

Abstract
Mandibuloacral dysplasia (MAD) is an autosomal recessive disorder characterized by hypoplasia of the mandible and clavicles, acro-osteolysis and lipodystrophy due to mutations in LMNA or ZMPSTE24. Only six MAD patients are reported so far with ZMPSTE24 mutations and limited phenotypic data are available for them. Here, we report on two brothers (4 years and 9 months old) with early onset MAD due to ZMPSTE24 mutations in whom thin skin was noted as early as 5 months of age. Both had micrognathia, mottled hyperpigmentation, and enlarged fontanelles but little evidence of lipodystrophy. There was no delay of mental development. The older brother showed small pinched nose, short clavicles, acro-osteolysis, stunted growth, joint stiffness, and repeated fractures. There was no evidence of renal disease. Both patients were compound heterozygotes harboring a previously reported missense ZMPSTE24 mutation, p.Pro248Leu and a novel null mutation, p.Trp450stop. These patients and the review of literature reveals that compared to MAD patients with LMNA mutations, those with ZMPSTE24 mutations develop manifestations earlier in life. Other distinguishing features in MAD due to ZMPSTE24 mutations may include premature birth, renal disease, calcified skin nodules, and lack of acanthosis nigricans. We conclude that in patients with MAD due to ZMPSTE24 mutations, the onset of disease manifestations such as thin skin and micrognathia occurs as early as 5 months of age. In these patients, skeletal phenotype presents earlier whereas lipodystrophy and renal disease may occur later in life.
Keywords: Lipodystrophy, ZMPSTE24, Lamin A/C, mandibuloacral dysplasia
INTRODUCTION
Mandibuloacral dysplasia (MAD; OMIM 248370 and 608612) is a rare, genetically and phenotypically heterogeneous, autosomal recessive disorder characterized by skeletal abnormalities including hypoplasia of the mandible and clavicles, acro-osteolysis, cutaneous atrophy, progeroid features and lipodystrophy [Garg 2004]. Using positional cloning and candidate gene approach, two loci have been identified for MAD. The first locus discovered was lamin A/C (LMNA) [Novelli et al., 2002] which encodes integral nuclear lamina proteins, lamins A and C, belonging to the intermediate filament family. The second locus, ZMPSTE24 [Agarwal et al., 2003], encodes a zinc metalloproteinase which is involved in post-translational processing of prelamin A to mature lamin A.
Based on in-depth evaluation of body fat distribution pattern, we had suggested two patterns of lipodystrophy in patients with MAD: type A (partial) type B (generalized) [Simha et al., 2003]. So far, approximately 28 MAD patients have been reported to harbor homozygous or compound heterozygous missense mutations in the C-terminal of lamin A/C [Novelli et al., 2002; Cao and Hegele 2003; Simha et al., 2003; Plasilova et al., 2004; Garg et al., 2005; Van Esch et al., 2006; Kosho et al., 2007; Lombardi et al., 2007; Agarwal et al., 2008; Zirn et al., 2008; Garavelli et al., 2009; Madej-Pilarczyk et al., 2009]. Nearly all of them have type A pattern of partial lipodystrophy. However, only 6 MAD patients have been reported with either compound heterozygous or homozygous mutations in ZMPSTE24 [Agarwal et al., 2003; Shackleton et al., 2005; Agarwal et al., 2006; Denecke et al., 2006; Miyoshi et al., 2008]. Of these, three patients had already died before their genetic basis was determined [Agarwal et al., 2003; Shackleton et al., 2005; Agarwal et al., 2006] and therefore, only limited information was available about the phenotype of these patients. We report in-depth phenotype analysis of two male siblings with early onset MAD due to compound heterozygous ZMPSTE24 mutations.
CLINICAL REPORTS
MAD 4700.3 (Patient 1): This 4-year-old boy was born to nonconsanguineous healthy parents of European descent. His mother went into preterm labor at 32 weeks gestation after spontaneous rupture of membranes. His birth weight was 1.94 kg and Apgar scores were 6 at 1 minute and 8 at 5 minutes. He required continuous positive airway pressure for the first 24 h of life and remained in the neonatal intensive care unit for 2–3 weeks because of jaundice and feeding difficulties.
At five months of age, his parents noted thin skin and stunted growth. Computed tomography of the head with 3D reformations at 7 months showed multiple Wormian bones on both the lambdoid sutures and plagiocephaly on the right (Fig. 1). Joint stiffness was noted at the age of 8 months.
Figure 1.
