


Abstract
Identification of rare variants by resequencing is important both for detecting novel variations and for screening individuals for known disease alleles. New technologies enable low-cost resequencing of target regions, although it is still prohibitive to test more than a few individuals. We propose a novel pooling design that enables the recovery of novel or known rare alleles and their carriers in groups of individuals. The method is based on a Compressed Sensing (CS) approach, which is general, simple and efficient. CS allows the use of generic algorithmic tools for simultaneous identification of multiple variants and their carriers. We model the experimental procedure and show via computer simulations that it enables the recovery of rare alleles and their carriers in larger groups than were possible before. Our approach can also be combined with barcoding techniques to provide a feasible solution based on current resequencing costs. For example, when targeting a small enough genomic region (∼100 bp) and using only ∼10 sequencing lanes and ∼10 distinct barcodes per lane, one recovers the identity of 4 rare allele carriers out of a population of over 4000 individuals. We demonstrate the performance of our approach over several publicly available experimental data sets.
INTRODUCTION
Genome-wide association studies (GWASs) (1) have been successfully used in recent years to detect associations between genotype and phenotype, and numerous new alleles have been found to be linked to various human traits (2–4). However, genotyping technologies are limited to only those variants that are pre-determined and prioritized for typing, which results in a bias towards typing of common alleles.
Although many common alleles were lately found to have statistically significant associations with different human traits, they were thus far shown to explain only a small fraction of most traits' heritability content. This, together with other theoretical and empirical arguments, raise the possibility that in fact rare alleles may play a significant role in the susceptibility of human individuals to many common diseases (5–8). Discovering and genotyping of rare alleles may, therefore, be of great bio-medical interest. However, such studies require genotyping of large human populations—a task considered infeasible until recently.
This state of affairs may change dramatically as we are currently witnessing a rapid revolution in genome sequencing due to emerging new technologies. Sequencing throughput at a given cost is growing at an exponential rate, in similar to Moore's law for computer hardware (9). Next-generation sequencing technologies (10–12) utilize massively parallel reading of short genomic fragments to achieve several orders of magnitude higher throughput at the same cost as previous Sanger sequencing machines (9). The availability of cheap, high-throughput rapid sequencing methods leads to a change in the way researchers approach various biological problems, as it enables addressing questions that were infeasible to be studied before.
Next-generation sequencing opens the possibility to obtain the genomic sequences of multiple individuals along specific regions of interest. This approach, often called resequencing, is likely to provide an extensive amount of novel information on human genetic variation. In particular, the ability to resequence a large number of individuals will enable the study of rare alleles in human populations. Resequencing of large populations can, thus, fill a gap in our knowledge by allowing us to discover and type these rare variants, often with frequencies well below 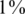 , at given pre-defined regions. Of particular interest are regions around genes or loci that have previously been established for the involvement in disease, as they can be resequenced across a large population to seek novel variations. A proof of principle of the approach was demonstrated for Diamond–Blackfan anemia (13) and breast cancer (14), with many more such studies likely to be conducted in the near future.
, at given pre-defined regions. Of particular interest are regions around genes or loci that have previously been established for the involvement in disease, as they can be resequenced across a large population to seek novel variations. A proof of principle of the approach was demonstrated for Diamond–Blackfan anemia (13) and breast cancer (14), with many more such studies likely to be conducted in the near future.
Another important application of interest is resequencing a set of specific known single nucleotide polymorphisms (SNPs) with low minor allele frequency, which are known or suspected to be important for a certain trait. In this case, we are interested in identifying individuals carrying these alleles out of a very large group of individuals. For example, this may assist in screening large populations for individuals carrying certain risk alleles for a potentially lethal disease such as Tay–Sachs or cystic fibrosis.
The two applications mentioned above can be treated in a unified framework: namely, identifying the genotypes of all individuals in pre-defined genomic loci. When detecting known alleles, carriers of the rare allele are, thus, identified. When identifying novel variants, some of the genotypes identified are different than the reference human genome in single-base genomic positions, and represent a discovery of new rare alleles. Our approach addresses both applications, and throughout the article the term `identifying rare-allele carriers' refers to both of them.
Current next-generation sequencing technologies provide throughput on the order of millions of reads in a single `run’ or `lane’ (9), where a sequence read is typically a short consecutive DNA fragment of a few dozens to a few hundreds nucleotides. In addition, novel experimental procedures enable targeted selection of pre-defined genomic regions prior to sequencing (15). When performing targeted sequencing for one or a few small regions, region enrichment can be achieved via the use of traditional PCR or novel technologies such as Rainstorm. When sequencing a larger number of relatively large regions, the use of `hybrid capture’ (or `hybrid selection') (15–18) enables significant enrichment of the DNA or RNA within the regions of interest, whose total combined length might be on the order of millions of nucleotides, and minimizes the number of reads `wasted’ on fragments residing outside these regions. Together, these high-throughput technologies have made the identification of carriers over a pre-defined region a feasible, yet still an expensive task.
A naive but costly option is to utilize one lane per individual. However, when considering a population of hundreds or thousands of individuals, such an approach is prohibitively expensive. Moreover, since resequencing is typically performed on targeted regions rather than the whole genome, throughput requirements to sequence an individual are much lower than the capacity of a single lane, thus the naive approach is also highly inefficient.
In such cases, ‘pooled’ sequence runs may offer a more feasible approach. In ‘pooled DNA’ experiments, DNA from several individuals is mixed and sequenced together on a single sequencing lane. Pooled genotyping has been used to quantify previously identified variations and study allele frequency distributions in populations (19–21). Given a measurement for each allele, it is possible to estimate the average frequency of the allele in those individuals participating in the pool. However, traditional pooled sequencing was used only to infer the frequency of rare alleles in a population, and did not give means to recover the identity of the rare-allele carriers. In this work, we focus on the latter task, of identifying rare-allele variants and their carriers by sequencing the pooled DNA.
The field of group testing (22) aims to tackle this problem of identifying individuals carrying a certain trait out of a group, by designing an efficient set of tests, i.e. pools. This field, which dates back to the mid-20th century has applications in several fields including molecular biology (22). Recently, several works have tried to use resequencing-based group testing methods in order to identify rare-allele carriers.
Prabhu and Pe'er (23) offered to use overlapping pools, elegantly designed based on error-correcting codes, to enable the recovery of a single rare-allele carrier from multiple pools. Individuals are represented in multiple pools, where the composition of different pools is constructed in a way that provides a unique pooling `signature’ for each individual. This carefully designed scheme enables the recovery of a rare-allele carrier by observing the presence of reads containing the rare allele in these `signature’ pools. Their design offers a significant saving in resources, as it enables the recovery of a single carrier out of N individuals, by using only a number of pools logarithmic in N. However, this method is limited to the case of a single rare-allele carrier within the group, and the problem of detecting multiple (albeit few) carriers remained unsolved. [The group testing literature does offer means of addressing the multiple carrier case, e.g. (24–26)].
In another approach by Erlich et al. (27), a clever barcoding scheme combined with pooling was used, in order to enable the identification of each sample's genotypes. When using barcoding, each sample is ‘marked’ by a unique short sequence identifier, i.e. barcode, thus upon sequencing one can identify the origin of each read according to its barcode, even when multiple samples are mixed in a single lane. Ideally, one could assign a different barcode to each individual sample, and then mix many samples in each lane while keeping the identity of each read based on its barcode. However, barcoding is a costly and laborious procedure, and one wishes to minimize the number of barcodes used. It was, therefore, suggested in (27) to barcode different pools of samples (rather than individual samples), thus allowing the barcode to identify the pool from which a certain read was obtained, but not the identity of the specific sample. Efficient algorithms based on the Chinese Remainder Theorem enable the accurate recovery of rare-allele carriers, where both the total number of pools and the number of individual samples participating in each pool were kept low—the identification of N individual genotypes was obtained by using 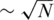 different pools with
different pools with 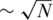 individuals per pool.
individuals per pool.
In this work, we present a different approach to recovering rare-allele variants and the identity of individuals carrying them, based on Compressed Sensing (CS). CS and group testing are intimately connected (28), and our work can be seen as an application of this approach in the context of rare-allele identification [a somewhat similar approach has independently been developed by Erlich et al. (29)]. Our work extends the idea of recovering the identity of rare-allele carriers using overlapping pools beyond the single carrier case analyzed in (23), and deals with heterozygous or homozygous rare alleles. The CS pooling approach enables testing of a larger cohort of individuals, thus identifying carriers of rarer SNPs. The CS paradigm also adapts naturally and efficiently to the addition of barcodes. We propose CS as a simple, generic and highly useful approach to identifying rare-allele carriers.
CS (30,31) is a new emerging and very active field of research, with foundations in statistics and optimization. New developments, updates and research papers in CS appear literally on a daily basis, in various websites (e.g. http://dsp.rice.edu/cs) and blogs (e.g. http://nuit-blanche.blogspot.com/). Applications of the CS theory can be found in many distantly related fields such as magnetic resonance imaging (32), single-pixel cameras (33), geophysics (34), astronomy (35) and multiplexed DNA microarrays (36).
In CS, one wishes to efficiently reconstruct an unknown vector of values x = (x1, … , xN), assuming that x is sparse, i.e. has at most s non-zero entries, for some s ≪ N. It has been shown that x can be reconstructed using k ≪ N basic operations termed ‘measurements', where a measurement is simply the output y of the dot-product of the (unknown vector) x with a known measurement vector m, y = m·x. By using the values of these k measurements and their corresponding m's, it is then possible to reconstruct the original sparse vector x.
Mapping of group testing into a CS setting is simple. The entries of x contain the genotype of each individual at a specific genetic locus and are non-zero only for minor allele carriers; thus, since we are interested in rare alleles x is indeed sparse. A measurement in our setting corresponds to sequencing the DNA of a pool of several individuals taken together, hence the measurement vector represents the individuals participating in a given pool and the output of the measurement is proportional to the total number of rare alleles in the pool. Our basic unit of operation is a single `run’ or `lane', which is used to sequence L pre-defined different loci in the genome, whether consecutive in one specific region, taken from different genomic regions or surrounding different SNPs. We treat each of the L different loci separately and reconstruct L different vectors x, thus the amount of computation increases linearly with the number of loci of interest.
Formulating the problem in terms of CS opens the door to utilizing this rich theory for our purposes. In particular, when designing a pooling experiment, one can use theoretical results such as CS bounds to estimate the number of samples and pools needed for successful reconstruction, and the robustness of the reconstruction to noise. At the data analysis stage, when trying to reconstruct the rare-allele carriers, we can apply numerous algorithms and techniques available for CS problems, and benefit from the development of faster and more accurate reconstruction algorithms as the state of the art is constantly improving (37). We thus argue that CS is a suitable approach for identifying rare-allele carriers, and hope that this article is merely a first step in this direction.
In this work, we present results of extensive simulations, which aim to explore the benefits and limitations of applying CS for the problem of identifying carriers of rare alleles in different scenarios. We provide a detailed model of the experimental procedure typical to next generation sequencing and find scenarios in which the benefit of applying CS is overwhelmingly large (up to over ∼70× improvement) compared to the naive one-individual-per-lane approach. We also show that our method can be used in addition to barcodes, to provide a significant improvement over applying either barcoding or CS solely.
In Appendices 1–3, we provide applications of the CS approach to experimental data. Appendix 1 is based on pooled data by Out et al. (38), which contains next generation sequencing of several SNPs in a single pool of 88 individuals. We have `transformed’ their data into the CS setting, which requires several pools, by performing a bootstrap simulation based on read statistics obtained from the actual data. This enabled us to perform a more realistic analysis, which approximates true pooling experimental data, and was shown to be in good accordance with our simulations’ results. In Appendix 2 we provide analysis of next generation sequences obtained in the Pilot 3 study of the 1000 Genomes Project. By using experimental reads to simulate pools, we show the applicability of the CS approach to large scale SNP identification using only a small fraction of the required sequencing lanes. Finally, we analyzed a data set that combines barcodes and pooling given in (27). Using this data set, Erlich et al. aimed to identify a large number of shRNA sequences via pooling experiments. Although this problem is different than the problem of rare-allele detection, we show in Appendix 3 that our CS approach can also be applied to this problem, thus demonstrating its success on a large-scale real-life pooling experiment.
The rest of the article is organized as follows: ‘Materials and Methods’ section presents CS in the context of identifying carriers of rare alleles. We discuss the specific details of our proposed pooling design, genotype reconstruction algorithm and present a noise model reflecting the pooled sequencing process. The ‘Results’ section presents simulations, and provides evidence for the efficiency of our approach along a wide range of parameters. Finally, the ‘Discussion’ section offers conclusions and outlines possible directions for future research.
MATERIALS AND METHODS
We first provide a short overview of CS, followed by a description of its application to our problem of identifying rare alleles, and the corresponding mathematical formulation including a noise model reflecting the sequencing process. Finally, we show how one performs reconstruction while utilizing barcoding.
The CS problem
In a standard CS problem, one wishes to reconstruct a sparse vector x of length N, by taking k different measurements yi = mi·x, i = 1, … , k. This may be represented as solving the following set of linear equations:
| (1) |
where M is a k × N measurement matrix or sensing matrix, whose rows are the different mi's (as a general rule, we use upper-case letters to denote matrices: M, E, … , lower boldface letters to denote vectors: x,y, … and lower case to denote scalars: x, y, xi, …).
Typically in CS problems, one wishes to reconstruct x from a small number of measurements, i.e. k ≪ N, hence the linear system (1) is under-determined; namely, there are ‘too few’ equations or measurements and x cannot be recovered uniquely. However, it has been shown that if x is sparse, and M has certain properties, the original vector x can be recovered uniquely from Equation (1) (30,31). More specifically, a unique solution is found in case 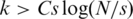 , where C is a constant and s the number of non-zero entries in x. This somewhat surprising result stems from the fact that the desired solution x is sparse, thus contains less ‘information’ than a general solution. Therefore, one can ‘compress’ the amount of measurements or ‘sensing’ operations required for the reconstruction of x.
, where C is a constant and s the number of non-zero entries in x. This somewhat surprising result stems from the fact that the desired solution x is sparse, thus contains less ‘information’ than a general solution. Therefore, one can ‘compress’ the amount of measurements or ‘sensing’ operations required for the reconstruction of x.
A sufficient condition for a sensing matrix to allow a correct reconstruction of x is satisfying a property known as ‘uniform uncertainty principle’ (UUP) or restricted isometry property (39,40). Briefly, UUP states that any subset of the columns of M of size 2s forms a matrix that is almost orthogonal (although since k < N the columns cannot be perfectly orthogonal), which, in practice, makes the matrix M ‘invertible’ for sparse vectors x. The construction of a ‘good’ sensing matrix is an easy task when one is able to use randomness. An example of a UUP matrix is a Bernoulli matrix; namely, a matrix whose entries are independent random variables set to be 1 or −1 with probability 0.5. It is known that a given instance of such a random matrix will satisfy UUP with an overwhelming probability (41–42) (the same is true when each entry in the matrix is a standard Gaussian random variable.)
Once M and y are given, CS aims to find the sparsest possible x, which obeys Equation (1). This can be written as the following optimization problem:
| (2) |
where the ℓ0 norm 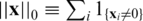 simply counts the number of non-zero elements in x.
simply counts the number of non-zero elements in x.
Problem (2) involves a non-convex ℓ0 term and can be shown to be computationally intractable in general (43). However, another impressive breakthrough of CS theory is that one can relax this constraint to the closest convex ℓp norm; namely, the ℓ1 norm, and still get a solution that, under certain conditions, is identical to the solution of Problem (2). Hence the problem is reformulated as the following ℓ1 minimization problem, which can be efficiently solved by convex optimization techniques:
| (3) |
In most realistic CS problems measurements are corrupted by noise, hence Equation (1) is replaced by 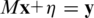 , where
, where  are the unknown errors in each of the k measurements, and the total measurement noise, given by the ℓ2 norm of
are the unknown errors in each of the k measurements, and the total measurement noise, given by the ℓ2 norm of 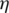 is assumed to be small. Therefore, the optimization problem is reformulated as follows:
is assumed to be small. Therefore, the optimization problem is reformulated as follows:
| (4) |
where ε > 0 is set to be the maximal level of noise we are able to tolerate, while still obtaining a sparse solution. It is known that CS reconstruction is robust to noise, thus adding the noise term ε does not cause a breakdown of the CS machinery, but merely leads to a possible increase in the number of required measurements k (40).
Many efficient algorithms are available for Problem (4) and enable a practical solution even for large matrices, with up to tens of thousands of rows. We have chosen to work with the commonly used gradient projection for sparse reconstrucion (GPSR) algorithm (44).
Rare-allele identification in a CS framework
We wish to reconstruct the genotypes of N individuals at a specific locus. The genotypes are represented by a vector x of length N, where xi represents the genotype of the i-th individual. We denote the reference allele by A and the alternative allele by B. The possible entries of xi are 0, 1 and 2, representing a homozygous reference allele (AA), a heterozygous allele (AB) and a homozygous alternative allele (BB), respectively. Hence, xi counts the number of (alternative) B alleles of the i-th individual, and since we are interested in rare minor alleles, most entries xi are zero. In classic CS the unknown variables are typically real numbers. The restriction on x in our case is expected to reduce the number of measurements needed for reconstruction and may also enable using faster reconstruction algorithms, as it is known that even a weaker restriction, namely, that all entries are positive, already simplifies the reconstruction problem (45).
The sensing matrix M is built of k different measurements represented by the rows of M. The entry mij is set to 1 if the j-th individual participates in i-th measurement, and zero otherwise. Each measurement includes a random subset of individuals, where the probability to include a certain individual is 0.5. Hence, M is equivalent to the Bernoulli matrix mentioned in the previous section, which is known to be a ‘good’ sensing matrix (another type of a sensing matrix, in which only 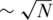 elements in each measurement are non-zero is considered in the ‘Results’ section).
elements in each measurement are non-zero is considered in the ‘Results’ section).
In practice, measurements are performed by taking equal amounts of DNA from the individuals chosen to participate in the specific pool, thus their contribution to the mixture is approximately equal. Then, the mixture is amplified using PCR, which ensures that the amplification bias generated by the PCR process affects all individuals equally (46). Finally, DNA of each pool is sequenced in a separate lane, and reads are mapped back to the reference genome [this may be performed using standard alignment algorithms such as MAQ (47)]. For each locus of interest, we record the number of reads containing the rare allele together with the total number of reads covering this locus in each pool, denoted by r. These numbers provide the measurement vector y representing the k frequencies obtained for this locus in the k different pools. The measurement process introduces various types of noise, which we model in the next section.
For each locus, our goal is to reconstruct the vector x, given the sensing matrix M and the measurement vector y, while realizing that some measurement error ε is present [see Equation (4)]. Our experimental design is illustrated in Figure 1, and the following section describes its mathematical formulation.
Figure 1.

