

Summary
Background
Large, rare chromosomal deletions and duplications known as copy number variants (CNVs) have been implicated in neurodevelopmental disorders similar to attention-deficit hyperactivity disorder (ADHD). We aimed to establish whether burden of CNVs was increased in ADHD, and to investigate whether identified CNVs were enriched for loci previously identified in autism and schizophrenia.
Methods
We undertook a genome-wide analysis of CNVs in 410 children with ADHD and 1156 unrelated ethnically matched controls from the 1958 British Birth Cohort. Children of white UK origin, aged 5–17 years, who met diagnostic criteria for ADHD or hyperkinetic disorder, but not schizophrenia and autism, were recruited from community child psychiatry and paediatric outpatient clinics. Single nucleotide polymorphisms (SNPs) were genotyped in the ADHD and control groups with two arrays; CNV analysis was limited to SNPs common to both arrays and included only samples with high-quality data. CNVs in the ADHD group were validated with comparative genomic hybridisation. We assessed the genome-wide burden of large (>500 kb), rare (<1% population frequency) CNVs according to the average number of CNVs per sample, with significance assessed via permutation. Locus-specific tests of association were undertaken for test regions defined for all identified CNVs and for 20 loci implicated in autism or schizophrenia. Findings were replicated in 825 Icelandic patients with ADHD and 35 243 Icelandic controls.
Findings
Data for full analyses were available for 366 children with ADHD and 1047 controls. 57 large, rare CNVs were identified in children with ADHD and 78 in controls, showing a significantly increased rate of CNVs in ADHD (0·156 vs 0·075; p=8·9×10−5). This increased rate of CNVs was particularly high in those with intellectual disability (0·424; p=2·0×10−6), although there was also a significant excess in cases with no such disability (0·125, p=0·0077). An excess of chromosome 16p13.11 duplications was noted in the ADHD group (p=0·0008 after correction for multiple testing), a finding that was replicated in the Icelandic sample (p=0·031). CNVs identified in our ADHD cohort were significantly enriched for loci previously reported in both autism (p=0·0095) and schizophrenia (p=0·010).
Interpretation
Our findings provide genetic evidence of an increased rate of large CNVs in individuals with ADHD and suggest that ADHD is not purely a social construct.
Funding
Action Research; Baily Thomas Charitable Trust; Wellcome Trust; UK Medical Research Council; European Union.
Introduction
Attention-deficit hyperactivity disorder (ADHD), a childhood-onset disorder, is characterised by severe and impairing inattention, motor hyperactivity, and impulsiveness.1,2 It affects around 2% of children and most of those affected continue to show impairment in adult life.1,2 Although ADHD is highly heritable, no specific susceptibility genes have been unequivocally identified.1,2 Despite high heritability, neurodevelopmental features, and associated learning difficulties, some persist in arguing that the disorder is mainly a social construct.3,4 There is now clear evidence that submicroscopic chromosomal structural abnormalities, often referred to as copy number variants (CNVs), are an important source of genetic variation,5 and that large, rare CNVs contribute to other neurodevelopmental disorders including intellectual disability, schizophrenia, and autism.6–11 Although some have postulated that rare CNVs might be involved in ADHD,12 no specific variant has yet been implicated in the disorder, and there are as yet no reports of an increased burden of CNVs in ADHD.
To investigate CNVs in ADHD, we studied a UK sample of carefully phenotyped children with ADHD and unrelated ethnically matched controls. Our first aim was to examine whether children with ADHD had an increased burden of CNVs compared with controls, and to test whether this increase was attributable to associated intellectual disability since ADHD, similarly to autism and schizophrenia, occurs with increased frequency in individuals with intellectual disability (IQ test score <70).13–15 Our second aim was to investigate whether identified CNVs in our ADHD sample were significantly enriched for a specific chromosomal locus or loci previously implicated in autism and schizophrenia.
Methods
Participants
This study was approved by Wales and northwest England multicentre research ethics committees. 410 children thought to have ADHD were consecutively recruited from 90 community child psychiatry and paediatric outpatient clinics across the UK between 2001 and 2009. All were of white UK origin (including parents and grandparents), aged 5–17 years, and met Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV or International Classification of Diseases (ICD)-10 criteria for ADHD or hyperkinetic disorder. Children with schizophrenia, Tourette's syndrome, autistic spectrum disorder, or a neurological disorder including epilepsy were excluded. Diagnoses were confirmed with the Child and Adolescent Psychiatric Assessment,13 a research diagnostic interview. Interviews were undertaken by trained psychologists who were supervised weekly by a child psychiatrist. Inter-rater reliability for a diagnosis of ADHD was perfect (κ=1·0). Symptoms and impairment at school are a diagnostic requirement of the disorder, and we confirmed the presence of these criteria by interviewing the children's teachers using the Child ADHD Teacher Telephone Interview.16 Of the 410 children recruited to the study, 375 were single cases, and 35 were known to have an affected sibling (only one case from each pair was included). All children were screened with the autism screening questionnaire and individuals with suspected autistic spectrum disorder were excluded.
