.7 | AGGTC–FFRRT | 0.4 | 0.6 | 0.4 | GCCAC–NRESA |
| 0.6 | 0.6 | 0.6 | AAACA–NSIRH | 0.2 | 0.2 | 0.8 | AGGTC–FRRTI | 0.4 | 0.4 | 0.6 | GCCAC–QSNRE |
| 0.5 | 0.5 | 0.6 | AAACA–PPYSY | 0.6 | 0.6 | 0.6 | AGGTC–GCKGF | 0.4 | 0.5 | 0.4 | GCCAC–RESAR |
| 0.4 | 0.4 | 0.6 | AAACA–PYSYI | 0.3 | 0.3 | 0.5 | AGGTC–GFFKR | 0.4 | 0.4 | 0.6 | GCCAC–RKQSN |
| 0.4 | 0.4 | 0.6 | AAACA–QNSIR | 0.4 | 0.4 | 0.5 | AGGTC–GFFRR | 0.4 | 0.4 | 0.5 | GCCAC–RLRKQ |
| 0.7 | 0.7 | 0.8 | AAACA–RHNLS | 0.3 | 0.3 | 0.6 | AGGTC–KGFFK | 0.4 | 0.4 | 0.5 | GCCAC–RRSRL |
| 0.5 | 0.5 | 0.8 | AAACA–SIRHN | 0.4 | 0.4 | 0.5 | AGGTC–KGFFR | 0.4 | 0.4 | 0.6 | GCCAC–RSRLR |
| 0.4 | 0.4 | 0.6 | AAACA–WQNSI | 0.4 | 0.4 | 0.9 | AGGTC–RNRCQ | 0.4 | 0.5 | 0.4 | GCCAC–SARRS |
| 0.4 | 0.4 | 0.6 | AACAA–HNLSL | 0.3 | 0.3 | 0.5 | AGGTC–TCEGC | 0.4 | 0.5 | 0.4 | GCCAC–SNRES |
| 0.3 | 0.3 | 0.6 | AACAA–IRHNL | 0.4 | 0.4 | 0.5 | AGGTC–VCGDK | 0.4 | 0.5 | 0.4 | GCCAC–SRLRK |
| 0.3 | 0.3 | 0.5 | AACAA–NSIRH | 0.2 | 0.2 | 0.5 | AGGTC–VVCGD | 0.6 | 0.6 | 0.8 | GGTCA–CEGCK |
| 0.3 | 0.3 | 0.7 | AACAA–PMNAF | 0.2 | 0.7 | 0.2 | ATTAA–FQNRR | 0.2 | 0.2 | 0.9 | GGTCA–CGDKA |
| 0.4 | 0.4 | 0.6 | AACAA–RHNLS | 0.2 | 0.6 | 0.2 | ATTAA–IWFQN | 0.5 | 0.5 | 0.6 | GGTCA–CKGFF |
| 0.3 | 0.3 | 0.7 | AACAA–RPMNA | 0.2 | 0.6 | 0.2 | ATTAA–KIWFQ | 0.3 | 0.3 | 0.9 | GGTCA–CQYCR |
| 0.3 | 0.3 | 0.7 | AACAA–SIRHN | 0.3 | 0.5 | 0.3 | ATTAA–NRRMK | 0.2 | 0.2 | 0.8 | GGTCA–CVVCG |
| 0.2 | 0.6 | 0.2 | AAGGT–CKGFF | 0.3 | 0.5 | 0.3 | ATTAA–QNRRM | 0.1 | 0.1 | 1 | GGTCA–DLVLD |
| 0.2 | 0.5 | 0.2 | AATTA–FQNRR | 0.2 | 0.7 | 0.2 | ATTAA–WFQNR | 0.5 | 0.5 | 0.8 | GGTCA–EGCKG |
| 0.3 | 0.3 | 0.3 | AATTA–NRRAK | 0.2 | 0.5 | 0.2 | CACCC–GEKPY | 0.2 | 0.2 | 0.8 | GGTCA–FFKRS |
| 0.4 | 0.4 | 0.5 | AATTA–QNRRA | 0.1 | 0.5 | 0.1 | CACCC–HTGEK | 0.2 | 0.2 | 0.8 | GGTCA–FFRRT |
| 0.3 | 0.3 | 0.7 | AATTA–QVWFQ | 0.1 | 0.5 | 0.1 | CACCC–TGEKP | 0.2 | 0.2 | 1 | GGTCA–FRRTI |
| 0.5 | 0.5 | 0.5 | AATTA–VWFQN | 0.5 | 0.5 | 0.5 | CCACG–ARRSR | 0.5 | 0.5 | 0.7 | GGTCA–GCKGF |
| 0.2 | 0.5 | 0.2 | AATTA–WFQNR | 0.5 | 0.5 | 0.6 | CCACG–ESARR | 0.2 | 0.2 | 0.5 | GGTCA–GFFKR |