Schematic description of the CS based procedure. Shown is a case of nine people, out of which one is a heterozygotic carrier of the rare SNP (marked green), and another one who is a homozygous alternative allele carrier (marked red.) Each sample is randomly assigned to a pool with probability 0.5, as described by the sensing matrix. For example, individuals 1,4,6,7 and 9 are assigned to the first pool. The DNA of the individuals participating in each pool is mixed, and the fraction of rare alleles in each pool is measured. For example, the first pool contains the two carriers, hence the frequency of the B′s is 1 + 2 out of the 2 × 9 alleles. The sensing matrix and the resulting frequencies are incorporated into an underdetermined set of linear constraints, from which the original rare SNP carriers are reconstructed.
Mathematical formulation of our model
The model presented here, including the range of parameters chosen, aims at reflecting the sequencing process by the Illumina technology (11), but may also be applied to other next generation technologies. It is similar, but not identical, to the model presented in (23). For clarity of presentation, we first describe our model while ignoring the different experimental noise factors, and these are added once the model is established.
Noiseless model
Let x be the unknown sparse genotypes vector, as described in the previous section. The fraction of individuals with the rare allele is denoted f, thus the vector x has s = fN non-zero elements. [Here and throughout the article we define the fraction of individuals, rather than alleles, as the `carrier frequency'—thus the fraction of alternative alleles, assuming Hardy–Weinberg (HW) equilibrium, is in fact 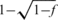 , which is approximately f/2 when f is small.] M is a k × N Bernoulli sensing matrix, and we denote by
, which is approximately f/2 when f is small.] M is a k × N Bernoulli sensing matrix, and we denote by 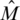 the normalized version of M whose entries represent the fraction of each individual's DNA in each pool
the normalized version of M whose entries represent the fraction of each individual's DNA in each pool
| (5) |
Assume that the mixing of DNA is perfect and unbiased, and that each DNA segment from each individual in a pool is equally likely to be read by the sequencing machinery. Suppose that a read from the i-th pool is drawn from a DNA segment covering our desired locus. It is then expected that this read will contain the B allele with probability 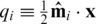 , where
, where 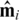 is the i-th row of
is the i-th row of 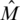 (the
(the  pre-factor is due to the fact that both alleles are sequenced for each individual). The vector of frequencies of the B allele, for each of the k pools is therefore
pre-factor is due to the fact that both alleles are sequenced for each individual). The vector of frequencies of the B allele, for each of the k pools is therefore
| (6) |
Had we been able to obtain a full and error-free coverage of the DNA present in the pool, our measurements would have provided us with the exact value of q. In practice, a specific position is covered by a limited number of reads, which we denote by r, and the number of reads from the rare alleles z out of the total number of reads r is binomially distributed zi ∼ Binomial(r,qi). Generally, one can only control the expected number of reads covering a specific locus, as r is also considered a random variable. Denoting R the total number of reads in a lane, the expected number of reads assigned to each locus is 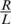 . The main cause for variation in r between different genomic regions is the different amplification biases for different genomic sequences, which are effected by properties such as a region's GC-content. The distribution of r over different loci depends on the experimental conditions, and was shown to follow a Gamma distribution in certain cases (23). We adopt this assumption, draw r for each locus from a Gamma distribution
. The main cause for variation in r between different genomic regions is the different amplification biases for different genomic sequences, which are effected by properties such as a region's GC-content. The distribution of r over different loci depends on the experimental conditions, and was shown to follow a Gamma distribution in certain cases (23). We adopt this assumption, draw r for each locus from a Gamma distribution 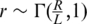 , and apply it to all k pools.
, and apply it to all k pools.
This binomial sampling process provides measurements that are close, yet not identical to the expected frequency of the rare allele rqi, and these fluctuations are regarded as sampling noise. Therefore, the CS problem formulation is given by [compare to Equation (4)]
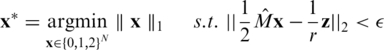 |
(7) |
Adding noise factors
The model described so far assumes that no noise or bias exist in our setting, besides sampling noise that is related to the limited number of reads used in practice. In a more realistic scenario, we do expect additional noise factors to be present due to imperfection in experimental procedures. We have modeled these factors by adding two more types of noise: sequencing read errors and DNA preparation errors (DP errors).
Read error models the noise factors introduced throughout the process of sequencing by next generation techniques such as Illumina, and reflects the fact that reads obtained from the sequencing machine may not match the DNA molecule sampled. This can be due to errors in certain bases present in the read itself, mis-alignment of a read to a wrong place in the genome, errors introduced by the PCR amplification process [which are known to introduce base substitutions in the replicated DNA (48)], or any other unknown factors. All of these can be modeled using a single parameter er, which represents the probability that the base read is different from the base of the measured sample's DNA at a given locus. The resulting base can be any of the other three different nucleotides; however, we conservatively assume that the errors will always produce the alternative allele B (if, e.g. the reference allele is `G’ and the alternative allele is `T', we assume that all erroneous reads produce `T'. In practice, it is likely that some reads will produce `A’ and `C', and these can be immediately discarded, thus, reducing the effective error rate). The probability of observing B at a certain read is, therefore, obtained by a convolution of the frequency of B alleles and the read error
| (8) |
The value of er may vary as a function of the sequencing technology, library preparation procedures, quality controls and alignment algorithms used. Typical values of er, which represent realistic values for Illumina sequencing (49), are in the range 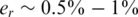 . We assume that er is known to the researcher, and that it is similar across different lanes. In this case, one can correct for the convolution in Equation (8) and obtain the following problem:
. We assume that er is known to the researcher, and that it is similar across different lanes. In this case, one can correct for the convolution in Equation (8) and obtain the following problem:
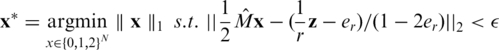 |
(9) |
Hence the measurement vector in our problem equals 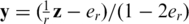 and the sensing matrix is
and the sensing matrix is 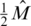 . If er is unknown, one may still estimate it, for example by running one lane with a single region on a single individual with known genotypes. Alternatively, we show in Appendix 4 how to incorporate the estimation of the read error term er within our CS framework from the overlapping pooled sequence data. The noise factors described thus far (including sampling noise and read errors) resemble the ones proposed previously in (23).
. If er is unknown, one may still estimate it, for example by running one lane with a single region on a single individual with known genotypes. Alternatively, we show in Appendix 4 how to incorporate the estimation of the read error term er within our CS framework from the overlapping pooled sequence data. The noise factors described thus far (including sampling noise and read errors) resemble the ones proposed previously in (23).
Finally, we add to our model one more source of noise, namely DP errors. This error term reflects the fact that in an experimental setting it is hard to obtain exactly equal amounts of DNA from each individual. The differences in the actual amounts taken result in noise in the measurement matrix M. While M is our original zero-one Bernoulli matrix, the actual measurement matrix M′ is obtained by adding DP errors to each non-zero entry. Hence, the true mixture matrix is M′ ≡ M + D, where the DP matrix D adds a centered Gaussian random variable to each non-zero entry of M
| (10) |
We consider values of σ in the range 0–0.05 reflecting up to 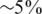 average noise on the DNA quantities of each sample. These values are consistent with errors encountered in real pooling experiments, and in fact a level of
average noise on the DNA quantities of each sample. These values are consistent with errors encountered in real pooling experiments, and in fact a level of 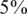 serves as a very conservative estimate. For example, in (50) it is shown that pooling variation is within the lower end of our considered range. Recent pooled sequencing experiments exhibit variation of at most
serves as a very conservative estimate. For example, in (50) it is shown that pooling variation is within the lower end of our considered range. Recent pooled sequencing experiments exhibit variation of at most 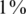 between individuals (M. Rivas, private communication). The matrix M′ is unknown and we only have access to M, hence the optimization problem in Equation (9) is unchanged in this case. M′ takes effect indirectly by modifying q, which effects z, the actual number of reads from the rare allele. As opposed to a classic CS problem in which the sensing matrix is usually assumed to be known exactly, DP effectively introduces noise into the matrix itself. We study this effect of DP errors in the ‘Results’ section, and show that a standard CS approach is robust to such noise.
between individuals (M. Rivas, private communication). The matrix M′ is unknown and we only have access to M, hence the optimization problem in Equation (9) is unchanged in this case. M′ takes effect indirectly by modifying q, which effects z, the actual number of reads from the rare allele. As opposed to a classic CS problem in which the sensing matrix is usually assumed to be known exactly, DP effectively introduces noise into the matrix itself. We study this effect of DP errors in the ‘Results’ section, and show that a standard CS approach is robust to such noise.
Targeted region length and coverage considerations
The expected number of reads from a certain locus is determined by the total number of reads in a lane R and the number of loci covered in a single lane L, and is given by 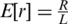 (we assume that the actual number of reads from each locus r follows a Gamma distribution with mean
(we assume that the actual number of reads from each locus r follows a Gamma distribution with mean 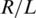 ).
).
L is determined by the number and size of the regions or the number of SNPs of interest in a given study, and by the ability of targeted selection techniques (16–18) to enrich for a given small set of regions. We consider L as a parameter and study its effect on the results. When interested in contiguous genomic regions, L should be interpreted as the length of the target region in reads, rather than nucleotides, since each read covers many consecutive nucleotides. Therefore, one should multiply L by the read length to get the total length of the targeted regions in base-pairs. For example, if our reads are of length 50 nt, and L is taken to be 100, we in fact cover a genomic region of length 5 kb. When we treat different isolated SNPs, L represents the number of SNPs we cover, as each read covers one SNP and the rest of the read is ‘wasted’ on nucleotides adjacent to the SNP of interest.
R is defined as the number of reads that were successfully aligned to our regions of interest. It is mostly determined by the sequencing technology, and is typically in the order of millions for modern sequencing machines. R is also greatly influenced by the number and length of the target regions and the targeted selection techniques used—since these techniques are not perfect, a certain fraction of reads might not originate from the desired regions and is thus `wasted'. The total number of reads varies according to experimental protocols, read length and alignment algorithms. Throughout this article we have fixed R to be R = 4 000 000, representing a rather conservative estimate of a modern Illumina genome analyzer's run [e.g. compare to (9)], and also assuming that targeted selection efficiency is high [it was reported to be as high as 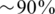 for relatively large genomic regions in (17)]. Other values of R may be easily dealt with using our simulation framework, thus adapting to a particular researcher's needs.
for relatively large genomic regions in (17)]. Other values of R may be easily dealt with using our simulation framework, thus adapting to a particular researcher's needs.
Another important and related parameter is the average coverage per individual per SNP, denoted c, which is given by
| (11) |
Our model does not directly use c and it is provided merely as a rough estimate for the coverage in the ‘Results’ section, as it can be easily interpreted and compared to coverage values quoted for single sample sequencing experiments. When the total number of reads in a pool r is given, the actual coverage obtained for each person in a pool has a distribution that is approximately Binomial(r,1/Npooled), where  is the number of individuals in the pool. Therefore, the average coverage per individual in a given pool is indeed approximately c.
is the number of individuals in the pool. Therefore, the average coverage per individual in a given pool is indeed approximately c.
Example
To visualize the effect of the three noise factors, i.e. sampling noise, read errors and DP errors, Figure 2 presents the measured values y in a specific scenario. We simulate an instance of N = 2000 individuals and carrier frequency 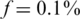 , tested over k = 50 pools. Hence, we have two heterozygotic carriers to be identified, and, in the absence of noise, the measurement in each pool should display three levels, which correspond to whether 0, 1 or 2 of the carriers are actually present in the specific pool.
, tested over k = 50 pools. Hence, we have two heterozygotic carriers to be identified, and, in the absence of noise, the measurement in each pool should display three levels, which correspond to whether 0, 1 or 2 of the carriers are actually present in the specific pool.
Figure 2.