Since CNVs occur with increased frequency in patients with unexplained intellectual disability,17 we needed to exclude the possibility that any increased rate of CNVs recorded for these disorders was not a result of comorbidity with intellectual disability. Therefore, assessments of intellectual ability were undertaken for the ADHD group. Intellectual disability was assessed with the Wechsler Intelligence Scale for Children-IV,18 and complete test scores were available for 396 children. In accordance with DSM-IV and ICD-10, individuals with IQ test scores lower than 70 were regarded as having intellectual disability.
Control genotype data were available for 1156 individuals (594 men, 562 women) born in the UK during one week in 1958 (the 1958 British Birth Cohort). All available data were included. The sample was genotyped by the Wellcome Trust Case-Control Consortium,19 from whom we obtained permission to use the data. Psychiatric data were not available for the control cohort. For disorders of low frequency (1–2% prevalence of ADHD), the effect of unscreened controls would be negligible. Over-representation of affected individuals in the control sample would only reduce power to detect association.
Replication was sought in an independent dataset of 825 Icelandic patients with ADHD (238 female and 587 male participants) from deCODE Genetics. All participants met DSM-IV criteria for ADHD (477 combined type, 250 inattentive type, 58 hyperactive-impulsive type, 40 unspecified), and those with schizophrenia and autism were excluded from the sample. Participants were recruited from outpatient paediatric, child, and adult psychiatry clinics in Iceland, and diagnoses had been made on the basis of standardised diagnostic assessments and had been reviewed by experienced clinicians.20,21 These participants were compared with 35 243 Icelandic controls, also from deCODE Genetics. CNV analysis in this sample has been previously described.22
Laboratory procedures
Single nucleotide polymorphisms (SNPs) were genotyped in the ADHD group with the Illumina (San Diego, CA, USA) Human660W-Quad BeadChip and in the control group with the HumanHap550 BeadChip. BeadStudio (version 2.0) was used to call genotypes, normalise the signal intensity data, and establish the log R ratio and B allele frequency at every SNP according to the standard Illumina protocols. All samples passed a standard SNP-based quality control procedure; all samples with a SNP call rate lower than 0·95, any duplicate or related samples (proportion identical by descent [IBD] >0·03), or any potentially contaminated samples (proportion IBD >0·03 with multiple samples) were excluded.
We undertook several analytic and validation measures to ensure that the results were not affected by variation in the performance of the two SNP arrays, including cross-platform analysis of CNV calls in 45 samples for which data were available for both BeadChips. We limited CNV analysis to 561 113 autosomal SNPs common to both SNP arrays. CNVs were defined by PennCNV (2009Aug27version).23 Called CNVs were required to span at least 15 consecutive informative SNPs; those with copy number calls lower than two were classed as deletions, and greater than two as duplications. As is customary for this type of analysis,10,11 samples for which high-quality data could not be obtained were excluded on the basis of either a high SD in their genome-wide log R ratio (>0·30) or because they carried more than 30 apparent CNVs larger than 100 kb. These exclusions left 366 patients with ADHD and 1047 controls for full analyses. These samples had minimum difference in the distribution of the mean SNP call rate (cases=0·999, SD=0·001; controls=0·992, SD=0·005) or the mean SD of the genome-wide log R ratio (cases=0·19, SD=0·03; controls=0·21, SD=0·03).
Large CNVs can be split by CNV calling algorithms; as is customary to overcome this issue, adjacent CNV calls were merged.10 We merged adjacent CNV calls larger than 200 kb that occurred in a single individual where the gap was less than 50% of the entire length of the newly merged CNV. The log R ratio and B allele frequency of SNPs spanning all CNVs generated from merging adjacent calls were manually inspected before acceptance. At loci where the log R ratio and B allele frequency did not support the presence of a split, adjacent CNVs were not merged (eg, heterozygote genotype calls between adjacent hemizygous CNVs). In keeping with previous studies,10 we excluded CNVs for which more than 50% of their length spanned known gaps of at least 200 kb in the SNP array, genomic segments containing more than 14 CNVs (ie, >1% frequency) in the combined patients and controls, known segmental duplications present in the March, 2006 human reference sequence (National Center for Biotechnology Information reference build 36.1, hg18), or known common CNVs defined by the Genome Structural Variation Consortium.
