

Abstract
Introduction
The genetic etiology of amyotrophic lateral sclerosis (ALS) is not well understood. Finland is a well-suited location for a genome-wide association study of ALS, as the incidence of the disease is one of the highest in the world, and because the genetic homogeneity of the Finnish population enhances the ability to detect risk loci.
Methods
We performed a genome-wide association study of 442 Finnish patients diagnosed with ALS, and 521 Finnish control subjects using Illumina genome-wide genotyping arrays. DNA was collected from patients attending an ALS specialty clinic that receives referrals from neurologists throughout Finland, whereas the control samples were obtained from a population-based study of elderly Finnish individuals. Individuals known to carry D90A alleles of the SOD1 gene (n = 40) were included in the final analysis as positive controls to determine if our GWAS was able to detect an association signal at this locus.
Findings
We identified two association peaks that exceeded genome-wide significance. One of these was located on chromosome 21q22 (rs13048019, p = 2·58×10−8) that corresponded to the known autosomal recessive D90A allele of the SOD1 gene. The other was detected in a 232kb block of linkage disequilibrium (rs3849942, p = 9·11×10−11) in a region of chromosome 9p that has been previously identified by linkage studies of ALS families. Within this region, we defined a 42-SNP haplotype that significantly increased risk of developing ALS (p = 4·2×10−33 among familial cases, odds ratio = 21·0, 95% CI = 11·2–39·1), and which overlapped with an association locus recently reported for fronto-temporal dementia (FTD). Based on the 93 familial ALS cases included in the analysis, population attributable risk percent for the chromosome 9p21 locus was 37.9% (95% CI, 27·7 – 48·1%), and for D90A homozygosity was 25·5% (95% CI, 16·9 – 34·1%).
Interpretation
In summary, we present evidence that the chromosome 9p21 ALS-FTD locus is a major cause of familial ALS in the Finnish population.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterised by progressive paralysis and death from respiratory failure, typically within three years of symptom onset.1 The etiology of the disease is not well understood, but genetic factors are thought to play an important role in its pathogenesis. To date, genome-wide association studies (GWAS) have failed to identify a single locus that clearly achieves significance after Bonferroni correction for multiple testing, and that successfully replicates in independent cohorts.2–9 The lack of success of GWAS in ALS most likely stems from the genetic and allelic heterogeneity associated with the disease.2 One approach to increase power to find a genetic locus in the face of such heterogeneity is to target isolated populations, where the genetic background is more homogeneous.10
Finland is an ideal population for genetic studies of ALS for several reasons. First, the incidence of ALS in Finland is the highest in the world with the disease occurring nearly twice as frequently compared to other European ancestry populations.11,12 Second, the small founder populations of Finland, together with the multiple population bottleneck events that have occurred during its history, have resulted in a remarkably genetically homogeneous population.13,14 This homogeneity greatly increases the power of GWAS to find genes, as it results in less polymorphisms (less allelic heterogeneity), fewer disease loci (less locus heterogeneity), and extended regions of high linkage disequilibrium among Finns.10 For example, the D90A allele of the SOD1 gene is known to occur with increased frequency in the Scandinavian population, and accounts for a portion, but not all, of the excess incidence of ALS observed in Finland.15
Here, we undertook a GWAS of 442 Finnish patients diagnosed with ALS and 521 Finnish controls using Human370 and Human1M SNP chips (Illumina, San Diego, CA). This GWAS was designed to identify genetic factors that increase risk of developing ALS in the Finnish population, and was initiated without an a priori hypothesis as to where these loci might exist in the genome. Strong association signals were detected on chromosome 21q22 corresponding to the known D90A allele of the SOD1 gene, and on chromosome 9p21.2 in a locus previously linked to autosomal dominant ALS.9,16–21 Together, these loci account for a large proportion of the increased ALS incidence observed in the Finnish population.