Example of measured values. The values measured for 50 different pools, for a specific case of N = 2000 individuals and carrier frequency 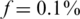 , thus two individuals carry the alternative allele. Shown are the measured carrier frequencies in each pool, for different coverage levels, (40, 400 and ∞ reads per pool), and different values of the read error (
, thus two individuals carry the alternative allele. Shown are the measured carrier frequencies in each pool, for different coverage levels, (40, 400 and ∞ reads per pool), and different values of the read error (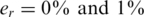 ). The data in all panels contain DP errors with σ = 0.05. The dashed lines represent the expected frequencies corresponding to 0,1,2 and 3 rare-allele carriers in a pool. For example, the frequency 10−3 corresponds to two carriers, calculated as the number of rare alleles (2) divided by twice the number of individuals per pool (1000). These frequencies (excluding the one corresponding to three carriers) are the values we would have obtained in the absence of read error, DP errors, and assuming that each pool contains exactly N/2 individuals. The coverage r (i.e. number of reads) is the most dominant factor causing deviations of the observed values from their expectancy.
). The data in all panels contain DP errors with σ = 0.05. The dashed lines represent the expected frequencies corresponding to 0,1,2 and 3 rare-allele carriers in a pool. For example, the frequency 10−3 corresponds to two carriers, calculated as the number of rare alleles (2) divided by twice the number of individuals per pool (1000). These frequencies (excluding the one corresponding to three carriers) are the values we would have obtained in the absence of read error, DP errors, and assuming that each pool contains exactly N/2 individuals. The coverage r (i.e. number of reads) is the most dominant factor causing deviations of the observed values from their expectancy.
To display the effect of sampling noise, we consider three values for the average coverage c, i.e. number of reads per individual per SNP: 40, 400 and an infinite number of reads, which corresponds to zero sampling noise. Each of these three values appears on a separate row in Figure 2. The panels on the left-hand side of Figure 2 correspond to read error 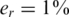 , while on the right-hand side there is no read error at all. The data in all panels contain DP errors with σ = 0.05. Each panel also displays the actual number of reconstruction errors in each case, namely the number of individuals whose genotype was incorrectly inferred, or mathematically speaking, the Hamming distance between the correct vector x and reconstructed vector x* obtained by solving Equation (9).
, while on the right-hand side there is no read error at all. The data in all panels contain DP errors with σ = 0.05. Each panel also displays the actual number of reconstruction errors in each case, namely the number of individuals whose genotype was incorrectly inferred, or mathematically speaking, the Hamming distance between the correct vector x and reconstructed vector x* obtained by solving Equation (9).
The effect of sampling noise is clearly visible in Figure 2. An infinite amount of reads (Figure 2e and f), causes the measurements to be very close to their expected frequency, where slight deviations are only due to DP errors and the fact that the pool size is not exactly N/2. For a moderate number of reads [c = 400—Figure 2c and d)], the measurements follow the expected frequency levels when there are no read errors (Figure 2d), but this rough quantization completely vanishes for 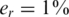 (Figure 2c). However, reconstruction was accurate even in this case, because our CS formulation [Equation (9)] takes these errors into account and aggregate the information from all pools to enable reconstruction. When the number of reads per person is small (c = 40—Figure 2a and b), the three levels disappear irrespective of the read error. Reconstruction is still accurate in the absence of read errors (Figure 2b), and there are three errors in the reconstructed genotype vector x* when
(Figure 2c). However, reconstruction was accurate even in this case, because our CS formulation [Equation (9)] takes these errors into account and aggregate the information from all pools to enable reconstruction. When the number of reads per person is small (c = 40—Figure 2a and b), the three levels disappear irrespective of the read error. Reconstruction is still accurate in the absence of read errors (Figure 2b), and there are three errors in the reconstructed genotype vector x* when 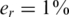 (Figure 2a), which probably implies that sampling noise is too high in this case (in Figure 2a, there are many pools for which the measurement reaches the level which corresponds to three carriers.) While a coverage of 40 reads per person is overwhelmingly sufficient when sequencing a single individual, it leads to errors in the reconstruction when pooling many individuals together.
(Figure 2a), which probably implies that sampling noise is too high in this case (in Figure 2a, there are many pools for which the measurement reaches the level which corresponds to three carriers.) While a coverage of 40 reads per person is overwhelmingly sufficient when sequencing a single individual, it leads to errors in the reconstruction when pooling many individuals together.
Reconstruction
We use the GPSR algorithm (44), to solve the optimization Problem (9). GPSR is designed to solve a slightly different, but equivalent, formulation as in Equation (9)
| (12) |
where the parameter τ provides the trade-off between the equations fit and the sparsity promoting factor, and is equivalent to specifying the maximal allowed error ε in Equation (9). It is often desirable in applications to let the parameter τ scale with ||MTy||∞ (51), where in our case 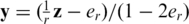 corresponds to the measurements. We have chosen to adopt this scaling throughout this article, and have set
corresponds to the measurements. We have chosen to adopt this scaling throughout this article, and have set 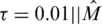 Ty||∞, although experimentation with different values of τ did not alter results significantly.
Ty||∞, although experimentation with different values of τ did not alter results significantly.
GPSR outputs a sparse vector x* with a few non-zero real entries, but does not use the fact that our variables are integers from the set {0,1,2}. We, therefore, perform a post-processing step in order to obtain such a solution. Simple rounding of the continuous results in x* may obtain such a vector. We chose an alternative common heuristic as a post-processing scheme, which yields better performance: we rank all non-zero values obtained by GPSR, and round the largest s non-zero values, setting to zero all other N−s values to get the vector x*s. We then compute an error term 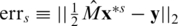 . Repeating this for different values of s, we select the vector x*s, which minimizes the error term errs. Thus, the final solution's sparsity s is always smaller or equal to the sparsity of the vector obtained by GPSR.
. Repeating this for different values of s, we select the vector x*s, which minimizes the error term errs. Thus, the final solution's sparsity s is always smaller or equal to the sparsity of the vector obtained by GPSR.
Utilizing barcodes
In this section, we describe how a CS-based method can be combined with a barcoding strategy resulting in improved performance. A barcode is obtained by attaching to the DNA in each sample, a unique DNA sequence of a few additional nucleotides, which enables the unique identification of this sample (52). Hence, samples with different barcodes can be mixed together into a single lane, and reads obtained from them can be uniquely attributed to the different samples. In a pooled-barcodes design (27), the DNA in each pool (as opposed to the DNA of a specific individual) is tagged using a unique barcode. If nbar different barcodes are available, we may apply nbar pools to a single lane and still identify the pool from which each read originated (although not the specific individual.)
Utilizing barcodes increases the number of pools per lane, while reducing the number of reads per pool. The usage of k lanes and nbar barcodes is simply translated into solving Problem (9) with k × nbar pools and R/nbar total reads per lane. Barcodes can, therefore, be combined easily with our CS framework so as to improve efficiency. We did not try to estimate the relative cost of barcodes and lanes as it may vary according to lab, timing and technology conditions. We, therefore, solve the CS problem for different (k,nbar) combinations, thus presenting the possible trade-offs.
Simulations
We have run extensive simulations in order to evaluate the performance of our approach. Various parameter ranges were simulated, where each set of parameters was tested in 500 instances. In each simulation, we have generated an input genotype vector x, applied measurements according to our mathematical model, and have tried to reconstruct x from these measurements. To evaluate the performance of our approach, one needs a measure of reconstruction accuracy, reflecting the agreement between the input vector x and the reconstructed CS vector x* (even when executing the naive and costly approach of sequencing each individual in a separate lane, one still expects possible disagreements between the original and reconstructed vectors due to insufficient coverage and technological errors.) Each entry i for which xi is different from 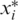 is termed a reconstruction error, and implies that the genotype for a certain individual was not reconstructed correctly, yielding either a false positive (
is termed a reconstruction error, and implies that the genotype for a certain individual was not reconstructed correctly, yielding either a false positive (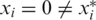 ) or a false negative (
) or a false negative (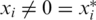 ). For simplicity, we have chosen to show a simple and quite restrictive measure of error: we distinguish between two `types’ of reconstructions—completely accurate reconstructions that have zero errors, and reconstructions for which at least one error occurred. A certain value of the problem's parameters (such as number of individuals, number of pools, read error, etc.) is termed ‘successful’ if at least
). For simplicity, we have chosen to show a simple and quite restrictive measure of error: we distinguish between two `types’ of reconstructions—completely accurate reconstructions that have zero errors, and reconstructions for which at least one error occurred. A certain value of the problem's parameters (such as number of individuals, number of pools, read error, etc.) is termed ‘successful’ if at least 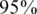 of its instances (i.e. 475 out of 500) had zero reconstruction errors, namely all individual genotypes were reconstructed correctly. Thus, even when testing for a few thousand individuals, we require that none of the reference allele carriers will be declared as a rare-allele carrier. In particular, this requirement guarantees that the false discovery rate of discovering rare-allele carriers will not exceed 0.05.
of its instances (i.e. 475 out of 500) had zero reconstruction errors, namely all individual genotypes were reconstructed correctly. Thus, even when testing for a few thousand individuals, we require that none of the reference allele carriers will be declared as a rare-allele carrier. In particular, this requirement guarantees that the false discovery rate of discovering rare-allele carriers will not exceed 0.05.
Performance is then measured in terms of Nmax, defined as the maximal number of individuals that allows for a ‘successful’ reconstruction, for certain values of the problem's parameters.
RESULTS
To explore the advantages of applying CS for efficiently identifying carriers of rare alleles, we performed various computer simulations of the experimental procedure described in the ‘Materials and Methods’ section (a Matlab implementation is available at www.broadinstitute.org/mpg/comseq/).
In each instance of a simulation, we followed the scheme in Figure 1. We grouped together N individuals, with a certain carrier frequency f, where f was chosen to be 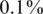 ,
, 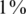 and
and 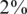 . Thus, we randomly selected s = Nf carriers, and this determined our input vector x. Since carrier frequency is low, we mostly considered the case of a heterozygous allele (AB), hence x is a binary vector, with 1's marking the carriers.The rare case of a homozygous alternative allele (BB), where x can also contain the value 2, was considered separately in a specific simulation.
. Thus, we randomly selected s = Nf carriers, and this determined our input vector x. Since carrier frequency is low, we mostly considered the case of a heterozygous allele (AB), hence x is a binary vector, with 1's marking the carriers.The rare case of a homozygous alternative allele (BB), where x can also contain the value 2, was considered separately in a specific simulation.
We then simulated k different pools, where in each pool ∼N/2 individuals were chosen at random, and their pooled DNA was sequenced in a separate lane. Sequencing results of the k pools were used to reconstruct x. The larger the number of pools (or lanes) k, the more information is available for reconstruction, and one can consider larger groups of individuals N.
Each of the k pools is designed to target L different loci where L was selected to be 1, 10, 100 or 500 (corresponding to targeted regions of length 100 bp to 50 kb, assuming each read is of length 100). The larger L the less reads are received from each targeted locus, thus reducing coverage and increasing sampling noise. The other noise factors were kept fixed, with read error 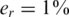 and DP error σ = 0.05, unless specified otherwise. Our simulations implicitly assume that coverage is sequence independent, i.e. each of the L loci is equally likely to be sequenced. We take this simplifying assumption for the sake of generality and clarity; however, the specific effects of coverage is considered in one of the examples.
and DP error σ = 0.05, unless specified otherwise. Our simulations implicitly assume that coverage is sequence independent, i.e. each of the L loci is equally likely to be sequenced. We take this simplifying assumption for the sake of generality and clarity; however, the specific effects of coverage is considered in one of the examples.
In the following section, we estimate the performance of CS given all relevant noise factors, and show that correct reconstruction may be performed in the presence of realistic or even highly pessimistic noise levels. In the ‘Noise Effects’ section we evaluate the individual effect of each of the three noise factors, i.e. sampling noise, read errors and DP errors. We then shortly present the effect of using a different sensing matrix in which only 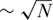 individuals participate in each pool, instead of ∼N/2. We show that each pooling scheme is advantageous in a different scenario. Finally, we present the effect of combining CS and barcodes, and display how barcodes boost CS performance.
individuals participate in each pool, instead of ∼N/2. We show that each pooling scheme is advantageous in a different scenario. Finally, we present the effect of combining CS and barcodes, and display how barcodes boost CS performance.
Performance of the ‘standard’ experimental setup
We analyzed the value of Nmax as a function of k, for different numbers of SNPs sequenced together on the same lane. The case 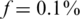 displayed a different behavior than
displayed a different behavior than 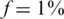 and
and 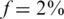 and is considered separately.
and is considered separately.
The case of 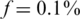
The advantages of CS appear most dramatically in the case of rare alleles, e.g. for 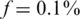 in Figure 3. Each panel in Figure 3 presents Nmax as a function of k, for different numbers of SNPs L. The number of rare-allele carriers tested in this case were 1, 2, … , 20, leading to N = 1000, 2000, … , 20000. The vertical right axis displays the corresponding average coverage c, obtained via Equation (11). The thick black line in each figure is simply the line y = x, demonstrating the performance of the naive approach of using a single lane per sample.
in Figure 3. Each panel in Figure 3 presents Nmax as a function of k, for different numbers of SNPs L. The number of rare-allele carriers tested in this case were 1, 2, … , 20, leading to N = 1000, 2000, … , 20000. The vertical right axis displays the corresponding average coverage c, obtained via Equation (11). The thick black line in each figure is simply the line y = x, demonstrating the performance of the naive approach of using a single lane per sample.
Figure 3.

