


Abstract
The 3′-peptidyl-tRNA conjugates that possess a hydrolysis-resistant ribose-3′-amide linkage instead of the natural ester linkage would represent valuable substrates for ribosomal studies. Up to date, access to these derivatives is severely limited. Here, we present a novel approach for the reliable synthesis of non-hydrolyzable 3′-peptidyl-tRNAs that contain all the respective genuine nucleoside modifications. In short, the approach is based on tRNAs from natural sources that are site-specifically cleaved within the TΨC loop by using DNA enzymes to obtain defined tRNA 5′-fragments carrying the modifications. After dephosphorylation of the 2′,3′-cyclophosphate moieties from these fragments, they are ligated to the respective 3′-peptidylamino-tRNA termini that were prepared following the lines of a recently reported solid-phase synthesis. By this novel concept, non-hydrolyzable 3′-peptidyl-tRNA conjugates possessing all natural nucleoside modifications are accessible in highly efficient manner.
INTRODUCTION
The chemical synthesis of 3′-peptidyl-tRNA conjugates containing the natural tRNA modifications represents a huge challenge from the synthetic chemist’s point of view. Such bioconjugates, in particular those which possess a hydrolysis-resistant ribose-3′-amide linkage instead of the natural ester linkage, would fill a missing gap with respect to ribosomal studies that focus on structural and functional characterization of pre- and postpeptidyl transfer states (1–3), of tRNA hybrid states (4–6), of translation termination (7), of ribosome stalling (8–9) but also of phenomena like macrolide antibiotic resistance (10–12), to mention a few examples.
To date, full-length tRNAs without the natural modification pattern are accessible either by in vitro transcription or by chemical RNA synthesis using phosphoramidite building blocks (13–15). More recent reports, however, favor the combined approach of enzymatic ligation of two chemically synthesized half-molecules to obtain tRNAs in multimilligram amounts with greatly reduced 3′-heterogeneity (compared to production via transcription by T7 RNA polymerase) (16–17). This approach also circumvents chemical synthesis of RNA larger than 50–60 nt, which is technically challenging.
The drawback of a total synthesis approach for the tRNA portion of our targets would be that by far not all naturally occurring tRNA nucleoside modifications could be taken into account (18–19). This is simply because most of the structurally complex modifications are not compatible with reaction conditions required for RNA solid-phase synthesis. Another aspect that we considered for our endeavors toward fully modified, non-hydrolyzable 3′-peptidyl-tRNAs was the enzymatic degradation of the tRNA 3′-terminus (..C74C75A76-3′) to yield intermediates with a truncated 3′-terminus (..C74C75-3′) for enzymatic attachment of 3′-amino-3′-deoxyadenosine using the corresponding triphosphate as precursor (20–21). The resulting tRNA would then provide a reactive 3′-amino group, which can be charged enzymatically with an amino acid using the cognate tRNA synthetase (20–23). This method was introduced in the 1970s (20) and has been occasionally applied ever since (21–23); however, the drawback of the approach is its restriction to the attachment of only one amino acid rather than a polypeptide. A further study from the early 1970s describes coupling of N-protected amino acid active esters and anhydrides directly to full-length tRNA (and also to 3′-amino-3′-deoxy tRNA) to achieve short peptidyl-tRNA derivatives (24–25). This approach, however, is not applicable in reliable manner to most tRNAs since these reagents react competitively with functional groups that are encountered in natural tRNA nucleoside modifications. Moreover, only weak ester activation can be used (because of competitive reactivity of the RNA 2′-hydroxyl groups) and thus the disadvantage of long reaction times together with incomplete coupling are further limitations that make this method unreliable.
Here, we present a novel approach for the efficient synthesis of non-hydrolyzable 3′-peptidyl-tRNAs that contain all natural tRNA nucleoside modifications. The basic concept is outlined in Figure 1. In short, we started from natural tRNAs that were cleaved within the TΨC loop by using DNA enzymes to obtain the tRNA 5′-fragment containing all modifications. After dephosphorylation of the 2′,3′-cyclophosphate moiety, this fragment fulfilled the structural requirements for enzymatic ligation to RNA-peptide conjugates that had been prepared according to a solid-phase synthesis approach elaborated previously in our laboratory (12).
Figure 1.

Hydrolysis-resistant 3′-peptidyl-tRNA conjugates. (a) Crucial structural feature of an exemplary target: The amide linkage (highlighted in red). (b) Cartoon presentation of a 3′-peptidyl-tRNA conjugate. Typical positions of natural chemical modifications are shown in red (PDB coordinates 1EVV: S. cerevisiae tRNAPhe). The 3′-terminal sequences (18 nt) of tRNAs are generally unmodified (blue); arrow (cyan) indicates the intended site of tRNA cleavage (by using DNA enzymes) and the site of ligation (to synthetic 3′-peptidylamino-RNA conjugates); (c) concept for the semi-synthesis of non-hydrolyzable 3′-peptidyl-tRNA conjugates (for explanation see main text). Modified nucleosides, N; cyclophosphate, cp; phosphate, p.