Analysis of eight children with ADHD and three controls genotyped in duplicate established perfect concordance between CNVs larger than 200 kb called in the duplicate samples (n=9). Analysis of CNVs called in 45 HapMap CEU samples (Utah residents with ancestry from northern and western Europe) that had been genotyped and supplied by Illumina with both the Human660W-Quad and HumanHap550 BeadChips also showed 100% concordance between the 12 rare CNVs larger than 200 kb identified. We noted no significant differences in the frequency of large rare CNVs between samples extracted from blood and those extracted from saliva, or in the frequency with which CNVs were called in male compared with female participants in either the ADHD or control groups (all data available from NMW). Finally, as a highly conservative measure we accepted for analysis only CNVs larger than 500 kb. Large CNVs are called with greatest accuracy and show good concordance across different platforms.11,24 Large CNVs also show the most robust evidence for association with neurodevelopmental disorders such as schizophrenia.9–11,22
As a further measure, all large CNVs identified in the ADHD group were additionally tested with custom Agilent Human Genome comparative genomic hybridisation (CGH) 44K microarrays (CA, USA) enriched for probes at each of the loci harbouring a CNV larger than 500 kb. This procedure was done at Oxford Gene Technologies (Oxford, UK) in accordance with standard protocols. CGH analysis failed for two samples, each carrying one CNV. Of the 57 rare CNVs larger than 500 kb with high-quality CGH data, only two were not validated. Of the controls, 940 (carrying 71 of the 78 CNVs identified) had previously undergone independent CNV analysis with Affymetrix 250K NspI and StyI arrays (CA, USA).11 68 of the 71 rare CNVs larger than 500 kb that we identified in these samples were also identified by the Affymetrix platform.11 To be conservative, we excluded the two CNVs in the ADHD group that were not confirmed by high-quality data on the second platform (leaving a total of 57), whereas we included all 78 CNVs in the control group including those that were not confirmed by the Affymetrix platform. Importantly, any bias arising from this approach will be against our hypothesis of an excess of CNVs in ADHD.
Statistical analysis
All analyses included rare CNVs larger than 500 kb. The genome-wide burden of CNVs was first assessed according to the average number of CNVs per sample. Significance of the burden comparisons was assessed via permutation (100 000 permutations, one-sided test) using PLINK (version 1.06)25 with analyses undertaken for all rare, large CNVs as well as stratified according to CNV type (deletion or duplication). For locus-specific tests of association, we first defined test regions according to the genomic boundaries for each CNV identified in the entire sample. When several CNVs identified in different samples overlapped, they were merged to create one locus that encompassed all overlapping CNVs. We then established the number of CNVs present within each test region in the patients and controls. Our analyses are based on rates of CNVs per patient, but to facilitate clinical interpretation, we also provide results for the percentage of individuals carrying large, rare CNVs.
To assess whether the CNVs identified in our ADHD cohort were significantly enriched for loci previously implicated in autism and schizophrenia, we first defined the genomic coordinates for a list of 14 loci enriched for CNVs associated with autism7,26–30 and six for schizophrenia.9,10,22,31 Apart from 16p11, all autism and schizophrenia loci were independent. After counting the number of CNVs larger than 500 kb in the patients and controls (conservatively, we did not stratify by CNV type) that overlapped with any of these test regions, we performed locus-specific tests of association using PLINK (10 000 permutations, one-sided). We also tested the overall significance of case-control comparisons for the total burden of CNVs at these loci using logistic regression analysis. To allow for the possibility that any significant overlap was caused by differences in the size of CNVs in the ADHD and control groups, we included CNV size as an independent variable.
Finally, we undertook locus-specific tests of association using PLINK25 again with significance assessed via permutation (1 000 000 permutations, one-sided).10 In view of the very small cell sizes, we calculated approximate odds ratios for locus-specific tests after the addition of 0·5 before computing the log odds-ratio estimate.10
Role of the funding source
The sponsors of the study had no role in the study design, data collection, data analysis, data interpretation, or writing of the report. NMW and AT had full access to all data in the study and had final responsibility for the decision to submit for publication. HS had full access to all Icelandic replication data.
Results
366 children with ADHD and 1047 controls with high-quality SNP array data were included in the final analysis. Children with ADHD (316 boys, 50 girls) had a mean age of 10·5 years (range 5–17; SD 2·72) and a mean IQ of 86 (range 43–123; SD 13·89). 33 participants with intellectual disability (IQ <70, n=33, mean IQ=60) were identified. After exclusion of common (minor allele frequency >0·01) CNVs, all association analyses included the remaining 135 rare CNVs larger than 500 kb (57 in patients and 78 in controls; table 1 and webappendix p 2).
Table 1.