Nmax as a function of the number of pools k for 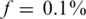 . The maximal number of individuals Nmax, which allow for a ‘successful’ reconstruction as a function of the number of pools used, for different numbers of loci treated simultaneously. A ‘successful’ reconstruction means that for a certain set of parameters at least 475 out of 500 simulations yield zero reconstruction errors. The black line is simply the line y = x, demonstrating the performance of the naive approach of using a single lane per sample. The vertical right axis displays the corresponding average coverage c for every value of Nmax, obtained via Equation (11). The insets in the top panels are zooming in on the region where the number of pools is small, which is at present the most realistic scenario (in the lower left panel Nmax was constant for low numbers of pools). The values of Nmax in this case were taken in units of 1000 individuals, which correspond to single carriers. Cases which appear to be missing, e.g. k < 200 for L = 500 simply mean that Nmax < 1000.
. The maximal number of individuals Nmax, which allow for a ‘successful’ reconstruction as a function of the number of pools used, for different numbers of loci treated simultaneously. A ‘successful’ reconstruction means that for a certain set of parameters at least 475 out of 500 simulations yield zero reconstruction errors. The black line is simply the line y = x, demonstrating the performance of the naive approach of using a single lane per sample. The vertical right axis displays the corresponding average coverage c for every value of Nmax, obtained via Equation (11). The insets in the top panels are zooming in on the region where the number of pools is small, which is at present the most realistic scenario (in the lower left panel Nmax was constant for low numbers of pools). The values of Nmax in this case were taken in units of 1000 individuals, which correspond to single carriers. Cases which appear to be missing, e.g. k < 200 for L = 500 simply mean that Nmax < 1000.
When the number of available pools was large, we were able to successfully identify the carriers in groups of up to 9000 or 20 000 individuals, for k = 500 pools, and L = 10 or 1, respectively (Figure 3 upper panels). In case the number of available pools was small, we could still identify a single carrier out of 1000 individuals with merely k = 20 pools, for L = 1 and 10. (inset in Figure 3a and b). With k = 30 (40) pools, we identified 2 (3) carriers in a group of 2000 (3000), for L = 1 (L = 10).
As evident from the four panels of Figure 3, Nmax decreased as a function of L. For example, 500 pools were sufficient to deal with 20 000 individuals for L = 1, but only with 1000 individuals for L = 500. This results from insufficient coverage that caused an increase in sampling noise. Increasing the number of pools can overcome this under-sampling as the value of Nmax increases almost linearly with k in most cases.
To quantify the advantage of applying CS, we defined an ‘efficiency score', presented in Figure 4, which is simply Nmax/k, i.e. the number of individuals for which reconstruction can be performed using the CS approach for a given number of pools, divided by the number of individuals that can be treated using the naive one-individual-per-lane approach. Therefore, the higher the score, the more beneficial it is to apply CS. The black line in each plot has a value of 1, which corresponds to the naive scenario of one individual per lane. When considering up to L = 100 SNPs, the efficiency score was around or above 10, and in some cases was as high as 70.
Figure 4.

Efficiency score of our approach for 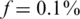 . The ratio Nmax/k between the number of individuals treated by our approach and the number treated in the naive approach (equal to the number of pools.) This represents the ratio of saved resources (pools.) Efficiency is highest when a few pools are used, and decreases gracefully as we use more and more pools. Efficiency is highest when the targeted number of loci is small, as in this case each lane provides very high coverage.
. The ratio Nmax/k between the number of individuals treated by our approach and the number treated in the naive approach (equal to the number of pools.) This represents the ratio of saved resources (pools.) Efficiency is highest when a few pools are used, and decreases gracefully as we use more and more pools. Efficiency is highest when the targeted number of loci is small, as in this case each lane provides very high coverage.
The axis on the right-hand side of Figure 3 displays the average number of reads per person, i.e. average coverage c, for the relevant Nmax. One important question is related to the optimal number of reads which allows for successful reconstruction: the smaller the coverage the more SNPs we can test on the same lane, yet we are more prone to (mostly sampling) noise. However, one can overcome the effects of low coverage by increasing the number of pools, hence it was interesting to test the performance for each combination of coverage c and number of pools k. In addition, such information may be important to evaluate the effect of sequence-dependent coverage. In reality, the expected coverage may follow a certain distribution depending on the sequence. Hence, it may be interesting to present reconstruction quality as a function of the expected coverage. In Figure 5, we present this performance for N = 2000 individuals and 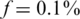 . For each pair of coverage c and number of pools k, we color coded the percentage of instances for which there were errors in CS reconstruction. An improvement in performance may be achieved both by increasing the coverage and by increasing the number of pools. The white line marks the
. For each pair of coverage c and number of pools k, we color coded the percentage of instances for which there were errors in CS reconstruction. An improvement in performance may be achieved both by increasing the coverage and by increasing the number of pools. The white line marks the 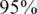 accuracy threshold. The transition between ‘successful’ and ‘unsuccessful’ reconstruction was rather sharp. For low coverage, e.g. lower than 100 reads per person, a very high number of pools was needed in order to overcome sampling noise.
accuracy threshold. The transition between ‘successful’ and ‘unsuccessful’ reconstruction was rather sharp. For low coverage, e.g. lower than 100 reads per person, a very high number of pools was needed in order to overcome sampling noise.
Figure 5.

Performance as a function of the number of pools k and the average coverage c. Shown is the percentage of runs in which (even a single) reconstruction error occurred. A rather sharp transition is shown, where above the white line we achieve a completely accurate reconstruction in at least 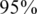 of the simulations. Notice the nonlinearity of scale in both axes.
of the simulations. Notice the nonlinearity of scale in both axes.
The case of 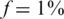 ,
, 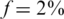
Figure 6 presents the results for 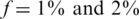 . In this case, the values of N tested were 100, 200, … , 4000 (no successful reconstruction according to our criteria was achieved for N > 4000). The resulting Nmax was lower than for the case of
. In this case, the values of N tested were 100, 200, … , 4000 (no successful reconstruction according to our criteria was achieved for N > 4000). The resulting Nmax was lower than for the case of 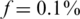 , although still much higher than in the naive approach. Results for L = 1 were similar to those of L = 10, namely increasing the coverage did not improve performance significantly in this case. The differences between results for
, although still much higher than in the naive approach. Results for L = 1 were similar to those of L = 10, namely increasing the coverage did not improve performance significantly in this case. The differences between results for 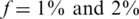 were rather small. The ‘efficiency score’ in this case was lower (see Figure 7), and was around 5, still offering a considerable saving compared to the naive approach.
were rather small. The ‘efficiency score’ in this case was lower (see Figure 7), and was around 5, still offering a considerable saving compared to the naive approach.
Figure 6.