| 0.5 | 0.5 | 0.7 | ACGTG–ARRSR | 0.3 | 0.3 | 0.7 | CCACG–KQSNR | 0.3 | 0.3 | 0.6 | GGTCA–GFFRR |
| 0.1 | 0.1 | 0.7 | ACGTG–ERELK | 0.2 | 0.2 | 0.6 | CCACG–LRKQA | 0.1 | 0.1 | 0.6 | GGTCA–GYHYG |
| 0.5 | 0.5 | 0.9 | ACGTG–ESARR | 0.6 | 0.6 | 0.6 | CCACG–NRESA | 0.1 | 0.1 | 1 | GGTCA–ITCEG |
| 0.2 | 0.2 | 0.8 | ACGTG–KQSNR | 0.3 | 0.3 | 0.6 | CCACG–QSNRE | 0.2 | 0.2 | 0.6 | GGTCA–KGFFK |
| 0.2 | 0.2 | 0.7 | ACGTG–LRKQA | 0.5 | 0.5 | 0.6 | CCACG–RESAR | 0.3 | 0.3 | 0.6 | GGTCA–KGFFR |
| 0.6 | 0.6 | 0.9 | ACGTG–NRESA | 0.2 | 0.2 | 0.7 | CCACG–RKQAE | 0.1 | 0.1 | 1 | GGTCA–NRCQY |
| 0.2 | 0.2 | 0.7 | ACGTG–QSNRE | 0.3 | 0.3 | 0.7 | CCACG–RKQSN | 0.1 | 0.1 | 1 | GGTCA–RCQYC |
| 0.5 | 0.5 | 0.9 | ACGTG–RESAR | 0.3 | 0.3 | 0.5 | CCACG–RLRKQ | 0.1 | 0.1 | 0.8 | GGTCA–RNQCQ |
| 0.1 | 0.1 | 0.7 | ACGTG–RKQAE | 0.3 | 0.3 | 0.6 | CCACG–RRSRL | 0.3 | 0.3 | 1 | GGTCA–RNRCQ |
| 0.2 | 0.2 | 0.8 | ACGTG–RKQSN | 0.3 | 0.3 | 0.6 | CCACG–RSRLR | 0.2 | 0.2 | 1 | GGTCA–SCEGC |
| 0.2 | 0.2 | 0.6 | ACGTG–RLRKQ | 0.5 | 0.5 | 0.6 | CCACG–SARRS | 0.1 | 0.1 | 0.5 | GGTCA–SGYHY |
| 0.2 | 0.2 | 0.7 | ACGTG–RRSRL | 0.5 | 0.5 | 0.6 | CCACG–SNRES | 0.3 | 0.3 | 0.6 | GGTCA–TCEGC |
| 0.2 | 0.2 | 0.8 | ACGTG–RSRLR | 0.4 | 0.4 | 0.4 | CCACG–SRLRK | 0.3 | 0.3 | 0.6 | GGTCA–VCGDK |
| 0.5 | 0.5 | 0.9 | ACGTG–SARRS | 0.5 | 0.5 | 0.5 | CGGAA–LRYYY | 0.2 | 0.2 | 0.7 | GGTCA–VVCGD |
| 0.5 | 0.5 | 0.9 | ACGTG–SNRES | 0.5 | 0.5 | 0.8 | CTTCC–LRYYY | 0.5 | 0.5 | 0.7 | GTCAA–KYGQK |
| 0.3 | 0.3 | 0.5 | ACGTG–SRLRK | 0.4 | 0.4 | 0.7 | CTTCC–LWQFL | 0.5 | 0.5 | 0.7 | GTCAA–RKYGQ |
| 0.6 | 0.7 | 0.6 | AGGTC–CEGCK | 0.4 | 0.7 | 0.4 | GATAA–CNACG | 0.5 | 0.5 | 0.7 | GTCAA–WRKYG |
| 0.3 | 0.3 | 0.8 | AGGTC–CGDKA | 0.4 | 0.7 | 0.4 | GATAA–LCNAC | 0.7 | 0.7 | 1 | TGACA–NWFIN |
| 0.6 | 0.7 | 0.6 | AGGTC–CKGFF | 0.6 | 0.7 | 0.6 | GATAA–NACGL |
Among the 131 pairs in Table 3, the TFBS DNA k-mers are quite conserved. There are only 15 distinct TFBS DNA k-mers. Each TFBS DNA k-mer forms pairs with 8.73 TF amino acid k-mers on average. One of the reasons may be the specificity of DNA residue, is lower in view of its alphabet size (4) as compared to the amino acid alphabet size (20).