MATERIALS AND METHODS
3′-Peptidylamino oligoribonucleotides
The peptide-RNA conjugates 6 were prepared by solid-phase synthesis following Ref. (12). We mention that this reference includes a significant improvement compared to the original concept introduced by Strazewski and colleagues (26), that is the alternative synthesis of the 3′-aminoacylamino-3′-deoxyadenosine key derivative together with the functionalization of an advanced solid-support material (GE Healthcare PS200 Support) resulting in significantly higher yields and being less time consuming.
DNA enzyme-mediated tRNA cleavage
Escherichia coli tRNAVal, E. coli tRNAPhe and Saccharomyces cerevisiae tRNAPhe were purchased from Sigma Aldrich. All DNA strands were obtained from IDT (grade: desalted) and directly used without further purification. For tRNA cleavage, the tRNA and the appropriate DNA enzyme (substrate recognition domains of 17 nt each) were lyophilized to dryness and dissolved in cleavage buffer [50 mM Tris–HCl (pH 7.5), 10 mM MgCl2; for tRNAs containing thio-modified nucleosides (e.g. s4U) 10 mM DTT was added]; ctRNA = 40 µM and cDNAzyme = 80 µM (substrate/enzyme = 1/2). Several rounds of thermal cycling using a heating block (ThermostatPlus; Eppendorf) were performed [S. cerevisiae tRNAPhe: 1 min at 90°C, 5 min at 60°C, 15 min at 25°C (three rounds); E. coli tRNAVal and tRNAPhe: 1 min at 90°C, 5 min at 60°C, 10 min at 35°C, 10 min at 25°C (three rounds), then 5 min at 60°C and 80 min at 25°C]. The reaction was stopped by freezing in liquid nitrogen.
Monitoring of the cleavage reaction was performed by anion-exchange chromatography on a Dionex DNAPac®PA-100 column (4 × 250 mm) at 60°C (80°C for E. coli tRNAVal). Flow rate: 1 ml/min; eluant A: 25 mM Tris–HCl (pH 8.0), 6 M urea; eluant B: 25 mM Tris–HCl (pH 8.0), 6 M urea, 500 mM NaClO4; gradient: 0–60% B in A within 45 min; UV detection at 260 nm. The obtained tRNA fragments were purified on a semi-preparative Dionex DNAPac®PA-100 column (9 × 250 mm) at 60°C (80°C for E. coli tRNAVal). Flow rate: 2 ml/min; gradient: 28–40% B in A within 20 min. Fractions containing the 5′-terminal tRNA fragment were loaded on a C18 SepPak®Plus cartridge (Waters/Millipore), washed with 0.1–0.2 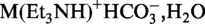 and eluted with H2O/CH3CN (1/1, v/v). The solvent was removed in vacuo and the purified RNA was dissolved in 1.0 ml of nanofiltered water. The yields of RNA were determined as units of optical density at 260 nm by UV spectroscopy (Varian Cary 100) at room temperature.
and eluted with H2O/CH3CN (1/1, v/v). The solvent was removed in vacuo and the purified RNA was dissolved in 1.0 ml of nanofiltered water. The yields of RNA were determined as units of optical density at 260 nm by UV spectroscopy (Varian Cary 100) at room temperature.
Dephosphorylation of 3′-ends
Aliquots of stock solutions that contained the 5′-terminal tRNA 2′,3′-cyclophosphates were lyophilized to dryness and dissolved in nanofiltered water (about two-third of the final total reaction volume, cRNAcp = 15 µM). The solution was heated to 90°C for 1 min and then allowed to cool to room temperature within 15 min. Subsequently, one-tenth of the final reaction volume of reaction buffer [700 mM Tris–HCl (pH 7.6), 100 mM MgCl2, 50 mM DTT = ‘10× reaction buffer’] was added to the mixture. The sample was vortexed and centrifuged before it was treated with T4 polynucleotide kinase (T4 PNK, Promega; 10 U/µl in storage solution) to give a concentration of 0.5 U/µl for S. cerevisiae tRNA ligation and 1.0 U/µl for E. coli tRNA ligation, respectively, in the final reaction volume. After addition of water to obtain the concentration of 15 µM of 5′-terminal tRNA 2′,3′-cyclophosphate, the reaction solution was vortexed again and incubated at 37°C for 6–24 h. Then, the reaction mixture was extracted twice with an equal volume of a phenol/chloroform/isoamyl alcohol solution (25/24/1, v/v/v) and twice with an equal volume of chloroform/isoamyl alcohol solution (24/1, v/v). All organic layers were rewashed once with an equal volume of nanofiltered water; the aqueous phases were combined and lyophilized in vacuo. Monitoring of the dephosphorylation reaction was performed by anion-exchange chromatography on a Dionex DNAPac®PA-100 column (4 × 250 mm) at 60°C under the same conditions as described above.
