

Abstract
Purpose
Both granular and lattice deposits are present in Avellino corneal dystrophy (ACD), primarily associated with R124H mutation of transforming growth factor β-induced protein (TGFBI). We investigated the presence of these deposits in other TGFBI mutations and the use of Thioflavin-T (ThT), a fluorescent amyloid stain for characterizing corneal amyloid deposits.
Methods
Surgical corneal specimens of three unrelated patients clinically diagnosed with ACD were studied. Corneal sections from normal and prior patients with lattice corneal dystrophy (LCD) were used as controls. Histochemical studies were performed with Congo red and Masson trichrome, and fluorescent imaging with scanning laser confocal microscopy was performed for ThT and an anti-TGFBI antibody staining.
Results
Clinical and histopathological findings supported the diagnoses of ACD in these three cases, in whom granular deposits stained with Masson trichrome and lattice deposits stained with ThT and Congo red showing birefringence and dichroism as expected. However, genotyping revealed a heterozygous R124C mutation in each case. In addition to classical stromal deposits, unique sub-epithelial TGFBI aggregates, that stain with neither ThT nor trichrome, were observed. In control LCD sections, stromal deposits stained with ThT but not with trichrome, confirming lack of granular deposits.
Conclusions
Our results demonstrate that both granular and lattice deposits can be associated with R124C mutation, other than R124H. An additional feature of non-hyaline, non-amyloid TGFBI sub-epithelial deposits might substantiate such cases be categorized as a variant form of LCD or ACD. This study further validates ThT staining for detection of amyloid TGFBI deposits.
Keywords: Avellino corneal dystrophy, keratoepithelin, TGFBI, amyloid, ThT
Introduction
Corneal epithelial and stromal dystrophies linked to the TGFBI gene on human chromosome 5q31 have thus far been classified according to their phenotypes 1, 2. Avellino corneal dystrophy (ACD) manifests both granular and lattice deposits, for which reason it is also referred to as “Combined granular-lattice corneal dystrophy” 3. In 1988, Folberg reported the histopathological findings of patients with this corneal dystrophy and traced the family origins of each patient to the Italian province of Avellino, from which the dystrophy gets its name 4. However, the subtle differences in clinical presentation of ACD and granular corneal dystrophy (GCD) were described long before ACD was linked to Avellino. In 1938, Bucklers described the phenotype of ACD, and using the same cases, Weidle sub-categorized GCD based on these differences 1, 5. The fact that ACD was known as a milder form of GCD for many decades, provides the historic reason as to why ACD is sub-classified today under GCDs as GCD type II. Histochemically, ACD is characterized by both hyaline and amyloid deposits, features of granular and lattice corneal dystrophies respectively. Hyaline and amyloid materials can be detected histochemically by using trichrome and Congo red staining respectively. Other than Congo red, amyloid materials also stain with fluorescent dye Thioflavin-T (ThT) 6.
A G→A mutation at nucleotide 418 in the TGFBI gene leading to an arginine to histidine substitution at codon 124 (R124H) has been correlated with the phenotype of ACD 7–13. Patients with homozygous mutations show more severity in phenotype compared to those with heterozygous mutations 14. ACD has frequently been misdiagnosed clinically as GCD I, which is usually associated with TGFBI R555W mutation 11, 15, 16. This is probably due to the late onset of lattice deposits in ACD cases, and unfamiliarity with icicle and star-shaped opacities, characteristic of ACD, when lattice lines may not be obvious 17, 18. For this reason, many investigators think that direct examination may be insufficient for the definite diagnosis of ACD and that mutation analysis confirming the R124H TGFBI mutation should be required 9, 19, 20. However, there have been reported cases of patients having a clinico-pathological diagnosis of ACD but have the TGFBI R555W mutation 20, 21. The current work describes three unrelated cases that were clinically and histopathologically diagnosed as ACD, but having the R124C TGFBI mutation, which is typically associated with lattice corneal dystrophy (LCD).
Materials and Methods
Clinical Specimens
Corneal specimens were obtained after therapeutic keratoplasty from three patients (one eye each) with a clinical diagnosis of ACD. The retrospective review was approved by Institutional Review Boards of Washington University and Eye Hospital of Ho Chi Minh City (EHHCMC), Vietnam, respectively. Deep anterior lamellar keratoplasty (DALK) was performed in one male patient (42 years of age) at Washington University (case 1) and penetrating keratoplasty was performed at EHHCMC in two unrelated male patients (27 and 35 years of age, respectively) (case 2 and 3). No known history of hereditary corneal diseases was noted in the immediate family members of these patients. However, the only son (15 years of age) of the patient from Washington University has been known to have symptoms of recurrent corneal erosions without evident corneal opacity. All three patients had no prior history of ocular surgery before keratoplasty and had severe symptoms of recurrent corneal erosion and severe decrease of vision in both eyes with dense central granular corneal opacities. The corneal buttons thus obtained from keratoplasty were fixed in 10% buffered formalin. Blood samples were collected from these three patients for genetic analysis after informed consents. Histopathology studies of all corneal specimens were performed at Washington University. Histological sections of normal corneas and three previously identified and unrelated cases of LCD (male 79y, female 84y, female 81y) from Washington University were used as negative and positive controls respectively, for ThT fluorescence microscopy. One out of the three LCD control cases manifested recurrent LCD (Female 84y). Normal corneal tissues for sections and protein extraction were also obtained from Mid-America Transplantation Services (Saint Louis, MO).