To act as a DNA binding protein, a TF needs to provide a basic interacting surface for the recognition of major/minor grooves as well as the phosphate backbone of DNA. Therefore, we searched through the set of pairs in Table 3 to count the occurring frequency for each residue. Interestingly, we found that the basic residues, lysine (50 times) and arginine (131 times), occur at the highest frequency among 131 pairs of TFBS–TF. On the other hand, the hydrophobic residues (38) such as isoleucine (15) and valine (13) occur at the lowest frequency. These results suggest the potential of the TF sequences for being the binding sequences between TFs and TFBSs. On the other hand, as the nucleotides of TFBSs are somehow negatively charged, it can be deduced that their binding amino acid residues of TFs should be positively charged. Thus the occurring frequencies were further examined. Among the 131 pairs, the positively charged residues: arginine (R) and lysine (K) occur 131 and 50 times, respectively. In contrast, the negatively charged residues aspartic acid (D) and glutamic acid (E) occur 8 and 30 times, respectively. Such discrepancy supports their potential for being the binding sequences between TFs and TFBSs.
Experimental analysis
This section follows the same approach in empirical analysis. The set of TFBS 5-mer–TF5mer pairs in Table 3 is selected for experimental analysis. Out of the 131 pairs, 5 of them were selected and analyzed. The first pair is GGTCA–CEGCK, which have been experimentally proved as binding sequences in Ref. (39). The TF amino acid k-mer (CEGCK) is considered part of P-box (CEGCKG) within the DNA binding domain of Bp-nhr-2, which is believed to bind the DNA k-mer (GGTCA). The second pair is AAACA–IRHNL mentioned in Ref. (40). Based on the corresponding PDB entry 3CO6, it is believed that the pair was the binding pair between a TF and a TFBS as shown in Figure 3. Similarly, the remaining pairs are GATAA–NACGL, GGTCA–GFFRR and CTTCC–LRYYY. They are found as binding pairs in PDB entries 3DFV (41), 3DZY (42) and 2NNY (43) as shown in Figure 3a, b and c, respectively. The above five pairs reveal that the pairs generated from the proposed approach have biological evidences in literatures. Among the previous figures, two of them (3CO6 and 2NNY) were further analyzed in terms of hydrogen bonding, which also means the specificity of the interaction between amino acids and the bases, as shown in Figure 4a and b. We have also highlighted the hydrogen bonds as black lines as well as the residues that make contact with the base (only predicted residues), which are the evidence of the significance and accuracy of the prediction of the TF–TFBS pairs. Nevertheless, as the proposed approach is applied on a large-scale database, such extensive and detailed analysis of all the binding core pairs discovered are not practical. Therefore, a scalable verification approach will be presented in the next section to verify the massive results generated.
Figure 3.

Four representative TF–TFBS pairs are shown in ribbon diagram. (a) AAACA–IRHNL pair in 3C06, (b) GATAA–NACGL pair in 3DFV, (c) GGTCA–GFFRR pair in 3DZY and (d) CTTCC–LRYYY pair in 2NNY are shown. The TF amino acids and TFBS nucleotides are highlighted in ball and stick format. The sequences of the TF–TFBS pairs are also labeled in the figures. The figures are generated using Protein Workshop (34).
Figure 4.

The interactions between the TF and TFBS of two representative pairs (a) AAACA–IRHNL in 3CO6 and (b) CTTCC–LRYYY in 2NNY are shown. The proteins are shown in ribbon diagram with the highlighted TF amino acids in ball and stick format. The helices and strands are colored in red and cyan, respectively. The amino acids that interact with the nucleotides are labeled. The hydrogen bonds are shown in dark line. The figures are generated using DS visualizer, Accelrys.
