

Summary
Background
Comparison of patients with coronary heart disease and controls in genome-wide association studies has revealed several single nucleotide polymorphisms (SNPs) associated with coronary heart disease. We aimed to establish the external validity of these findings and to obtain more precise risk estimates using a prospective cohort design.
Methods
We tested 13 recently discovered SNPs for association with coronary heart disease in a case-control design including participants differing from those in the discovery samples (3829 participants with prevalent coronary heart disease and 48 897 controls free of the disease) and a prospective cohort design including 30 725 participants free of cardiovascular disease from Finland and Sweden. We modelled the 13 SNPs as a multilocus genetic risk score and used Cox proportional hazards models to estimate the association of genetic risk score with incident coronary heart disease. For case-control analyses we analysed associations between individual SNPs and quintiles of genetic risk score using logistic regression.
Findings
In prospective cohort analyses, 1264 participants had a first coronary heart disease event during a median 10·7 years' follow-up (IQR 6·7–13·6). Genetic risk score was associated with a first coronary heart disease event. When compared with the bottom quintile of genetic risk score, participants in the top quintile were at 1·66-times increased risk of coronary heart disease in a model adjusting for traditional risk factors (95% CI 1·35–2·04, p value for linear trend=7·3×10−10). Adjustment for family history did not change these estimates. Genetic risk score did not improve C index over traditional risk factors and family history (p=0·19), nor did it have a significant effect on net reclassification improvement (2·2%, p=0·18); however, it did have a small effect on integrated discrimination index (0·004, p=0·0006). Results of the case-control analyses were similar to those of the prospective cohort analyses.
Interpretation
Using a genetic risk score based on 13 SNPs associated with coronary heart disease, we can identify the 20% of individuals of European ancestry who are at roughly 70% increased risk of a first coronary heart disease event. The potential clinical use of this panel of SNPs remains to be defined.
Funding
The Wellcome Trust; Academy of Finland Center of Excellence for Complex Disease Genetics; US National Institutes of Health; the Donovan Family Foundation.
Introduction
Coronary heart disease is complex in origin, with contributions from lifestyle and genetic factors.1 Family history of premature coronary heart disease is an independent risk factor, suggesting that inherited DNA sequence variants contribute to risk of the disease. Using a case-control design, genome-wide association studies have identified single nucleotide polymorphisms (SNPs) at 13 genomic regions associated (p<5×10−8 with coronary heart disease, myocardial infarction, or both.2 In discovery studies, each copy of the risk allele at these loci was estimated to increase risk of myocardial infarction by 12–92%.
Discovery genome-wide association studies for myocardial infarction or coronary heart disease have ascertained cases on the basis of early age of disease onset or affected family members, and as such the reported effect estimates might not be representative of the general population. Although efficient for discovery, cross-sectional and case-control designs have the potential for several types of bias, whereas the prospective cohort study is regarded as the gold standard in epidemiological investigations.3 Therefore, we set out to answer two questions: first, are the reported genetic association findings externally generalisable in studies differing from the discovery studies; and second, can more precise risk estimates be obtained with a prospective cohort design?
Methods
Study populations
We tested the 13 recently discovered SNPs for association with coronary heart disease in two designs: a case-control design including participants differing from those in the discovery samples (3829 participants with prevalent coronary heart disease and 48 897 controls free of the disease); and a prospective cohort design including 30 725 participants free of cardiovascular disease from Finland and Sweden. Coronary heart disease was defined as myocardial infarction, unstable angina pectoris, coronary revascularisation (coronary artery bypass graft or percutaneous transluminal coronary angioplasty), or death due to coronary heart disease. Cardiovascular disease included coronary heart disease and ischaemic stroke events. Detailed case definitions are described in the webappendix.
Participants from seven cohorts were included in our analyses (table 1). The FINRISK 1992, 1997, and 2002 cohorts consist of a representative random sample selected from inhabitants of different regions in Finland aged 25–74 years. The survey included a mailed questionnaire and a clinical examination at which a blood sample was drawn. The study protocol has been described previously.4 23 036 individuals participated in these cohorts and genotype data was available from 20 927 participants.
Table 1.
Background characteristics of the seven study populations
| FR 1992 (n=5353) | FR 1997 (n=7939) | FR 2002 (n=7635) | Health 2000 (n=5796) | MDC-CC (n=5104) | COROGENE (n=6015) | MPP (n=14 884) | ||
|---|---|---|---|---|---|---|---|---|
| Sex | ||||||||
| Male | 2449 (46%) | 4006 (50%) | 3526 (46%) | 2697 (47%) | 2141 (42%) | 4192 (70%) | 9773 (66%) | |
| Female | 2904 (54%) | 3933 (50%) | 4109 (54%) | 3099 (53%) | 2963 (58%) | 1823 (30%) | 5111 (34%) | |
| Age (years) | 44·5 (11·3) | 49 (13·4) | 48·4 (13·1) | 50·8 (13·0) | 57·4 (5·9) | 66·5 (10·9) | 45·3 (7·0) | |
| Cholesterol | ||||||||
| Total (mmol/L) | 5·6 (1·1) | 5·5 (1·1) | 5·6 (1·1) | 5·9 (1·1) | 6·2 (1·1) | NA | 5·6 (1·0) | |
| LDL (mmol/L) | 3·5 (1·0) | 3·5 (0·9) | 3·4 (0·9) | 3·8 (1·2) | 4·2 (1·0) | NA | NA | |
| HDL (mmol/L) | 1·4 (0·3) | 1·4 (0·4) | 1·5 (0·4) | 1·3 (0·4) | 1·4 (0·4) | NA | NA | |
| Blood pressure | ||||||||
| Systolic (mm Hg) | 135·3 (19·4) | 136·2 (20·1) | 135·0 (20·0) | 133·7 (20·6) | 141·1 (18·9) | NA | 127·7 (14·4) | |
| Diastolic (mm Hg) | 81·2 (11·9) | 82·4 (11·3) | 79·1 (11·4) | 82·1 (11·2) | 86·9 (9·5) | NA | 84·2 (8·8) | |
| Body-mass index (kg/m2) | 26·1 (4·4) | 26·7 (4·5) | 26·9 (4·7) | 26·9 (4·7) | 25·9 (4·9) | NA | 24·4 (3·4) | |
| Current smoker | 1498 (28%) | 1846 (23%) | 1993 (26%) | 1728 (30%) | 1307 (26%) | NA | 5639 (38%) | |
| Current drug therapy | ||||||||
| Lipid-lowering | 82 (2%) | 280 (4%) | 563 (7%) | 1396 (24%) | 109 (2%) | NA | NA | |
| Antihypertensive | 496 (9%) | 1056 (13%) | 1095 (14%) | 1306 (23%) | 641 (13%) | NA | 645 (4%) | |
| Diabetes mellitus | 198 (4%) | 481 (6%) | 433 (6%) | 460 (8%) | 399 (8%) | NA | 617 (4%) | |
| Prevalent cases | ||||||||
| CHD | 64 (1%) | 222 (3%) | 198 (3%) | 59 (1%) | NA | NA | NA | |
| CVD | 77 (1%) | 276 (3%) | 264 (3%) | 378 (7%) | 107 (2%) | NA | 1749 (12%) | |
| MI | 50 (1%) | 161 (2%) | 119 (2%) | 28 (<1%) | 63 (1%) | 2101 (35%) | 1122 (8%) | |
| Incident cases | ||||||||
| CHD | 296 (6%) | 419 (5%) | 132 (2%) | 151 (3%) | NA | NA | NA | |
| CVD | 416 (8%) | 599 (8%) | 186 (2%) | 201 (3%) | 468 (9%) | NA | NA | |
| MI | 157 (3%) | 230 (3%) | 65 (1%) | 112 (2%) | 266 (5%) | NA | NA | |
Data are mean (SD) or number (%). FR=FINRISK. MDC-CC=Malmö Diet and Cancer Cardiovascular cohort. MPP=Malmö Preventive Project. NA=not available. CHD=coronary heart disease. CVD=cardiovascular disease. MI=myocardial infarction.