Enzymatic ligation with T4 RNA ligase
Ligation without DNA splint
Equimolar amounts of acceptor and donor strands (final RNA concentration = 40 µM each strand) were mixed in nanofiltered water (about two-third of the final total reaction volume), the solution was heated to 90°C for 2 min and then allowed to cool to room temperature within 15 min. One-tenth of the final reaction volume of reaction buffer [500 mM HEPES–NaOH (pH 8.0), 100 mM MgCl2, 100 mM DTT = ‘10× ligation buffer’], ATP and BSA from stock solutions were added to the mixture to give concentrations of 1 mM ATP and 0.1 mg/ml BSA (in the final reaction volume). The sample was vortexed and centrifuged before it was treated with T4 RNA ligase (Fermentas; 10 U/µl in storage solution) to give a concentration of 0.5 U/µl (in the final reaction volume). After addition of water to obtain the final reaction volume, the reaction solution was vortexed again and incubated at 37°C for 3 h.
Ligation with DNA splint
Equimolar amounts of acceptor, donor and DNA strands (final concentration = 40 µM each strand) were mixed in nanofiltered water (about two-third of the final total reaction volume), the solution was heated to 90°C for 2 min and then allowed to cool to room temperature within 15 min. One-tenth of the final reaction volume of reaction buffer [500 mM HEPES–NaOH (pH 8.0), 100 mM MgCl2, 100 mM DTT = ‘10× ligation buffer’], ATP and BSA from stock solutions were added to the mixture to give concentrations of 0.5 mM ATP and 0.1 mg/ml BSA (in the final reaction volume). The sample was vortexed and centrifuged before it was treated with T4 RNA ligase (Fermentas; 10 U/µl in storage solution) to give a concentration of 0.25 U/µl (in the final reaction volume). After addition of water to obtain the final reaction volume, the reaction solution was vortexed again and incubated at 37°C for 3 h.
General work-up
The reaction mixture was extracted twice with an equal volume of a phenol/chloroform/isoamyl alcohol solution (25/24/1, v/v/v) and twice with an equal volume of chloroform/isoamyl alcohol solution (24/1, v/v). All organic layers were rewashed once with an equal volume of nanofiltered water; the aqueous phases were combined and reduced by ∼50%. Monitoring of the ligation reaction and purification of the product were performed by anion-exchange chromatography on a Dionex DNAPac®PA-100 column (4 × 250 mm or 9 × 250 mm; gradient for purification: 31–44% B in A within 20 min) at 60°C under the same conditions as described above.
LC-ESI mass spectrometry
All experiments were performed on a Finnigan LCQ Advantage MAX ion trap instrumentation (Thermo Fisher Scientific) connected to an Amersham Ettan Micro HPLC system (GE Healthcare). Oligonucleotides were analyzed in the negative-ion mode with a potential of −4 kV applied to the spray needle. Oligonucleotides (100–160 pmol) in stock solution were dried in vacuo and redissolved in 30 µl of 20 mM EDTA solution; column (XTerra®MS, C18, particle size 2.5 µm, 1.0 × 50 mm; Waters) at 21°C; flow rate: 30 µl/min; eluant A: 8.6 mM triethylamine, 100 mM 1,1,1,3,3,3-hexafluoroisopropanol in H2O (pH 8.0); eluant B: methanol; gradient: 0–100% B in A within 20 min; UV detection at 254 nm. Prior to each injection, column equilibration was performed by buffer A for 30 min at a flow rate of 30 µl/min.
RESULTS AND DISCUSSION
tRNA cleavage in the TΨC loop by DNA enzymes
For the first step of our undertaking, namely the DNA enzyme catalyzed cleavage of tRNA, we chose natural tRNA together with synthetic 10–23 type DNA enzymes (27). The site of tRNA cleavage was placed between A58 and U59 for two reasons. First, these nucleosides are highly conserved and represent an established dinucleotide sequence for cleavage by 10–23 DNA enzymes (5′-..AU..-3′) (28,29) and second, they provide ideal nucleobase termini (purine at the 3′-end of the acceptor strand and pyrimidine at the 5′-end of the donor strand) for high-yield enzymatic ligation using T4 RNA ligase later on (30,31). Furthermore, this cleavage site has the advantage that the resulting 58-nt tRNA 5′-fragment contains all natural modifications, while the remaining 18-nt tRNA 3′-fragment that is generally unmodified in tRNA can be disposed. The A58/U59 site provides another convenience with respect to the ligation of 3′-amino-3′-deoxyribose-linked RNA-peptide conjugates: from previous studies (12), we experienced that the RNA portion should contain about the same number of nucleoside units (or more) compared to the number of amino acids of the peptide portion to make these compounds unrestrictedly water soluble. In this sense, we were confident to be able to attach the peptides of our interest to tRNA, which are in particular dipeptides (of varying sequence) and macrolide-resistance peptides (pentapeptides) (10,11).