VERIFICATIONS
In this section, we try to verify the discovered pairs with external data sources, in particular the 3D protein-DNA complex structures experimentally determined from PDB. Homology modeling has also been done for further verifications.
Verification by PDB
In this article, PDB is selected for providing 3D protein–DNA complex data for 3D structural verification. The PDB data were downloaded from RCSB PDB (http://www.pdb.org) from 16 September 2009 to 22 September 2009, where the protein–DNA complexes were selected based on the entry-type list provided in ftp://ftp.wwpdb.org/.
For each set of pairs in Supplementary Table S2, each pair is independently evaluated as shown in Figure 5. For each pair, its TF k-mer is used to query which PDB chain has the TF k-mer. Once the corresponding set of PDB chains has been identified and returned, its redundancy is removed by BLASTClust using 90% sequence identity (32). The removal is to ensure that redundant PDB chains are not double counted. After the removal, the pair is evaluated for binding in the 3D space:
A TFBS k-mer–TF k-mer pair is considered binding for a PDB chain if and only if an atom of the TFBS k-mer and an atom of the TF k-mer are close to each other. Two atoms are considered close if and only if their distance is <3.5 Å (25,28).
Figure 5.

Flowchart of 3D verification for each set of pairs.
With the pair evaluated in its PDB chains, its PDB chains can be classified into the following three categories:
PDB chains only having the TF k-mer (a)
- PDB chains having both TF k-mer and TFBS k-mer
- The pair binds together (b)
- The pair does not bind together (c)
Thus the number of chains in each category is counted and converted into the following performance metrics:
TFBS prediction score=(b + c)/(a + b + c)
TFBS binding prediction score =b/(a + b + c)
Binding prediction score=b/(b + c)
Given the resultant PDB chains queried by a TF k-mer, TFBS prediction score measures the proportion of PDB chains that contain the corresponding TFBS k-mer. In other words, it measures the backward confidence of a pair in PDB. TFBS binding prediction score is a more stringent metric. It measures the proportion of PDB chains that have the corresponding TFBS k-mer binding with the queried TF k-mer. Lastly, binding prediction score is the most important metric. It measures the proportion of PDB chains in which the pair is really binding. To verify the cases when (b + c) = 0 (i.e. the pairs do not appear in PDB), homology modeling is also performed.
For each setting, we have a set of pairs. For each pair, the above performance metrics are calculated. The overall results are averaged and summarized in Supplementary Tables S9–S11. For each setting, we also have a set of merged pairs. For each merged pair, the above performance metrics are also calculated. The overall results are averaged and summarized in Supplementary Tables S12–S14. Note that the most conservative calculation has been used for each performance metric for each pair. If a performance metric of a pair does not have enough PDB data for calculation, a value of zero will be given to the performance metric of the pair. For instance, the cases when (b + c) = 0 or (a + b + c) = 0. Despite the above setting, the performance metrics of the pairs still have reasonable performances. They are shown to be significantly better than the maximal performance of 50 random runs in a later section.
Nevertheless, although the above metrics can capture the performance of a pair quantitatively, the most important point is to know how many generated pairs could be verified [with at least one binding evidence in PDB data (b > 0)]. To gain more insights, the number of pairs with at least one related PDB chain [(b + c) > 0] are tabulated in Supplementary Tables S15 and S16. Correspondingly, the percentage of verified pairs ((Number of pairs with b > 0/Number of pairs with (b + c) > 0)) are calculated and tabulated in Supplementary Tables S17 and S18. In the tables, the percentage of verified pairs is high enough to justify that the proposed approach has produced pairs proven to be binding in PDB. For instance, the statistics for the TFBS 5-mer–TF 5-mer pairs is extracted in Table 4 and Figure 6. Among the 80 TFBS 5-mer–TF 5-mer pairs with at least one related PDB chain [(b + c) > 0] when the confidence level = 0.0, more than 81% of them have at least one binding evidence (b > 0).
Table 4.