The Health 2000 study was based on a stratified two-stage cluster sampling from the National Population Register to represent the total Finnish population aged 30 years and older.5 The survey included an interview about medical history, health-related lifestyle habits, and a clinical examination at which a blood sample was drawn. 6200 people participated in the study. After exclusion of individuals older than 80 years and without sufficient genotype data, the final dataset consisted of 5796 participants. A detailed methodology report is available online.6
The Malmö Diet and Cancer (MDC) study was a community-based prospective epidemiological cohort of 28 449 people recruited for a baseline examination between 1991 and 1996.7 From this cohort, 6103 people were randomly selected to participate in the Cardiovascular Cohort (MDC-CC), which sought to investigate risk factors for cardiovascular disease. All participants underwent a medical history, physical examination, and laboratory assessment for cardiovascular risk factors, as described previously.8 Final data with genotypes were available for 5104 participants.
During follow-up of the FINRISK and HEALTH 2000 cohorts, data for admission to hospital and mortality were obtained from the Finnish National Hospital Discharge Register and the Finnish National Causes-of-Death Register. These registers have excellent validity and coverage.9,10 Follow-up ended on Dec 31, 2007. Follow-up of the MDC-CC is as previously described.11
The Malmö Preventive Project (MPP) is a cohort from southern Sweden that was set up in 1974. 33 346 individuals were screened during 1974–92. Information concerning lifestyle factors and medical history was obtained from a questionnaire. All participants underwent physical examination and biochemical analyses. Of individuals who participated in the baseline examinations, 17 284 were rescreened during 2002–06. The final data with genotypes included 14 884 individuals.
For the COROGENE cohort, initially, all consecutive Finnish patients undergoing coronary angiogram between June, 2006, and March, 2008 (n=5330), in the Helsinki University Central Hospital were included and a questionnaire, information about previous medical conditions and cardiovascular risk factors, hospital records for patients' history, laboratory measurements, electrocardiogram, echocardiogram, and medication were obtained. Of these patients, 2118 (53%) had acute coronary syndrome and were selected as COROGENE cases. The controls for COROGENE cases were selected from the Helsinki-Vantaa region participants of FINRISK 1997, 2002, and 2007 by risk set sampling.12 For each case, two controls matched by sex and birth year and free of acute coronary syndrome were sampled. In total, 2101 cases and 3914 controls (of which 1453 were unique) formed the final genotyped COROGENE case-control sample.
The FINRISK 1992, 1997, 2002 and Health 2000 study protocols were approved by the ethics committee of the National Institute for Health and Welfare, the MDC-CC and MPP study protocols by the ethics committee of Lund University, and the COROGENE study protocol by the ethics committee of Helsinki University Hospital, Internal Medicine. All participants provided written informed consent.
SNP selection and genotyping
We selected SNPs from genome-wide association studies published before June, 2009 in which phenotypes studied were myocardial infarction or coronary heart disease, and association between a SNP and myocardial infarction or coronary heart disease exceeded a genome-wide association threshold (p<5×10−8). 13 SNPs from seven reports13–19 met these criteria, including 1q41 in MIA3, 1p32 near PCSK9, 1p13 near CELSR2–PSRC1–SORT1, 2q33 in WDR12, 6p24 in PHACTR1, 9p21 near CDKN2A–CDKN2B, 10q11 near CXCL12, 19p13 near LDLR, 21q22 near SLC5A3–MRPS6–KCNE2, 3q22 in MRAS, 6q26 in LPA, 12q24 near HNF1A, and 12q24 in SH2B3.
Samples were genotyped with the Sequenom platform (iPlex MassARRAY, San Diego, CA, USA) at the Institute for Molecular Medicine Finland FIMM (FINRISK 1992 and 2002), the Wellcome Trust Sanger Institute, UK (Health 2000), or the Broad Institute, USA (FINRISK 1997 and MDC-CC), and with Sequenom or Taqman (Applied Biosystems, Foster City, CA, USA) platforms at Lund University, Sweden (MDC-CC and MPP). COROGENE was genotyped with Illumina 610K chip (Illumina HumanHap 610-Quad SNP array, San Diego, CA, USA) at the Sanger Institute. Genotypes were manually curated with call rates above 97%.