Materials
Western C molecular weight protein markers, 7.5% tris-HCl precast gels, 0.2 μm PVDF blotting paper, and StrepTactin-horseradish peroxidase conjugate were purchased from Biorad (Hercules, CA). Affinity purified rabbit anti-TGFBI antibody was obtained from Proteintech Group (Chicago, IL). Recombinant full-length TGFBI and monoclonal anti-TGFBI antibody were purchased from R & D Systems (Minneapolis, MN). ECL horseradish peroxidase linked donkey-anti-rabbit IgG antibody and ECL Plus western blotting detection system were purchased from GE Amersham (Piscataway, NJ). SYTOX Orange nucleic acid fluorescent stain, Alexa633 conjugated-goat-anti-rabbit secondary antibody, Image-iT FX signal enhancer and ProLong Gold antifade reagent were obtained from Invitrogen Molecular Probes (Carlsbad, CA). Antigen unmasking solution for antigen retrieval was purchased from Vector Laboratories (Burlingame, CA). Phenylmethyl sulfonyl fluoride (PMSF) was purchased from Amresco (Solon, Ohio). Puromycin was purchased from Clonetech (Mountain View, CA) and Fugene 6 transfecting agent was obtained from Roche Applied Sciences (Indianapolis, IN). HEK293FT cells and all other cell-culture reagents were obtained from Invitrogen (Carlsbad, CA). HisPur Cobalt resin was obtained from Pierce (Rockford, IL). Citrosolv solvent and clearing agent for histology, ThT, tris base, urea, and bromophenol blue were obtained from Fisher Scientific (Pittsburg, PA). Sodium dodecyl sulphate (SDS), β-mercaptoethanol, glycine, glycerol and all other chemicals were purchased from Sigma Aldrich (Saint Louis, MO).
Purification of recombinant TGFBI
The human TGFBI cDNA was PCR amplified from an I.M.A.G.E. clone (clone ID 2957915, Genbank accession number BE206112) and inserted into pIRES.puro2 for stable expression in HEK293FT mammalian cells. A hexa-histidine tag and a Strep II tag were inserted after the signal peptide and before the RGD motifs of TGFBI, respectively. Cells were transfected using Fugene 6 transfecting reagent, after which they were maintained in DMEM (10% FBS and 3 μg/ml puromycin). Individual clones were selected by monitoring TGFBI expression levels measured by dot-blot assays. Clones stably expressing TGFBI were cultured in DMEM (10% FBS, 3 μg/ml puromycin). For purification of TGFBI, media was slowly replaced with serum-free media FreeStyle 293 over the course of three passages. Secreted TGFBI (amino acids 24-641) was purified by metal affinity chromatography using HisPur Cobalt resin, after which it was dialyzed against phosphate buffer and concentrated by filtration (50Kda centrifuge filters) to lyophilize and store at −80°C until needed. Lyophilized TGFBI was dissolved in 10 M urea (10mM phosphate buffer, pH 7.4) for 1 h at room temperature and then gel filtered into phosphate buffer. It was then allowed to aggregate in PBS at 37°C.
Western blotting
Normal cornea buttons were homogenized in protein lysis buffer as previously described 22. In brief, half a normal cornea button stored in liquid nitrogen was homogenized on ice, after which 200 μl of lysis buffer A (100 mM glycine, 5.5 mM tris pH 8.2, 2.7% SDS, 3.45 M urea, 12% β-mercaptoethanol and 0.014% bromophenol blue) or B (1%SDS, 10% glycerol, 1% β-mercaptoethanol and 0.001% bromophenol blue) was added to it. Following addition of protease inhibitor PMSF, mixtures were homogenized again and kept at room temperature for 1 h. The mixture was then centrifuged at 16000g for 5 min. The supernatants were stored at −20°C until used as the protein lysis samples. Proteins were separated on a SDS-polyacrylamide gel by electrophoresis and transferred onto PVDF membranes. Recombinant TGFBI (amino acids 24-641) purified from transfected HEK293FT cells and full-length recombinant TGFBI (amino acids 1-683) purchased from R&D systems were used as controls on the same gel. Membranes were blocked with 5% non-fat dry milk in tris-buffered saline-tween (TBS-T) and then incubated with TGFBI antibody (1:200 dilution) overnight at 4°C. Three TGFBI antibodies (R&D Systems, Proeintech Group, and one rabbit anti-TGFBI custom-made for our lab) were used on separate blots. Blots were washed with TBS-T and probed with donkey-anti-rabbit horseradish conjugated secondary antibody (1: 100,000 dilution) for 1h at room temperature. Western C molecular weight protein markers were developed using StrepTactin-horseradish peroxidase conjugate, separately from the lanes containing TGFBI samples. Blots were developed using electro-chemifluorescence ECL reagent and imaged using the STORM PhosphorImager system (GE Amersham, NJ) with a 450nm excitation and 520LP emission filter. Resultant images from the scanner control software were analyzed using NIH ImageJ software.
Immunofluorescence and Histological Staining
Normal and dystrophic corneas were fixed in 10% buffered formalin at room temperature, dehydrated in ethanol and embedded in paraffin. For immunofluorescence, 4 μm sections were de-paraffinized by incubation in Cirosolv for 10 minutes, and subsequently re-hydrated using a series of aqueous ethanol solutions (100% to 30% v/v). After washing with phosphate buffer saline (PBS, pH 7.4), antigen retrieval was carried out using citrate buffer based Antigen Unmasking solution (pH 6.0). Tissue sections were then blocked using Image-iT FX signal enhancer for 30–50 min at room temperature and incubated with TGFBI antibody (1:200 in PBS, antibody from Proteintech Group) overnight at 4°C. Two other antibodies specific for TGFBI were used for immunofluorescence staining of dystrophic and non-dystrophic cornea sections. One of them was monoclonal mouse antibody (R&D Systems) and other a polyclonal affinity purified rabbit antibody (purified in-house). Following fixation with 0.4% paraformaldehyde for 15 min, goat-anti-rabbit Alexa 633 secondary antibody (1:500) and SYTOX Orange nuclear stain (1:1000) were applied for 1 h at room temperature. Tissue sections re-fixed with 0.4% paraformaldehyde were then incubated in 200 μM ThT solution for 1 h and mounted using ProLong Gold antifade reagent. Three days after mounting, slides were sealed using nail polish and viewed using a Zeiss LSM 510 confocal microscope (Carl Zeiss Microimaging, NY) and a 20× objective. The 458 nm, 543 nm and 633 nm lasers were used to excite ThT, SYTOX Orange and Alexa 633 secondary antibody respectively. The resultant fluorescence was detected under multi-track mode using 475–525 nm, 560–615 nm and LP650 emission filters respectively.