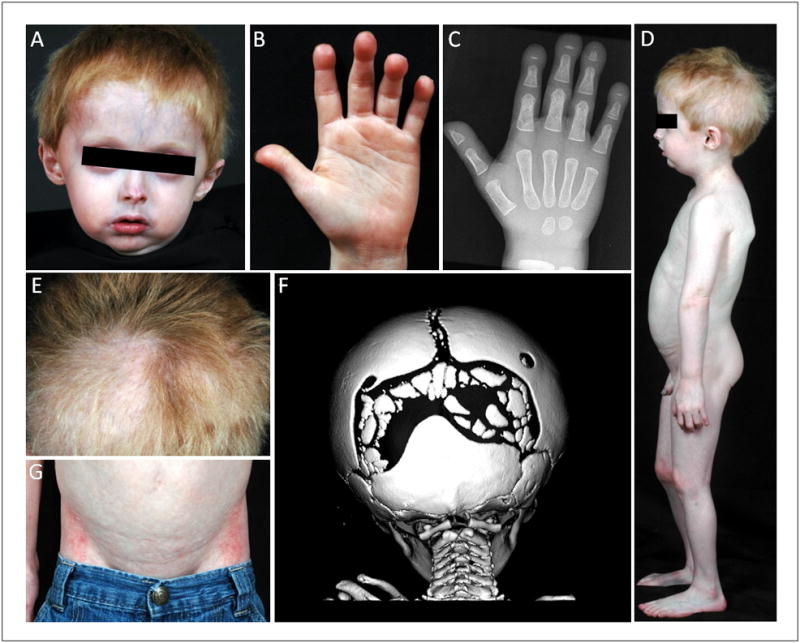
Clinical Features of Patient MAD4700.3. A, The patient has a small nose with telangiectasias, prominent cheeks, prominent superficialveins, low-set ears, micrognathia, and slight double chin. B, Bulbous appearance of distal fingers consistent with acro-osteolysis. C, Radiograph of the hand at 13 months showing irregularity of the tufts of all terminal phalanges, consistent with acro-osteolysis. D, Lateral view of the patient showing near normal body fat distribution and lack of overt lipodystrophy. E, Dry, straight, bright red hair with areas of hair thinning and alopecia particularly on the back of scalp and on the left parietal and frontal region. F, Computerized tomography of the head at 7 months with 3-Dreformations showing multiple Wormian bones in both lambdoid sutures. G, Mottled pigmentation in the abdomen and inguinal areas.
At 13 months of age, he presented to the Genetics Center at Children’s Hospital Philadelphia for evaluation of failure to thrive, mixed tone and delayed motor milestones. Radiographs of the hands showed deformity and irregularity of the tufts of all the digits consistent with acro-osteolysis (Fig. 1), and radiographs of the feet showed irregularity of the distal phalanges.
He started walking at 14 months, but continues to have difficulty going up and down the stairs. Dentition has been delayed: the first teeth erupted at 15 months, and the teeth were crowded. A skeletal survey at 19 months was remarkable for widening of the cranial sutures, extensive Wormian bones along the lambdoid sutures bilaterally, and short clavicles. Hair loss started at 1.5-2 years of age at the vertex. Hair shaft analysis revealed twisted hair suggestive of pili torti and the patient was started on biotin supplementation. There has been no delay of cognitive, social, emotional or language milestones. Evaluation of growth retardation included a gastric emptying study at 3 years of age showing decreased rate of gastric emptying. At 4 years of age, a zinc level was 56 μg/dL [normal range 60–120] and he was started on zinc supplementation.
His clinical course has also been remarkable for multiple fractures including buckle fractures of the right distal radius and ulna at 2 years of age and a skull fracture at 30 months. Dual energy X-ray absorptiometry (Hologic Densitometer, Waltham, MA) at the age of 3 years showed a lumbar-spine bone mineral density of 0.38 g/cm2 with a Z-score of −1.8 (“low bone density for chronological age” is defined as lumbar spine Z score less than or equal to −2.0 in children [Writing Group for the International Society for Clinical Densitometry Position Development Conference 2004; Baim et al., 2008]). He received cyproheptadine for appetite stimulation from 6 months of age to 8 months. Initially his appetite increased but later cyproheptadine did not have any effect. It was restarted at the age of 3 years and parents noted that he had increased weight by 0.45 kg. He was also taking erythromycin for delayed gastric emptying; polyethylene glycol 3350 and lactulose for constipation; and loratadine as needed.
He weighed 11.1 kg (3rd centile) and was 90 cm tall (3rd centile). He had low-set ears, prominent cheeks, micrognathia, and slight double chin. He had a small, thin, overhanging nose with telangiectasias. He had 7 maxillary teeth and 4 mandibular teeth. Posterior occipital fontanelle and left parietal fontanelle were both open while anterior fontanelle and right parietal fontanelle were closed. A hematoma was noted at the vertex. He had bright red hair with areas of hair thinning and alopecia particularly on the back of scalp and on the left side. The hair seemed dry and straight. No loss of eyebrows or eyelashes was noted. He had normal male external genitalia. The Tanner stage was A0P1G1. Musculoskeletal examination revealed narrow sloping shoulders and reduced mobility in the ankles with dorsiflexion to 20 degrees. Hips could be abducted to only 25 degrees with limited internal and external rotation. Acro-osteolysis of the fingers was observed. The feet were narrow. He walked with heel-to-toe gait. He had prominent veins on the scalp, chin, and nose. There was no overt loss of fat from the extremities, but he seemed to have relatively decreased fat in the lower neck, lower legs and feet, and mid-back.
Laboratory data revealed total cholesterol of 119 mg/dL (normal range 125-170), high density lipoprotein cholesterol of 55 mg/dL (reference range not established), triglycerides of 76 mg/dL (normal range 30–104), and low density lipoprotein cholesterol of 49 mg/dL (normal range < 110 mg/dL). Lactate dehydrogenase was high at 565 U/L (normal range 155–345). Fasting glucose was 83 mg/dL (normal range 65–99). Creatinine was 0.26 mg/dL (normal range 0.29–0.68) and blood urea nitrogen was 14 mg/dL (normal range 7–20). He had a slightly high aspartate aminotransferase 47 U/L (normal range 20–39) with normal alanine aminotransferase 20 U/L (normal range 8–30) and alkaline phosphatase 220 U/L (normal range 93–309). Creatine phosphokinase was normal at 111 U/L (normal range < 160). Complete blood counts were unremarkable. Hemoglobin A1C was 5.2%. Urinalysis was unremarkable without any proteinuria. An electrocardiogram was normal.