Statistical analysis
We tested associations between SNPs and incident cardiovascular events using Cox proportional hazards models adjusted for traditional risk factors: sex, LDL and HDL cholesterol, current cigarette smoking, body-mass index, systolic and diastolic blood pressure, blood pressure treatment, and prevalent type 2 diabetes. Age was used as the baseline timescale in the Cox models. The proportional hazards assumption was met when tested with scaled Schoenfeld residuals.20
We constructed a multilocus genetic risk score for each individual by summing the number of risk alleles (0/1/2) for each of the 13 SNPs weighted by their estimated effect sizes in the discovery sample (table 2 shows SNP specific weights). Missing genotype values were imputed with the cohort-specific averages of risk allele frequencies. Estimates of association between the genetic risk score divided into quintiles and time to coronary heart disease, cardiovascular disease, and myocardial infarction were calculated with Cox proportional hazards models. For each cohort we calculated 95% CIs for hazard ratios (HRs) and tested the null hypothesis of no linear effect over the quintiles using 1 df Wald test.
Table 2.
Association between SNPs and incident coronary heart disease, cardiovascular disease, and myocardial infarction*
| Region | Candidate gene(s) | Weight† | Reference† | Risk allele | Risk allele frequency | Other allele |
Coronary heart disease (total n=19 790) |
Cardiovascular disease (total n=24 894) |
Myocardial infarction (total n=24 894) |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pooled HR (95% CI)‡ | p value | Pooled HR (95% CI)‡ | p value | Pooled HR (95% CI)‡ | p value | ||||||||
| rs17465637 | 1q41 | MIA3 | 1·14 | 15 | C | 0·75 | A | 0·99 (0·87–1·12) | 0·854 | 1·03 (0·94–1·12) | 0·546 | 1·03 (0·91–1·18) | 0·624 |
| rs11206510 | 1p32 | PCSK9 | 1·15 | 15 | T | 0·84 | C | 0·94 (0·81–1·09) | 0·431 | 0·97 (0·87–1·07) | 0·515 | 1·04 (0·89–1·22) | 0·636 |
| rs646776 | 1p13 | CELSR2–PSRC1–SORT1 | 1·19 | 15 | T | 0·79 | C | 0·96 (0·84–1·09) | 0·512 | 0·98 (0·89–1·07) | 0·587 | 0·94 (0·82–1·08) | 0·378 |
| rs6725887 | 2q33 | WDR12 | 1·17 | 15 | C | 0·11 | T | 1·14 (0·96–1·35) | 0·126 | 1·00 (0·89–1·12) | 0·960 | 0·96 (0·80–1·14) | 0·629 |
| rs9818870 | 3q22 | MRAS | 1·15 | 16 | T | 0·10 | C | 0·88 (0·73–1·06) | 0·174 | 0·92 (0·81–1·03) | 0·150 | 0·86 (0·71–1·03) | 0·097 |
| rs3798220 | 6q26 | LPA | 1·68 | 18 | C | 0·01 | T | 2·07 (1·39–3·09) | 3·8×10−5 | 1·63 (1·22–2·17) | 0·001 | 1·80 (1·19–2·72) | 0·005 |
| rs9349379 | 6p24 | PHACTR1 | 1·12 | 15 | C | 0·44 | T | 1·16 (1·04–1·29) | 0·008 | 1·10 (1·02–1·19) | 0·014 | 1·11 (0·99–1·24) | 0·075 |
| rs4977574 | 9p21 | CDKN2A–CDKN2B | 1·29 | 15 | G | 0·43 | A | 1·21 (1·08–1·34) | 0·001 | 1·16 (1·08–1·25) | 9·3×10−6 | 1·19 (1·06–1·33) | 0·002 |
| rs1746048 | 10q11 | CXCL12 | 1·17 | 15 | C | 0·84 | T | 1·13 (0·97–1·33) | 0·113 | 1·01 (0·91–1·12) | 0·846 | 1·11 (0·95–1·31) | 0·198 |
| rs2259816 | 12q24 | HNF1A | 1·08 | 16 | T | 0·36 | G | 1·02 (0·91–1·14) | 0·774 | 1·00 (0·92–1·08) | 0·932 | 1·01 (0·90–1·14) | 0·836 |
| rs3184504 | 12q24 | SH2B3 | 1·13 | 17 | T | 0·40 | C | 1·03 (0·92–1·15) | 0·568 | 1·10 (1·02–1·19) | 0·011 | 1·15 (1·03–1·29) | 0·012 |
| rs1122608 | 19p13 | LDLR | 1·15 | 15 | G | 0·79 | T | 1·00 (0·87–1·14) | 0·988 | 1·02 (0·93–1·12) | 0·693 | 0·98 (0·86–1·12) | 0·774 |
| rs9982601 | 21q22 | SLC5A3–MRPS6–KCNE2 | 1·20 | 15 | T | 0·14 | C | 1·29 (1·07–1·57) | 0·009 | 1·11 (0·98–1·26) | 0·092 | 1·24 (1·04–1·48) | 0·016 |
SNP=single nucleotide polymorphism. HR=hazard ratio.
Association tested with Cox proportional hazards model adjusted for sex, LDL and HDL cholesterol, smoking, body-mass index, systolic and diastolic blood pressure, blood pressure treatment, and diabetes; age was used as the timescale.
SNP specific weights for genetic risk score calculation; weights are effect sizes for the SNPs from the studies referenced.
Results from FINRISK 1992, 1997, and 2002, Health 2000, and Malmö Diet and Cancer Cardiovascular Cohort were combined with fixed effects meta-analysis.21
For prevalent case-control analyses, we analysed individual SNP and quintiles of genetic risk score associations using a logistic regression model adjusted for age and sex. COROGENE data were analysed with conditional logistic regression. Each cohort was analysed separately, and the estimates weighted on the inverse of their standard errors were combined across cohorts with fixed effects meta-analysis.21
To evaluate the potential value of genetic risk score in risk prediction, we used two cohorts (FINRISK 1992 and 1997) and up to 10-year follow-up. First, we compared the receiver operating characteristic (ROC) curves22 of models with and without genetic risk score. The statistical significance of change in the area under the ROC curve (AUC) between models was tested with the correlated C-index approach.23 Second, we calculated net reclassification improvement (NRI) and clinical NRI24 using the Kaplan-Meier approach with bootstrap-based p values,25 and integrated discrimination improvement (IDI).26 The model calibration was tested with Hosmer-Lemeshow goodness-of-fit test.