Paraffin sections of 5 μm from dystrophic and normal corneas were also stained routinely with Masson trichrome stains and/or Congo red and imaged using an Olympus BX60 microscope (Olympus, Japan) and a RT Slider Spot camera.
Genotyping
The genotyping of our three index cases was conducted by the John and Marcia Carver Nonprofit Genetic Testing Laboratory at the University of Iowa.
Results
Genotyping
Automated DNA sequencing of the coding sequence of the TGFBI gene from cases 1, 2 and 3 revealed the following disease-causing variation and other non-disease-causing variations:
Case 1: One probably high penetrance sequence of disease-causing variation and four other non-disease-causing variations in the TGFBI gene: R124C (CGC>TGC) heterozygous-EPP=3; L472L (CTC>CTT) homozygous-EPP=0; F540F (TTT>TTC) homozygous-Epp=0; IVS10-43 A>G homozygous-EPP=0; IVS12+23 G>A heterozygous-EPP=0.
Case 2: One probably high penetrance sequence of disease-causing variation and two other non-disease-causing variations in the TGFBI gene: R124C (CGC>TGC) heterozygous-EPP=3; F540F (TTT>TTC) heterozygous-Epp=0; IVS11-30 A>G heterozygous-EPP=0.
Case 3: One probably high penetrance sequence of disease-causing variation and two other non-disease-causing variations in the TGFBI gene: R124C (CGC>TGC) heterozygous-EPP=3; F540F (TTT>TTC) heterozygous-Epp=0; IVS11-30 A>G heterozygous-EPP=0.
Clinical and Histopathological findings
Slit lamp examination of all index cases showed bilateral central corneal opacity with snowflake-like or crumb-like granular deposits (arrows in Figure 1A, case 1) and lattice-line or arborizing deposits in both eyes (* in Figure 1A, case 1), supporting a clinical diagnosis of ACD. Masson trichrome staining of all the index tissue sections showed trichrome-stained hyaline aggregates in the anterior stroma, confirming presence of granular deposits (red colored deposits, Figure 1B, case 2). Also present were multiple sub-epithelial deposits (Figure 1B, arrows) and fusiform stromal lesions in the mid- to deep stroma (Figure 1B, arrowheads), neither of which stained with trichrome. When these sub-Bowman trichrome-stained deposits are present underneath the sub-epithelial deposits, they are found to be larger compared to those located under normal epithelium or in deeper stroma. Congo red staining of tissue sections showed presence of amyloid deposits under normal and polarized light (Figures 1C and 1D, from case 2). The fusiform deposits in the mid- to deep stroma stain with Congo red and show characteristic yellow-green birefringence under polarized light (arrowheads), indicating that these are amyloid in nature, as found in LCD. There are also some Congo red-stained birifringent deposits in the sub Bowman or anterior stroma (underneath the sub-epithelial deposits). Interestingly, the sub-epithelial deposits (arrows in Figures 1B to 1D) did not stain with Congo red. Tissue sections from cases 1 and 3 also showed similar Congo red staining patterns (data not shown). Approximately one year after DALK in case 1, recurrent granular deposits were noted in the corneal raft slights off the visual axis (Figure 1E) with a best corrected visual acuity of 20/25. Taken together, the clinical findings of granular and lattice-like deposits by slit-lamp examination and presence of both trichrome-stained hyaline material and Congo red-stained amyloid material in tissue sections in all our index cases support the diagnosis of ACD clinically and histopathologically.
FIGURE 1.

Clinical and histopathological findings confirming ACD. A, Slit lamp image of case 1. Arrows: granular deposits; *lattice lesions. A inset, Magnified image of confluent granular deposits. B, Masson trichrome stained tissue of case 2; sub-Bowman’s deposits are trichrome stained; sub-epithelial (arrows) and stromal fusiform deposits (arrowheads) do not stain with trichrome. C, Light microscopy of Congo red stained tissue from case 2. D, Same section as in C under polarized light. Stromal fusiform deposits stain with Congo red (C) and show birefringence under polarized light (D) (arrows). There is also some anterior stromal staining by Congo red. Sub-epithelial deposits do not stain with Congo red (arrows). E, Slit lamp photo of case 1 approximately 1 year after DALK shows multiple recurrent granular deposits (arrows) in the corneal graft.
Specificity of TGFBI antibody
Although the TGFBI antibody obtained from Protein Tech Group has been shown to be specific in staining of cell lysates and lung tissue sections 23, to establish its specificity for corneal TGFBI, crude protein extracts from the cornea were separated by SDS electrophoresis and probed with this antibody. The immunoblot in Figure 2 shows that this antibody detects bands of both full-length (a.a. 1-683, lane 1) or almost full-length TGFBI (a.a 24-641, lane 2) in protein extracts (lanes 3–6). These bands migrated as ~68kda proteins on the gel, same as recombinant TGFBI (lane 1, full-length TGFBI, a.a. 1-681) obtained from R&D Systems and recombinant TGFBI purified in our lab (lane 2, a.a 24-641; purified from HEK293FT cells). The protein bands of around 140 kda seen in both protein extracts and recombinant TGFBI samples correspond to dimers of TGFBI. The two other TGFBI antibodies (R&D systems and that custom made for our lab) showed similar staining patterns on the normal corneal tissue as well as on immunoblots of protein extracts (data not shown), further confirming that our findings are consistent in corneal tissues.
FIGURE 2.