To evaluate our concept we chose three commercially available tRNAs, namely E. coli tRNAVal, E. coli tRNAPhe and S. cerevisiae tRNAPhe. Figure 2 shows the exemplary cleavage of E. coli tRNAVal 1 by the 10–23 DNA enzyme 2 with 17-nt long recognition arms. Temperature cycles were required to push tRNA cleavage almost toward completeness within 3 h (for details see ‘Materials and Methods’ section). The tRNA 5′-fragment 3 was purified by anion-exchange chromatography under denaturing conditions and the correct molecular weight was confirmed by LC-ESI mass spectrometry. Also for E. coli tRNAPhe and S. cerevisiae tRNAPhe, cleavage by the corresponding 10–23 DNA enzymes was very efficient (see Supplementary Data). It is noteworthy that S. cerevisiae tRNAPhe possesses N1-methyladenosine as a modified nucleoside directly at the site of cleavage, which is tolerated by the DNA enzyme.
Figure 2.

Example for the cleavage of a natural tRNA by DNA enzymes. (a) Secondary structure of E. coli tRNAVal 1 and 10–23 DNA enzyme 2. The desired tRNA 5′-fragment 3 contains all modified nucleosides and possesses a 2′,3′-cyclophosphate terminus. (b) Anion-exchange HPLC analysis of the cleavage reaction (88% yield according to peak area integration) and the purified fragment; reaction conditions: cDNAzyme = 80 µM; ctRNA = 40 µM; 50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 10 mM DTT, temperature cycles (25–90°C); anion-exchange HPLC: for conditions see ‘Materials and Methods’ section. (c) LC-ESI MS analysis of 3: m.w. (calcd) = 19113, m.w. (found) = 19112 ± 10. For structures and abbreviations of modified nucleosides, see Supplementary Data.
Dephosphorylation
The tRNA 5′-fragments that were obtained by 10–23 DNA enzyme cleavage carried 2′,3′-cyclophosphate groups. These termini cannot be used as substrates for the intended enzymatic ligation to synthetic 3′-peptidylamino-RNA conjugates using T4 RNA ligase. Therefore, we applied T4 PNK that is known to have 3′-phosphatase activity to release the cyclophosphate and to generate terminal ribose moieties with free 2′-OH and 3′-OH groups (32). The enzymatic reaction proceeded smoothly within 6 h for the tRNA 5′-fragments 3 of E. coli tRNAPhe (Figure 3) and S. cerevisiae tRNAPhe 2′,3′-cyclophosphates (Supplementary Data), but required longer reaction times (24 h) and higher enzyme concentrations for E. coli tRNAVal derived fragment 3 (Supplementary Data). We speculate that a significantly stable misfold of E. coli tRNAVal fragment 3 could be the reason to retard recognition by the enzyme. The tRNA 5′-fragments 5 were purified by phenol/chloroform extraction and the correct molecular weight was confirmed by LC-ESI mass spectrometry. We mention that for all three types of tRNA tested, the difference in HPLC retention times between educt 3 and product 5 was either very slight or could not be resolved at all. Therefore, we recommend MS analysis for product verification in any case (Figure 3 and Supplementary Data).
Figure 3.

Example for enzymatic dephosphorylation of the 2′,3′-cyclophosphate at the tRNA 5′-fragment. (a) Structure of the 5′-fragment from E. coli tRNAPhe 2′,3′-cyclophosphate 3 and product 5 after exposure to T4 PNK. (b) The PNK reaction was monitored by anion-exchange HPLC analysis. The difference in retention time between 3 and 5 is marginal; reaction conditions: T4 PNK (0.5 U/µl; cRNA = 15 µM; 70 mM Tris–HCl (pH 7.6), 10 mM MgCl2, 5 mM DTT, 37°C. (c) LC-ESI MS analysis of 5: m.w. (calcd) = 19045, m.w. (found) = 19046 ± 10. Anion-exchange HPLC: for conditions see ‘Materials and Methods’ section. For structures and abbreviations of modified nucleosides see Supplementary Data.
Enzymatic ligation of 3′-peptidylamino-tRNA
Having the desired tRNA 5′-fragments 5 with all natural modifications in our hands, we moved on to the final step, namely enzymatic ligation with synthetic 3′-peptidylamino-RNA conjugates 6 to yield the target compounds 8. The usage of T4 RNA ligase seemed appropriate since the 18-nt RNA portion of the synthetic conjugate was expected to form a perfectly base paired and sufficiently stable preligation complex to bring the 5′-phosphate of the donor strand 6 (synthetic conjugate) in close proximity to the 3′-OH group of the acceptor strand 5 (58-nt tRNA fragment). This is a prerequisite for efficient ligation with T4 RNA ligase (30,31,33–36).