Since each of the 13 reported SNPs has previously been associated with coronary heart disease or myocardial infarction at significance levels exceeding a stringent genome-wide threshold, in this report we regarded an association to be significant if a two-sided p value was less than 0·05 (for the same risk allele in the same direction as in the original report). The R statistical package (version 2.11.1) was used for all analyses.
Role of the funding source
The sponsors had no role in the conduct or interpretation of the study. The corresponding author had full access to all data in the study and had final responsibility for the decision to submit for publication.
Results
52 726 participants in FINRISK 1992, 1997, and 2002, Health 2000, MDC-CC, MPP, and COROGENE were included in our analysis of prevalent cases versus controls. The total number of prevalent cases of coronary heart disease was 3829 (7%). 30 725 participants from five cohorts (FINRISK 1992, 1997, and 2002, Health 2000, and MDC-CC) were included in the prospective cohort analyses. Median follow-up was 10·7 years (IQR 6·7–13·6). 1264 (4%) incident cases of coronary heart disease occurred during follow-up. Table 1 shows background characteristics along with risk factor distributions for the cohorts.
In single SNP analyses for prevalent cases, 9p21 near CDKN2A–CDKN2B, 21q22 near SLC5A3–MRPS6–KCNE2, and 1q41 in MIA3 were associated with coronary heart disease, cardiovascular disease, and myocardial infarction, and 19p13 near LDLR was associated with prevalent coronary heart disease (webappendix p 2). In analysis of incident cases, 6q26 in LPA and 9p21 near CDKN2A–CDKN2B were associated with all three endpoints. Additionally, 6p24 in PHACTR1 was associated with incident coronary heart disease and cardiovascular disease, 12q24 in SH2B3 with cardiovascular disease and myocardial infarction, and 21q22 near SLC5A3–MRPS6–KCNE2 with coronary heart disease and myocardial infarction (table 2). Overall, seven of the 13 variants were associated in at least one analysis.
Genetic risk score was strongly associated with incident coronary heart disease, cardiovascular disease, and myocardial infarction when adjusted for age and sex (webappendix p 3) and traditional risk factors (table 3). Adjustment for traditional risk factors did not substantially change the estimates from the model adjusted for age and sex only. Participants in the top quintile of genetic risk score were estimated to have 1·66-times increased risk of coronary heart disease compared with those in the bottom quintile (95% CI 1·35–2·04, p value for linear trend across the quintiles=7·3×10−10), 1·50-times increased risk of cardiovascular disease (95% CI 1·29–1·75, p=1·9×10−10), and 1·46-times increased risk of myocardial infarction (95% CI 1·15–1·86, p=2·8×10−5).
Table 3.
Association between genetic risk score and incident coronary heart disease, cardiovascular disease, and myocardial infarction*
|
Genetic risk score quintile |
p value for trend | |||||
|---|---|---|---|---|---|---|
| 1 (reference) | 2 | 3 | 4 | 5 | ||
| HR (95% CI) for CHD (total n=25 243) | ||||||
| FR 1992 | 1·00 | 0·97 (0·65–1·45) | 1·07 (0·71–1·60) | 1·60 (1·10–2·34) | 1·54 (1·06–2·25) | 0·001 |
| FR 1997 | 1·00 | 1·02 (0·72–1·44) | 1·17 (0·84–1·62) | 1·32 (0·95–1·83) | 1·76 (1·28–2·41) | 1·1×10−5 |
| FR 2002 | 1·00 | 1·06 (0·56–1·99) | 1·18 (0·65–2·15) | 1·43 (0·79–2·58) | 1·82 (1·03–3·22) | 0·019 |
| Health 2000 | 1·00 | 0·93 (0·51–1·68) | 1·41 (0·81–2·45) | 1·13 (0·62–2·06) | 1·51 (0·87–2·62) | 0·087 |
| Pooled† | 1·00 | 1·00 (0·80–1·25) | 1·17 (0·94–1·46) | 1·39 (1·12–1·72) | 1·66 (1·35–2·04) | 7·3×10−10 |
| HR (95% CI) for CVD (total n=29 318) | ||||||
| FR 1992 | 1·00 | 1·03 (0·74–1·43) | 1·10 (0·79–1·54) | 1·35 (0·98–1·87) | 1·55 (1·14–2·12) | 0·001 |
| FR 1997 | 1·00 | 0·88 (0·66–1·18) | 1·12 (0·86–1·46) | 1·20 (0·92–1·58) | 1·54 (1·18–1·99) | 1·1×10−5 |
| FR 2002 | 1·00 | 1·18 (0·70–2·00) | 1·43 (0·87–2·36) | 1·33 (0·79–2·23) | 2·01 (1·24–3·25) | 0·004 |
| Health 2000 | 1·00 | 1·03 (0·63–1·69) | 1·31 (0·81–2·12) | 1·30 (0·80–2·12) | 1·70 (1·08–2·68) | 0·009 |
| MDC-CC | 1·00 | 0·83 (0·57–1·19) | 0·82 (0·57–1·18) | 0·75 (0·51–1·10) | 1·13 (0·80–1·58) | 0·511 |
| Pooled† | 1·00 | 0·95 (0·80–1·12) | 1·10 (0·93–1·29) | 1·16 (0·99–1·36) | 1·50 (1·29–1·75) | 1·9×10−10 |
| HR (95% CI) for MI (total n=29 318) | ||||||
| FR 1992 | 1·00 | 1·00 (0·57–1·73) | 1·00 (0·57–1·77) | 1·60 (0·95–2·69) | 1·45 (0·86–2·44) | 0·039 |
| FR 1997 | 1·00 | 1·02 (0·63–1·65) | 1·36 (0·87–2·11) | 1·32 (0·84–2·08) | 1·87 (1·21–2·87) | 0·002 |
| FR 2002 | 1·00 | 1·22 (0·50–2·95) | 0·89 (0·35–2·26) | 1·16 (0·48–2·81) | 2·05 (0·92–4·60) | 0·095 |
| Health 2000 | 1·00 | 0·89 (0·44–1·81) | 1·68 (0·89–3·17) | 1·00 (0·48–2·05) | 1·35 (0·70–2·62) | 0·320 |
| MDC-CC | 1·00 | 0·93 (0·58–1·51) | 0·78 (0·47–1·28) | 0·89 (0·55–1·46) | 1·03 (0·65–1·64) | 0·891 |
| Pooled† | 1·00 | 0·99 (0·76–1·27) | 1·11 (0·87–1·43) | 1·19 (0·93–1·53) | 1·46 (1·15–1·86) | 2·8×10−5 |
HR=hazard ratio. CHD=coronary heart disease. FR=FINRISK. CVD=cardiovascular disease. MDC-CC=Malmö Diet and Cancer Cardiovascular cohort. MI=myocardial infarction.