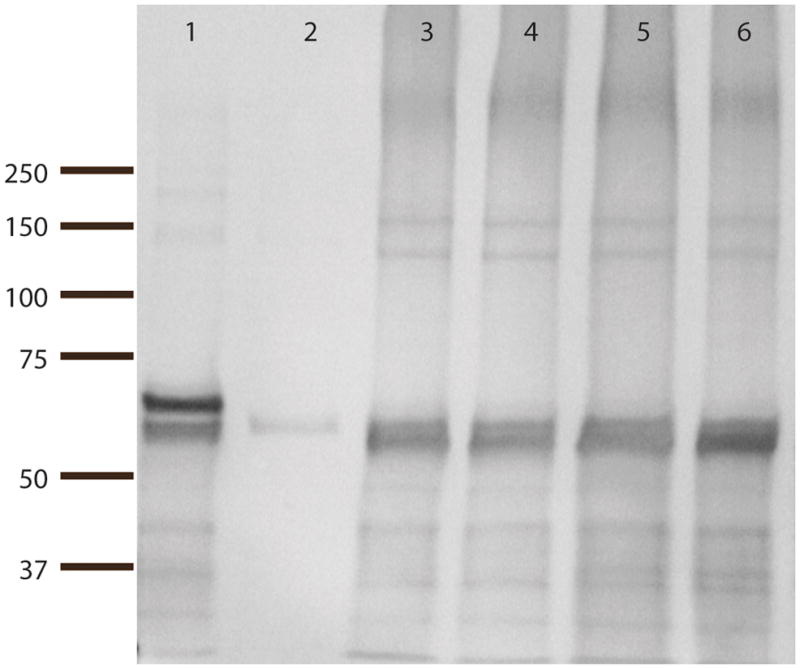
Western blot of crude protein extracts from normal cornea using a TGFBI antibody (Proteintech Group). Lane 1: commercially available recombinant TGFBI (a.a. 1-683). Lane 2: recombinant TGFBI purified from transfected mammalian cells (24-641). Lane 3 and 4: 1 μl and 0.5 μl corneal protein extract A, respectively. Lanes 5 and 6: 1 μl and 0.5 μl corneal protein extract B, respectively.
Amyloid and Non-amyloid deposits of TGFBI
To establish if the observed amyloid deposits in corneal tissue consist of TGFBI, tissue sections were immunofluorescently co-stained with TGFBI antibody and amyloid-specific fluorescent probe ThT. A representative image of a cornea section from case 2 stained with TGFBI antibody (in green), ThT (in red) and nuclear stain SYTOX Orange (in blue) is shown in Figure 3. Interestingly, the sub-epithelial deposits that stained with neither trichrome nor Congo red in histopathological sections (Figure 1, arrows), stained positively with TGFBI antibody (as seen in green in Figure 3A and Figure 3C, arrows), but not with ThT. A dense band of amyloid deposits (stained with ThT in red, in Figure 3B and 3C) were noted in the anterior stroma just beneath the sub-epithelial deposits. Although TGFBI was present throughout the entire stroma, focal areas of TGFBI aggregates with higher fluorescent intensity can be observed (Figure 3A–C, arrowheads). These aggregates correspond to the fusiform deposits in Figure 1, which also stained positively with ThT, indicating that they contain TGFBI and are amyloid in nature. It is evident from Figure 3C that the sub-epithelial deposits consist of TGFBI and are non-amyloid in nature. In contrast, the deposits in the anterior stroma and smaller fusiform lesions (arrowheads) in the mid- to deep stroma contain TGFBI and are amyloid in nature. Furthermore, these ThT-stained deep stromal deposits were dichroic and birifringent by Congo red staining under polarized light (Figure 1C, 1D), corroborating that they are indeed amyloid in nature.
FIGURE 3.

Corneal tissue section from case 2. A, Stained with TGFBI antibody (green) and nuclear stain SYTOX Orange (blue); shows fusiform stromal deposits of TGFBI (arrowheads), corresponding to Congo red-stained lesions (arrowheads), and sub-epithelial TGFBI deposits (arrows). B, Stained with ThT (red) and nuclear stain SYTOX Orange (blue). C, Superimposition of TGFBI and ThT staining. Fusiform TGFBI stromal deposits that stained with Congo red, stain with ThT (arrowheads). Sub-epithelial deposits that did not stain with either Congo red or trichrome, stain with TGFBI antibody, but not with ThT.
Similar three-color florescence composite images of dystrophic tissue from cases 1 and 3 are shown in Figure 4A and 4B, respectively. Although there is thin epithelium secondary to recurrent erosions in the tissue from case 1, it is evident in Figure 4A and 4B that the TGFBI deposits in the anterior stroma are amyloid in nature. The smaller and deeper fusiform lesions are also ThT-stained in tissue sections from cases 1 and 3 (Figure 4A and 4B), similar to those in case 2. Moreover, none of the sub-epithelial TGFBI deposits observed in these three cases of ACD were amyloid in nature (Figures 3C, 4A and 4B, arrows). We used cornea tissue sections from patients with LCD as positive controls for ThT staining (Figure 4C). These sections exhibited similar staining patterns as our ACD patients with positive ThT staining in both anterior and deeper stroma, except that no sub-epithelial TGFBI deposits were observed in LCD tissues. No focal TGFBI deposits or ThT staining was observed in normal tissues (Figure 4D). However, we also observed TGFBI staining in the interlamellar junctions of the stroma in normal tissue, as previously described by Streeten et al. 24.
FIGURE 4.

Tissue sections stained with TGFBI antibody (green), ThT (red) and nuclear stain SYTOX Orange (blue) from A, Case 1; shows ThT stained deposits in the anterior stroma and stromal fusiform deposits in deeper stroma that stain with ThT, and more brightly with TGFI antibody compared to stromal background staining for TGFBI. B, Case 3; shows TGFBI deposits in the deeper stroma stained also with ThT, and some TGFBI-positive and ThT positive deposits in anterior stroma. B inset, different area of same section as in B; shows sub-epithelial TGFBI deposits that do no stain with ThT. C, LCD positive control. C inset, LCD positive control from another patient; shows larger stromal ThT-stained deposits. D, normal cornea; no evident ThT staining. (Bars in insets are 50 μm).