Figure 4 depicts the ligation set-up for E. coli tRNAPhe. The two strands 5 and 6 aligned properly and efficient ligation was observed after 3 h. The full-length tRNA-peptide conjugate 8 was purified by anion-exchange chromatography and the correct molecular weight was confirmed by LC-ESI mass spectrometry. Unexpectedly, the analogous experimental set-up did not work for E. coli tRNAVal and S. cerevisiae tRNAPhe (Figure 5). However, when using the non-modified tRNA sequence homologues of 5 in combination with synthetic conjugates 6, ligation worked efficiently. To rationalize this behavior two scenarios are conceivable: in their natural counterparts, the nucleoside modifications of these particular tRNA fragments 5 either stabilize structures (of 5), which render proper alignment with 6 impossible, or the binary preligation complex of 5 and 6 can form but is stabilized by the modifications in such a manner (tRNA tertiary fold) that the ‘nicked’ tRNA is no longer accessible to the ligase. As a resort, we considered splint-supported ligation with T4 RNA ligase. Indeed, when we applied the DNA template 7 to the ligation reaction with S. cerevisiae tRNAPhe sequences, we observed ligation of the two fragments with reasonable yields (Figure 5c). The same holds true for E. coli tRNAVal (Supplementary Data).
Figure 4.

Example for enzymatic ligation of fully modified tRNA 5′-fragments to synthetic 3′-peptidylamino-RNA conjugates. (a) Structures of the 5′-fragment from E. coli tRNAPhe 5 and the dipeptide-RNA conjugate 6 to form a preligation complex that allows T4 RNA ligation of the full-length tRNA-peptide conjugate 8. (b) The ligation reaction was monitored by anion-exchange HPLC analysis: 83% yield was achieved after 3 h; reaction conditions: T4 RNA ligase (0.5 U/µl; cRNA = 40 µM each strand; donor/acceptor = 1/1), 50 mM HEPES–NaOH (pH 8.0), 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 0.1 mg/ml BSA, 37°C. (c) Purified 3′-peptidyl-tRNA; (d) LC-ESI MS analysis of 8: m.w. (calcd) = 25030, m.w. (found) = 25029 ± 10. Anion-exchange HPLC: for conditions see ‘Materials and Methods’ section. For structures and abbreviations of modified nucleosides see Supplementary Data.
Figure 5.

Example for splint-assisted enzymatic ligation of fully modified tRNA 5′-fragments to synthetic 3′-peptidylamino-RNA conjugates. (a) Structures of the 5′-fragment from S. cerevisiae tRNAPhe 5 and the dipeptide-RNA conjugate 6 to form a preligation complex that allows T4 RNA ligation of the full-length tRNA-peptide conjugate 8. (b) Without splint 7 only marginal amounts of product 8 were formed; reaction conditions: T4 RNA ligase (0.5 U/µl; cRNA = 40 µM each strand; donor/acceptor = 1/1), 50 mM HEPES–NaOH (pH 8.0), 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 0.1 mg/ml BSA, 37°C. (c) Ligation promoted by splint 7 resulted in 75% yield of 8. The reaction was monitored by anion-exchange HPLC (for conditions see ‘Materials and Methods’ section); an unidentified, unreactive impurity is marked by an asterisk; reaction conditions: T4 RNA ligase (0.25 U/µl; cRNA = 40 µM each strand; cDNA = 40 µM; donor/acceptor/splint = 1/1/1), buffer as in (b) and 0.5 mM ATP, 37°C. For structures and abbreviations of modified nucleosides see Supplementart Data.
The DNA splint 7 was designed to bring the nucleotide termini of 5 and 6 in close proximity by forming a stable ternary preligation complex (Figure 5a), in analogy to two reports on RNA ligation in the literature (37,38). We also tested splints that form a fully base paired, nicked duplex at the site of ligation, which should be appropriate for ligation using T4 DNA ligase. However, in the case of S. cerevisiae tRNAPhe, no ligation product was detectable, most likely because N1-methyladenine-58 prevented the adoption of Watson–Crick base pair geometry at the site of ligation, which is required for this ligase type (39,40). Another peculiarity that we observed only for ligations concerning S. cerevisiae tRNAPhe was the mass spectrometric detection of a 3′-peptidylamino-tRNA byproduct that most likely carried an A-5′-pp-5′-G(1) terminus (Supplementary Data). The formation of such a byproduct can be explained by the T4 RNA ligase activity (31,41). Consuming ATP, the enzyme transfers 5′-p-A to 5′-phosphorylated RNA and thereby generates an adenylated RNA donor intermediate. This activated species then reacts with the ribose 3′-OH of the acceptor RNA strand. If no acceptor strand is available (or if it reacts slowly), the A-5′-pp-5′-RNA intermediate can be isolated. In our experiments, the formation of the S. cerevisiae A-5′-pp-5′-tRNAPhe byproduct was significantly reduced when using only a small excess of ATP during the ligation reaction and this observation also supports the suggested structure of the byproduct (Supplementary Data).