Nmax as a function of k for 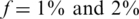 . The maximal number of individuals Nmax as a function of the number of pools. Similar to Figure 3 but with a higher carrier frequency,
. The maximal number of individuals Nmax as a function of the number of pools. Similar to Figure 3 but with a higher carrier frequency, 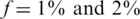 . The number of individuals Nmax achieved decreases as we increase the carrier frequency, but we are still able to treat a much larger sample size than the naive approach. For example, one can use 40 pools for L = 100 and recover four rare-allele carriers out of 400 (
. The number of individuals Nmax achieved decreases as we increase the carrier frequency, but we are still able to treat a much larger sample size than the naive approach. For example, one can use 40 pools for L = 100 and recover four rare-allele carriers out of 400 ( 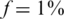 , zoomed-in view in lower left panel). The case
, zoomed-in view in lower left panel). The case 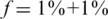 corresponds to
corresponds to 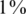 AB and
AB and 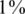 of BB alleles.
of BB alleles.
Figure 7.

Efficiency score of our approach for 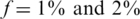 . Efficiency score of our approach. Similar to Figure 4 but with carrier frequency of
. Efficiency score of our approach. Similar to Figure 4 but with carrier frequency of 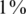 and
and 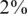 . As expected, efficiency is decreased as carrier frequency increases. We still reach up to 13× and 7× improvement over the naive approach for carrier frequencies of
. As expected, efficiency is decreased as carrier frequency increases. We still reach up to 13× and 7× improvement over the naive approach for carrier frequencies of 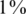 and
and 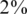 , respectively.
, respectively.
All former simulations considered the case of identifying carriers of a heterozygous allele (AB). To study the possibility of also identifying homozygous alternative alleles (BB) via CS we simulated the following case: 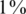 of the individuals were BB in addition to
of the individuals were BB in addition to 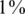 which were AB (this yields a vastly higher frequency of BB than is expected to be encountered in practice, and was taken as an extreme case to test the robustness of our reconstruction results). The results were marked as ‘
which were AB (this yields a vastly higher frequency of BB than is expected to be encountered in practice, and was taken as an extreme case to test the robustness of our reconstruction results). The results were marked as ‘ ’ in Figures 6 and 7. Our CS framework dealt with this scenario in exactly the same way as the other AB cases, although the results were, as expected, slightly worse than those obtained for
’ in Figures 6 and 7. Our CS framework dealt with this scenario in exactly the same way as the other AB cases, although the results were, as expected, slightly worse than those obtained for 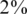 heterozygous carriers.
heterozygous carriers.
The effect of noise
Figures 8 and 9 present the effects of three types of noise in the specific case of 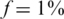 . In both figures, the reference was the ‘standard’ performance of
. In both figures, the reference was the ‘standard’ performance of 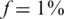 , which appeared before in Figure 6, and included sampling noise, read error (
, which appeared before in Figure 6, and included sampling noise, read error (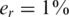 ) and DP errors (σ = 0.05). We considered the case of sampling error separately from the other two sources, as its impact was different.
) and DP errors (σ = 0.05). We considered the case of sampling error separately from the other two sources, as its impact was different.
Figure 8.

Effect of sampling error. The dashed line represent results obtained in the limit where the number of reads goes to infinity, thus sampling error is zero. The solid line represents the realistic scenario with the current number of reads used. Sampling error is seen to be a significant factor when we treat many loci together in the same lane (L = 100 or more), while for a few loci (L = 10 or less) we already have enough coverage to make sampling error negligible.
Figure 9.

Effect of read errors and DP errors. The two dashed lines represent results obtained when assuming that reads are perfect and DP errors is zero, respectively. The solid line represents a realistic scenario with a read error of 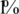 and DP errors of σ = 0.05. While read error appears to have a significant factor in reducing Nmax, the effect of DP errors is negligible.
and DP errors of σ = 0.05. While read error appears to have a significant factor in reducing Nmax, the effect of DP errors is negligible.
Sampling error
Figure 8 compares the ‘standard’ performance to the case where an infinite number of reads were available (although read error and DP errors were still present). Differences between the cases appeared only when the number of SNPs L was high, thus the number of reads per person c was insufficient. In these cases Nmax was reduced by a factor of 2 to 4 with respect to an infinite coverage. When the number of SNPs L was small, and coverage was high, there was no difference between the ‘standard’ performance and the infinite read case.
Read errors and DP errors
Figure 9 compares the ‘standard’ performance to two cases: one in which er = 0 and another in which σ = 0. In the absence of read errors, Nmax may be twice as large as when 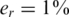 . Read errors made a significant effect on performance only when L was large (100 or 500), since when coverage was high read errors were compensated for [see Equation (9)]. In all cases, we have studied the results were very robust to DP errors, thus noise introduced by realistic pooling protocols should be easily overcome by the CS reconstruction.
. Read errors made a significant effect on performance only when L was large (100 or 500), since when coverage was high read errors were compensated for [see Equation (9)]. In all cases, we have studied the results were very robust to DP errors, thus noise introduced by realistic pooling protocols should be easily overcome by the CS reconstruction.
Modifying the sensing matrix
In all simulations presented so far, we have considered the case where each pool includes approximately N/2 individuals. It may be desirable to minimize the number of individuals per pool (27), as this can lead to a faster and cheaper preparation of each pool. Here, we shortly present the possibility of modifying M into a sparse sensing matrix, thus accommodating the requirement of having few individuals per pool.
Figure 10 presents the results of using only 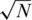 individuals in each pool, for the case
individuals in each pool, for the case 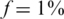 (marked as
(marked as 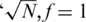 ’). For a small number of loci taken together (L = 1 or 10) the former dense Bernoulli(0.5) sensing matrix achieved higher Nmax values. However, when the number of loci was large (L = 100 or 500) and for large number of pools, it was preferable to use sparse pools of size
’). For a small number of loci taken together (L = 1 or 10) the former dense Bernoulli(0.5) sensing matrix achieved higher Nmax values. However, when the number of loci was large (L = 100 or 500) and for large number of pools, it was preferable to use sparse pools of size 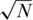 . The same qualitative behavior was observed for
. The same qualitative behavior was observed for 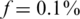 and
and 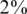 . The success of sparse matrices in recovering the true genotypes is not surprising given theoretical and experimental evidence (53). Further research is needed in order to determine the optimal sparsity of the sensing matrix for a given set of parameters.
. The success of sparse matrices in recovering the true genotypes is not surprising given theoretical and experimental evidence (53). Further research is needed in order to determine the optimal sparsity of the sensing matrix for a given set of parameters.
Figure 10.

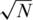 versus N/2 sensing matrix. The effect of applying pools of
versus N/2 sensing matrix. The effect of applying pools of 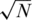 or N/2 individuals (on average) for the case of
or N/2 individuals (on average) for the case of 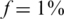 . Overall, results are comparable, yet each pooling design is preferable for different settings of pools and loci. The sparse (
. Overall, results are comparable, yet each pooling design is preferable for different settings of pools and loci. The sparse (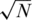 ) design is more beneficial for large number of pools and longer target regions. The average coverage per individual on the right axis of each panel corresponds to the N/2 case. The coverage in the
) design is more beneficial for large number of pools and longer target regions. The average coverage per individual on the right axis of each panel corresponds to the N/2 case. The coverage in the 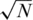 case is much larger since the total number of reads R is divided among a smaller number of individuals.
case is much larger since the total number of reads R is divided among a smaller number of individuals.
Combining barcodes and CS
Barcodes may also be combined with CS so as to improve efficiency and further reduce the number of required lanes. The DNA in each pool (as opposed to the DNA of a specific individual) may be tagged using a unique barcode (see ‘Materials and Methods’ section). Hence, in case we have nbar different barcodes available, we apply nbar pools to a single lane, with the price being that each pool contains only R/nbar reads. Figure 11 displays Nmax as a function of the number of lanes, for different values of nbar, and for different carrier frequencies.
Figure 11.