Global burden of large, rare CNVs
|
All CNVs |
Deletions only |
Duplications only |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ADHD | Controls | Ratio | p value | ADHD | Controls | Ratio | p value | ADHD | Controls | Ratio | p value | |
| All ADHD and controls* | ||||||||||||
| Number of CNVs | 57 | 78 | .. | .. | 15 | 13 | .. | .. | 42 | 65 | .. | .. |
| Rate | 0·156 | 0·075 | 2·09 | 8·9×10−5 | 0·041 | 0·012 | 3·30 | 0·0018 | 0·115 | 0·062 | 1·85 | 0·0016 |
| ADHD (IQ ≥70) and controls†‡ | ||||||||||||
| Number of CNVs | 40 | 78 | .. | .. | 10 | 13 | .. | .. | 30 | 65 | .. | .. |
| Rate | 0·125 | 0·075 | 1·68 | 0·0077 | 0·031 | 0·012 | 2·58 | 0·031 | 0·094 | 0·062 | 1·51 | 0·0387 |
| ADHD (IQ <70) and controls†§ | ||||||||||||
| Number of CNVs | 14 | 78 | .. | .. | 4 | 13 | .. | .. | 10 | 65 | .. | .. |
| Rate | 0·424 | 0·075 | 5·69 | 2·0×10−6 | 0·121 | 0·012 | 10·10 | 0·0012 | 0·303 | 0·062 | 4·88 | 0·00020 |
Rate is the average number of CNVs per person. p values are empirical and one-sided. CNV=copy number variant. ADHD=attention-deficit hyperactivity disorder.
366 participants with ADHD, 1047 controls.
IQ data were unavailable for 14 participants with ADHD.
319 participants with ADHD, 1047 controls.
33 participants with ADHD and intellectual disability, 1047 controls.
We identified a highly significant excess of large, rare CNVs in children with ADHD compared with control participants, with the average number of CNVs per child with ADHD being 2·09 times higher than that in controls (p=8·9×10−5 table 1). 50 (14%) affected children were shown to carry a CNV larger than 500 kb, compared with 75 (7%) controls. In each of the ADHD and control samples, the rates of CNVs did not differ between male and female participants (data not shown; results available from NMW).
The enrichment of large, rare CNVs was greatest in the children with ADHD and intellectual disability, in whom the average number of CNVs per patient was 5·69 times higher (p=2·0×10−6) than that in the control group (table 1). 12 (36%) children with ADHD and intellectual disability carried a CNV larger than 500 kb, compared with 38 (11%) of those without intellectual disability. Nevertheless, despite a significant difference in the rates of CNVs in the children with ADHD with and without intellectual disability (p=0·00099, two-tailed test), large, rare CNVs were still significantly enriched by 1·68 times (p=0·0077) in those without intellectual disability (n=319, mean IQ=89) when compared with the control group (table 1). Irrespective of the presence or absence of intellectual disability, children with ADHD had a significant excess of both deletions and duplications (table 1). Finally, when genomic DNA was available for both parents, we examined the inheritance of each large, rare CNVs (12 ADHD probands without intellectual disability). Of 15 CNVs, four were de novo, six were inherited from the mother, and five from the father (webappendix p 2).