Association tested with Wald test with a Cox proportional hazards model adjusted for sex, LDL and HDL cholesterol, smoking, body-mass index, systolic and diastolic blood pressure, blood pressure treatment and diabetes; age was used as the timescale.
Results were combined with fixed effects meta-analysis.21
Results were broadly similar when the genetic risk score was divided into tertiles. In models adjusted for traditional risk factors, participants in the top tertile of the genetic risk score were estimated to have a 1·56-times increased risk of coronary heart disease compared with those in the bottom tertile (95% CI 1·33–1·83, p value for linear trend across tertiles=2·8×10−8), 1·40-times increased risk of cardiovascular disease (95% CI 1·24–1·58, p=1·4×10−8), and 1·34-times increased risk of myocardial infarction (95% CI 1·12–1·62, p=0·0015).
The genetic risk score conferred risk comparable to other established risk factors such as plasma LDL cholesterol (HR 2·08, 95% CI 1·57–2·76, for top vs bottom quintile of LDL cholesterol in FINRISK studies), systolic blood pressure (HR 1·66, 95% CI 1·19–2·30, for top vs bottom quintile of systolic blood pressure in FINRISK studies), or plasma C-reactive protein (HR 1·79, 95% CI 1·15–2·80, for top vs bottom quintile in FINRISK studies). Although the group means were statistically different, the distribution of each quantitative risk factor between those who went on to develop coronary heart disease and those who did not was broadly overlapping (figure).
Figure.
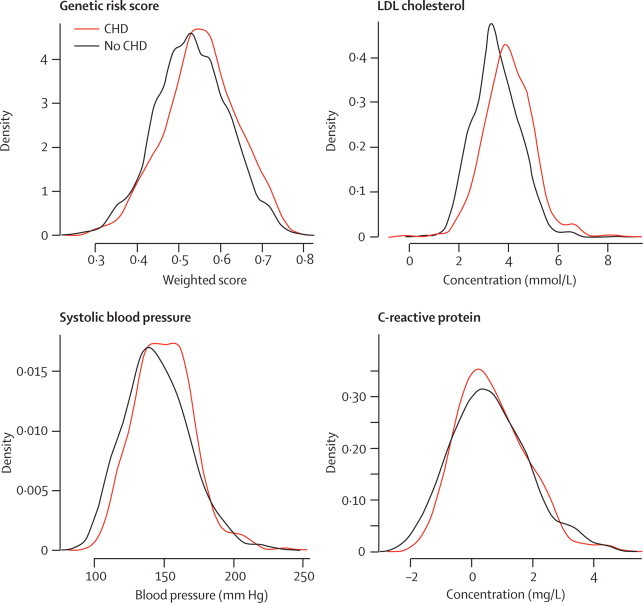
Distributions at baseline of genetic risk score, LDL cholesterol, systolic blood pressure, and log-transformed C-reactive protein by 10-year incident coronary heart disease event status in FINRISK 1992 and 1997 cohorts
Data for C-reactive protein only available in FINRISK 1997. CHD=coronary heart disease.
Table 4 shows results for prevalent events. The odds ratio for coronary heart disease between the highest and lowest quintile group was 1·63 (95% CI 1·24–2·15, p=4·8×10−5), for cardiovascular disease was 1·30 (95% CI 1·15–1·47, p=2·6×10−8), and for myocardial infarction was 1·56 (95% CI 1·38–1·76, p=1·2×10−15).
Table 4.
Association between genetic risk score and prevalent coronary heart disease, cardiovascular disease, and myocardial infarction*
|
Genetic risk score quintile |
p value for trend | |||||
|---|---|---|---|---|---|---|
| 1 (reference) | 2 | 3 | 4 | 5 | ||
| OR (95% CI) for CHD (total n=29 318) | ||||||
| FR 1992 | 1·00 | 2·30 (0·93–5·69) | 1·22 (0·43–3·40) | 2·67 (1·10–6·46) | 2·13 (0·84–5·36) | 0·117 |
| FR 1997 | 1·00 | 0·95 (0·62–1·47) | 0·86 (0·56–1·33) | 0·86 (0·55–1·35) | 1·32 (0·87–2·01) | 0·341 |
| FR 2002 | 1·00 | 1·01 (0·60–1·71) | 1·55 (0·96–2·51) | 1·32 (0·79–2·19) | 2·03 (1·26–3·25) | 0·002 |
| Health 2000 | 1·00 | 1·56 (0·76–3·22) | 1·67 (0·80–3·47) | 1·96 (0·97–3·99) | 1·58 (0·78–3·23) | 0·184 |
| Pooled† | 1·00 | 1·15 (0·86–1·53) | 1·20 (0·90–1·59) | 1·27 (0·95–1·70) | 1·63 (1·24–2·15) | 4·8×10−5 |
| OR (95% CI) for CVD (total n=47 179) | ||||||
| FR 1992 | 1·00 | 1·7 (0·76–3·78) | 0·95 (0·38–2·38) | 2·49 (1·17–5·29) | 1·69 (0·76–3·80) | 0·090 |
| FR 1997 | 1·00 | 0·89 (0·6–1·32) | 0·87 (0·59–1·28) | 0·81 (0·54–1·21) | 1·26 (0·87–1·84) | 0·396 |
| FR 2002 | 1·00 | 1·10 (0·71–1·69) | 1·36 (0·90–2·05) | 1·17 (0·76–1·81) | 1·63 (1·09–2·46) | 0·022 |
| Health 2000 | 1·00 | 0·97 (0·71–1·33) | 1·12 (0·82–1·53) | 1·04 (0·76–1·42) | 1·16 (0·86–1·57) | 0·275 |
| MDC-CC | 1·00 | 1·78 (0·89–3·54) | 2·32 (1·20–4·48) | 1·39 (0·68–2·87) | 1·58 (0·79–3·17) | 0·502 |
| MPP | 1·00 | 0·97 (0·82–1·15) | 1·11 (0·94–1·30) | 1·34 (1·14–1·57) | 1·27 (1·08–1·49) | 1·0×10−5 |
| Pooled† | 1·00 | 1·01 (0·89–1·15) | 1·13 (1·00–1·28) | 1·24 (1·09–1·40) | 1·30 (1·15–1·47) | 2·6×10−8 |
| OR (95% CI) for MI (total n=53 741) | ||||||
| FR 1992 | 1·00 | 2·83 (1·00–7·97) | 1·07 (0·30–3·73) | 2·51 (0·88–7·16) | 2·79 (0·98–7·94) | 0·120 |
| FR 1997 | 1·00 | 0·95 (0·58–1·56) | 0·76 (0·46–1·27) | 1·08 (0·67–1·76) | 1·01 (0·61–1·68) | 0·807 |
| FR 2002 | 1·00 | 0·97 (0·49–1·93) | 1·88 (1·03–3·43) | 1·52 (0·80–2·90) | 1·82 (0·98–3·38) | 0·023 |