Discussion
In 1938, Bucklers described a family of patients who suffered from what we now refer to as ACD, as a milder version of GCD. These cases were used 50 years later by Weidle to sub-classify variants of GCD 1, 5. In 1988, Folberg et al. described a series of four unrelated ACD patients as atypical granular corneal dystrophy with pathologic features of lattice-like amyloid deposits. The anterior stroma was reported to contain granular deposits amongst which amyloid matter was also shown to be present by Congo red staining. In addition to these amyloid deposits located in the superficial stroma, amyloid was also noted in the mid- to deep stromal fusiform lesions 4. These findings have been confirmed by many other investigators 3, 8, 11, 15, 24. Thus, ACD can be histopathologically defined as having both trichrome-stained hyaline deposits (corresponding to granular deposits) and Congo red or ThT-stained amyloid deposits (corresponding to lattice deposits). Classic clinical signs of ACD include anterior stromal discrete grayish-white deposits, lattice lesions in the mid to posterior stroma and anterior stromal haze 25. Using these definitions of ACD, it is clear that the index cases of corneal dystrophy described in this study should be clinically and histopathologically categorized as ACD. However, to our surprise, all three of our index cases showed R124C TGFBI mutation, which is usually associated with LCD.
The corneal dystrophy described in this study also show similar TGFBI antibody staining characteristics as those observed by other investigators. Using TGFBI antibodies raised against amino acids 426-682 (K15) and 69-364 (K2), Konishi et al. have shown ACD corneas with R124H TGFBI mutation to have sub-epithelial TGFBI deposits (trichrome-stained), superficial stromal TGFBI deposits (trichrome-stained), and fusiform TGFBI deposits in the mid- to deep stroma (Congo red-stained) 26. The major difference between their ACD cases and our series is that the sub-epithelial deposits observed in our study did not stain with trichrome or Congo red. In addition, there was amyloid deposition in the anterior stroma (by Congo red and ThT staining) amongst trichrome stained deposits in our tissues. The granular deposits of ACD are frequently described to break through Bowman’s membrane and extend into the corneal surface 18, 24, 27. Konishi et al. showed that tissues from GCD patients with R555W mutation had only such trichrome-stained sub-epithelial deposits. Interestingly, tissues from five of their seven LCD patients with R124C mutation showed trichrome-stained deposits between the epithelium and Bowman’s layer. However, those sub-epithelial deposits did not with Congo red. To the best of our knowledge, our report is the first one of R124C-related corneal dystrophy tissue to contain sub-epithelial TGFBI deposits that do not stain with trichrome along with trichrome-stained deposits in the anterior stroma. In fact, these sub-epithelial deposits do not contain amyloid either, as indicated by lack of Congo red birefringence under polarized light or ThT fluorescence. Such sub-epithelial deposits that stain with neither trichrome nor Congo red or ThT were observed in a patient with the H626P TGFBI mutation 28, suggesting that this phenomenon of non-hyaline and non-amyloid sub-epithelial aggregation of TGFBI is not limited to the R124C TGFBI mutation, as shown in our index cases. Precisely, Wheeldon et al. described the H626P TGFBI mutation carrying patient to have phenotypic features of both type I and type II corneal dystrophies of the Bowman’s layer (CDB), also known as Reis-Buckler and Thiel-Behnke (or honeycomb) corneal dystrophies, respectively. In fact, it has been shown that such undulating sub-epithelial deposits that do no stain with either Congo red or trichrome, but do contain TGFBI, are a classical feature of Thiel-Behnke corneal dystrophy, which is associated with R555Q TGFBI mutation 24, 29, 30. This fibrocellular layer that replaces the Bowman’s layer is shown to consist of curly fibers by transmission electron microcopy (TEM). As there were no tissue specimens from our index cases remaining for TEM processing, we are unable to confirm that the sub-epithelial deposits observed in the cases described here in have the same ultra-structure as those present in Thiel-Behnke corneal dystrophy tissues. However, since no amyloid deposits are associated with Thiel-Behnke corneal dystrophy, our index cases would most appropriately be classified as a variant form of ACD: one that manifests amyloid and hyaline deposits, as in classical ACD, but it also shows TGFBI non-amyloid and non-hyaline sub-epithelial deposits, as those found in Thiel-Behnke corneal dystrophy. It is possible that these sub-epithelial deposits are likely a result of hyper-productivity of TGFBI in hyper-proliferating epithelial cells.
After Vassar and Culling demonstrated the use of ThT to identify amyloid deposits in various organs such as the kidney in 1959 6, ThT had been used to identify amyloid deposits in the cornea using UV filters in the late 1960s 27, 31, 32. It was pointed out that amyloid deposits are more sharply defined with ThT staining compared to other amyloid dyes such as Congo red, although the technique did not become widely adapted in routine histopathology probably due to photo-bleaching and the limited availability of fluorescent microscopes at the time 6, 33. With the advent of fluorescence anti-photobleaching agents, the means to take and store digital images and the availability of fluorescence microscopes, these initial disadvantages of ThT are largely resolved. We have imaged ThT stained cornea sections up to several months after staining without problems of fluorescence photobleaching. In a recent study in which both ThT and Congo red were used to detect amyloid deposits in corneal dystrophies, Congo red stained the stroma but was not birigringent under polarized light 34. Such a finding suggests that although Congo red bound to the material, it was not amyloid in nature due to the lack of Congo red molecule alignment, which is responsible for the birifringent property of the dye. The ThT signal was negative since it is a fluorescent dye that has a high Stokes shift when bound only to amyloid, making it an ideal fluorescent reporter for amyloid. Although ThT fluorescence has been shown to depend on the morphology of amyloid fibrils 35, it is not yet known to bind to native and partially folded conformations other than amyloid fibrils, as does Congo red 36.