MAD 4700.4 (Patient 2): This 9-month-old boy was born at 32 weeks gestation after premature rupture of membranes. He remained in the neonatal intensive care unit for 3 weeks due to difficulties with oxygenation, feeding, jaundice, and low birth weight. At about 5 months of age, his parents noted thin tight skin, similar to his older brother, prompting them to seek further evaluation. He was taking cyproheptadine since age 6 months for appetite stimulation, nizatidine for reflux, and lactulose for constipation.
He was an alert and interactive child. Height was 64 cm (3rd centile) and weight was 4.2 kg (3rd centile). He had normal hair on the scalp, eyebrows, and eyelashes. Anterior fontanelle was open. Eyes and cheeks were prominent. He also had micrognathia (Fig. 2), and no teeth were present. Examination of his fingers did not reveal any overt signs of clubbing or acro-osteolysis although they appeared slightly bulbous. He had thin tight skin with prominent superficial veins on the legs and the scalp anteriorly. There was no obvious decrease in subcutaneous fat. A urinalysis was normal.
Figure 2.
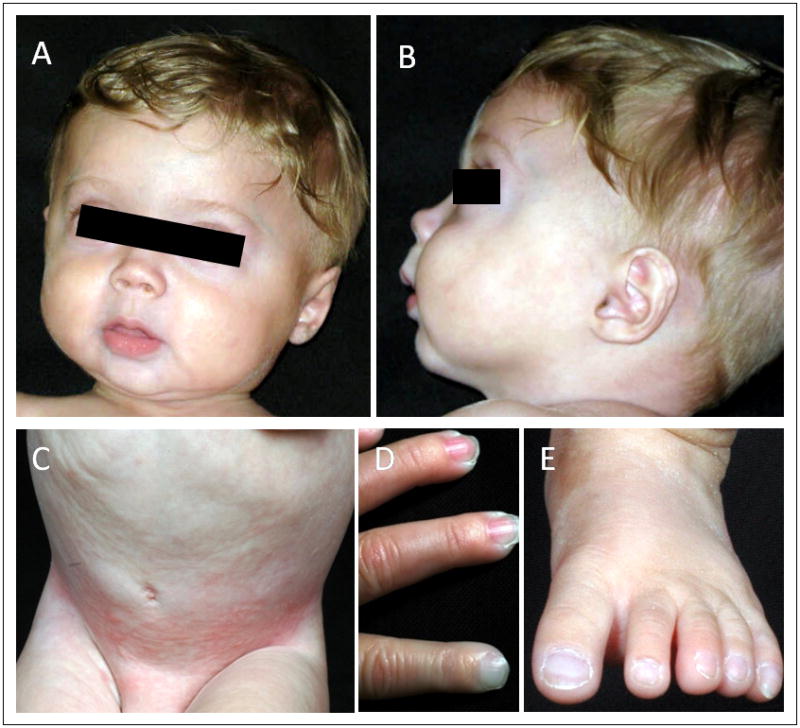
Clinical Features of Patient MAD4700.4. A, B, The patient has low-set ears, prominent cheeks, and micrognathia. Skin on the face was thin and superficial veins were seen on the forehead. No hair loss from the scalp, eyebrows or eyelids. C, Mottled pigmentation in the abdomen and inguinal areas. D, Base of the fingers appeared slightly bulbous but there were no overt signs of acro-osteolysis. E, Normal appearance of the toes.
MATERIALS AND METHODS
Both the patients and their family members were evaluated at the Clinical and Translational Research Center at the UT Southwestern. A written informed consent was obtained from both the parents, and the study was approved by the Institutional Review Board of UT Southwestern.
Height and body weight were measured by standard procedures. Skinfold thickness was measured with a Lange caliper (Cambridge Scientific Industries, Cambridge, Maryland) at five truncal sites (chest, mid-axillary, abdomen, subscapular, and suprailiac), six peripheral sites (biceps, triceps, forearm, hip, thigh and calf) on the right side of the body, and at the chin.
Whole body and regional fat in the head, trunk, upper and lower extremities were determined using a dual-energy x-ray absorptiometry (DEXA) scan with a multiple detector fan-beam Hologic QDR-2000 densitometer (Hologic, Inc., Waltham, MA).
Lipids, lipoproteins, chemistries, and blood hemoglobin A1C were analyzed as part of a systematic multichannel analysis (Synchron CX9 ALX Clinical System, Beckman, Fullerton, CA).
Mutational analysis was performed by PreventionGenetics, Marshfield, WI, for ZMPSTE24 on both the patients and their parents. In addition, MAD 4700.3 was screened for LMNA mutations. Using genomic DNA extracted from the patients’ cells, all coding regions as well as ~50 bases of flanking non-coding sequences were amplified and sequenced. The patients’ sequences were then aligned and compared to the reference sequences. For sequence analysis, Applied Biosystems software as well as Mutation Surveyor software was used. The sequence electropherograms were also manually reviewed.
RESULTS
Sequencing of LMNA in MAD 4700.3 revealed no disease causing variants. Compound heterozygous mutations, however, were found in both the patients at exon 6 (c.743C>T; p.Pro248Leu) and exon 10 (c.1349G>A; p.Trp450stop) of ZMPSTE24 gene. The patients’ father carried the heterozygous variant, p.Pro248Leu, while the mother carried the heterozygous variant, p.Trp450stop.