| Health 2000 | 1·00 | 1·44 (0·54–3·85) | 1·28 (0·45–3·61) | 2·70 (1·09–6·66) | 1·58 (0·61–4·09) | 0·142 |
| MDC-CC | 1·00 | 2·35 (0·90–6·18) | 2·39 (0·92–6·22) | 1·51 (0·53–4·27) | 2·97 (1·18–7·50) | 0·092 |
| MPP | 1·00 | 1·04 (0·85–1·28) | 1·05 (0·86–1·29) | 1·38 (1·13–1·67) | 1·33 (1·10–1·62) | 1·1×10−5 |
| COROGENE | 1·00 | 1·23 (1·03–1·46) | 1·36 (1·14–1·62) | 1·40 (1·18–1·67) | 1·75 (1·48–2·08) | 8·8×10−12 |
| Pooled† | 1·00 | 1·16 (1·03–1·31) | 1·22 (1·08–1·38) | 1·40 (1·24–1·58) | 1·56 (1·38–1·76) | 1·2×10−15 |
OR=odds ratio. CHD=coronary heart disease. FR=FINRISK. CVD=cardiovascular disease. MDC-CC=Malmö Diet and Cancer Cardiovascular Cohort. MPP=Malmö Preventive Project. MI=myocardial infarction.
Association tested with Wald test with a logistic regression adjusted for age and sex; COROGENE data with matched case-control design was analysed with conditional logistic regression.
Results were combined with fixed effects meta-analysis.21
Additionally, we investigated whether adjustment for a history of early-onset myocardial infarction among first-degree relatives changed genetic risk score estimates in the FINRISK studies. Family history was significantly associated with incident events (HR for coronary heart disease 1·40, 95% CI 1·20–1·64; webappendix p 4), but the effect of the genetic risk score did not change after adjustment for family history (webappendix p 5).
We then investigated whether the genetic risk score association was dominated by rs4977574 at 9p21 near CDKN2B–CDKN2A, which is the strongest myocardial infarction locus reported to date. After adjustment for rs4977574, the HR between the highest and lowest genetic risk score quintile was 1·51 (95% CI 1·19–1·91) for coronary heart disease. Thus, other variants in the genetic risk score seem to have predictive power beyond the 9p21 locus (webappendix p 5).
The AUC estimates for coronary heart disease, cardiovascular disease, and myocardial infarction models with traditional risk factors and genetic risk score were 0·872, 0·853, and 0·881, respectively, and they were not significantly higher than the estimates from the models with only traditional risk factors (0·871, p=0·19; 0·853, p=0·48; and 0·880, p=0·35, for coronary heart disease, cardiovascular disease, and myocardial infarction, respectively).
Table 5 and table 6 show risk reclassification results for coronary heart disease. When participants were classified into four risk categories (0–5%, 5–10%, 10–20%, and >20%) on the basis of their 10-year predicted risk, 22 (13%) participants with coronary heart disease classified at 10–20% risk category in the model with traditional risk factors changed their risk category into the greater than 20% category when genetic risk score was included in the model. Similarly, 54 (13%) of the participants without incident coronary heart disease were reclassified from the greater than 20% to the 10–20% category. IDI was significant for coronary heart disease (IDI 0·004, p=0·0006), cardiovascular disease (IDI 0·004, p=0·0004), and for myocardial infarction (IDI 0·003, p=0·03). For coronary heart disease, overall NRI was not significant (NRI 2·2%, p=0·182), but there was a significant improvement in reclassification of participants at intermediate risk (clinical NRI 9·7%, p=3×10−6). The calibration of the models with (p=0·52) and without (p=0·47) genetic risk score was good.
Table 5.
Reclassification of individuals in the FINRISK 1992 and 1997 cohorts on the basis of 10-year predicted risk of coronary heart disease with and without genetic risk score
| Genetic risk score 0–5% | Genetic risk score 5–10% | Genetic risk score 10–20% | Genetic risk score >20% | |
|---|---|---|---|---|
| Cases, by predicted risk* | ||||
| 0–5% | 82 (93%) | 6 (7%) | 0 | 0 |
| 5–10% | 10 (8%) | 100 (84%) | 9 (8%) | 0 |
| 10–20% | 0 | 9 (5%) | 136 (81%) | 22 (13%) |
| >20% | 0 | 0 | 9 (7%) | 129 (93%) |
| Non-cases, by predicted risk* | ||||
| 0–5% | 9224 (99%) | 121 (1%) | 0 | 0 |
| 5–10% | 179 (12%) | 1149 (79%) | 131 (9%) | 0 |
| 10–20% | 0 | 120 (14%) | 687 (80%) | 49 (6%) |
| >20% | 0 | 0 | 54 (13%) | 351 (87%) |
10-year predicted risk on the basis of traditional risk factors only.
Table 6.