Subjects and Methods
Samples
Demographics and clinical features of the case and control cohorts are shown in table 1. DNA was collected from patients attending an ALS specialty clinic that receives referrals from neurologists throughout Finland since 1994. All patients included in the study had been diagnosed with ALS according to the El Escorial criteria22 by a neurologist specializing in ALS (HL). Both familial and sporadic ALS cases were included in the analysis. Individuals known to carry D90A alleles of the SOD1 gene (n = 40) were included in the final analysis as positive controls to determine if our GWAS was able to detect an association signal at this locus. Personnel performing the genotyping were blinded to familial and sporadic status, and to D90A allele status prior to analysis. The TDP-43, FUS, SETX, and OPTN genes were not sequenced for mutations in either the case or control cohorts. Control samples were obtained from a population-based study of elderly Finnish individuals that were collected as part of a separate project.23 Thus, the control cohort used in our GWAS represents convenience samples that were matched for ethnicity, but were not age- or gender-matched to the cases. Written informed consent for genetic analysis was obtained from each individual. This study has been approved by the Ethics Committees for Ophthalmology, Otorhinolaryngology, Neurology and Neurosurgery (Decision 68/2002) and Internal Medicine (Dnro 401/13/03/01/09) in the Hospital District of Helsinki and Uusimaa, and by the Institutional Review Board of the National Institute on Aging (protocol number 2008-146).
Table 1.
Demographics and clinical details of 405 Finnish ALS cases and 497 Finnish controls included in genome-wide association analysis*
| Controls (n = 497) |
Cases (n = 405) |
Cases with homozygous D90A SOD1 alleles (n = 40) |
Cases with chromosome 9p21 risk haplotype (n = 102) |
Cases without homozygous D90A SOD1 allele or 9p21 risk haplotype (n = 266)† |
|
|---|---|---|---|---|---|
| Mean age (range)‡ | 88·6 (85·0 – 104·0) | 56·8 (23·0 – 85·0) | 47·9 (23 – 79) | 54·2 (30 – 77) | 59·0 (32 – 85) |
| Male (%) | 104 (20·9%) | 199 (49·1%) | 17 (42·5%) | 49 (48·0%) | 134 (50·4%) |
| Female (%) | 393 (79·1%) | 206 (50·9%) | 23 (57·5%) | 53 (52·0%) | 132 (49·6%) |
| Site of symptom onset:# | |||||
| Bulbar (%) | Not applicable | 75 (23·9%) | 0 | 21 (26·6%) | 54 (26·3%) |
| Spinal (%) | Not applicable | 220 (70·1%) | 32 (100%) | 57 (71·4%) | 134 (65·4%) |
| Generalised (%) | Not applicable | 15 (4·8%) | 0 | 2 (2·0%) | 13 (6·3%) |
| Respiratory (%) | Not applicable | 4 (1·3%) | 0 | 0 | 4 (2·0%) |
| Cognitive impairment (%) | Not applicable | 14 (3·5%) | 0 | 7 (6·9%) | 7 (2·6%) |
| Family history of ALS (%) | Not applicable | 93 (23·0%) | 27 (67·5%) | 44 (43·1%) | 25 (9·4%) |
The genotype status of the 40 cases that were homozygous for the D90A allele was known at the start of the current study, whereas the 102 cases carrying the chromosome 9p21 risk haplotype were identified as a consequence of the GWAS reported here
3 cases carrying the chromosome 9p21 risk haplotype were also homozygous for the D90A SOD1 allele
age was not available for 22 cases
site of symptom onset data were not available for 91 cases
Genotyping
853 samples were genotyped using Infinium Human370 BeadChips, which assay 345,111 SNPs across the genome, whereas 110 samples were assayed using Infinium Human1M BeadChips, which assay 1,154,691 SNPs. Initial analyses were confined to the 329,355 autosomal SNPs that were common across platforms. Bonferroni correction for multiple testing yielded a threshold p-value of 1·52×10−7 for genome-wide significance (α = 0·05/329,355 autosomal SNPs).