According to the International Committee for Classification of Corneal dystrophies (IC3D), corneal dystrophies have been categorized based on genetic, clinical and histopathological findings, including the shape, distribution and localization of deposits within the cornea 1. Moreover, TGFBI-related corneal dystrophies have been correlated with specific gene mutations 37. The phenotype of ACD (GCD II) has been shown to correlate with the TGFBI mutation R124H 7–13, 19 and can be distinguished as either homozygous or heterozygous 14. Homozygous ACD has been further classified phenotypically as Type I or Type II depending on whether the anterior stromal opacity is spot-like or reticular 38. However, there have been reports of cases that manifest the phenotype of ACD but have other mutations of TGFBI other than R124H 20, 21. A D123H TGFBI mutation has been associated with ACD in Vietnamese patients 7. Solari et al. described a Brazilian patient diagnosed with ACD, but carried the R555W TGFBI mutation 21. Kocak-Altintas et al. investigated a Turkish family, whose members were affected by corneal dystrophy, describing the corneal deposits to be similar to those in GCD 20. They pointed out that with increased age, “Lattice-type phenotype with fine, subtle, and linear or branching deposits were also observed”. The genotype of one such elderly family member was found to carry the R555W TGFBI mutation. In both these reports, the authors assumed that since the patients did not have the R124H TGFBI mutation, which is classically associated with ACD, there was a misdiagnosis. However, it is possible that these cases, showing clear lattice lines in the cornea, are in fact a variant of ACD stemming from a non-R124H TGFBI mutation, as in the cases described herein. Furthermore, before genotyping to search for TGFBI mutations was routine practice, or that it was known that TGFBI was a component of deposits in what we now refer to as TGFBI-related corneal dystrophies, there have been reports of amyloid deposits in tissue from what seemed to be cases of GCD 27, 31, 39–42. We can assume that these cases were probably those of ACD, mis-diagnosed as GCD. However, in the pre-genotyping era, investigators have also reported trichrome-positive deposits in otherwise typical cases of LCD I 16, 43, 44. Taken together, these observations suggest that abnormal TGFBI protein has, in most cases, some tendency toward amyloid aggregation. This notion is consistent with our previous in vitro findings and those of others that native and mutant TGFBI can all form amyloid aggregates 45, 46. Data in the current study further supports the idea that TGFBI may also have an intrinsic tendency to form hyaline aggregates under certain circumstances.
In summary, this study demonstrates the potential use of ThT in co-staining with immunofluorescence to detect amyloid deposits of TGFBI. We have also confirmed that granular and lattice deposits can be associated with R124C TGFBI mutation and shown that corneal tissues from such patients contain sub-epithelial TGFBI deposits that are neither hyaline nor amyloid in nature, an additional feature that might substantiate such cases be categorized as a variant form of ACD or LCD. As we work towards understanding the etiology and molecular mechanisms of corneal dystrophies related to TGFBI mutations, we have made progress in correlating genotype with phenotype 1, 2. It has been proposed that the classification of TGFBI-related corneal dystrophies be based on the mutation alone 47, 48. Our study, however, demonstrates that a single TGFBI mutation such as R124C can lead to multiple phenotypes, some of which may be generally correlated with other mutations such as R124H previously.
Acknowledgments
We thank Dr. Morton Smith for his expert advice and providing histological sections of previously identified and unrelated cases of LCD at Washington University. We also thank Dr. Hunter Cherwick of Orbis International for coordinating collection of tissue. This work has been funded by the grants from National Institutes of Health R01EY017852 (AJWH), NRSA 5-T32-EY13360-09 (DAP), a RPB Physician Scientist Award (AJWH), and an unrestricted grant from Horncrest Foundation (AJWH). This work was also supported in part by awards to the Department of Ophthalmology and Visual Sciences at Washington University from a Research to Prevent Blindness, Inc. unrestricted grant, and the NIH Vision Core Grant P30 EY02687.
Sources of Support that require acknowledgement: NIH R01EY017609, RPB Physician Scientist Award and an unrestricted grant from Horncrest Foundation, Inc. (AJWH), NRSA 5-T32-EY13360-09 (DAP). This work was also supported in part by awards to the Department of Ophthalmology and Visual Sciences at Washington University from a Research to Prevent Blindness, Inc. unrestricted grant, and the NIH Vision Core Grant P30 EY 02687.
Footnotes
Disclosure of funding received: refer to sources of support that require acknowledgement above.