Combining CS and barcodes. Results obtained by combining the CS approach with barcoding for L = 1. Barcoding improves results by allowing several pools to be tested on the same lane (although these lanes contain a smaller number of reads.) The effect of adding a large number of barcodes is more pronounced for high minor-allele frequency. For example, at 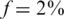 and with 7 lanes, we can treat roughly 300 individuals with 10 barcodes, but around 2500 individuals with 200 barcodes. The increase in power is sub-linear, as is seen by the fact that when we add more barcodes, the performance becomes closer to that of the naive approach (shown in black) which increases linearly with the number of lanes. Still, only at a very high number of barcodes the naive approach can perform as well as the CS design.
and with 7 lanes, we can treat roughly 300 individuals with 10 barcodes, but around 2500 individuals with 200 barcodes. The increase in power is sub-linear, as is seen by the fact that when we add more barcodes, the performance becomes closer to that of the naive approach (shown in black) which increases linearly with the number of lanes. Still, only at a very high number of barcodes the naive approach can perform as well as the CS design.
The black line in each figure represents the ‘naïve’ capacity, which is simply k × nbar. Incorporating even a small number of barcodes into our CS framework resulted in a dramatic increase in Nmax. For the same problem parameters, but without using barcodes, we could not recover the minimal possible number of individuals Nmax = 1000 for 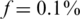 and could not reach more than Nmax = 100 for
and could not reach more than Nmax = 100 for 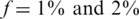 (see Figures 3 and 6). Similarly to the non-barcodes case, the advantage over the naive approach was most prominent for
(see Figures 3 and 6). Similarly to the non-barcodes case, the advantage over the naive approach was most prominent for 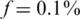 , but was still significant for
, but was still significant for 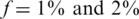 . As the number of barcodes increased, the difference in performance between different sparsity values f became smaller. As long as the coverage was kept high, it was still beneficial to increase the number of barcodes, as it effectively increased linearly the number of lanes. At a certain point, when many different barcodes were present in a single lane, coverage dropped and sampling error became significant, hence the advantage of adding more barcodes began to diminish.
. As the number of barcodes increased, the difference in performance between different sparsity values f became smaller. As long as the coverage was kept high, it was still beneficial to increase the number of barcodes, as it effectively increased linearly the number of lanes. At a certain point, when many different barcodes were present in a single lane, coverage dropped and sampling error became significant, hence the advantage of adding more barcodes began to diminish.
Experimental results
In Appendices 1–3, we present applications of the CS approach to three experimental data sets. Appendices 1 and 2 compare between the performance of reconstruction which is based on experimental data, and reconstruction in the fully simulated case. In both examples, there is very good accordance between the two cases. In Appendix 3, we show an application of CS reconstruction to the experimental data of Erlich et al. (27). We map this different, yet related, problem to a CS approach, and achieve comparable reconstruction results to Erlich et al. Together, these examples display the applicability and effectiveness of the CS approach to real-world sequencing data.
DISCUSSION
We have presented a method for identifying rare alleles and their carriers via CS-based group testing. The method naturally deals with all possible scenarios of multiple carriers and heterozygous or homozygous rare alleles. Our results display the advantages of the approach over the naive one-individual-per-lane scenario: it is particularly useful for the case of a large number of individuals and low carrier frequencies. We have also shown that our method can benefit from the addition of barcodes for different pools (27), and still improve upon `standard’ barcoding.
As for practical aspects, our approach may also prove to be advantageous over the naive approach. The overall costs of an experiment stem from the procedures related to sample preparation (e.g. PCR, performing gel extraction if needed, etc.), and from direct sequencing costs. While our approach requires an additional pooling step, both sample preparation procedures and sequencing are then performed over the pools and not on single specimens, as in the naive approach. Hence, costs reduction in both steps is proportional to the efficiency score defined in the ‘Results’ section.
We view our main contribution as outlining a generic approach that puts together sequencing and CS for solving the problem of identifying novel variations and carrier screening of known SNPs. Following this mapping, we may apply the vast amount of CS literature and benefit from any advancement in this rapidly growing field. We believe this is a major advantage over more `tailored’ approaches, e.g. (23,27), although these methods may be superior to ours in specific cases.
Our method is simple in the sense that it applies CS in the most straightforward fashion. We have used an off-the-shelf CS solver and did not try to optimize any CS related parameter for the different scenarios—all parameters were kept fixed for all types of simulations, thus optimizing reconstruction algorithms is likely to improve our results. Moreover, the method's performance as detailed in the ‘Results’ section may be further improved, since our formulation of the CS problem has incorporated only part of the available information at hand. Using additional information may reduce the number of pools or total reads needed to achieve a certain accuracy, as well as enable faster algorithms for reconstruction, thus allowing us to deal with larger sample sizes and larger regions.
The information that the input signal is trinary (0,1,2) was considered only in the post-processing step after running GPSR, although it may be incorporated into the optimization procedure itself. Since most alleles are approximately distributed according to the HW equilibrium, the frequency of 2's, derived from the frequency of 1's, is very low, and the input vector is most often Boolean. Therefore, we could also modify the optimization so as to `punish’ for deviations from this pattern. Another possible direction may be to apply techniques borrowed from the recent work on Bayesian CS (54,55) and estimate the posterior probability of possible genotypes. This may assist in further reducing the number of pools needed for reconstruction.
In addition, we have treated each rare allele independently, although in case alleles originate from the same genomic region, one could use linkage disequilibrium information and reconstruct haplotypes. For example, if two individuals share a rare allele at position i, they are also likely to share an allele, probably rare, at position i + 1. It would be a challenge to add these constraints and enable the reconstruction of several adjacent loci simultaneously. Another improvement may stem from more accurate modeling of specific errors of sequencing machines, including quality scores that provide an estimate of error probability of each sequenced base (49,56). Such careful modeling typically results in far smaller error rates than the ones we have conservatively used (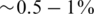 .)
.)
Our method is non-adaptive, in the sense that the pooling strategy is designed in advance. A natural extension one might consider is to apply adaptive group testing, namely to decide whether to use pool k, based on reconstruction results of pools 1, … , k–1. In principle, such an approach enables an adaptation of the number of samples to unknown sparsity by simply generating pools one by one, each time solving the CS problem and checking if we get a sparse and robust solution. Once the solution stabilizes, we can stop our experiments, thus not `wasting’ unnecessary lanes (it may also be beneficial to change the pooling design of the next measurement and allow deviations from the randomized construction we have shown, once statistical information regarding the likely carriers is starting to accumulate). This approach may be problematic, however, since we typically deal with several SNPs simultaneously. If the carriers of rare alleles are different for the relevant SNPs, performing such an adaptive reconstruction is not straightforward as the optimal pooling strategy would be different across loci.
Our approach is most useful when one wishes to sequence a relatively small region over a large number of individuals, as opposed to the naive approach that aims at sequencing large regions in a single individual. We envision that this scenario will be of great importance, as many genes known to contribute to certain diseases will be sequenced in large populations. While the scope of our approach, i.e. the size of the targeted region, may be limited, the rapid increase in sequencing capacity of a single lane may further increase the region size (and population size) for which our approach is beneficial.
Our method does not require any prior knowledge about rare allele frequency, which is the case when considering novel variants, or when screening a new population for known SNPs. The only relevant question is whether a certain number of pools is sufficient to provide accurate reconstruction. Hence, one needs to perform an ad hoc estimate of allele frequency, which may be approximated by averaging over allele frequencies found in all pools. Based on this estimate, one can check whether the relevant number of individuals is higher or lower than Nmax for the current number of pools and estimated allele frequency, and add pools if necessary. Our simulations also indicate that while an insufficient number of pools may lead to incorrect reconstruction, in such cases we still correctly report allele frequency. Also, in such cases reconstruction is of worse quality thus our algorithm can detect and report the need for more pools (data not shown).
The drawbacks of our method stem from the limitations of CS and sequencing technologies. First, in case carrier frequency is high enough, the sparsity assumption at the heart of the CS theory breaks down, and it may be problematic for CS to reconstruct the signal. The highest frequency possible for CS to perform well in this application was not determined, but one should expect a certain frequency above which it is no longer beneficial to apply CS and the naive one-individual-per-lane approach is preferable. The simulations we have performed estimate this frequency to be over 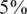 in most cases, thus taking effect only for the case of common alleles. In general, it is reasonable to assume that unknown variants typically have low frequencies, usually <
in most cases, thus taking effect only for the case of common alleles. In general, it is reasonable to assume that unknown variants typically have low frequencies, usually <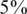 (for otherwise they would have been identified already). It is, therefore, likely that our approach would be well suited for identifying novel variants.
(for otherwise they would have been identified already). It is, therefore, likely that our approach would be well suited for identifying novel variants.
Another possible difficulty in our approach is related to the issue of randomness of the sensing matrix M. This randomness may be discarded by simply fixing a certain instance of the sensing matrix, although randomness in this case may be viewed as an advantage of CS—almost any (random) matrix would enable reconstruction, as opposed to intricate pooling schemes which need to be carefully designed.
The last drawback we should mention is related to the fact that each pool contains approximately half the individuals in the group. This may be problematic in cases where pooling preparation might be slow and costly, and one needs to minimize the number of individuals in each pool (27). In this case, it may be interesting to apply a sparser pooling design. As shown in the ‘Results’ section there are scenarios in which it is advantageous to assign only 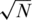 individuals to a pool. Therefore, one needs to optimize the pool design together with other parameters, e.g. number of loci and lanes considered. This issue remains for future study.
individuals to a pool. Therefore, one needs to optimize the pool design together with other parameters, e.g. number of loci and lanes considered. This issue remains for future study.
While we have tried to model the different sources of error encountered in next generation-pooled sequencing, a complete validation of the approach still requires large scale pooled CS experiments. As far as we know such an experiment has not been performed yet. Therefore, in the appendices we present analysis of three experimental data sets, which capture the main aspects of the approach. Using the data of Out et al. we directly test the linearity assumption, which is at the heart of the CS approach, and show that performance is similar to the predicted performance on simulated mixtures. In a second example, reads from the Pilot 3 study of the 1000 Genomes Project are used to simulate pools by randomly sampling reads collected when sequencing each individual. We show that performance of the CS approach using these experimental reads is similar to performance obtained using simulated reads sampled by our model. In a third example, the CS method is applied to the data of Erlich et al., showing an implementation of the CS approach to a large scale, pooled and barcoded experiment.
Finally, while we have demonstrated the benefits of CS-based group testing approach for genotyping, any genetic or epigenetic variant is amenable to our approach, as long as it can be detected by next generation sequencing technology and is rare in the population of interest. For example, copy number variations (CNVs), important for studying both normal population variations and alterations occurring in cancerous tissues, provide a natural extension to our framework. In this case, the number of reads serves as a proxy to the copy number at a given locus, and the vector to be reconstructed contains the (integer) copy number levels of each individual, rather than their genotypes. Another example is given by rare translocations, often present in various tumor types—where an evidence for a translocation may be provided by a read whose head is mapped to one genomic region and whose tail is mapped to another distal region (or by two paired-end reads, each originating from a different genomic region). Carriers of a particular rare translocation may be discovered using this approach. The extension of our method to these and perhaps other novel applications provides an exciting research direction we plan to pursue in the future.
FUNDING
Funding for open access charge: XXX
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Y. Erlich and G.J. Hannon for kindly providing the Arabidopsis experimental data. We also thank M. Rivas for useful discussions and Eytan Domany for his continuous support. We are grateful to the anonymous referees for their important suggestions.
APPENDIX 1
Carrier identification based on pooled experimental data
We applied our approach to the data described by Out et al. (38). Briefly, a region of ∼5.7 kb of human chromosome 1 containing the MUTYH gene, was amplified and sequenced (using Sanger sequencing) in each of 88 breast cancer patients. In addition, the authors created a single pool consisting of equal amounts of DNA from these 88 individuals, and performed targeted sequencing by the Illumina GAI. Using these two methods, the authors detected a set of about 25 heterozygous SNPs present in the 88 individuals. Carrier identity for each of the SNPs was determined by the Sanger sequencing data, which served as `ground truth’ for the genotypes of each individual. Only 9 SNPs out of the 25 were present at a carrier frequency below 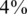 : 5 SNPs with a single carrier among the 88 individuals, 3 SNPs with 2 carriers, and a single SNP with 3 carriers. The rest of the SNPs tested had higher carrier rate, thus were discarded from our analysis. Figure A1 compares the estimated allele frequencies via next generation sequencing to the Sanger-based frequencies. The former frequencies were estimated from sequencing the pool as f = rrare/rtotal, where rrare and rtotal were the number of reads from the rare allele and the total number of reads from the relevant SNP, respectively. For most SNPs, sequencing the pools provides a good estimate of rare alleles’ frequencies.
: 5 SNPs with a single carrier among the 88 individuals, 3 SNPs with 2 carriers, and a single SNP with 3 carriers. The rest of the SNPs tested had higher carrier rate, thus were discarded from our analysis. Figure A1 compares the estimated allele frequencies via next generation sequencing to the Sanger-based frequencies. The former frequencies were estimated from sequencing the pool as f = rrare/rtotal, where rrare and rtotal were the number of reads from the rare allele and the total number of reads from the relevant SNP, respectively. For most SNPs, sequencing the pools provides a good estimate of rare alleles’ frequencies.
Figure A1.

Allele frequency—Illumina versus ground truth. Frequency of rare alleles in 88 individuals for nine SNPs as sequenced by (38). These frequencies are compared to their expected value based on Sanger sequencing each of these 88 individuals. Some of the five single-carrier-SNPs have very similar measured values, and are almost overlapping in the graph.
The experimental data at hand consists of sequencing many SNPs over a single pool. We have used this data in a bootstrap approach to simulate many different in silico pools required for our CS framework. The simulation was performed by first grouping the SNPs according to their carrier rate within the 88 individuals. We then considered each such group of equal rate SNPs as if it represented measuring a single SNP over several pools. For example, in order to simulate a single carrier SNP, we considered the five single carrier SNPs of Out et al. as if they originated from five different pools targeting a single simulated SNP. Hence, in order to simulate a pool, which happens to contains a single carrier, we randomly sampled one of the five single carrier SNPs and used its measured value f for the allele frequency in that pool.
The CS simulation was performed as follows: we created a genotype vector x of length 2 × 88 = 176, which contained either 1, 2 or 3 non-zero entries, corresponding to the carriers. We then selected a random sensing matrix M of k rows and 176 columns, which contained exactly 88 non-zero entries in each row, representing the 88 individuals present in each pool (notice this is slightly different from the Bernoulli matrix described in the main text, which would contain 88 individuals on average; however, this has no practical importance). Hence, by dividing each row of M by 176, the measurements y = Mx correspond to the frequencies of the rare allele in the k `pools'. Ideally, these values should be 0,1,2 or 3 divided by 176, but we replaced them by sampling the relevant experimental values of f. For example, in case the i-th pool contained two carriers, i.e. 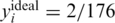 , we randomly selected one of the three SNPs which had two carriers, and set the measurement to be
, we randomly selected one of the three SNPs which had two carriers, and set the measurement to be 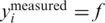 of that specific SNP. Similarly, in case the pool contained a single carrier, we randomly selected the value f of one of the five relevant SNPs, while if the pool consisted of three carriers the measurements were based on a single SNP (pools which did not include any carrier were taken as f = 0).
of that specific SNP. Similarly, in case the pool contained a single carrier, we randomly selected the value f of one of the five relevant SNPs, while if the pool consisted of three carriers the measurements were based on a single SNP (pools which did not include any carrier were taken as f = 0).
For a given genotype vector, different pools may have different numbers of carriers. Thus, for example, when x contains three carriers, 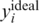 may have four ‘levels’ corresponding to whether 0,1,2 or 3 carriers participated in the actual pool. Therefore, the bootstrap simulation may use values of f from different SNPs for different pools, based on the carrier frequencies observed in the pool. All nine values of f, corresponding to the nine different SNPs, may potentially enter the simulation. Finally, we used CS to reconstruct the genotype vector x based on this `experimental’ case, and compared it to the `true’ genotype vector obtained by Sanger sequencing.
may have four ‘levels’ corresponding to whether 0,1,2 or 3 carriers participated in the actual pool. Therefore, the bootstrap simulation may use values of f from different SNPs for different pools, based on the carrier frequencies observed in the pool. All nine values of f, corresponding to the nine different SNPs, may potentially enter the simulation. Finally, we used CS to reconstruct the genotype vector x based on this `experimental’ case, and compared it to the `true’ genotype vector obtained by Sanger sequencing.
To compare reconstruction performance based on `experimental’ data to reconstruction based on the model suggested in the main text, we also simulated this experimental setting of 176 individuals. For each genotype vector x to be reconstructed, we performed an analogous simulation, using exactly the same x, sensing matrix M and values of rtotal. To apply our model as detailed in the Section `Mathematical Formulation', we set the read errors as 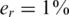 and DP errors (
and DP errors (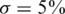 ). For each pool, we considered the relevant value of rtotal as the expected number of reads, and sampled the number of reads according to Γ(rtotal,1). We then sampled the reads according to a Binomial distribution.
). For each pool, we considered the relevant value of rtotal as the expected number of reads, and sampled the number of reads according to Γ(rtotal,1). We then sampled the reads according to a Binomial distribution.
Figure A2 presents the percentage of zero-error reconstructions out of 1000 trials for the `experimental’ and simulated cases (black and red lines, respectively). Figure A2a–c correspond to different carrier rates. There is very good similarity between the `experimental’ data and our simulations, especially in the case of one and two carriers (Figure A2a–b, respectively.) However, there are rather large differences in the case of three carriers. The latter case is, in part, based on a single SNP measurement in the `experimental’ data, which happened to be noisy (see Figure A2). To check whether these results originate from this single noisy measurement, we generated a different sensing matrix M, containing no more than two carriers in each pool, by randomly eliminating one of the three carriers from the pool, whenever a pool contained all the three. Hence, we tried to reconstruct genotypes in the case of three carriers, via `pools’ each containing two carriers at most. As a result the differences between `experimental’ and simulated data were much smaller (Figure A2d), thus indicating that indeed the single noisy experimental measurement of three carriers caused most of the differences.
Figure A2.