Statistical analysis
Standard quality control procedures (i.e. exclusion of samples with low call rates, non-European ancestry, and cryptic relatedness defined as identity-by-descent proportion of inheritance (pi_hat, from PLINK) > 0·1; and exclusion of SNPs with low call rates, minor allele frequency (MAF) < 0·01 in controls, and Hardy-Weinberg equilibrium p < 0·001 in controls) were applied to the data (see webappendix p 1 – 6 for detailed description). The cryptic relatedness threshold (i.e. pi_hat) led to the exclusion of individuals sharing more than 10% of their genome, meaning that related individuals down to 3rd or 4th degree relatives were not included in the final analysis. Association testing was performed within the PLINK software toolset (version 1.06).24 P-values are based on the Cochrane-Armitage trend test, unless otherwise stated, adjusted for genomic inflation. Odds ratios (OR) and upper and lower bound 95% confidence intervals (CI) were computed for each SNP’s minor allele. Assuming a MAF of 0·15 and an adjusted level of significance of ~2·0×10−7, the study had roughly 90% power to detect an associated SNP with odds ratios of around 2 under the additive model. Haplotypes were viewed and analyzed using Haploview 4.2, which uses an accelerated EM algorithm to estimate phased haplotypes based on the maximum likelihood determined from the unphased input.25 QQ plots suggested no significant population stratification in our study cohort (webappendix p 7, genomic inflation factor = 1·093). The most significantly associated SNPs identified in the genome-wide scan are listed in webappendix p 8. Population attributable risk percent (PAR%) was calculated using the formula: PAR% = p(r-1)/p(r-1)+1, where p is the proportion of the population exposed to the risk, and r is the relative risk.26
Sequence analysis
Sequencing was performed using the Big-Dye Terminator v3.1 sequencing kit (Applied Biosystems Inc., Foster City, CA, USA), run on an ABI 3730xl genetic analyzer, and analyzed using Sequencher software version 4.2 (Gene Codes Corp., Ann Arbor, MI, USA).
Public availability of genotype data
Individual level genotype data have been made available for the Finnish ALS cohort on the dbGAP web portal.
Role of the funding source
Study sponsors had no role in study design, in the collection, analysis and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication.
Results
After quality control filters were applied, 318,167 SNPs in 405 cases and 497 controls were available for genome-wide association analysis. This identified two peaks that exceeded the Bonferroni threshold for significance (table 2 and figure 1). One was located on chromosome 21q22 (rs13048019, p = 2·58×10−8, OR for recessive carrier status = 4·14, 95% CI = 2·32 – 7·40). This signal corresponded to the known autosomal recessive D90A mutation of the SOD1 gene. The other signal was detected in a previously identified autosomal dominant ALS locus on chromosome 9p21.2 (rs3849942, p = 9·11×10−11, OR = 2·16, 95% CI = 1·72 – 2·70).
Table 2.
SNPs that achieve genome-wide significance (p-value less than 1·52×10−7) based on Cochrane-Armitage trend test of 405 Finnish ALS cases and 497 Finnish controls.
| Rank | SNP | Chr | Position | Minor allele |
Major allele |
Risk allele |
MAF Cases |
MAF Controls |
MAF CEU |
Odds Ratio (95% CI) |
p |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | rs3849942 | 9 | 27,533,281 | A | G | A | 0·295 | 0·163 | 0·183 | 2·16 (1·72 – 2·70) | 9·11×10−11 |
| 2 | rs2814707 | 9 | 27,526,397 | A | G | A | 0·295 | 0·163 | 0·192 | 2·14 (1·71 – 2·69) | 1·18×10−10 |
| 3 | rs774359 | 9 | 27,551,049 | C | T | C | 0·298 | 0·177 | 0·225 | 1·98 (1·58 – 2·47) | 3·64×10−9 |
| 4 | rs13048019 | 21 | 31,840,165 | T | C | T | 0·291 | 0·169 | 0·150 | 2·02 (1·61 – 2·53) | 2·58×10−8 |
| 5 | rs2225389 | 9 | 27,399,264 | C | A | C | 0·180 | 0·089 | 0·100 | 2·26 (1·70 – 3·00) | 2·88×10−8 |
Chr, chromosome; SNP position is based on the Human Genome Build 36·3; MAF, minor allele frequency; MAF CEU is the frequency of the minor allele of this SNP in the HapMap Caucasian reference population consisting of 60 unrelated individuals (release 23, see www.hapmap.org for further details); 95% CI, 95% confidence interval.
Figure 1.