Table 1 summarizes a selection of 3′-peptidyl-tRNA conjugates synthesized in this study and provides their total (isolated) amounts. We mention that the average overall yield is estimated to ∼20% starting from the natural tRNA species and considering each of the three steps of preparation after purification.
Table 1.
Selection of 3′-peptidylamino-tRNA conjugates synthesizeda
| Conjugate sequencea | Yieldb (pmol) |
|---|---|
| E. coli tRNAPhe-3′-NH-FA-NH2 | 210 |
| S. cerevisiae tRNAPhe-3′-NH-FA-NH2 | 2800 |
| E. coli tRNAVal-3′-NH-VFLVM-NH2 | 510 |
| S. cerevisiae tRNAPhe-3′-NH-FLLVF-NH2 | 380 |
aFor the chemical structure of the RNA-peptide linkage see Figure 1.
bAfter purification.
Conclusions
With the approach presented here, we have laid the basis for convenient chemical modification and labeling of the 3′-terminus of natural tRNAs. The use of DNA enzymes as a tool for tRNA manipulation has been demonstrated by Helm and coworkers (42,43) for analytical purposes; however, to the best of our knowledge, DNA enzymes have not been integrated into synthetic concepts for tRNA derivatives before. We also mention that our approach is promising to be applicable to many kinds of tRNA, which are of different amino acid specificity and from various organisms. From the three examples shown here, it becomes evident that finetuning of the conditions, e.g. with respect to the length of the recognition arms of the DNA enzymes or the length of the splint for ligation might be necessary to optimize yields.
Moreover, the present approach is not restricted to the type of conjugates shown here, but basically appropriate for any kind of chemical modification that can be attached and introduced via the 18-nt tRNA terminal sequence. This will, for example, open up a variety of new positions for fluorescent labeling of natural tRNAs that have been largely inaccessible to date. Such tRNAs are of growing interest for the investigation of ribosome translation kinetics using single-molecule fluorescence techniques (5).
With the combined synthetic and enzymatic approach, we have abolished the preparative bottleneck for hydrolysis-resistant 3′-peptidyl-tRNA that are highly requested for ribosomal studies to achieve detailed molecular insights into structure and dynamics of pre- and postpeptidyl transfer states and of tRNA hybrid states, but also of translation termination and ribosome stalling. Our own research will integrate these conjugates into investigations on macrolide antibiotic resistance induced by short resistance peptides (10–12).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Austrian Science Foundation Fonds zur Förderung der wissenschaftlichen Forschung (P21641, I317 to R.M.; Y315 to N.P.); Ministry of Science and Research (GenAU project ‘Non-coding RNAs’, P0726-012-012 to R.M. and P110420-012-012 to N.P.). Funding for open access charge: Austrian Science Foundation Fonds zur Förderung der wissenschaftlichen Forschung.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Wilson DN, Nierhaus KH. The ribosome through the looking glass. Angew. Chem. Int. Ed. Engl. 2003;42:3464–3486. doi: 10.1002/anie.200200544. [DOI] [PubMed] [Google Scholar]
- 2.Voorhees RM, Weixlbaumer A, Loakes D, Kelley AC, Ramakrishnan V. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat. Struct. Mol. Biol. 2009;16:528–533. doi: 10.1038/nsmb.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lang K, Erlacher M, Wilson DN, Micura R, Polacek N. The role of 23S ribosomal RNA residue A2451 in peptide bond synthesis revealed by atomic mutagenesis. Chem. Biol. 2008;15:485–492. doi: 10.1016/j.chembiol.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, Dunkle JA, Cate JH. Structures of the ribosome in intermediate states of ratcheting. Science. 2009;325:1014–1017. doi: 10.1126/science.1175275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munro JB, Altman RB, O’Connor N, Blanchard SC. Identification of two distinct hybrid state intermediates on the ribosome. Mol. Cell. 2007;25:505–517. doi: 10.1016/j.molcel.2007.