Carrier identification in an experimental mixture: `experimental data’ versus simulated. The percentage of zero-error reconstructions out of 1000 trials, as a function of the number of pools, for reconstruction based on (38) (black) and simulated data (red). (a–c) Correspond to 1,2 and 3 carriers, respectively. (d) corresponds to reconstructing three carriers, based on pools which contain two carrier at most. The dashed line in each panel corresponds to our zero-error 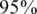 threshold.
threshold.
Figure A3.

Number of SNPs correctly reconstructed based on the 1000 Genomes Project data. Number of SNPs with zero-error reconstructions as a function of the number of pools. Each panel corresponds to a different coverage level. The black line corresponds to the standard reconstruction based on generating pools from the 1000 Genomes Project's actual reads, and the red line corresponds to reconstruction based on sampling reads according to our statistical model. The green line corresponds generating pools by adjusting the 1000 Genomes Project's actual reads to account for different read errors for each individual (see text in Appendix 2).
To conclude, although the number of SNPs covered by Out et al. is rather limited, it does show that the linearity assumption, which is at the heart of CS holds, i.e. the measurements’ values are approximately linear in the number of rare alleles in the pool. Our simulations are in good agreement with reconstruction based on actual read counts. Our predictions as to the quality of reconstruction and our estimate of the number of pools required to reach the level 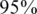 are rather accurate. Therefore, there is reason to believe that our model and our predictions regarding larger cohorts of individuals, presented in the article, would also prove to be as accurate.
are rather accurate. Therefore, there is reason to believe that our model and our predictions regarding larger cohorts of individuals, presented in the article, would also prove to be as accurate.
APPENDIX 2
Decoding mixtures based on the 1000 genomes project
We applied the CS approach to a subset of the Pilot 3 data from the 1000 Genomes Project (www.1000genomes.org), in which exons of roughly 1000 genes were resequenced with high coverage in a few hundred individuals. The following sections present the data, the CS setting, and the results of carrier detection based on this experimental data.
Data set
The Pilot 3 data set, as of May 2010, consists of sequences of approximately 700 individuals, from which we used the 364 individuals sequenced by an Illumina machine. Using the Genome Analysis toolkit (57), we calculated the number of rare-allele reads and the total number of reads for each individual, for each of the 3489 loci listed in the Northern and Western European ancestry SNP list (CEU variant call format file).
Individuals for which the total number of reads for a certain SNP was lower than 40, were excluded from the analysis of that specific SNP. Minor allele frequencies below 0.1 were labeled as reference homozygous (AA), frequencies between 0.35 and 0.85 were labeled as heterozygous (AB), and frequencies above 0.85 were taken as rare homozygous (BB). Cases in which minor allele frequency was in the range 0.1–0.35 were considered as ambiguous and were excluded. We then calculated carrier frequency for each SNP, and analyzed 633 SNPs whose carrier frequency was lower or equal than 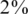 . Coverage was different for different people, and the number of individuals per SNP varied across the 633 SNPs, with mean and SD of 185 and 55 individuals per SNP, respectively. Twenty of the SNPs included rare heterozygous (BB) carriers.
. Coverage was different for different people, and the number of individuals per SNP varied across the 633 SNPs, with mean and SD of 185 and 55 individuals per SNP, respectively. Twenty of the SNPs included rare heterozygous (BB) carriers.
The CS Setting
The CS approach was independently applied to each of the 633 SNPs. The genotype vector x to be reconstructed was determined according to the above classification, and the sensing matrix M was randomly selected as in the main text. We simulated the pooling process and the resulting measurements vector according to the following procedure; For each pool, we set the total number of reads for a given SNP to rtotal = c × n, where n is the number of individuals in the pool, and c being the coverage. Each read was sampled by randomly selecting one of the n individuals, and then drawing uniformly one of the reads covering the relevant SNP for this individual in the 1000 Genomes data set. This sampling procedure was repeated rtotal times to obtain rtotal reads and the measurement was taken as f = rrare/rtotal, where rrare is the number of reads from the rare allele. The same procedure was repeated for different values of coverage c and for a variable number of pools.
Since read error is unknown for this data set, we applied the modified reconstruction algorithm that appears in Appendix 4. This algorithm simultaneously reconstructs the genotype and infers the read error, without prior knowledge of the true error by just assuming that it is constant across different pools, i.e. lanes.
To compare reconstruction performance based on `experimental’ data to reconstruction based on our statistical model, we also simulated measurements according to our statistical model (‘Methods’ section). For each SNP, we performed an analogous simulation, using exactly the same sensing matrix M and value of rtotal. We applied our model as detailed in the Section `Mathematical Formulation', with read error set 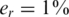 and DP error set
and DP error set 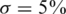 . We considered the relevant value of rtotal as the expected number of reads, and sampled the number of reads according to Γ(rtotal,1). We then sampled the reads according to a Binomial distribution.
. We considered the relevant value of rtotal as the expected number of reads, and sampled the number of reads according to Γ(rtotal,1). We then sampled the reads according to a Binomial distribution.
Carrier detection
Figure A3 presents the number of zero-error reconstructions out of the 633 SNPs as a function of the number of pools. Figure A3a–d correspond to different values of the coverage c. The black and red lines correspond to reconstruction based on experimental reads (`exp'-standard) and simulated reads, respectively. The performance according to `exp'-standard was in good accordance with the fully simulated model: 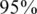 of the SNPs are reconstructed with zero errors, when considering 100 pools and with coverage c = 80, as opposed to
of the SNPs are reconstructed with zero errors, when considering 100 pools and with coverage c = 80, as opposed to 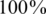 of the SNPs in the completely simulated case. However, when considering lower coverage and number of pools there were larger differences between reconstruction results, and performance for the experimental reads was poorer.
of the SNPs in the completely simulated case. However, when considering lower coverage and number of pools there were larger differences between reconstruction results, and performance for the experimental reads was poorer.
A possible factor contributing to these differences is that data for the 1000 Genomes Project were generated by sequencing on several Illumina machines and at different laboratories, which may lead to different levels of read errors for different individuals. This setting is different from the one suggested by our reconstruction model, which assumes a constant read error for all pools (see Appendix 4). Hence in order to overcome this variability, we estimated the read error for each individual separately, in the following way: for each individual i, we identified the SNPs for which it was classified as reference homozygous (AA) out of all 3489 SNPs, and estimated the read error for that sample er(i) as the total number of B reads for those SNPs divided by their total number of reads. We assume that SNP j in individual i had rrare(i,j) reads from the rare allele when sequenced in the 1000 Genomes Project, and its total number of reads for SNP j was rtotal(i,j). Then when sampling the reads for SNP j in individual i in our resampling-based simulated pools, the probability of selecting a rare allele was taken as rrare(i,j)/rtotal(i,j)−er(i), instead of the `standard’ probability rrare(i,j)/rtotal(i,j). The same value er(i) was subtracted from all SNPs of individual i. The rest of the settings were the same as in `exp'-standard. Reconstruction results using these `adjusted’ read counts appear as the green line in Figure A3, and show better correspondence to the simulated results. For example, in the case of c = 80 and 100 pools, >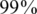 of the SNPs are reconstructed without errors.
of the SNPs are reconstructed without errors.
We conclude that our simulations are in very good accordance with the simulations based on experimental reads from the 1000 Genome Project, which may substantiate our model. These results are based on simulating pools, and not on real experimental pools, while the previous Appendix described small-scale results based on a true experimental pool. Together, the two datasets highlight the great potential of the approach—one may be able to identify carriers of hundreds of SNPs using only a very small number of pools.
APPENDIX 3
Decoding a library of microRNAs
Our CS framework can also be applied to a different, yet related, decoding problem described in (27). The aim in this problem was to determine the sequences of a large number of clones via group testing.
The experimental data consisted of N = 40 320 wells, each containing a single shRNA clone (targeting Arabidopsis genes), whose sequence we wish to determine. The wells were grouped into pools based on a Chinese Remainder Theorem design, where each pool consisted of samples from ∼100 wells, thus providing a rather sparse pooling design which is similar to the 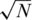 design we have examined. Each pool was then marked by one of nbar = 384 different barcodes, thus allowing up to 384 different pools to be applied to the same lane. Five different lanes of an Illumina GAII machine were used, resulting in k≅5 × 384 = 1920 pools in total (the actual number of pools was slightly smaller due to the Chinese Remainder Theorem setting). The total number of sequence reads from those 5 lanes was 28 million. As a first pre-processing step, we determined the set of unique sequences appearing in the wells by marking u = 27 276 unique sequences si, i = 1, … , 27 276, of length 30 bp each, which had more than 10 reads in at least one of the pools (this number is lower than the number of wells, which probably means that the same sequence may be present in more than one well): the goal was to assign to each well one of the sequences si, thus we wish to reconstruct an N × u sparse binary matrix providing the assignment. Ideally, each row in this matrix should contain a single non-zero entry.
design we have examined. Each pool was then marked by one of nbar = 384 different barcodes, thus allowing up to 384 different pools to be applied to the same lane. Five different lanes of an Illumina GAII machine were used, resulting in k≅5 × 384 = 1920 pools in total (the actual number of pools was slightly smaller due to the Chinese Remainder Theorem setting). The total number of sequence reads from those 5 lanes was 28 million. As a first pre-processing step, we determined the set of unique sequences appearing in the wells by marking u = 27 276 unique sequences si, i = 1, … , 27 276, of length 30 bp each, which had more than 10 reads in at least one of the pools (this number is lower than the number of wells, which probably means that the same sequence may be present in more than one well): the goal was to assign to each well one of the sequences si, thus we wish to reconstruct an N × u sparse binary matrix providing the assignment. Ideally, each row in this matrix should contain a single non-zero entry.
To solve this problem using the compressed se(que)nsing approach, we considered each of the u sequences independently. We essentially reduce the task to a SNP detection problem, where the reconstructed vector xi is a binary vector of dimension N = 40 320, which is non-zero for those wells that contain the sequence si. Thus, the sparsity condition for xi in this case is exemplified by the fact that each unique sequence is contained in only one, or a few wells (by design the purpose was to assign a different sequence to each well thus ensuring sparsity s = 1, yet it is often the case that a few wells contain the same sequence.) The sensing matrix M in this case is a 1920 × 40 320 sparse binary matrix indicating which wells participate in each pool, and is the same for all different sequences. The measurements vector yi is a binary vector of length k = 1920, indicating whether si was observed in each measurement. Hence, using this representation the decoding task in (27) is similar to the SNP detection problem, with three noticeable differences: first, the sensing matrix M is a specific structured and deterministic matrix, as opposed to our random sensing matrices. Second, the measurement vector yi is binary, while in our rare-allele detection task y contains frequencies of the rare allele in the pool. Lastly, in this task we have to solve many (rather than one) CS reconstruction problem, all sharing the same sensing matrix M. Given these differences our CS approach was completely robust to these modifications, and performed comparably well to the approach in (27) as described below.