Measurement of skinfold thickness at truncal sites of MAD 4700.3 revealed chest 7 mm, mid-axillary 4.5 mm, abdomen 11 mm, subscapular 4.5 mm (normal values,10th to 90th centile: 4.2–8.1 mm [McDowell et al., 2005]), and suprailiac 10 mm. At the peripheral sites, biceps skinfold was 5 mm, triceps 12 mm (normal values, 10th to 90th centile: 6.3 – 12.4 mm [McDowell et al., 2005]), forearm 11 mm, hip 44.5 mm, thigh 14 mm, and calf 10 mm. His skinfold at the chin measured 5 mm. A whole body DEXA scan revealed total body fat of 27.5% (mean ± SD for 12 white 3–5 year old boys with BMI 15.7 ± 0.9 kg/m2 was 17.6 ± 2.0 %, [Ellis, 1997]). Fat in the upper extremities was 31.7%, in the lower extremities was 45.2%, and in the trunk was 21.8%.
Measurement of skinfold thickness at truncal sites of MAD 4700.4 revealed chest 8 mm, mid-axillary 6 mm, abdomen 9 mm, subscapular 6 mm (normal values, 15th to 85th centile: 5.9–9.1 mm [McDowell et al., 2005]), and suprailiac 11 mm. At the peripheral sites, biceps skinfold was 8 mm, triceps 12 mm (normal values,15th to 85th centile: 8.4–13.1 mm [McDowell et al., 2005]), forearm 9 mm, hip 36.75 mm, thigh 12 mm, and calf 1 mm. Skinfold thickness at the chin was 6 mm.
DISCUSSION
We report on two brothers (4 years and 9 months old) with MAD harboring a previously reported missense ZMPSTE24 mutation, p.Pro248Leu, and a novel null mutation, p.Trp450stop. Similar to previously reported MAD/Progeria-like cases due to mutations of the same gene [Agarwal et al., 2003; Shackleton et al., 2005; Agarwal et al., 2006; Denecke et al., 2006; Miyoshi et al., 2008], our patients demonstrated early onset of symptoms, postnatal growth retardation, feeding difficulty, delayed dentition, micrognathia, delayed closure of cranial sutures, joint stiffness, mottled hyperpigmentation, and thin skin with prominent superficial vasculature. The older brother also demonstrated a thin nose, short clavicles, acro-osteolysis, hair loss, Wormian bones, and dental overcrowding while the younger one did not demonstrate hair loss or a thin nose.
Serum chemistry values in the older brother did not show any evidence of metabolic disturbances, and the DEXA study did not show decreased total body fat. Total body fat values using DEXA have been reported in only one previous ZMPSTE24 MAD patient, MAD3300.3 [Miyoshi et al., 2008]. At the age of 7 years, she had total body fat of 18.3% and was noted to have lipodystrophy that excluded the cheeks, arms and thigh. Skinfold thickness was measured in both of our patients; subscapular and triceps skin folds were normal. Normal values for very young children under the age of 4 years are not available for the other sites measured; however, the younger brother had calf skinfold thickness of only 1 mm, suggesting a lack of subcutaneous fat.
In contrast to previously reported MAD cases due to ZMPSTE24 mutations, our patients did not show generalized lipodystrophy, suggesting that this two finding may manifest later in childhood (Fig. 3). In fact, a previously reported Japanese patient was not noted to have any loss of subcutaneous fat until the age of 7 years [Miyoshi et al., 2008]. Also, two previous patients have been reported to develop focal sclerosing glomerulosclerosis at age 19 [Agarwal et al., 2006] and 25 [Agarwal et al., 2003] years, and urinalysis in both the patients from Japan showed microhematuria at ages 7 and 3.5 years [Miyoshi et al., 2008] (Fig. 3). Our patients so far did not show any early signs of renal dysfunction.
Figure 3.

Onset of disease manifestations in ZMPSTE24 MAD/Progeria-like cases. Each patient is represented by a different symbol.
When compared to MAD patients with LMNA mutations, patients with ZMPSTE24 mutations develop clinical manifestations earlier in life, are premature at birth and can develop focal segmental glomerulosclerosis and calcified skin nodules later during adulthood. The differential diagnosis between MAD due to LMNA and ZMPSTE24 mutations may be difficult especially in infancy, because the main clinical features are common (Table I), the type of lipodystrophy can be similar and the classical patterns named type A (partial) and type B (generalized) may not be evident in childhood. Telltale features of MAD in infancy may be micrognathia, overhanging nasal tip, thin nose and bulbous distal phalanges. Radiographic examination of the hands may confirm the presence of acro-osteolysis, and mutational analysis of LMNA and ZMPSTE24 may confirm the diagnosis. Acanthosis nigricans and other metabolic disturbances have not been reported so far in MAD due to ZMPSTE24 mutations. The early diagnoses of the two conditions, MAD due to ZMPSTE24 or LMNA mutations, are of clinical significance since the late complications are different: severe progressive glomerulopathy in the former and metabolic complications due to insulin resistance and diabetes in the latter.
TABLE I.