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker SE, Shoji S, Pan D, Cooperman BS, Fredrick K. Role of hybrid tRNA-binding states in ribosomal translocation. Proc. Natl Acad. Sci. USA. 2008;105:9192–9197. doi: 10.1073/pnas.0710146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weixlbaumer A, Jin H, Neubauer C, Voorhees RM, Petry S, Kelley AC, Ramakrishnan V. Insights into translational termination from the structure of RF2 bound to the ribosome. Science. 2008;322:953–956. doi: 10.1126/science.1164840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seidelt B, Innis CA, Wilson DN, Gartmann M, Armache JP, Villa E, Trabuco LG, Becker T, Mielke T, Schulten K, et al. Structural insight into nascent polypeptide chain-mediated translational stalling. Science. 2009;326:1412–1415. doi: 10.1126/science.1177662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhushan S, Gartmann M, Halic M, Armache JP, Jarasch A, Mielke T, Berninghausen O, Wilson DN, Beckmann R. Alpha-helical nascent polypeptide chains visualized within distinct regions of the ribosomal exit tunnel. Nat. Struct. Mol. Biol. 2010;17:313–317. doi: 10.1038/nsmb.1756. [DOI] [PubMed] [Google Scholar]
- 10.Mankin AS. Macrolide myths. Curr. Opin. Microbiol. 2008;11:414–421. doi: 10.1016/j.mib.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tenson T, Mankin AS. Short peptides conferring resistance to macrolide antibiotics. Peptides. 2001;22:1661–1668. doi: 10.1016/s0196-9781(01)00501-0. [DOI] [PubMed] [Google Scholar]
- 12.Moroder H, Steger J, Graber D, Fauster K, Trappl K, Marquez V, Polacek N, Wilson DN, Micura R. Non-hydrolyzable RNA-peptide conjugates: a powerful advance in the synthesis of mimics for 3′-peptidyl tRNA termini. Angew. Chem. Int. Ed. Engl. 2009;48:4056–4060. doi: 10.1002/anie.200900939. [DOI] [PubMed] [Google Scholar]
- 13.Ogilvie KK, Usman N, Nicoghosian K, Cedergren RJ. Total chemical synthesis of a 77-nucleotide-long RNA sequence having methionine-acceptance activity. Proc. Natl Acad. Sci. USA. 1988;85:5764–5768. doi: 10.1073/pnas.85.16.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perreault JP, Pon RT, Jiang MY, Usman N, Pika J, Ogilvie KK, Cedergren R. The synthesis and functional evaluation of RNA and DNA polymers having the sequence of Escherichia coli tRNA(fMet) Eur. J. Biochem. 1989;186:87–93. doi: 10.1111/j.1432-1033.1989.tb15181.x. [DOI] [PubMed] [Google Scholar]
- 15.Chaix C, Duplaa AM, Molko D, Téoule R. Solid phase synthesis of the 5′-half of the initiator t-RNA from B. subtilis. Nucleic Acids Res. 1989;17:7381–7393. doi: 10.1093/nar/17.18.7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sherlin LD, Bullock TL, Nissan TA, Perona JJ, Lariviere FJ, Uhlenbeck OC, Scaringe SA. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA. 2001;7:1671–1678. [PMC free article] [PubMed] [Google Scholar]
- 17.Miller JT, Khvorova A, Scaringe SA, Le Grice SF. Synthetic tRNALys,3 as the replication primer for the HIV-1HXB2 and HIV-1Mal genomes. Nucleic Acids Res. 2004;32:4687–4695. doi: 10.1093/nar/gkh813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grosjean H, Benne R, editors. Modification and Editing of RNA. Washington, DC: ASM Press; 1998. [Google Scholar]
- 19.Brückl T, Globisch D, Wagner M, Müller M, Carell T. Parallel isotope-based quantification of modified tRNA nucleosides. Angew. Chem. Int. Ed. Engl. 2009;42:7932–7934. doi: 10.1002/anie.200902740. [DOI] [PubMed] [Google Scholar]
- 20.Fraser TH, Rich A. Synthesis and aminoacylation of 3′-amino-3'-deoxy transfer RNA and its activity in ribosomal protein synthesis. Proc. Natl Acad. Sci. USA. 1973;70:2671–2675. doi: 10.1073/pnas.70.9.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sprinzl M, Faulhammer HG. Participation of X47-fluorescamine modified E. coli tRNAs in in vitro protein biosynthesis. Nucleic Acids Res. 1978;5:4837–4840. doi: 10.1093/nar/5.12.4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramesh V, Mayer C, Dyson MR, Gite S, RajBhandary UL. Induced fit of a peptide loop of methionyl-tRNA formyltransferase triggered by the initiator tRNA substrate. Proc. Natl Acad. Sci. USA. 1999;96:875–880. doi: 10.1073/pnas.96.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayer C, RajBhandary UL. Conformational change of Escherichia coli initiator methionyl-tRNA(fMet) upon binding to methionyl-tRNA formyl transferase. Nucleic Acids Res. 2002;30:2844–2850. doi: 10.1093/nar/gkf411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lapidot Y, de Groot N. The chemical synthesis and the biochemical properties of peptidyl-tRNA. Prog. Nucleic Acid Res. Mol. Biol. 1972;12:189–228. doi: 10.1016/s0079-6603(08)60663-7. [DOI] [PubMed] [Google Scholar]