Using our CS approach, we carried u = 27 276 independent reconstructions for each of the sequences present in the data. In each reconstruction, we have solved the standard CS problem with the GPSR equivalent formulation
| (A1) |
where each of the 27 276 sequences provides a different measurement vector yi (and thus usually different solution xi.) The value of τ was set as described in the ‘Results’ section.
Aggregating the results for all sequences, we obtained potential assignments for 34 623 out of the 42 320 wells, while the rest of the wells were not predicted to contain any of the sequences, i.e. their corresponding entry in the vector xi was zero for all reconstruction problems. Out of the wells with potential assignments, 25 991 were `unambiguous', i.e. predicted to contain a single sequence. To validate the sequence-to-well assignments, the sequences of 2760 randomly selected wells were determined independently using a conventional sequencing experiment. In 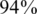 of these cases, our CS approach correctly matched the sequence in each well. These results are comparable to those reported in (27). Erlich et al. have also applied several clever heuristics, which filtered and weighed lower quality reads, thus achieving better results than ours. We have not used these heuristics, as our goal was merely to examine the robustness of our CS approach in this problem, applied as a `black-box’ solution.
of these cases, our CS approach correctly matched the sequence in each well. These results are comparable to those reported in (27). Erlich et al. have also applied several clever heuristics, which filtered and weighed lower quality reads, thus achieving better results than ours. We have not used these heuristics, as our goal was merely to examine the robustness of our CS approach in this problem, applied as a `black-box’ solution.
APPENDIX 4
Coping with unknown read error
We assume that the read error er is unknown to the researcher, but is constant across all lanes. One can introduce a slight modification to our procedure, which enables the learning of er from our pooling data. We replace z in Equation (7) by the convolution:
| (A2) |
The additive factor er−2zer is different for different values of z, but its dominant part is er. We can approximate it by 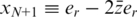 , obtained from averaging the term 2zer over all z values (i.e.
, obtained from averaging the term 2zer over all z values (i.e.  is the mean value of the vector z). We can now reformulate the CS problem by adding one extra variable. Specifically, the unknown vector x is replaced by x′ = (x,xN + 1) and Equation (7) is replaced by
is the mean value of the vector z). We can now reformulate the CS problem by adding one extra variable. Specifically, the unknown vector x is replaced by x′ = (x,xN + 1) and Equation (7) is replaced by
| (A3) |
where M′ is built from M by adding a constant column to its right as its N + 1's column with all its values set to −1.
REFERENCES
- 1.Hirschhorn J, Daly M. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 2.Klein R, Zeiss C, Chew E, Tsai J, Sackler R, Haynes C, Henning A, SanGiovanni J, Mane S, Mayne ST, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burton P, Clayton D, Cardon L, Craddock N, Deloukas P, Duncanson A, Kwiatkowski D, McCarthy M, Ouwehand W, Samani N, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 5.Cohen J, Kiss R, Pertsemlidis A, Marcel Y, McPherson R, Hobbs H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 6.McClellan J, Susser E, King M. Schizophrenia: a common disease caused by multiple rare alleles. Br. J. Psychiatry. 2007;190:194–199. doi: 10.1192/bjp.bp.106.025585. [DOI] [PubMed] [Google Scholar]
- 7.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li B, Leal S. Discovery of rare variants via sequencing: implications for the design of complex trait association studies. PLoS Genet. 2009;5:e1000481. doi: 10.1371/journal.pgen.1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mardis E. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. 2008. [DOI] [PubMed] [Google Scholar]
- 10.Margulies M, Egholm M, Altman W, Attiya S, Bader J, Bemben L, Berka J, Braverman M, Chen Y, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gunderson K, Kruglyak S, Graige M, Garcia F, Kermani B, Zhao C, Che D, Dickinson T, Wickham E, Bierle J, et al. Decoding randomly ordered DNA arrays. Genome Res. 2004;14:870–877. doi: 10.1101/gr.2255804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris T, Buzby P, Babcock H, Beer E, Bowers J, Braslavsky I, Causey M, Colonell J, DiMeo J, Efcavitch J, et al. Single-molecule DNA sequencing of a viral genome. Science. 2008;320:106–109. doi: 10.1126/science.1150427. [DOI] [PubMed] [Google Scholar]
- 13.Martinez B, Eriksson O, Badhai J, Frjmark A-S, Bongcam-Rudloff E, Dahl N, Schuster J. Targeted resequencing and analysis of the Diamond-Blackfan anemia disease locus RPS 19. PLoS ONE. 2009;4:e6172. doi: 10.1371/journal.pone.0006172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosa-Rosa J, Gracia-Aznrez F, Hodges E, Pita G, Rooks M, Xuan Z, Bhattacharjee A, Brizuela L, Silva J, Hannon G, et al. Deep sequencing of target linkage assay-identified regions in familial breast cancer: methods, analysis pipeline and troubleshooting. PLoS ONE. 2010;5:e9976. doi: 10.1371/journal.pone.0009976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mamanova L, Coffey A, Scott C, Kozarewa I, Turner E, Kumar A, Howard E, Shendure J, Turner D. Target-enrichment strategies for next-generation sequencing. Nat. Meth. 2009;110:471–478. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- 16.Albert T, Molla M, Muzny D, Nazareth L, Wheeler D, Song X, Richmond T, Middle C, Rodesch M, Packard C, et al. Direct selection of human genomic loci by microarray hybridization. Nat. Meth. 2007;4:903–905. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- 17.Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust E, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng S, Turner E, Robertson P, Flygare S, Bigham A, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler E, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norton N, Williams N, Williams H, Spurlock G, Kirov G, Morris D, Hoogendoorn B, Owen M, O'Donovan M. Universal, robust, highly quantitative SNP allele frequency measurement in DNA pools. Hum. Genet. 2002;110:471–478. doi: 10.1007/s00439-002-0706-6. [DOI] [PubMed] [Google Scholar]
- 20.Yang H, Fann C. Association mapping using pooled DNA. Meth. Mol. Biol. 2007;376:161–175. doi: 10.1007/978-1-59745-389-9_12. [DOI] [PubMed] [Google Scholar]
- 21.Shaw S, Carrasquillo M, Kashuk C, Puffenberger E, Chakravarti A. Allele frequency distributions in pooled DNA samples: applications to mapping complex disease genes. Genome Res. 1998;8:111–123. doi: 10.1101/gr.8.2.111. [DOI] [PubMed] [Google Scholar]
- 22.Du D, Hwang F. Combinatorial group testing and its applications. River Edge, NJ: World Scientific Pub; 2000. [Google Scholar]
- 23.Prabhu S, Pe'er I. Overlapping pools for high-throughput targeted resequencing. Genome Res. 2009;19:1254–1261. doi: 10.1101/gr.088559.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D'yachkov AG, Macula A, Torney D, Vilenkin P, Yekhanin S. Proceedings of International Workshop on Algebraic and Combinatorial Coding Theory (ACCT) Bansko, Bulgaria: 2000. New results in the theory of superimposed codes; pp. 126–136. [Google Scholar]
- 25.Ericson T, Gyorfi L. Superimposed codes in Rn. IEEE Trans. Inf. Theory. 1988;34:877–880. [Google Scholar]
- 26.Dai W, Milenkovic O. Weighted superimposed codes and constrained integer compressed sensing. IEEE Trans. Inf. Theory. 2009;55:2215–2229. [Google Scholar]
- 27.Erlich Y, Chang K, Gordon A, Ronen R, Navon O, Rooks M, Hannon G. DNA Sudoku-harnessing high-throughput sequencing for multiplexed specimen analysis. Genome Res. 2009;19:1243–1253. doi: 10.1101/gr.092957.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilbert A, Iwen M, Strauss M. In 42nd Asilomar Conference on Signals, Systems, and Computers. 2008. Group testing and sparse signal recovery. Monterey, CA. IEEE Signal Processing Society, Piscataway, NJ, USA. [Google Scholar]
- 29.Erlich Y, Gordon A, Brand M, Hannon G, Mitra P. Compressed Genotyping. Arxiv preprint Quantitative Biology/0909.3691. 2009 [Google Scholar]
- 30.Candes E. In Proceedings of the International Congress of Mathematics. Madrid, Spain: 2006. Compressive sampling; pp. 1433–1452. [Google Scholar]
- 31.Donoho D. Compressed sensing. IEEE Trans. Inf. Theory. 2006;52:1289–1306. [Google Scholar]
- 32.Lustig M, Donoho D, Pauly J. Sparse MRI: the application of compressed sensing for rapid MR imaging. Magn. Reson. Med. 2007;58:1182–1195. doi: 10.1002/mrm.21391. [DOI] [PubMed] [Google Scholar]
- 33.Duarte M, Davenport M, Takhar D, Laska J, Sun T, Kelly K, Baraniuk R. Single-pixel imaging via compressive sampling. IEEE Signal Process. Mag. 2008;25:83–91. [Google Scholar]
- 34.Lin T, Herrmann F. Compressed wavefield extrapolation. Geophysics. 2007;72:SM77–SM93. [Google Scholar]
- 35.Bobin J, Starck J, Ottensamer R. Compressed sensing in astronomy. J. Sel. Top. Signal Process. 2008;2:718–726. [Google Scholar]
- 36.Dai W, Sheikh M, Milenkovic O, Baraniuk R. Compressive sensing DNA microarrays. EURASIP J. Bioinform. Syst. Biol. 2009;2009:162824. doi: 10.1155/2009/162824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Becker S, Bobin J, Candes E. NESTA: a fast and accurate first-order method for sparse recovery. Arxiv preprint arXiv:0904.3367. 2009 [Google Scholar]
- 38.Out A, van Minderhout I, Goeman J, Ariyurek Y, Ossowski S, Schneeberger K, Weigal D, van Galen M, Taschner P, Tops C, et al. Deep sequencing to reveal new variants in pooled DNA samples. Hum. Mutat. 2009;30:1703–1712. doi: 10.1002/humu.21122. [DOI] [PubMed] [Google Scholar]
- 39.Candes E, Tao T. Decoding by linear programming. IEEE Trans. Inf. Theory. 2005;51:4203–4215. [Google Scholar]
- 40.Candes E, Romberg J, Tao T. Stable signal recovery from incomplete and inaccurate measurements. Arxiv preprint math/0503066. 2005 [Google Scholar]
- 41.Donoho D. For most large underdetermined systems of linear equations the minimal l1-norm solution is also the sparsest solution. Commun. Pure Appl. Math. 2006;59:797–829. [Google Scholar]
- 42.Candes E, Tao T. Near-optimal signal recovery from random projections: universal encoding strategies? IEEE Trans. Inf. Theory. 2006;52:5406–5425. [Google Scholar]
- 43.Candes E, Rudelson M, Tao T, Vershynin R. In Annual Symposium on Foundations of Computer Science. Vol. 46. Hoboken, USA: Wiley-IEEE Computer Society Press; 2005. Error correction via linear programming; pp. 295–308. [Google Scholar]
- 44.Figueiredo M, Nowak R, Wright S. Gradient projection for sparse reconstruction: application to compressed sensing and other inverse problems. IEEE J. Sel. Top. Signal Process. 2007;1:586–597. [Google Scholar]
- 45.Donoho D, Tanner J. Sparse nonnegative solution of underdetermined linear equations by linear programming. Proc. Natl Acad. Sci. USA. 2005;102:9446–9451. doi: 10.1073/pnas.0502269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ingman M, Gyllensten U. SNP frequency estimation using massively parallel sequencing of pooled DNA. Eur. J. Hum. Genet. 2009;17:383–386. doi: 10.1038/ejhg.2008.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Freeman W, Walker S, Vrana K. Quantitative RT-PCR: pitfalls and potential. Biotechniques. 1999;26:112–125. doi: 10.2144/99261rv01. [DOI] [PubMed] [Google Scholar]
- 49.Kao W, Stevens K, Song Y. BayesCall: a model-based base-calling algorithm for high-throughput short-read sequencing. Genome Res. 2009;19:1884–1895. doi: 10.1101/gr.095299.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Macgregor S. Most pooling variation in array-based DNA pooling is attributable to array error rather than pool construction error. Eur. J. Hum. Genet. 2007;15:501–504. doi: 10.1038/sj.ejhg.5201768. [DOI] [PubMed] [Google Scholar]
- 51.Kim S, Koh K, Lustig M, Boyd S, Gorinevsky D. An interior-point method for large-scale l1-regularized least squares. IEEE J. Sel. Top. Signal Process. 2007;1:606–617. [Google Scholar]
- 52.Hamady M, Walker J, Harris J, Gold N, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Meth. 2008;5:235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berinde R, Indyk P. Sparse recovery using sparse random matrices. preprint. 2008 [Google Scholar]
- 54.Ji S, Xue Y, Carin L. Bayesian compressive sensing. IEEE Trans. Signal Process. 2008;56:2346–2356. [Google Scholar]
- 55.Baron D, Sarvatham S, Baraniuk R. Bayesian compressive sensing via belief propagation. IEEE Trans. Signal Process. 2010;58:269–280. [Google Scholar]
- 56.Brockman W, Alvarez P, Young S, Garber M, Giannoukos G, Lee W, Russ C, Lander E, Nusbaum C, Jaffe D. Quality scores and SNP detection in sequencing-by-synthesis systems. Genome Res. 2008;18:763. doi: 10.1101/gr.070227.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McKenna AH, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010 doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]