Clinical features of two types of MAD/Progeria-like syndromes due to ZMPSTE24 or LMNA mutations, APS, and HGPS
| Clinical Feature | ZMPSTE24 MAD cases (n=8)* [Agarwal et al., 2003; Shackleton et al., 2005; Agarwal et al., 2006; Denecke et al., 2006; Miyoshi et al., 2008] | LMNA MAD/Progeria- like syndrome cases (n=28) [Novelli et al., 2002; Cao and Hegele 2003; Simha et al., 2003; Plasilova et al., 2004; Garg et al., 2005; Van Esch et al., 2006; Kosho et al., 2007; Lombardi et al., 2007; Agarwal et al., 2008; Zirn et al., 2008; Garavelli et al., 2009; Madej-Pilarczyk et al., 2009] | APS (n=26) [Garg et al., 2009] [Caux et al., 2003; Chen et al., 2003; Csoka et al., 2004; Jacob et al., 2005; Kirschner et al., 2005; Mory et al., 2008; Doh et al., 2009; McPherson et al., 2009; Renard et al., 2009] | HGPS** (n=41) A= [Gordon et al., 2007] B= [Merideth et al., 2008] | HGPS***(n=142) [Hennekam 2006]* |
|---|---|---|---|---|---|
| Birth ≤ 33 wk gestation | 3/7 (43%) | 0 | 0 | NA | 12/86 (14%) |
| Post-natal growth retardation | 4/5 (80%) | 20/24 (83%) | 4/6 (67%) | 15/15 (100%) [B] | 75–100% |
| Median Age of onset of symptoms | 4 months (range: birth – 18 months) | 7 years (range: birth - 30 years) | 6.4 years (range: 5 months – 17 years) | NA | NA |
| Dental overcrowding | 5/5 (100%) | 21/23 (91%) | 7/8 (88%) | 7/7 (100%) [A] | NA |
| Delayed closure of cranial sutures | 6/6 (100%) | 16/20 (80%) | 0 | NA | 50–75% |
| Sparse/absent hair or alopecia | 4/5 (80%) | 13/21 (62%) | 8/9 (89%) | 15/15 (100%)[A] | 75–100% |
| Mottled hyperpigmentation | 7/7 (100%) | 22/23 (96%) | 7/7 (100%) | 15/15 (100%) [B] | NA |
| Acanthosis nigricans | 0/4 (0%) | 15/17 (88%) | 2/8 (25%) | NA | NA |
| Calcified skin nodules | 2/4 (50%) | 0 | 0 | 0 | 0 |
| Acro-osteolysis | 6/7 (86%) | 25/26 (96%) | 5/12 (42%) | 14/14 (100%) [A] | NA |
| Dysplastic/hypoplastic clavicles | 6/7 (86%) | 24/27 (89%) | 4/11 (36%) | 15/17 (88%) [A] | NA |
| Joint stiffness or contractures | 6/7 (86%) | 25/27 (93%) | 10/10 (100%) | 19/19 (100%) [A] | 75–100% |
| Fractures | 3/4 (75%) | 3/7 (43%) | 0 | 1/19 (5%) [A] | 10 families |
| Focal sclerosing glomerulosclerosis | 2/4 (50%) | 0 | 0 | 0 | NA |
| Lipodystrophy | 5/7 (71%) | 27/27 (100%) | 19/22 (86%) | 15/15 (100%) [B] | 75–100% |
NA, not available;
MAD, mandibuloacral dysplasia
APS, atypical progeroid syndrome
HGPS, Hutchinson-Gilford Progeria Syndrome
Includes the two patients described in this paper.
These studies had overlapping patients with HGPS-causing G608G LMNA mutations.
Not all patients were confirmed to have a HGPS causing LMNA mutation.
Interestingly, the features of MAD due to ZMPSTE24 mutations show significant clinical overlaps with both Hutchinson-Gilford progeria syndrome (HGPS; OMIM 176670), characterized by precocious development of aging-like phenotypes, and atypical progeroid syndrome (APS) (Table I). Both HGPS and APS are usually caused by de novo heterozygous LMNA mutations. In comparison to MAD due to ZMPSTE24 mutations, lipodystrophy, prominent subcutaneous vasculature, and pinched nose are more prominent in HGPS [Hennekam 2006], while acro-osteolysis, clavicular hypoplasia, and mandibular hypoplasia are less prominent in APS [Garg et al., 2009]. Hennekam [2006] had reported slowly progressive lipodystrophy in patients with “autosomal recessive, non-classical progeria” with reported survival into adulthood and severe osteolysis resulting in increased risk of fractures. Since the molecular basis of “autosomal recessive, non-classical progeria” has not been elucidated, it is likely that some of them may have had MAD with severe progeroid manifestations due to LMNA or ZMPSTE24 mutations as reported by us and others [Plasilova et al., 2004; Shackleton et al., 2005; Denecke et al., 2006; Agarwal et al., 2008; Miyoshi et al., 2008].
ZMPSTE24 mutations can also cause restrictive dermopathy (RD; OMIM 275210, [Navarro et al., 2004; Moulson et al., 2005; Navarro et al., 2005] and when compared to MAD due to ZMPSTE24 mutations, both disorders manifest with prematurity, micrognathia, small pinched nose, sparse or absent hair, enlarged fontanelles, dysplastic clavicles, and acro-osteolysis. However, RD is lethal within the newborn period and has more profound manifestations including intrauterine growth retardation (IUGR), birth weight < 1500 g, fixed facial expression, mouth in the “o” position, skin erosions and denudations, and contractures [Morais et al., 2009]. Nearly all reported cases of RD harbored homozygous or compound heterozygous null ZMPSTE24 mutations [Smigiel et al., 2010]. In contrast, patients with compound heterozygous ZMPSTE24 mutations with a null mutation on one allele and a missense mutation on the other allele have always presented with MAD phenotype [Agarwal et al., 2003; Shackleton et al., 2005; Agarwal et al., 2006; Miyoshi et al., 2008]. Thus, those with RD have almost no detectable ZMPSTE24 enzymatic activity whereas those with MAD may have some residual ZMPSTE24 enzymatic activity. In fact, in a yeast halo assay, the mutant p.Pro248Leu, as seen in our patients, was nearly as active as the wild-type ZMPSTE24 construct [Miyoshi et al., 2008]. As such, since the pathophysiological mechanisms in MAD and RD involve defective processing of prelamin A by ZMPSTE24, the variable manifestations of the two disorders can be explained by varying amounts of prelamin A accumulation.