Number of the TFBS 5-mer–TF 5-mer pairs verified across different confidence levels
| Confidence | 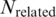 |
 |
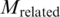 |
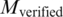 |
|---|---|---|---|---|
| 0.0 | 80 | 65 | 19 | 16 |
| 0.1 | 80 | 65 | 19 | 16 |
| 0.2 | 71 | 59 | 15 | 13 |
| 0.3 | 50 | 44 | 15 | 13 |
| 0.4 | 32 | 28 | 12 | 11 |
| 0.5 | 19 | 17 | 7 | 6 |
| 0.6 | 9 | 9 | 5 | 5 |
| 0.7 | 2 | 2 | 1 | 1 |
| 0.8 | 0 | 0 | 0 | 0 |
| 0.9 | 0 | 0 | 0 | 0 |
| 1.0 | 0 | 0 | 0 | 0 |
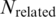 , number of the TFBS 5-mer–TF 5-mer pairs with at least one related PDB chain [
, number of the TFBS 5-mer–TF 5-mer pairs with at least one related PDB chain [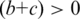 ];
]; 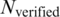 , number of the TFBS 5-mer–TF 5-mer pairs with at least one PDB chain as a binding evidence [
, number of the TFBS 5-mer–TF 5-mer pairs with at least one PDB chain as a binding evidence [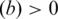 ];
]; 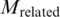 , number of the TFBS 5-mer–TF 5-mer merged pairs with at least one related PDB chain [
, number of the TFBS 5-mer–TF 5-mer merged pairs with at least one related PDB chain [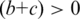 ];
]; 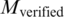 , number of the TFBS 5-mer–TF 5-mer merged pairs with at least one PDB chain as a binding evidence [
, number of the TFBS 5-mer–TF 5-mer merged pairs with at least one PDB chain as a binding evidence [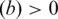 )].
)].
Figure 6.

Percentage of the TFBS 5-mer–TF 5-mer pairs verified across different confidence levels.
The TFBS–TF pairs that we found to have binding evidences in the PDB show typical structural features of DNA–protein interactions. Such features include the 'recognition helix' of the DNA–binding protein making base contacts in the major groove and direct hydrogen bonds between the side chains and the bases. These interactions play the crucial role in the DNA recognition and site-specific binding, respectively (44). Interestingly, the nucleotides of TFBS are located in the major groove of the DNA, which are close to, and make contacts with the amino acids of the ‘recognition helix' of the TF (as for example shown in Figure 3).
The verification is considered satisfactory since those pairs not found in PDB [(b + c) = 0] may be unannotated discovery as shown in the following verification by homology modeling.
Verification by homology modeling
Regarding the pairs without any related PDB chain [(b + c) = 0], there is no PDB data for us to verify them. Thus, we have taken the most conservative approach to assign zero to their performance metrics in the aforementioned evaluations. Nevertheless, we believe that most of those pairs are true and our approach can be used as an effective protein–DNA binding discovery tool. Thus 6 TFBS 5-mer–TF 5-mer pairs were taken and merged. The resultant pair ACGTG-SNRESARRSR was analyzed by homology modeling as follows:
The model of DNA–protein complex was built by homology modeling (INSIGHT II, MSI) based on the structure of the GCN4–DNA complex (1YSA) (45). Briefly, three amino acids (R234S, T236R and A238S) and two nucleotides (T29C and A31T) were mutated in the original structure. The side chains of the mutated amino acids were chosen from the rotamer database and examined using the Ramachandran plots to prevent any steric effect. The interactions between the amino acids and the nucleotides were searched based on the distance of the hydrogen bond.
Figure 7.

The pair ACGTG–SNRESARRSR using homology modeling.
As shown in Figure 3, we found that the pair ACGTG-SNRESARRSR exists in plant as the basic leucine-zipper (bZIP) transcription factor which binds to G-box binding factors (GBF) of DNA (46). Moreover, the ACGTG sequence is the consensus sequence, which is defined as G-box core and locates at the major groove of the double-stranded DNA. It is believed that the G-box core is the DNA sequence of GBF that provides the specificity of the binding to bZIP proteins. In order to further understand the interactions between the TF–TFBS, we built a model by using homology modeling based on the structure of GCN4–DNA (1YSA) complex (45). As shown in the model, the protein helix fits into the major groove of the DNA very well and forms extensive interactions (black lines) between the amino acids and the nucleotides. Interestingly, the mutations of the protein (R234S, T236R and A238S) as well as nucleotides (T29C and A31T) increases the number of hydrogen bonds compared with the original structure (1YSA), suggesting the binding specificity between this pair of TF–TFBS. In conclusion, we believe that the protein–DNA binding sequence patterns found using association rule mining on the large-scale database reveal real TF–TFBS pairs in physiologically relevant situation and this method could guide us to discover new and undescribed TF–TFBS pairs in the future.