In an analysis restricted to children with ADHD without intellectual disability, eight of 40 CNVs larger than 500 kb identified in the ADHD group overlapped with a locus previously implicated in autism,7,26–30 compared with only one of 78 in controls (p=0·0095; table 2). No specific single autism locus showed a significant excess of CNVs in children with ADHD (table 2). We also found that of the CNVs identified in ADHD, nine of 40 overlapped with a locus previously implicated in schizophrenia (p=0·010).9,10,22,31 Locus-specific tests revealed that this finding was largely due to the 16p13.11 region (p=0·0012), where we identified six duplications in participants with ADHD (table 2; figure 1 and figure 2). Logistic regression analysis showed no significant association between CNV size and overlap with autism (two-sided p=0·41) or schizophrenia loci (p=0·90).
Table 2.
Overlap between CNVs identified in ADHD (IQ>70) and loci implicated in autism and schizophrenia
| Description |
UK |
Iceland |
|||||
|---|---|---|---|---|---|---|---|
| ADHD | Controls | p value* | ADHD | Controls | p value† | ||
| Loci implicated in autism | |||||||
| chr1:174.1–175.1 | 1q25.1‡7 | 0 | 0 | 1 | 0 | 0 | 1 |
| chr2:13.12–13.16 | 2p24.3§7 | 0 | 0 | 1 | 0 | 0 | 1 |
| chr2:49.99–51.12 | NRXN129 | 0 | 0 | 1 | 0 | 1 | 1 |
| chr3:2.11–3.08 | CNTN428 | 2 | 0 | 0·14 | 1 | 15 | 0·41 |
| chr3:4.37–4.49 | SUMF17 | 2 | 0 | 0·14 | 0 | 0 | 1 |
| chr3:122.83–122.87 | 3q13.33‡7 | 0 | 0 | 1 | 0 | 0 | 1 |
| chr3:174.59–175.49 | NLGN17 | 1 | 0 | 0·61 | 0 | 2 | 1 |
| chr4:144.85–144.85 | 4q31.21§7 | 0 | 0 | 1 | 0 | 0 | 1 |
| chr6:161.68–163.07 | PARK27 | 0 | 0 | 1 | 0 | 13 | 1 |
| chr7:68.69–69.88 | AUTS230 | 1 | 0 | 0·61 | 0 | 3 | 1 |
| chr10:87.33–88.12 | GRID17 | 0 | 0 | 1 | 1 | 1 | 0·063 |
| chr15:23.12–23.24 | UBE3A7 | 1 | 0 | 0·61 | 0 | 2 | 1 |
| chr16:29.55–30.08 | 16p11.2‡27 | 1 | 1 | 0·87 | 1 | 22 | 0·53 |
| chr22:49.44–49.52 | SHANK326 | 0 | 0 | 1 | 0 | 0 | 1 |
| Total CNVs overlapping | .. | 8 | 1 | 0·0095¶ | 3 | 59 | 0·32† |
| Total CNVs not overlapping | .. | 32 | 77 | .. | 60 | 1816 | .. |
| Loci implicated in schizophrenia | |||||||
| chr1:144.94–146.29 | 1q21.1‡9,10 | 0 | 1 | 1 | 3 | 18 | 0·028 |
| chr15:20.31–20.78 | 15q11.2‡9 | 0 | 0 | 1 | 10 | 245 | 0·31 |
| chr15:28.72–30.3 | 15q13.2-13.3‡9,10 | 2 | 0 | 0·064 | 0 | 18 | 1 |
| chr16:15.38–16.20 | 16p13.11‡22 | 6 | 1 | 0·0012 | 4 | 36 | 0·038 |
| chr16:29.55–30.08 | 16p11.231 | 1 | 1 | 0·87 | 1 | 22 | 0·53 |
| chr22:17.5–20.0 | 22q11.21‡9,10 | 0 | 1 | 1 | 3 | 18 | 0·028 |
| Total CNVs overlapping | .. | 9 | 4 | 0·010¶ | 21 | 367 | 0·0081† |
| Total CNVs not overlapping | .. | 31 | 74 | .. | 42 | 1508 | .. |
CNV=copy number variant. ADHD=attention-deficit hyperactivity disorder. NRXN1=neurexin 1. CNTN4=contactin 4. SUMF1=sulphatase modifying factor 1. NLGN1=neuroligin 1. PARK2=parkin. AUTS2=autism susceptibility candidate 2. GRID1=glutamate receptor. UBE3A=ubiquitin ligase E3A. SHANK3=SH3 and multiple ankyrin repeat domains 3.
Empirical (one-tailed).
Fisher's exact test (one-tailed).
Locus spans a contiguous set of genes.
Locus contains no known genes.
Logistic regression correcting for CNV size.
Figure 1.
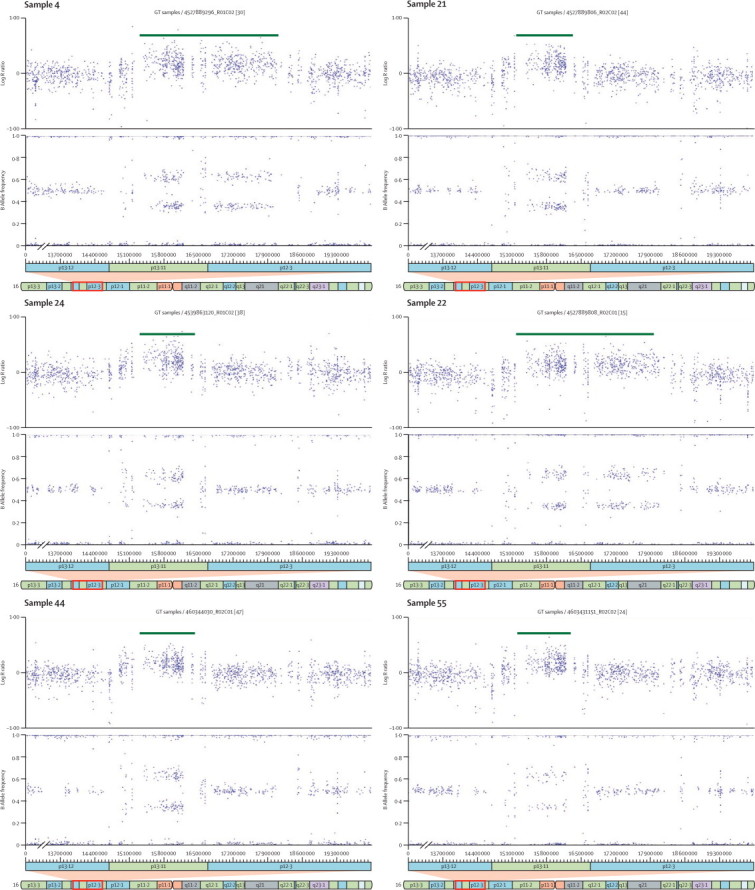
CNVs identified at chromosome 16p13.11
Log R ratio and B allele frequency plots of the six copy number variants (CNVs; all duplications) larger than 500 kb identified at the chromosome 16p13.11 region in participants with attention-deficit hyperactivity disorder.
Figure 2.