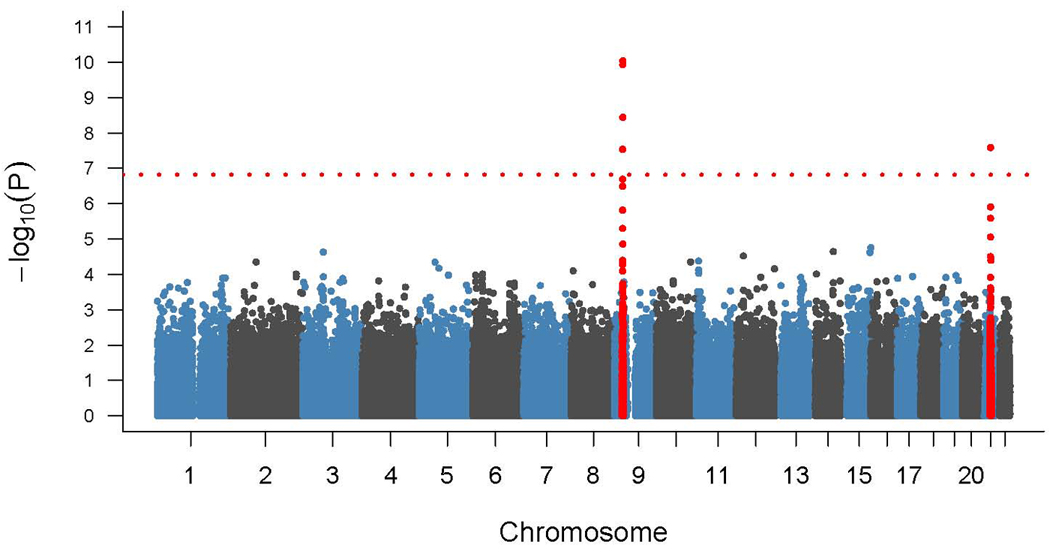
Graphical representation of p-values from genome-wide association study of 405 Finnish ALS cases and 497 elderly Finnish controls. P-values are log transformed (y-axis) and plotted against chromosomes (x-axis). The red dotted line indicates the Bonferroni threshold. Signals indicated in red are on chromosome 9p21 and chromosome 21q22 and surpass the Bonferroni threshold for genome-wide significance.
Both the chromosome 21q22 and 9p21 associations were largely driven by the 93 previously known familial cases included in the analysis, and the association signals increased when analysis was restricted to the familial group. The p-value for rs13048019 on chromosome 21 was 7·0×10−15 (figure 2A). The p-value for rs3849942 on chromosome 9p21 was 2·85×10−11, whereas the peak on 9p21 was obtained with rs2225389 (p = 2·23×10−12). Both signals were not genome-wide significant in patients without a known family history (p-value for rs13048019 on 21q22 = 6·05×10−4 and for rs3849942 on 9p21 = 3·94×10−7, figure 2B), though rs3849942 remained the most significant signal observed in the sporadic cases.
Figure 2.
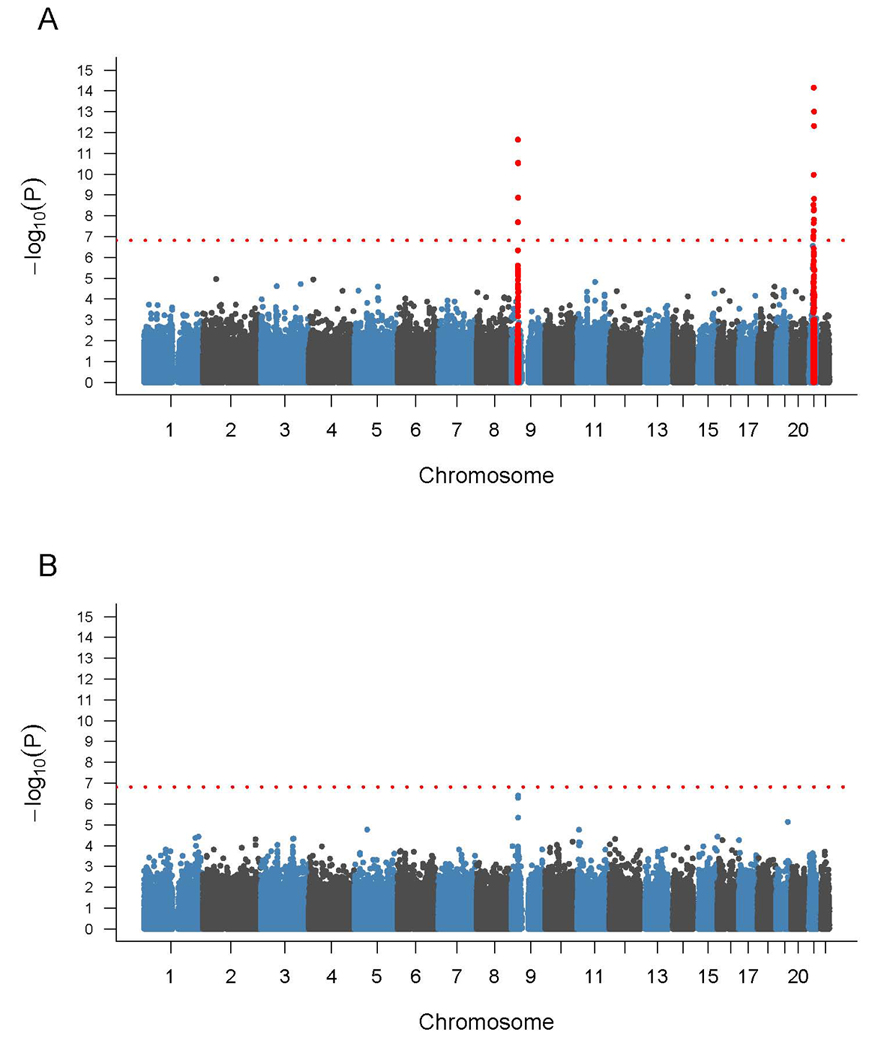
Graphical representation of p-values from genome-wide association study of (A) 93 familial ALS cases and 497 Finnish controls, and (B) 312 sporadic ALS cases and 497 Finnish controls. P-values are log transformed (y-axis) and plotted against chromosomes (x-axis). The red dotted line indicates the Bonferroni threshold. Signals indicated in red are on chromosome 9p21 and chromosome 21q22 and surpass the Bonferroni threshold for genome-wide significance.