The early diagnosis and slowly progressive nature of MAD due to ZMPSTE24 in our patients offers an opportunity to prevent the disease’s complications, such as renal disease. Thus far, no such therapeutic option is available, but in the Zmpste24−/− mice, farnesyl-transferase inhibitors (FTIs) and the combination of statins with bisphosphonates have been tried to inhibit accumulation of farnesylated prelamin A [Fong et al., 2006; Varela et al., 2008]. Fong et al [2006] administered an FTI inhibitor, ABT-100, to Zmpste24−/− mice and reported significant improvement in grip strength, reduced rib fractures, increased body weight and improved survival (although all mice were killed at 20 weeks). However, benefits of FTIs may be limited because prelamin A may be alternatively prenylated by geranylgeranyltransferase I. Varela et al [2008] explored the treatment of Zmpste24−/− mice with an aminobisphosphonate, zoledronate, and a statin, pravastatin, with the rationale of inhibiting both farnesylation and geranylgeranylation. Although no survival benefit was seen with either pravastatin or zoledronate alone, the combination therapy improved body weight, increased subcutaneous fat, reduced kyphosis and alopecia, improved bone density, and increased median survival of mice from 101 days to 179 days. Thus, which therapeutic approach will be more beneficial for patients with MAD due to ZMPSTE24 deficiency remains to be determined.
In conclusion, we describe two young children with MAD who were found to harbor compound heterozygous mutations in ZMPSTE24. The report of these patients confirms early onset of disease manifestations such as thin skin, micrognathia, small pinched noses, mottled hyperpigmentation, and enlarged fontanelles as early as 5 months of age. It appears that in patients affected with MAD due to ZMPSTE24 mutations, the skeletal phenotype presents earlier whereas evident lipodystrophy and renal disease may occur later in life.
Acknowledgments
We thank Dr. Geral Dietz for reviewing the radiographs of the patient and Claudia Quittner for her assistance during patient evaluations. We also thank Sarah Masood and Crystal Kittisopikul for help with illustrations and mutational screening. This work was supported by the National Institutes of Health grants R01-DK54387, CTSA Grant UL1 RR024982 and Southwest Medical Foundation.
References
- Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Kazachkova I, Ten S, Garg A. Severe mandibuloacral dysplasia-associated lipodystrophy and progeria in a young girl with a novel homozygous Arg527Cys LMNA mutation. J Clin Endocrinol Metab. 2008;93:4617–4623. doi: 10.1210/jc.2008-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal AK, Zhou XJ, Hall RK, Nicholls K, Bankier A, Van Esch H, Fryns JP, Garg A. Focal segmental glomerulosclerosis in patients with mandibuloacral dysplasia owing to ZMPSTE24 deficiency. J Investig Med. 2006;54:208–213. doi: 10.2310/6650.2006.05068. [DOI] [PubMed] [Google Scholar]
- Baim S, Binkley N, Bilezikian JP, Kendler DL, Hans DB, Lewiecki EM, Silverman S. Official Positions of the International Society for Clinical Densitometry and executive summary of the 2007 ISCD Position Development Conference. J Clin Densitom. 2008;11:75–91. doi: 10.1016/j.jocd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Cao H, Hegele RA. LMNA is mutated in Hutchinson-Gilford progeria (MIM 176670) but not in Wiedemann-Rautenstrauch progeroid syndrome(MIM 264090) J Hum Genet. 2003;48:271–274. doi: 10.1007/s10038-003-0025-3. [DOI] [PubMed] [Google Scholar]
- Caux F, Dubosclard E, Lascols O, Buendia B, Chazouilleres O, Cohen A, Courvalin JC, Laroche L, Capeau J, Vigouroux C, Christin-Maitre S. A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. J Clin Endocrinol Metab. 2003;88:1006–1013. doi: 10.1210/jc.2002-021506. [DOI] [PubMed] [Google Scholar]
- Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg A, Hanson NB, Martin GM, Mian IS, Kennedy BK, Oshima J. LMNA mutations in atypical Werner’s syndrome. Lancet. 2003;362(9382):440–445. doi: 10.1016/S0140-6736(03)14069-X. [DOI] [PubMed] [Google Scholar]
- Csoka AB, Cao H, Sammak PJ, Constantinescu D, Schatten GP, Hegele RA. Novel lamin A/C gene (LMNA) mutations in atypical progeroid syndromes. J Med Genet. 2004;41:304–308. doi: 10.1136/jmg.2003.015651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denecke J, Brune T, Feldhaus T, Robenek H, Kranz C, Auchus RJ, Agarwal AK, Marquardt T. A homozygous ZMPSTE24 null mutation in combination with a heterozygous mutation in the LMNA gene causes Hutchinson-Gilford progeria syndrome (HGPS): insights into the pathophysiology of HGPS. Hum Mutat. 2006;27:524–531. doi: 10.1002/humu.20315. [DOI] [PubMed] [Google Scholar]