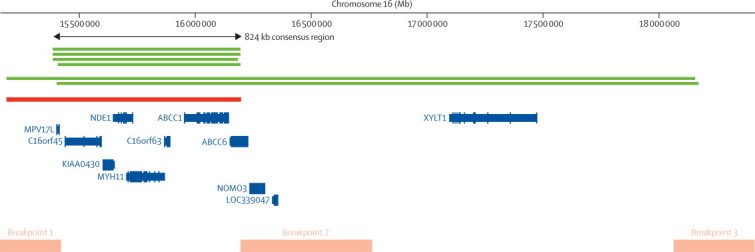
Positions of CNVs identified at chromosome 16p13.11
The positions of rare copy number variants (CNVs) larger than 500 kb identified at the chromosome 16p13.11 region. Green lines show the six duplications identified in patients with attention-deficit hyperactivity disorder and the red line shows the deletion identified in a single control. The consensus region that is spanned by all CNVs is shown by the arrow. Orange bars show known segmental duplications and therefore the most likely location of the breakpoints for the CNVs identified. The relative locations of genes are based on National Center for Biotechnology Information reference build 36.1 in the University of California Santa Cruz (UCSC) Genome Browser.
To ensure that subsequent findings related to ADHD rather than comorbid intellectual disability, we restricted genome-wide locus-specific tests to participants without intellectual disability. A locus on chromosome 16p13.11 (chr16:15156431-18174650) showed a significant excess of rare CNVs larger than 500 kb in children with ADHD (p=0·0008 corrected for multiple testing of all CNV loci by permutation; OR 13·88, 95% CI 2·3–82·2). Post-hoc analysis revealed that the excess of CNVs at this locus was due to duplications (corrected p=0·0001), which spanned an 824 kb consensus region (chr16:15 385 551–16 209 503; figure 2). Analysis of DNA from both parents for two ADHD probands carrying 16p13.11 duplications revealed one to be maternally inherited and the other to be de novo (webappendix p 2). No duplications in this region were identified in our control sample. This consensus region is flanked by segmental duplications, spans only seven genes, and has been implicated in schizophrenia.22
We next sought replication of our finding of an excess of chromosome 16p13.11 duplications in an independent dataset of 825 Icelandic patients with ADHD and 35 243 Icelandic controls.22 In this second sample, there was a significant excess (p=0·031 after correction for relatedness and potential population stratification32) of chromosome 16p13.11 duplications in the ADHD group (n=4; frequency, 0·48%) compared with controls (n=36; frequency, 0·09%). Analysis of the Icelandic data also revealed that the CNVs identified in the ADHD sample were significantly enriched for loci previously implicated in schizophrenia (p=0·0081), but not autism (p=0·32; table 2).
Discussion
Using a well characterised sample, we show that children with ADHD have a significantly increased burden of large, rare CNVs that include both duplications and deletions. Importantly, since CNV burden is increased in people with unexplained intellectual disability, autism, and schizophrenia, our clinical assessment procedure allowed us to conclude that the CNV burden is not attributable to comorbidity with these disorders. Although ADHD is one of the most heritable psychiatric disorders, with a heritability estimate of 76%,1,2 genome-wide association and linkage studies have so far failed to identify common genetic risk variants. Our results suggest that further investigation of rare CNVs in ADHD is likely to be fruitful. Up to now, there has been only one reported investigation of CNVs in ADHD, which did not detect a significantly increased burden of rare CNVs in 335 patients with the disorder.12 However, that study examined CNVs of all sizes rather than large CNVs, which are the ones that are most likely to be deleterious24 and are particularly enriched in neurodevelopmental disorders such as schizophrenia.9–11
We also showed that large, rare CNVs identified in our ADHD cohort were significantly enriched for chromosomal loci previously implicated in autism and schizophrenia. ADHD is currently thought to be entirely separate from these disorders. However, there is some overlap between ADHD and autism in terms of clinical symptoms and cognitive deficits.33 Autistic traits and ADHD behaviours in the general population (not clinical disorder) also seem to be affected by shared heritability.34 Our results suggest that there could also be a shared biological basis to these two childhood-onset disorders. So far, possibly because of the dearth of relevant studies, there are no clinical or genetic data clearly pointing to overlap between ADHD and schizophrenia. In view of the strong evidence for association between duplications at 16p13.11 and schizophrenia,22 we note with particular interest that our ADHD cohort was significantly enriched for duplications at the same locus, a finding that was independently replicated in the Icelandic population. Moreover, further analysis of two duplications at 16p13.11 revealed that one was de novo, adding further support that this locus is functionally relevant to ADHD. Future studies analysing the segregation patterns of familial CNVs will be needed to estimate disease penetrance.