Mutational screening confirmed that the signal on chromosome 21q22 was due to 27 familial cases and 13 apparently sporadic cases that were homozygous carriers of the SOD1 D90A allele. The p-value for association of D90A allele status with ALS in the total cohort of 405 cases and 497 controls was 1·60×10−13 based on the recessive model. The signal on chromosome 9p21 was being driven by 44 of the 66 (66·7%) familial cases without D90A homozygosity, and 58 of the 312 (18·6%) apparently sporadic cases. Analysis of these cases using Haploview software showed that they all shared a 42-SNP haplotype across the locus. This haplotype was also found in 18 (3·6%) of 497 controls (p-value for haplotype association among familial cases = 7·47×10−33, haplotype is listed in webappendix p 11). The odds ratio among familial ALS cases associated with carrier status of this risk haplotype was 21·0 (95% CI, 11·2 – 39·1). Furthermore, comparison of the 42-SNP haplotype identified in the Finnish ALS population with the risk haplotype in the same region of chromosome 9 reported in a recent GWAS of fronto-temporal dementia (FTD) indicated at least partial overlap between these two haplotypes involving the same three most associated SNPs in the Finnish ALS cohort, namely rs3849942, rs2814707, and rs774359 (webappendix p 11).27 In contrast, an association signal was not observed within the Finnish ALS dataset for the chromosome 7p21.3 locus, which was the most associated region observed in the FTD GWAS published by van Deerlin et al (minimum p-value under the additive model = 0·021).27
The chromosome 9p21 signal was located in a 232Kb genomic region that harbors four genes: MOBKL2b, LOC100288294, IFNK and C9orf72 (figure 3). Mutational screening of these genes did not reveal any amino acid changing mutations that were present in cases, but not in controls (webappendix p 12).
Figure 3.