Net reclassification improvement of genetic risk score for coronary heart disease in the FINRISK 1992 and 1997 cohorts*
|
Individuals reclassified intable 5 |
NRI |
Clinical NRI† |
||||||
|---|---|---|---|---|---|---|---|---|
| Up | Down | Value | 95% CI | p value | Value | 95% CI | p value | |
| Cases | 37 | 28 | 0·018 | −0·014 to 0·051 | 0·278 | 0·041 | 0·001 to 0·081 | 0·042 |
| Non-cases | 301 | 353 | 0·004 | 0·0001 to 0·008 | 0·038 | 0·056 | 0·044 to 0·069 | 5×10−18 |
| Total | .. | .. | 0·022 | −0·010 to 0·055 | 0·182 | 0·097 | 0·056 to 0·140 | 3×10−6 |
NRI=net reclassification improvement.
Comparison of models with and without genetic risk score, adjusted for sex, LDL and HDL cholesterol, smoking, body-mass index, systolic and diastolic blood pressure, blood pressure treatment, and diabetes; age was used as the timescale.
NRI for the subset of participants in 5–20% risk category in the model without genetic risk score, with risk classes 0–5%, 5–20%, and >20%.23
Discussion
Using case-control and prospective cohort samples independent from the discovery samples, we sought to validate recently discovered genetic risk factors for coronary heart disease and to estimate the magnitude of risk conferred by these genetic risk factors in the population setting. We found that a genetic risk score including 13 SNPs associated with coronary heart disease or myocardial infarction was associated with risk of prevalent and incident coronary heart disease (even after we accounted for traditional risk factors), and that the 20% of individuals of European ancestry who carry the most risk alleles have a roughly 1·7-times increased risk of coronary heart disease when compared with those in the lowest quintile.
These findings allow us to draw several conclusions. First, the results from case-control discovery samples do seem to generalise to independent samples, including those from prospective cohorts. Second, the magnitude of effect conferred by genetic risk score (roughly 1·7-times increased risk in our study) is attenuated when compared with the discovery reports (about 2·2-times in one report9). Third, even though family history of early-onset cardiovascular disease raised the risk of cardiovascular events by 25–40%, adjustment for family history had no effect on the risk estimates because of the genetic risk score. In view of measurement error for family history, and since genetic variants only account for a small proportion of familial risk, this finding might not be unexpected.
Finally, although strongly associated with risk of incident coronary heart disease, genetic risk score did not improve risk discrimination when assessed by the C index. This finding of a biomarker being associated with incident disease but yet not improving risk discrimination has been seen with several other biomarkers, including C-reactive protein and B-type natriuretic peptide, among others.27 Since some have argued that the C index might be an insensitive measure of risk discrimination, newer approaches have been developed, including IDI, NRI, and clinical NRI. The tested genetic risk score slightly improved risk prediction for coronary heart disease and myocardial infarction when assessed by IDI and clinical NRI. Overall, these results emphasise the challenge of risk prediction for complex traits on the basis of any single factor.28
Our combined results show much larger risk differences between the tails of genetic risk score than in a recently reported follow-up study of women enrolled in a clinical trial.29 The difference cannot be accounted for by the inclusion of men in our study, since our estimates and predictions are similar for both sexes. Also, nine of the genetic loci in our genetic risk score are the same as in the cardiovascular disease score reported by Paynter and colleagues,29 and the statistical models are comparable. We suggest three potential reasons for the differing results. Paynter and co-workers included a range of endpoints such as strokes (203 of the 777 outcomes) that have not been associated with the genetic variants studied. The inclusion of such outcomes might have diminished their effect estimates. Second, Paynter and colleagues had a small number of myocardial infarction events (199 events) and thus lower statistical power might account for the absence of association in that study. Finally, the women studied were a selected set of health professional participants who volunteered for the clinical trial and as such might not represent the full range of risk seen in the general population.
Although we present the largest effort to date to study the association between genetic risk score and risk of incident cardiovascular disease, our results should be interpreted in the context of several potential limitations. Our SNP panel could be incomplete. For example, we did not systematically evaluate SNPs related to cardiovascular risk markers, as Paynter and colleagues did.29 Whereas hundreds of blood biomarkers have been shown to be associated with cardiovascular disease in observational epidemiology research, few have been proven to causally relate to the disease; plasma LDL cholesterol and lipoprotein(a) are notable exceptions. Therefore, we did not consider SNPs that are only associated with cardiovascular risk markers. Our study was undertaken in individuals of Swedish and Finnish descent and hence the results might not be generalisable to others in Europe or to other ancestries. We divided the continuous genetic risk score into five groups and compared risk between the top and bottom quintiles. Since alternative categorisations are possible we also generated comparisons of the top and bottom third of the distribution, and these results were similar to those seen with quintiles.
In conclusion, a genetic risk score based on 13 SNPs from genome-wide association studies for myocardial infarction and coronary heart disease was associated with a first coronary heart disease event, with a relative risk estimate of 1·7 between the highest and lowest quintiles of genetic risk score. Genetic risk score improved risk reclassification in participants who were at intermediate risk on the basis of traditional risk factors. Whether this genetic risk score will have clinical usefulness remains to be defined in future studies.
Acknowledgments
Acknowledgments
We wish to dedicate this paper to the memory of Leena Peltonen, who passed away on March 11, 2010. This work was supported by the Wellcome Trust (WT089062/Z/09/Z, WT089061/Z/09/Z), US National Institutes of Health (R01 HL087676), and the Donovan Family Foundation. We thank David Altshuler for guidance and helpful suggestions regarding study design and analyses. LP was supported by the Center of Excellence for Complex Disease Genetics of the Academy of Finland (grants 213506, 129680), the Biocentrum Helsinki Foundation, and The Nordic Center of Excellence in Disease Genetics. VS is supported by the Academy of Finland (grant number 129494), the Finnish Foundation for Cardiovascular Research, and the Sigrid Juselius Foundation. JS was funded by Finnish Foundation for Cardiovascular Research, Special governmental subsidy for health sciences research (EVO). MP was supported by Finnish Foundation for Cardiovascular Research and Finnish Academy Salve Program, grant number 129322
Contributors
SR, OM, VS, LP, and SK are all senior authors, and planned and managed the project. SR led the data analysis. ET, MO-M, and ASH prepared the data and did the data analyses. CG took part in genotyping. M-LL, JS, and MSN provided COROGENE data. MP and AJ provided clinical guidance and Finnish data. AS took part in data analysis for the Swedish cohorts. KS participated in study planning, did literature searches, and took part in genotyping. SR, ET, ASH, MO-M, VS, and SK wrote the report (with significant contributions from other authors).