The consensus duplicated region at 16p13.11 spans only seven genes and is flanked by segmental duplications that predispose to recurrent chromosomal rearrangements (figure 2). The genes mapping within 16p13.11 therefore provide a specific focus for further research into the neurobiology of ADHD. Among the genes spanned by the CNV, NDE1 (nuclear distribution gene E homologue 1) is of particular interest because of its role in neurodevelopment and interaction with DISC1 (disrupted in schizophrenia 1), a gene implicated in schizophrenia and other major psychiatric disorders that encodes a protein also involved in neurodevelopmental processes.35,36 Other investigators have detected CNVs spanning this region; deletions have been strongly associated with intellectual disability,17,37 whereas duplications have been detected in patients with autism,38 intellectual disability,17,38 and schizophrenia,10,11 again suggesting that the same large, rare CNVs might contribute to several, phenotypically different neurodevelopmental disorders.
Our findings have important clinical and research implications. First, our results emphasise that further investigation of CNVs in ADHD is a priority for research into this disorder. We do not suggest, however, that the search for common genetic variants using SNPs should be abandoned because, up to now, SNP-based studies1 have not had sufficient power to allow realistic assessment of the role of that class of variant in ADHD. Moreover, with the application of appropriate precautions such as we have undertaken, studies analysing SNPs and large, rare CNVs can be undertaken simultaneously. Key measures that allowed us to undertake such an analysis despite the use of two different SNP arrays were: that we limited our analysis to SNPs overlapping between platforms, we undertook quality-control checks in samples genotyped with both platforms, we validated the CNVs with independent platforms, and crucially, as our own cross-platform validation data show, we focused on CNVs large enough to be detected at high sensitivity and specificity irrespective of SNP array.
Second, the findings allow us to refute the hypothesis that ADHD is purely a social construct, which has important clinical and social implications for affected children and their families. Finally, although the number of children with intellectual disability in our sample is small, more than a third carried a large, rare CNV. None of these participants had been assessed for this type of mutation by clinical services. Microarray-based comp-arative genomic hybridisation enables the accurate detection of submicroscopic CNVs and is increasingly being used to investigate patients with intellectual disability or congenital abnormalities in some clinical settings. Our results suggest that routine referral to clinical geneticists and screening for such mutations could be helpful for children with ADHD and intellectual disability.
Acknowledgments
Acknowledgments
We thank Charlotte Davies, Emma Evans, Rachel Roberts, Sharifah Syed, Alka Ahuja, Janet Robinson, Neil Buttigieg, and Evangelia Stergyiakouli for sample collection and laboratory support, and the families and clinicians who have supported this study. Funding was received from Action Research, Baily Thomas, Wellcome Trust, and MRC Centre Funding (UK). This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of investigators who contributed to generation of the data is available from the Consortium's website, and funding for the project was provided by the Wellcome Trust under award 076113. The deCODE replication was supported by funding from the European Union (LSHM-CT-2006-037761 [Project SGENE] and HEALTH-F2-2009-223423 [Project PsychCNVs]).
Contributors
AT, NMW, MJO, and MOD designed the overall study. AT oversaw sample collection and recruitment and supervised all clinical aspects. NMW supervised all laboratory work and did the genetics analysis. PH advised on statistical analysis. AM, KL, PM, and OOG undertook the clinical data analysis and collected the clinical data. KS, HS, RF, and OG contributed to data analysis. IZ and KM undertook all the laboratory work. AT, NMW, KL, PH, MJO, and MOD interpreted the results. All authors contributed to the report.
Conflicts of interest
We declare that we have no conflicts of interest.
Contributor Information
Nigel M Williams, Email: williamsnm@cf.ac.uk.
Anita Thapar, Email: thapar@cf.ac.uk.
Web Extra Material
References
- 1.Franke B, Neale BM, Faraone SV. Genome-wide association studies in ADHD. Hum Genet. 2009;126:13–50. doi: 10.1007/s00439-009-0663-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thapar A, Langley K, Owen MJ, O’Donovan MC. Advances in genetic findings on attention deficit hyperactivity disorder. Psychol Med. 2007;37:1681–1692. doi: 10.1017/S0033291707000773. [DOI] [PubMed] [Google Scholar]
- 3.Timimi S, Taylor E. ADHD is best understood as a cultural construct. Br J Psychiatry. 2004;184:8–9. doi: 10.1192/bjp.184.1.8. [DOI] [PubMed] [Google Scholar]
- 4.Amaral OB. Psychiatric disorders as social constructs: ADHD as a case in point. Am J Psychiatry. 2007;164:1612. doi: 10.1176/appi.ajp.2007.07060942. [DOI] [PubMed] [Google Scholar]
- 5.Wain LV, Armour JAL, Tobin MD. Genomic copy number variation, human health, and disease. Lancet. 2009;374:340–350. doi: 10.1016/S0140-6736(09)60249-X. [DOI] [PubMed] [Google Scholar]