- 25.Shiloach J, Lapidot Y, de Groot N, Sprinzl M, Cramer F. The specificity of peptidyl-tRNA hydrolase from. E. coli. FEBS Lett. 1975;57:130–133. doi: 10.1016/0014-5793(75)80700-9. [DOI] [PubMed] [Google Scholar]
- 26.Terenzi S, Biała E, Nguyen-Trung NQ, Strazewski P. Amphiphilic 3′-peptidyl-RNA conjugates. Angew. Chem. Int. Ed. Engl. 2003;42:2909–2912. doi: 10.1002/anie.200350926. [DOI] [PubMed] [Google Scholar]
- 27.Santoro SW, Joyce GF. A general purpose RNA-cleaving DNA enzyme. Proc. Natl Acad. Sci. USA. 1997;94:4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Höbartner C, Silverman SK. Recent advances in DNA catalysis. Biopolymers. 2007;87:279–292. doi: 10.1002/bip.20813. [DOI] [PubMed] [Google Scholar]
- 29.Silverman SK, Baum DA. Use of deoxyribozymes in RNA Research. Meth. Enzymol. 2009;469:95–117. doi: 10.1016/S0076-6879(09)69005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Persson T, Willkomm DK, Hartmann RK. T4 RNA ligase. In: Hartmann RK, Bindereif A, Schön A, Westhof E, editors. Handbook of RNA Biochemistry. Weinheim, Germany: Wiley-VCH; 2005. pp. 53–74. [Google Scholar]
- 31.Arn EA, Abelson J. RNA ligases: function, mechanism, and sequence conservation. In: Simons RW, Grunberg-Manago M, editors. RNA Structure and Function. New York, USA: CSHL Press; 1998. pp. 695–726. [Google Scholar]
- 32.Schürer H, Lang K, Schuster J, Mörl M. A universal method to produce in vitro transcripts with homogeneous 3′ ends. Nucleic Acids Res. 2002;30:e56. doi: 10.1093/nar/gnf055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lang K, Micura R. The preparation of site-specifically modified riboswitch domains as an example for enzymatic ligation of chemically synthesized RNA fragments. Nat. Protoc. 2008;3:1457–1466. doi: 10.1038/nprot.2008.135. [DOI] [PubMed] [Google Scholar]
- 34.Höbartner C, Micura R. The chemical synthesis of selenium-modified oligoribonucleotides and their enzymatic ligation leading to an U6 snRNA stem-loop segment. J. Am. Chem. Soc. 2004;126:1141–1149. doi: 10.1021/ja038481k. [DOI] [PubMed] [Google Scholar]
- 35.Höbartner C, Rieder R, Kreutz C, Puffer B, Lang K, Polonskaia A, Serganov A, Micura R. Syntheses of RNAs with up to 100 nucleotides containing site-specific 2′-methylseleno labels for use in X-ray crystallography. J. Am. Chem. Soc. 2005;127:12035–12045. doi: 10.1021/ja051694k. [DOI] [PubMed] [Google Scholar]
- 36.Rieder R, Höbartner C, Micura R. Enzymatic ligation strategies for the preparation of purine riboswitches with site-specific chemical modifications. Riboswitches. Meth. Mol. Biol. 2009;540:15–24. doi: 10.1007/978-1-59745-558-9_2. [DOI] [PubMed] [Google Scholar]
- 37.Stark MR, Pleiss JA, Deras M, Scaringe SA, Rader SD. An RNA ligase-mediated method for the efficient creation of large, synthetic RNAs. RNA. 2006;12:2014–2019. doi: 10.1261/rna.93506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bain JD, Switzer C. Regioselective ligation of oligoribonucleotides using DNA splints. Nucleic Acids Res. 1992;20:4372. doi: 10.1093/nar/20.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moore MJ, Query CC. Joining of RNAs by splinted ligation. Meth. Enzymol. 2000;317:109–123. doi: 10.1016/s0076-6879(00)17009-0. [DOI] [PubMed] [Google Scholar]
- 40.Kurschat W, Müller J, Wombacher R, Helm M. Optimizing splinted ligation of highly structured small RNAs. RNA. 2005;11:1909–1914. doi: 10.1261/rna.2170705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruce AG, Uhlenbeck OC. Reactions at the termini of tRNA with T4 RNA ligase. Nucleic Acids Res. 1978;5:3665–3677. doi: 10.1093/nar/5.10.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hengesbach M, Meusburger M, Lyko F, Helm M. Use of DNAzymes for site-specific analysis of ribonucleotide modifications. RNA. 2008;14:180–187. doi: 10.1261/rna.742708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hengesbach M, Voigts-Hoffmann F, Hofmann B, Helm M. Formation of a stalled early intermediate of pseudouridine synthesis monitored by real-time FRET. RNA. 2010;16:610–620. doi: 10.1261/rna.1832510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.