- Doh YJ, Kim HK, Jung ED, Choi SH, Kim JG, Kim BW, Lee IK. Novel LMNA gene mutation in a patient with Atypical Werner’s Syndrome. Korean J Intern Med. 2009;24:68–72. doi: 10.3904/kjim.2009.24.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis KJ. Body composition of a young, multiethnic, male population. Am J Clin Nutr. 1997;66:1323–1331. doi: 10.1093/ajcn/66.6.1323. [DOI] [PubMed] [Google Scholar]
- Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, Young SG. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–1623. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- Garavelli L, D’Apice MR, Rivieri F, Bertoli M, Wischmeijer A, Gelmini C, De Nigris V, Albertini E, Rosato S, Virdis R, Bacchini E, Dal Zotto R, Banchini G, Iughetti L, Bernasconi S, Superti-Furga A, Novelli G. Mandibuloacral dysplasia type A in childhood. Am J Med Genet A. 2009;149A(10):2258–64. doi: 10.1002/ajmg.a.33005. [DOI] [PubMed] [Google Scholar]
- Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350(12):1220–34. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- Garg A, Cogulu O, Ozkinay F, Onay H, Agarwal AK. A novel homozygous Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia. J Clin Endocrinol Metab. 2005;90(9):5259–64. doi: 10.1210/jc.2004-2560. [DOI] [PubMed] [Google Scholar]
- Garg A, Subramanyam L, Agarwal AK, Simha V, Levine B, D’Apice MR, Novelli G, Crow Y. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab. 2009;94:4971–4983. doi: 10.1210/jc.2009-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, McCarten KM, Giobbie-Hurder A, Machan JT, Campbell SE, Berns SD, Kieran MW. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics. 2007;120:824–833. doi: 10.1542/peds.2007-1357. [DOI] [PubMed] [Google Scholar]
- Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140A:2603–2264. doi: 10.1002/ajmg.a.31346. [DOI] [PubMed] [Google Scholar]
- Jacob KN, Baptista F, dos Santos HG, Oshima J, Agarwal AK, Garg A. Phenotypic heterogeneity in body fat distribution in patients with atypical Werner’s syndrome due to heterozygous Arg133Leu lamin A/C mutation. J Clin Endocrinol Metab. 2005;90:6699–6706. doi: 10.1210/jc.2005-0939. [DOI] [PubMed] [Google Scholar]
- Kirschner J, Brune T, Wehnert M, Denecke J, Wasner C, Feuer A, Marquardt T, Ketelsen UP, Wieacker P, Bonnemann CG, Korinthenberg R. p.S143F mutation in lamin A/C: a new phenotype combining myopathy and progeria. Ann Neurol. 2005;57:148–151. doi: 10.1002/ana.20359. [DOI] [PubMed] [Google Scholar]
- Kosho T, Takahashi J, Momose T, Nakamura A, Sakurai A, Wada T, Yoshida K, Wakui K, Suzuki T, Kasuga K, Nishimura G, Kato H, Fukushima Y. Mandibuloacral dysplasia and a novel LMNA mutation in a woman with severe progressive skeletal changes. Am J Med Genet A. 2007;143A:2598–2603. doi: 10.1002/ajmg.a.31983. [DOI] [PubMed] [Google Scholar]
- Lombardi F, Gullotta F, Columbaro M, Filareto A, D’Adamo M, Vielle A, Guglielmi V, Nardone AM, Azzolini V, Grosso E, Lattanzi G, D’Apice MR, Masala S, Maraldi NM, Sbraccia P, Novelli G. Compound heterozygosity for mutations in LMNA in a patient with a myopathic and lipodystrophic mandibuloacral dysplasia type A phenotype. J Clin Endocrinol Metab. 2007;92:4467–4471. doi: 10.1210/jc.2007-0116. [DOI] [PubMed] [Google Scholar]
- Madej-Pilarczyk A, Rosinska-Borkowska D, Rekawek J, Marchel M, Szalus E, Jablonska S, Hausmanowa-Petrusewicz I. Progeroid syndrome with scleroderma-like skin changes associated with homozygous R435C LMNA mutation. Am J Med Genet A. 2009;149A:2387–2392. doi: 10.1002/ajmg.a.33018. [DOI] [PubMed] [Google Scholar]
- McDowell MA, Fryar CD, Hirsch R, Ogden CL. Anthropometric reference data for children and adults: U.S. population, 1999–2002. Adv Data. 2005:1–5. [PubMed] [Google Scholar]
- McPherson E, Turner L, Zador I, Reynolds K, Macgregor D, Giampietro PF. Ovarian failure and dilated cardiomyopathy due to a novel lamin mutation. Am J Med Genet A. 2009;149A:567–572. doi: 10.1002/ajmg.a.32627. [DOI] [PubMed] [Google Scholar]
- Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, Reynolds JC, Gropman A, Yanovski JA, Gerhard-Herman M, Collins FS, Nabel EG, Cannon RO, 3rd, Gahl WA, Introne WJ. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358:592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi Y, Akagi M, Agarwal AK, Namba N, Kato-Nishimura K, Mohri I, Yamagata M, Nakajima S, Mushiake S, Shima M, Auchus RJ, Taniike M, Garg A, Ozono K. Severe mandibuloacral dysplasia caused by novel compound heterozygous ZMPSTE24 mutations in two Japanese siblings. Clin Genet. 2008;73:535–544. doi: 10.1111/j.1399-0004.2008.00992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais P, Magina S, Ribeiro Mdo C, Rodrigues M, Lopes JM, Thanh Hle T, Wehnert M, Guimaraes H. Restrictive dermopathy--a lethal congenital laminopathy. Case report and review of the literature. Eur J Pediatr. 2009;168:1007–1012. doi: 10.1007/s00431-008-0868-x. [DOI] [PubMed] [Google Scholar]