Conflicts of interest
SK has received consultancy fees from Merck and Daiichi Sankyo and grants from Alnylam and Pfizer. All other authors declare that they have no conflicts of interest.
Web Extra Material
References
- 1.Kannel WB, Dawber TR, Kagan A, Revotskie N, Stokes J., 3rd Factors of risk in the development of coronary heart disease—six year follow-up experience. The Framingham Study. Ann Intern Med. 1961;55:33–50. doi: 10.7326/0003-4819-55-1-33. [DOI] [PubMed] [Google Scholar]
- 2.Musunuru K, Kathiresan S. Genetics of coronary artery disease. Annu Rev Genomics Hum Genet. 2010;11:91–108. doi: 10.1146/annurev-genom-082509-141637. [DOI] [PubMed] [Google Scholar]
- 3.Healy DG. Case-control studies in the genomic era: a clinician's guide. Lancet Neurol. 2006;5:701–707. doi: 10.1016/S1474-4422(06)70524-5. [DOI] [PubMed] [Google Scholar]
- 4.Vartiainen E, Laatikainen T, Peltonen M. Thirty-five-year trends in cardiovascular risk factors in Finland. Int J Epidemiol. 2009;12:1–15. doi: 10.1093/ije/dyp330. [DOI] [PubMed] [Google Scholar]
- 5.Kattainen A, Salomaa V, Härkänen T. Coronary heart disease: from a disease of middle-aged men in the late 1970s to a disease of elderly women in the 2000s. Eur Heart J. 2006;27:296–301. doi: 10.1093/eurheartj/ehi630. [DOI] [PubMed] [Google Scholar]
- 6.Heistaro S, editor. Methodology report. Health 2000 survey. National Public Health Institute; Helsinki, Finland: 2008. http://www.terveys2000.fi/doc/methodologyrep.pdf (accessed Feb 16, 2010). [Google Scholar]
- 7.Berglund G, Elmstähl S, Janzon L, Larsson SA. The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med. 1993;233:45–51. doi: 10.1111/j.1365-2796.1993.tb00647.x. [DOI] [PubMed] [Google Scholar]
- 8.Persson M, Hedblad B, Nelson JJ, Berglund G. Elevated Lp-PLA2 levels add prognostic information to the metabolic syndrome on incidence of cardiovascular events among middle-aged nondiabetic subjects. Arterioscler Thromb Vasc Biol. 2007;27:1411–1416. doi: 10.1161/ATVBAHA.107.142679. [DOI] [PubMed] [Google Scholar]
- 9.Pajunen P, Koukkunen H, Ketonen M. The validity of the Finnish Hospital Discharge Register and Causes of Death Register data on coronary heart disease. Eur J Cardiovasc Prev Rehabil. 2005;12:132–137. doi: 10.1097/00149831-200504000-00007. [DOI] [PubMed] [Google Scholar]
- 10.Tolonen H, Salomaa V, Torppa J, Sivenius J, Immonen-Räihä P, Lehtonen A. The validation of the Finnish Hospital Discharge Register and Causes of Death Register data on stroke diagnoses. Eur J Cardiovasc Prev Rehabil. 2007;14:380–385. doi: 10.1097/01.hjr.0000239466.26132.f2. [DOI] [PubMed] [Google Scholar]
- 11.Kathiresan S, Melander O, Anevski D. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008;358:1240–1249. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- 12.Langholz B, Goldstein L. Risk set sampling in epidemiologic cohort studies. Stat Sci. 1996;11:35–53. [Google Scholar]
- 13.Helgadottir A, Thorleifsson G, Manolescu A. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 14.Samani N, Erdmann J, Hall S. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:1–11. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Myocardial Infarction Genetics Consortium Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erdmann J, Grobhennig A, Braund P. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41:280–282. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gudbjartsson D, Bjornsdottir U, Halapi E. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet. 2009;41:342–347. doi: 10.1038/ng.323. [DOI] [PubMed] [Google Scholar]
- 18.Clarke R, Peden J, Hopewell J. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 19.McPherson R, Pertsemlidis A, Kavaslar N. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoenfeld D. Residuals for the proportional hazards regression model. Biometrika. 1982;69:239–241. [Google Scholar]
- 21.Hedges L, Olkin I. Statistical methods for meta-analysis. Academic Press; London: 1985. [Google Scholar]
- 22.Heagerty PJ, Lumley T, Pepe MS. Time dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56:337–344. doi: 10.1111/j.0006-341x.2000.00337.x. [DOI] [PubMed] [Google Scholar]
- 23.Antolini L, Nam BH, D'Agostico RB. Inference on correlated discrimination measures in survival analysis: A nonparametric approach. Commun Stat Theory Methods. 2004;33:2117–2135. [Google Scholar]
- 24.Cook NR. Comments on “Evaluating the added predictive ability of a new marker: From area under the ROC curve to reclassification and beyond” by M. J. Pencina et al., Statistics in Medicine (DOI:10.1002/sim.2929) Stat Med. 2008;27:191–195. doi: 10.1002/sim.2987. [DOI] [PubMed] [Google Scholar]
- 25.Steyerberg EW, Pencina MJ. Reclassification calculations for persons with incomplete follow-up. Ann Intern Med. 2010;152:195–196. doi: 10.7326/0003-4819-152-3-201002020-00019. [DOI] [PubMed] [Google Scholar]
- 26.Pencina M, D'Agostino RS, D'Agostino RJ, Vasan R. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172. doi: 10.1002/sim.2929. [DOI] [PubMed] [Google Scholar]
- 27.Melander O, Newton-Cheh C, Almgren P. Novel and conventional biomarkers for prediction of incident cardiovascular events in the community. JAMA. 2009;302:49–57. doi: 10.1001/jama.2009.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ware J. The limitations of risk factors as prognostic tools. N Engl J Med. 2006;355:2615–2617. doi: 10.1056/NEJMp068249. [DOI] [PubMed] [Google Scholar]
- 29.Paynter NP, Chasman DI, Paré G. Association between a literature-based genetic risk score and cardiovascular events in women. JAMA. 2010;303:631–637. doi: 10.1001/jama.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.