Verification by random analysis
For each set of pairs in Supplementary Table S1, we use a random process to generate a random set with the same number of pairs. Within a random set, its pairs were randomly sampled from all the combinations of the k-mers used in the proposed approach. Fifty random runs were performed. The maximal performance metrics of the 50 random runs are summarized in Supplementary Tables S19–S21. In a comparison to the proposed approach, their performance has been depicted in Figure 8. It can be observed that the performance of the proposed approach is significantly better than the best one of the 50 random runs. For instance, the binding prediction score of the 131 TFBS 5-mer–TF 5-mer pairs generated is 0.36±0.39 on average, whereas the maximal binding prediction score over 50 random runs is only 0.00509±0.06492 on average. Similar observation can also be drawn for their merged pairs in Supplementary Tables S22–S24. It can be concluded that the performance of the proposed approach is very unlikely to happen purely by chance in PDB.
Figure 8.

Performance Comparison for PDB verifications. (a) TFBS prediction score, (b) TFBS binding prediction score, (c) binding prediction score (d) TFBS prediction score (merged pairs), (e) TFBS binding prediction score (merged pairs) and (f) binding prediction score (merged pairs) are shown.
DISCUSSION
In this article, we have proposed a framework based on association rule mining with Apriori algorithm to discover associated TF–TFBS binding sequence patterns in the most explicit and interpretable form from TRANSFAC. With downward closure property, the algorithm guarantees the exact and optimal performance to generate all frequent TFBS k-mer TF k-mer pairs from TRANSFAC. The approach relies merely on sequence information without any prior knowledge in TF binding domains or protein–DNA 3D structure data. From comprehensive evaluations, statistics of the discovered patterns are shown to reflect meaningful binding characteristics. According to external literatures, PDB data and homology modeling, a good number of TF–TFBS binding patterns discovered have been verified by experiments and annotations. They exhibit atomic-level bindings between the respective TF binding domains and specific nucleotides of the TFBS from experimentally determined protein–DNA 3D structures. In fact, most of the pairs discovered are actually the binding cores from the TF binding domains and TFBS, respectively.
The proposed approach has great potential for discovering intuitive and interpretable rules of TF–TFBS binding mechanisms. Such rules are able to reveal TF binding domains, detailed interactions between amino acids and nucleotides, accurate TFBS sequence motifs, and help better understanding and deciphering of protein–DNA interactions. It also offers strategic help to reduce the labor and costs involved in wet-lab experiments. With increasing computational power and more sophisticated mining approaches, the proposed methodology can be further improved for discovering more intriguing TF–TFBS binding patterns and rules.
In the future, approximate associations will be considered to handle the experimental and biological noises, although the inevitable computational burden needs to be carefully handled, and much more efforts are needed to distinguish real signals from the large number of false positives introduced by loosening the pattern matching and clustering. Combinatorial associations between multiple TF and TFBS k-mers will also be another challenging topic. We will also seek further real applications of the approach on experimentally verifiable TF–TFBS bindings.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Research Grants Council of the Hong Kong SAR, China (project numbers: CUHK41407 and CUHK414708. Funding for open access charge: Block Grant Project of the Chinese University of Hong Kong, ref #2150591; Focused Investment Scheme D on Hong Kong Bioinformatics Centre, The Chinese University of Hong Kong (project number: 1904014).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to the anonymous reviewers for their valuable comments. They would also like to thank Shaoke Lou, Kin-Cheung Ling and Leung-Yau Lo for their valuable help on preparing the study and the manuscript.