- Mory PB, Crispim F, Kasamatsu T, Gabbay MA, Dib SA, Moises RS. Atypical generalized lipoatrophy and severe insulin resistance due to a heterozygous LMNA p.T10I mutation. Arq Bras Endocrinol Metabol. 2008;52:1252–1256. doi: 10.1590/s0004-27302008000800008. [DOI] [PubMed] [Google Scholar]
- Moulson CL, Go G, Gardner JM, van der Wal AC, Smitt JH, van Hagen JM, Miner JH. Homozygous and compound heterozygous mutations in ZMPSTE24 cause the laminopathy restrictive dermopathy. J Invest Dermatol. 2005;125:913–919. doi: 10.1111/j.0022-202X.2005.23846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro CL, Cadinanos J, De Sandre-Giovannoli A, Bernard R, Courrier S, Boccaccio I, Boyer A, Kleijer WJ, Wagner A, Giuliano F, Beemer FA, Freije JM, Cau P, Hennekam RC, Lopez-Otin C, Badens C, Levy N. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum Mol Genet. 2005;14:1503–1513. doi: 10.1093/hmg/ddi159. [DOI] [PubMed] [Google Scholar]
- Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I, Boyer A, Genevieve D, Hadj-Rabia S, Gaudy-Marqueste C, Smitt HS, Vabres P, Faivre L, Verloes A, Van Essen T, Flori E, Hennekam R, Beemer FA, Laurent N, Le Merrer M, Cau P, Levy N. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13:2493–503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D’Apice MR, Massart C, Capon F, Sbraccia P, Federici M, Lauro R, Tudisco C, Pallotta R, Scarano G, Dallapiccola B, Merlini L, Bonne G. Mandibuloacral dysplasia is caused by a mutation in LMNA-encodinglamin A/C. Am J Hum Genet. 2002;71:426–431. doi: 10.1086/341908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasilova M, Chattopadhyay C, Pal P, Schaub NA, Buechner SA, Mueller H, Miny P, Ghosh A, Heinimann K. Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson-Gilford progeria syndrome. J Med Genet. 2004;41:609–614. doi: 10.1136/jmg.2004.019661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard D, Fourcade G, Milhaud D, Bessis D, Esteves-Vieira V, Boyer A, Roll P, Bourgeois P, Levy N, De Sandre-Giovannoli A. Novel LMNA mutation in atypical Werner syndrome presenting with ischemic disease. Stroke. 2009;40(2):e11–4. doi: 10.1161/STROKEAHA.108.531780. [DOI] [PubMed] [Google Scholar]
- Shackleton S, Smallwood DT, Clayton P, Wilson LC, Agarwal AK, Garg A, Trembath RC. Compound heterozygous ZMPSTE24 mutations reduce prelamin A processing and result in a severe progeroid phenotype. J Med Genet. 2005;42(6):e36. doi: 10.1136/jmg.2004.029751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simha V, Agarwal AK, Oral EA, Fryns JP, Garg A. Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J Clin Endocrinol Metab. 2003;88(6):2821–4. doi: 10.1210/jc.2002-021575. [DOI] [PubMed] [Google Scholar]
- Smigiel R, Jakubiak A, Esteves-Vieira V, Szela K, Halon A, Jurek T, Levy N, De Sandre-Giovannoli A. Novel frameshifting mutations of the ZMPSTE24 gene in two siblings affected with restrictive dermopathy and review of the mutations described in the literature. Am J Med Genet A. 2010;152A:447–452. doi: 10.1002/ajmg.a.33221. [DOI] [PubMed] [Google Scholar]
- Van Esch H, Agarwal AK, Debeer P, Fryns JP, Garg A. A homozygous mutation in the lamin A/C gene associated with a novel syndrome of arthropathy, tendinous calcinosis, and progeroid features. J Clin Endocrinol Metab. 2006;91:517–521. doi: 10.1210/jc.2005-1297. [DOI] [PubMed] [Google Scholar]
- Varela I, Pereira S, Ugalde AP, Navarro CL, Suarez MF, Cau P, Cadinanos J, Osorio FG, Foray N, Cobo J, de Carlos F, Levy N, Freije JM, Lopez-Otin C. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008;14:767–772. doi: 10.1038/nm1786. [DOI] [PubMed] [Google Scholar]
- Writing Group for the International Society for Clinical Densitometry Position Development Conference. Diagnosis of osteoporosis in men, premenopausal women, and children. J Clin Densitom. 2004;7:17–26. doi: 10.1385/jcd:7:1:17. [DOI] [PubMed] [Google Scholar]
- Zirn B, Kress W, Grimm T, Berthold LD, Neubauer B, Kuchelmeister K, Muller U, Hahn A. Association of homozygous LMNA mutation R471C with new phenotype: mandibuloacral dysplasia, progeria, and rigid spine muscular dystrophy. Am J Med Genet. 2008;A146A:1049–1054. doi: 10.1002/ajmg.a.32259. [DOI] [PubMed] [Google Scholar]