- 6.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glessner JT, Wang K, Cai G. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cook EH, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 9.Stefansson H, Rujescu D, Cichon S. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirov G, Grozeva D, Norton N. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet. 2009;18:1497–1503. doi: 10.1093/hmg/ddp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elia J, Gai X, Xie HM. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry. 2009;15:637–646. doi: 10.1038/mp.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Angold A, Costello EJ. The Child and Adolescent Psychiatric Assessment (CAPA) J Am Acad Child Adolesc Psychiatry. 2000;39:39–48. doi: 10.1097/00004583-200001000-00015. [DOI] [PubMed] [Google Scholar]
- 14.Einfeld SL, Piccinin AM, Mackinnon A. Psychopathology in young people with intellectual disability. JAMA. 2006;296:1981–1989. doi: 10.1001/jama.296.16.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Einfeld S, Emerson E. Intellectual disability. In: Rutter M, Bishop D, Pine D, editors. Rutter's child and adolescent psychiatry. Blackwell Publishing; Oxford, UK: 2008. pp. 820–840. [Google Scholar]
- 16.Holmes J, Lawson D, Langley K. The Child Attention-Deficit Hyperactivity Disorder Teacher Telephone Interview (CHATTI): reliability and validity. Br J Psychiatry. 2004;184:74–78. doi: 10.1192/bjp.184.1.74. [DOI] [PubMed] [Google Scholar]
- 17.Mefford HC, Cooper GM, Zerr T. A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Genome Res. 2009;19:1579–1585. doi: 10.1101/gr.094987.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wechsler D. WISC-IV administration and scoring manual. The The Psychological Corporation; San Antonio, TX, USA: 2003. [Google Scholar]
- 19.The Wellcome Trust Case-Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magnússon P, Smári J, Grétarsdóttir H, Prándardóttir H. Attention-deficit/hyperactivity symptoms in Icelandic schoolchildren: assessment with the Attention Deficit/Hyperactivity Rating Scale-IV. Scand J Psychol. 1999;40:301–306. doi: 10.1111/1467-9450.404130. [DOI] [PubMed] [Google Scholar]
- 21.Magnússon P, Smári J, Sigurdardóttir D. Validity of self-report and informant rating scales of adult ADHD symptoms in comparison with a semistructured diagnostic interview. J Atten Disord. 2006;9:494–503. doi: 10.1177/1087054705283650. [DOI] [PubMed] [Google Scholar]
- 22.Ingason A, Rujescu D, Cichon S. Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol Psychiatry. 2009 doi: 10.1038/mp.2009.101. published online Sept 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang K, Li M, Hadley D. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itsara A, Cooper GM, Baker C. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell S, Neale B, Todd-Brown K. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moessner R, Marshall CR, Sutcliffe JS. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weiss LA, Shen Y, Korn JM. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 28.Roohi J, Montagna C, Tegay DH. Disruption of contactin 4 in three subjects with autism spectrum disorder. J Med Genet. 2009;46:176–182. doi: 10.1136/jmg.2008.057505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim H, Kishikawa S, Higgins AW. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalscheuer VM, FitzPatrick D, Tommerup N. Mutations in autism susceptibility candidate 2 (AUTS2) in patients with mental retardation. Hum Genet. 2007;121:501–509. doi: 10.1007/s00439-006-0284-0. [DOI] [PubMed] [Google Scholar]
- 31.McCarthy SE, Makarov V, Kirov G. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Devlin B, Roeder K, Wasserman L. Genomic control, a new approach to genetic-based association studies. Theor Popul Biol. 2001;60:155–166. doi: 10.1006/tpbi.2001.1542. [DOI] [PubMed] [Google Scholar]
- 33.Sinzig J, Walter D, Doepfner M. Attention deficit/hyperactivity disorder in children and adolescents with autism spectrum disorder: symptom or syndrome? J Atten Disord. 2009;13:117–126. doi: 10.1177/1087054708326261. [DOI] [PubMed] [Google Scholar]
- 34.Ronald A, Simonoff E, Kuntsi J, Asherson P, Plomin R. Evidence for overlapping genetic influences on autistic and ADHD behaviours in a community twin sample. J Child Psychol Psychiatry. 2008;49:535–542. doi: 10.1111/j.1469-7610.2007.01857.x. [DOI] [PubMed] [Google Scholar]
- 35.Bradshaw NJ, Ogawa F, Antolin-Fontes B. DISC1, PDE4B, and NDE1 at the centrosome and synapse. Biochem Biophys Res Commun. 2008;377:1091–1096. doi: 10.1016/j.bbrc.2008.10.120. [DOI] [PubMed] [Google Scholar]
- 36.Bradshaw NJ, Christie S, Soares D. NDE1 and NDEL1: multimerisation, alternate splicing and DISC1 interaction. Neurosci Lett. 2009;449:228–233. doi: 10.1016/j.neulet.2008.10.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hannes FD, Sharp AJ, Mefford HC. Recurrent reciprocal deletions and duplications of 16p13. 11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. BMJ. 2009;46:223. doi: 10.1136/jmg.2007.055202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ullmann R, Turner G, Kirchhoff M. Array CGH identifies reciprocal 16p13. 1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat. 2007;28:674. doi: 10.1002/humu.20546. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.