Appendix 1
| Algoritham 1 Pseudocode of Apriori algorithm (29) |
|---|
| data: A dataset of itemsets |
| Ln: Frequent n-itemsets |
| Cn: Candidate n-itemsets |
| x : An itemset |
| minsupport: Minimum Support |
| i ← 1; |
| Scan data to get Li; |
while
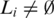 do
do
|
| Ci + 1 ← Extend (Li); |
| Li + 1 ← ∅; |
| For x∈Ci + 1 do |
If
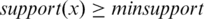 then
then
|
| Li + 1 ← Li + 1∩x; |
| end if |
| end for |
| i ← i + 1; |
| end while |
| Notes: |
| Extend(Li) is the function ‘Candidate itemset generation procedure' stated in (29). Support(x) returns the support (30) of the itemset x. A frequent n-itemset is the n-itemset support is higher than minsupport. |
REFERENCES
- 1.Luscombe NM, Thornton JM. Protein-DNA interactions: amino acid conservation and the effects of mutations on binding specificity. J. Mol. Biol. 2002;320:991–1009. doi: 10.1016/s0022-2836(02)00571-5. [DOI] [PubMed] [Google Scholar]
- 2.Luscombe NM, Austin SE, Berman HM, Thornton JM. An overview of the structures of protein-DNA complexes. Genome Biol. 2000;1:REVIEWS001. doi: 10.1186/gb-2000-1-1-reviews001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galas DJ, Schmitz A. DNAse footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 1987;5:3157–3170. doi: 10.1093/nar/5.9.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garner MM, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 1981;9:3047–3060. doi: 10.1093/nar/9.13.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith AD, Sumazin P, Das D, Zhang MQ. Mining ChIP-chip data for transcription factor and cofactor binding sites. Bioinformatics. 2005;21(Suppl 1):i403–i412. doi: 10.1093/bioinformatics/bti1043. [DOI] [PubMed] [Google Scholar]
- 6.MacIsaac KD, Fraenkel E. Practical strategies for discovering regulatory DNA sequence motifs. PLoS Comput. Biol. 2006;2:e36. doi: 10.1371/journal.pcbi.0020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu XS, Brutlag DL, Liu JS. An algorithm for finding protein–DNA binding sites with applications to chromatinimmunoprecipitation microarray experiments. Nat. Biotechnol. 2002;20:835–839. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- 8.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:108–110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hulo N, Bairoch A, Bulliard V, Cerutti L, Cuche BA, de Castro E, Lachaize C, Langendijk-Genevaux PS, Sigrist CJA. The 20 years of PROSITE. Nucleic Acids Res. 2008;36(Suppl.1):D245–D249. doi: 10.1093/nar/gkm977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, GrifRths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer ELL, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kel AE, Goessling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;31:3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stormo GD. Computer methods for analyzing sequence recognition of nucleic acids. Annu. Rev. Biochem. 1988;17:241–263. doi: 10.1146/annurev.bb.17.060188.001325. [DOI] [PubMed] [Google Scholar]
- 14.Jensen ST, Liu XS, Zhou Q, Liu JS. Computational discovery of gene regulatory binding motifs: a Bayesian perspective. Statistical Science. 2004;19:188–204. [Google Scholar]
- 15.Tompa M, Li N, Bailey TL, Church GM, Moor BD, Eskin E, Favorov AV, Frith MC, Fu Y, Kent, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- 16.Sandve GK, Abul O, Walseng V, Drablos F. Improved benchmarks for computational motif discovery. BMC Bioinformatics. 2007;8:193. doi: 10.1186/1471-2105-8-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones S, van Heyningen P, Berman HM, Thornton JM. Protein-DNA interactions: a structural analysis. J. Mol. Biol. 1999;287:877–896. doi: 10.1006/jmbi.1999.2659. [DOI] [PubMed] [Google Scholar]
- 18.Luscombe NM, Laskowski RA, Thornton JM. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishna SS, Majumdar I, Grishin NV. Structural classification of zinc fingers: survey and summary. Nucleic Acids Res. 2003;31:532–550. doi: 10.1093/nar/gkg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones S, Shanahan HP, Berman HM, Thornton JM. Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins. Nucleic Acids Res. 2003;31:7189–7198. doi: 10.1093/nar/gkg922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mandel-Gutfreund Y, Schueler O, Margalit H. Comprehensive analysis of hydrogen bonds in regulatory protein DNA-complexes: in search of common principles. J. Mol. Biol. 1995;253:370–382. doi: 10.1006/jmbi.1995.0559. [DOI] [PubMed] [Google Scholar]