References
- 1.Weiss JS, moller HU, Lisch W, et al. The IC3D Classification of the Corneal Dystrophies. Cornea. 2008;27:S1–S42. doi: 10.1097/ICO.0b013e31817780fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aldave AJ, Sonmez B. Elucidating the molecular genetic basis of the corneal dystrophies: are we there yet? Arch Ophthalmol. 2007;125:177–186. doi: 10.1001/archopht.125.2.177. [DOI] [PubMed] [Google Scholar]
- 3.Kennedy SM, McNamara M, Hillery M, Hurley C, Collum LM, Giles S. Combined granular lattice dystrophy (Avellino corneal dystrophy) Br J Ophthalmol. 1996;80:489–490. doi: 10.1136/bjo.80.5.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Folberg R, Alfonso E, Croxatto JO, et al. Clinically atypical granular corneal dystrophy with pathologic features of lattice-like amyloid deposits. A study of these families. Ophthalmology. 1988;95:46–51. doi: 10.1016/s0161-6420(88)33226-4. [DOI] [PubMed] [Google Scholar]
- 5.Weidle E. Granular corneal dystrophy: two variants. In: Olivera LFd., editor. Ophthalmology Today. Elsevier; 1988. pp. 617–619. [Google Scholar]
- 6.Vassar PS, Culling CF. Fluorescent stains, with special reference to amyloid and connective tissues. Arch Pathol. 1959;68:487–498. [PubMed] [Google Scholar]
- 7.Cung le X, Ha NT, Chau HM, et al. Mutation analysis of the TGFBI gene in Vietnamese with granular and Avellino corneal dystrophy. Jpn J Ophthalmol. 2004;48:12–16. doi: 10.1007/s10384-003-0009-z. [DOI] [PubMed] [Google Scholar]
- 8.El-Ashry MF, Abd El-Aziz MM, Larkin DF, et al. A clinical, histopathological, and genetic study of Avellino corneal dystrophy in British families. Br J Ophthalmol. 2003;87:839–842. doi: 10.1136/bjo.87.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huerva V, Velasco A, Sanchez MC, Matias-Guiu X. Role of BIGH3 R124H mutation in the diagnosis of Avellino corneal dystrophy. Eur J Ophthalmol. 2008;18:345–350. doi: 10.1177/112067210801800305. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi A, Fujiki K, Fujimaki T, Murakami A, Sugiyama K. In vivo laser confocal microscopic findings of corneal stromal dystrophies. Arch Ophthalmol. 2007;125:1168–1173. doi: 10.1001/archopht.125.9.1168. [DOI] [PubMed] [Google Scholar]
- 11.Meallet MA, Affeldt JA, McFarland TJ, et al. An unusual clinical phenotype of Avellino corneal dystrophy associated with an Arg124His beta iG-H3 mutation in an African-American woman. Am J Ophthalmol. 2004;137:765–767. doi: 10.1016/j.ajo.2003.09.062. [DOI] [PubMed] [Google Scholar]
- 12.Moon JW, Kim SW, Kim TI, Cristol SM, Chung ES, Kim EK. Homozygous granular corneal dystrophy type II (Avellino corneal dystrophy): natural history and progression after treatment. Cornea. 2007;26:1095–1100. doi: 10.1097/ICO.0b013e3181484013. [DOI] [PubMed] [Google Scholar]
- 13.Stewart HS, Ridgway AE, Dixon MJ, Bonshek R, Parveen R, Black G. Heterogeneity in granular corneal dystrophy: identification of three causative mutations in the TGFBI (BIGH3) gene-lessons for corneal amyloidogenesis. Hum Mutat. 1999;14:126–132. doi: 10.1002/(SICI)1098-1004(1999)14:2<126::AID-HUMU4>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 14.Okada M, Yamamoto S, Inoue Y, et al. Severe corneal dystrophy phenotype caused by homozygous R124H keratoepithelin mutations. Invest Ophthalmol Vis Sci. 1998;39:1947–1953. [PubMed] [Google Scholar]
- 15.Alavi A, Elahi E, Rahmati-Kamel M, Karimian F, Rezaei-Kanavi M. Mutation screening of TGFBI in two Iranian Avellino corneal dystrophy pedigrees. Clin Experiment Ophthalmol. 2008;36:26–30. doi: 10.1111/j.1442-9071.2007.01649.x. [DOI] [PubMed] [Google Scholar]
- 16.Santos LN, Fernandes BF, de Moura LR, et al. Histopathologic study of corneal stromal dystrophies: a 10-year experience. Cornea. 2007;26:1027–1031. doi: 10.1097/ICO.0b013e318123f298. [DOI] [PubMed] [Google Scholar]
- 17.Holland EJ, Daya SM, Stone EM, et al. Avellino corneal dystrophy. Clinical manifestations and natural history. Ophthalmology. 1992;99:1564–1568. doi: 10.1016/s0161-6420(92)31766-x. [DOI] [PubMed] [Google Scholar]
- 18.Rosenwasser GO, Sucheski BM, Rosa N, et al. Phenotypic variation in combined granular-lattice (Avellino) corneal dystrophy. Arch Ophthalmol. 1993;111:1546–1552. doi: 10.1001/archopht.1993.01090110112035. [DOI] [PubMed] [Google Scholar]
- 19.Konishi M, Mashima Y, Yamada M, Kudoh J, Shimizu N. The classic form of granular corneal dystrophy associated with R555W mutation in the BIGH3 gene is rare in Japanese patients. Am J Ophthalmol. 1998;126:450–452. doi: 10.1016/s0002-9394(98)00105-6. [DOI] [PubMed] [Google Scholar]
- 20.Kocak-Altintas AG, Kocak-Midillioglu I, Akarsu AN, Duman S. BIGH3 gene analysis in the differential diagnosis of corneal dystrophies. Cornea. 2001;20:64–68. doi: 10.1097/00003226-200101000-00013. [DOI] [PubMed] [Google Scholar]
- 21.Solari HP, Ventura MP, Perez AB, Sallum JM, Burnier MN, Jr, Belfort R., Jr TGFBI gene mutations in Brazilian patients with corneal dystrophy. Eye. 2007;21:587–590. doi: 10.1038/sj.eye.6702264. [DOI] [PubMed] [Google Scholar]
- 22.Takacs L, Boross P, Tozser J, Modis L, Jr, Toth G, Berta A. Transforming growth factor-beta induced protein, betaIG-H3, is present in degraded form and altered localization in lattice corneal dystrophy type I. Exp Eye Res. 1998;66:739–745. doi: 10.1006/exer.1998.0471. [DOI] [PubMed] [Google Scholar]