- 22.Mandel-Gutfreund Y, Margalit H. Quantitative parameters for amino acid-base interaction: implications for prediction of protein-DNA binding sites. Nucleic Acids Res. 1998;26:2306–2312. doi: 10.1093/nar/26.10.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarai A, Kono H. Protein-DNA recognition patterns and predictions. Annu. Rev. Biophys. Biomol. Struct. 2005;34:379–398. doi: 10.1146/annurev.biophys.34.040204.144537. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Q, Liu JS. Extracting sequence features to predict protein-DNA interactions: a comparative study. Nucleic Acids Res. 2008;36:4137–4148. doi: 10.1093/nar/gkn361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmad S, Gromiha MM, Sarai A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics. 2004;20:477–486. doi: 10.1093/bioinformatics/btg432. [DOI] [PubMed] [Google Scholar]
- 26.Ahmad S, Keskin O, Sarai A, Nussinov R. Protein-DNA interactions: structural, thermodynamic and clustering patterns of conserved residues in DNA-binding proteins. Nucleic Acids Res. 2008;36:5922–5932. doi: 10.1093/nar/gkn573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pham TH, Clemente JC, Satou K, Ho TB. Computational discovery of transcriptional regulatory rules. Bioinformatics. 2005;21:101–107. doi: 10.1093/bioinformatics/bti1117. [DOI] [PubMed] [Google Scholar]
- 28.Ofran Y, Mysore V, Rost B. Prediction of DNA-binding residues from sequence. Bioinformatics. 2007;23:i347–i353. doi: 10.1093/bioinformatics/btm174. [DOI] [PubMed] [Google Scholar]
- 29.Agrawal R, Imieliński T, Swami A. SIGMOD '93: Proceedings of the 1993 ACM SIGMOD international conference on Management of data. 1993. Mining association rules between sets of items in large databases; pp. 207–216. http://portal.acm.org/citation.cfm?id=170072. [Google Scholar]
- 30.Hipp J, Güntzer U, Nakhaeizadeh G. Algorithms for association rule mining—a general survey and comparison. SIGKDD Explor. Newsl. 2000;2:58–64. [Google Scholar]
- 31.May KO. A set of independent necessary and sufficient conditions for simple majority decision. Econometrica. 1952;20:680–684. [Google Scholar]
- 32.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 33.Geng L, Hamilton HJ. Interestingness measures for data mining: a survey. ACM Comput. Surv. 2006;38:9. [Google Scholar]
- 34.Moreland JL, Gramada A, Buzko OV, Zhang Q, Bourne PE. The Molecular Biology Toolkit (MBT): a modular platform for developing molecular visualization applications. BMC Bioinformatics. 2005;6:21. doi: 10.1186/1471-2105-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guilford JP. Psychometric Methods. New York: McGraw-Hill; 1936. [Google Scholar]
- 36.Brin S, Motwani R, Ullman JD, Tsur S. Dynamic itemset counting and implication rules for market basket data. SIGMOD Rec. 1997;26:255–264. [Google Scholar]
- 37.Wilson D, Charoensawan V, Kummerfeld SK, Teichmann SA. DBD taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res. 2008;36:D88–D92. doi: 10.1093/nar/gkm964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Privalov PL, Gill SJ. Stability of protein structure and hydrophobic interaction. Adv. Protein Chem. 1988;39:191–234. doi: 10.1016/s0065-3233(08)60377-0. [DOI] [PubMed] [Google Scholar]
- 39.Moore J, Devaney E. Cloning and characterization of two nuclear receptors from the filarial nematode Brugia pahangi. Biochem. J. 1999;344(Pt 1):245–252. [PMC free article] [PubMed] [Google Scholar]
- 40.Brent MM, Anand R, Marmorstein R. Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure. 2008;16:1407–1416. doi: 10.1016/j.str.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bates DL, Chen Y, Kim G, Guo L, Chen L. Crystal structures of multiple GATA zinc fingers bound to DNA reveal new insights into DNA recognition and self-association by GATA. J. Mol. Biol. 2008;381:1292–1306. doi: 10.1016/j.jmb.2008.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F. Structure of the intact PPAR-gamma-RXR-alpha nuclear receptor complex on DNA. Nature. 2008;456:350–356. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamber EP, Vanhille L, Textor LC, Kachalova GS, Sieweke MH, Wilmanns M. Regulation of the transcription factor Ets-1 by DNA-mediated homo-dimerization. EMBO J. 2008;27:2006–2017. doi: 10.1038/emboj.2008.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pabo CO, Sauer RT. Transcription Factors: structural families and Principles of DNA recognition. Annu. Rev. Biochem. 1992;61:1053–1095. doi: 10.1146/annurev.bi.61.070192.005201. [DOI] [PubMed] [Google Scholar]
- 45.Ellenberger TE, Brandl CJ, Struhl K, Harrison SC. The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted alpha helices: crystal structure of the protein-DNA complex. Cell. 1992;71:1223–1237. doi: 10.1016/s0092-8674(05)80070-4. [DOI] [PubMed] [Google Scholar]
- 46.Sibe'ril Y, Doireau P, Gantet P. Plant bZIP G-box binding factors. Modular structure and activation mechanisms. Eur. J. Biochem. 2001;268:5655–5666. doi: 10.1046/j.0014-2956.2001.02552.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.