- 23.Ma C, Rong Y, Radiloff DR, et al. Extracellular matrix protein betaig-h3/TGFBI promotes metastasis of colon cancer by enhancing cell extravasation. Genes Dev. 2008;22:308–321. doi: 10.1101/gad.1632008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Streeten BW, Qi Y, Klintworth GK, Eagle RC, Jr, Strauss JA, Bennett K. Immunolocalization of beta ig-h3 protein in 5q31-linked corneal dystrophies and normal corneas. Arch Ophthalmol. 1999;117:67–75. doi: 10.1001/archopht.117.1.67. [DOI] [PubMed] [Google Scholar]
- 25.Ferry AP, Benson WH, Weinberg RS. Combined granular-lattice (‘Avellino’) corneal dystrophy. Trans Am Ophthalmol Soc. 1997;95:61–77. [PMC free article] [PubMed] [Google Scholar]
- 26.Konishi M, Yamada M, Nakamura Y, Mashima Y. Immunohistology of kerato-epithelin in corneal stromal dystrophies associated with R124 mutations of the BIGH3 gene. Curr Eye Res. 2000;21:891–896. doi: 10.1076/ceyr.21.5.891.5536. [DOI] [PubMed] [Google Scholar]
- 27.Garner A. Histochemistry of corneal granular dystrophy. Br J Ophthalmol. 1969;53:799–807. doi: 10.1136/bjo.53.12.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wheeldon CE, de Karolyi BH, Patel DV, Sherwin T, McGhee CN, Vincent AL. A novel phenotype-genotype relationship with a TGFBI exon 14 mutation in a pedigree with a unique corneal dystrophy of Bowman’s layer. Mol Vis. 2008;14:1503–1512. [PMC free article] [PubMed] [Google Scholar]
- 29.Kuchle M, Green WR, Volcker HE, Barraquer J. Reevaluation of corneal dystrophies of Bowman’s layer and the anterior stroma (Reis-Bucklers and Thiel-Behnke types): a light and electron microscopic study of eight corneas and a review of the literature. Cornea. 1995;14:333–354. doi: 10.1097/00003226-199507000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Ridgway AE, Akhtar S, Munier FL, et al. Ultrastructural and molecular analysis of Bowman’s layer corneal dystrophies: an epithelial origin? Invest Ophthalmol Vis Sci. 2000;41:3286–3292. [PubMed] [Google Scholar]
- 31.Smith ME, Zimmerman LE. Amyloid in corneal dystrophies. Differentiation of lattice from granular and macular dystrophies. Arch Ophthalmol. 1968;79:407–412. doi: 10.1001/archopht.1968.03850040409008. [DOI] [PubMed] [Google Scholar]
- 32.Stafford WR, Fine BS. Amyloidosis of the cornea. Report of a case without conjunctival involvement. Arch Ophthalmol. 1966;75:53–56. doi: 10.1001/archopht.1966.00970050055010. [DOI] [PubMed] [Google Scholar]
- 33.Smith ME, Zimmerman LE. Amyloidosis of the eyelid and conjunctiva. Arch Ophthalmol. 1966;75:42–51. doi: 10.1001/archopht.1966.00970050044009. [DOI] [PubMed] [Google Scholar]
- 34.Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. Am J Ophthalmol. 2008;145:656–661. doi: 10.1016/j.ajo.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murakami K, Irie K, Morimoto A, et al. Neurotoxicity and physicochemical properties of Abeta mutant peptides from cerebral amyloid angiopathy: implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer’s disease. J Biol Chem. 2003;278:46179–46187. doi: 10.1074/jbc.M301874200. [DOI] [PubMed] [Google Scholar]
- 36.Khurana R, Uversky VN, Nielsen L, Fink AL. Is Congo red an amyloid-specific dye? J Biol Chem. 2001;276:22715–22721. doi: 10.1074/jbc.M011499200. [DOI] [PubMed] [Google Scholar]
- 37.Munier FL, Frueh BE, Othenin-Girard P, et al. BIGH3 mutation spectrum in corneal dystrophies. Invest Ophthalmol Vis Sci. 2002;43:949–954. [PubMed] [Google Scholar]
- 38.Tsujikawa K, Tsujikawa M, Watanabe H, et al. Allelic homogeneity in Avellino corneal dystrophy due to a founder effect. J Hum Genet. 2007;52:92–97. doi: 10.1007/s10038-006-0083-4. [DOI] [PubMed] [Google Scholar]
- 39.Akiya S, Brown SI. Granular dystrophy of the cornea. Characteristic electron microscopic lesion. Arch Ophthalmol. 1970;84:179–192. doi: 10.1001/archopht.1970.00990040181011. [DOI] [PubMed] [Google Scholar]
- 40.Iwamoto T, Stuart JC, Srinivasan BD, et al. Ultrastructural variation in granular dystrophy of the cornea. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1975;194:1–9. doi: 10.1007/BF00408270. [DOI] [PubMed] [Google Scholar]
- 41.Stuart JC, Mund ML, Iwamoto T, Troutman RC, White H, DeVoe AG. Recurrent granular corneal dystrophy. Am J Ophthalmol. 1975;79:18–24. doi: 10.1016/0002-9394(75)90451-1. [DOI] [PubMed] [Google Scholar]
- 42.Yanoff M, Fine BS, Colosi NJ, Katowitz JA. Lattice corneal dystrophy. Report of an unusual case. Arch Ophthalmol. 1977;95:651–655. doi: 10.1001/archopht.1977.04450040117018. [DOI] [PubMed] [Google Scholar]
- 43.Folberg R, Stone EM, Sheffield VC, Mathers WD. The relationship between granular, lattice type 1, and Avellino corneal dystrophies. A histopathologic study. Arch Ophthalmol. 1994;112:1080–1085. doi: 10.1001/archopht.1994.01090200086027. [DOI] [PubMed] [Google Scholar]
- 44.Lanier JD, Fine M, Togni B. Lattice corneal dystrophy. Arch Ophthalmol. 1976;94:921–924. doi: 10.1001/archopht.1976.03910030461004. [DOI] [PubMed] [Google Scholar]
- 45.Kim JE, Park RW, Choi JY, et al. Molecular properties of wild-type and mutant betaIG-H3 proteins. Invest Ophthalmol Vis Sci. 2002;43:656–661. [PubMed] [Google Scholar]
- 46.Yuan C, Berscheit HL, Huang AJ. Identification of an amyloidogenic region onkeratoepithelin via synthetic peptides. FEBS Lett. 2007;581:241–247. doi: 10.1016/j.febslet.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 47.Klintworth GK. Advances in the molecular genetics of corneal dystrophies. Am J Ophthalmol. 1999;128:747–754. doi: 10.1016/s0002-9394(99)00358-x. [DOI] [PubMed] [Google Scholar]
- 48.Stone EM, Mathers WD, Rosenwasser GO, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nat Genet. 1994;6:47–51. doi: 10.1038/ng0194-47. [DOI] [PubMed] [Google Scholar]