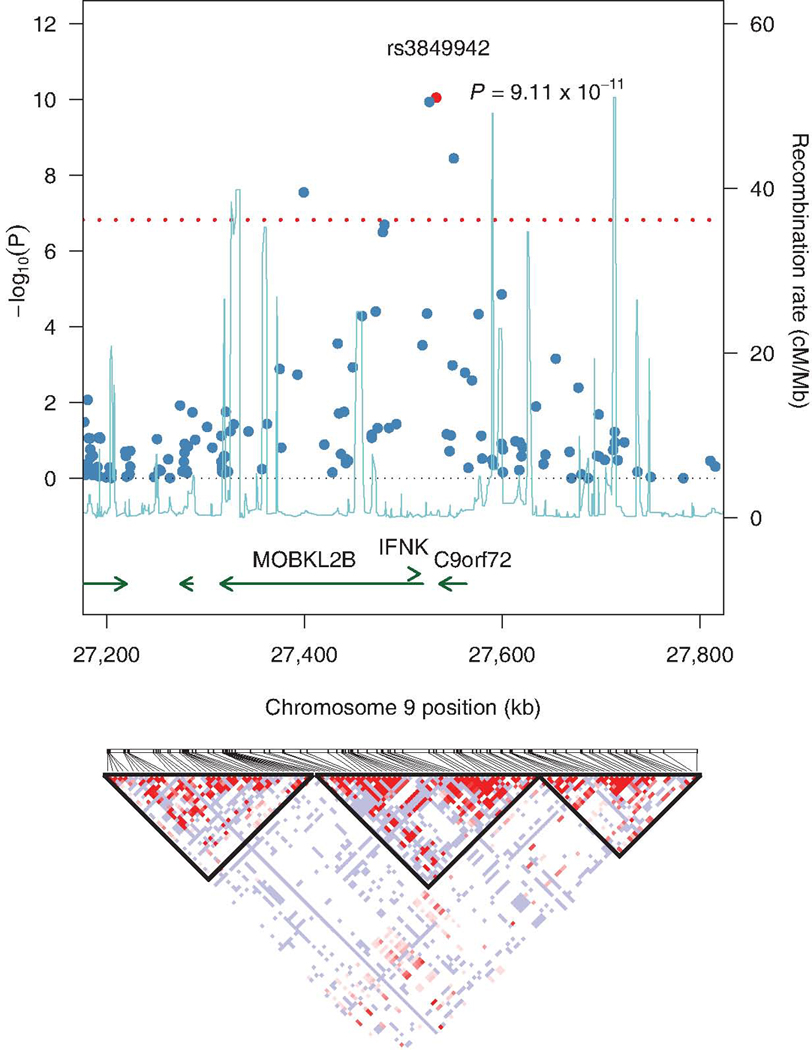
Graphical representation of the association results, recombination rates and linkage disequilibrium (LD) across the chromosome 9p21 locus in the Finnish cohort. −log10 p-values are shown for genotyped SNPs (blue). Recombination rates from phase 2 CEPH samples of HapMap are represented by the blue line. Annotated transcripts are shown across the bottom of the plot. Red dotted line indicates the threshold for genome-wide significance. LD structure in the region is shown on the bottom and was generated using data from the Finnish ALS cases carrying the 42-SNP risk haplotype.
Based on our referral-clinic case cohort, the population attributable risk percent (PAR%) for D90A homozygosity among the familial cohort was 25·5% (95% CI, 16·9 – 34·1%, calculated based on 27 of 93 Finnish familial ALS cases and 1 of 497 controls that were homozygous D90A carriers). The chromosome 9p21 locus accounted for a higher proportion of familial cases than the known D90A SOD1 allele (PAR% = 37·9%, 95% CI, 27·7 – 48·1%, based on 41 of 93 Finnish familial ALS cases and 18 of 497 controls that carried the 9p21 risk haplotype and were not homozygous D90A carriers). Together, D90A homozygosity and the chromosome 9p21 locus accounted for nearly one fifth of all cases (PAR% = 19·6%, 95% CI, 15·8 – 23·4%), and nearly two thirds of the familial cases included in our study (64·9%, 95% CI, 54·6 – 75·2%, table 3).
Table 3.
Population attributable risk percent and odds ratios associated with the chromosome 9p21 risk haplotype carrier status and with homozygous D90A SOD1 allele status among the Finnish population.*
| Familial ALS samples (n = 93) |
Sporadic ALS samples (n = 312) |
All ALS samples (n = 405) |
||||
|---|---|---|---|---|---|---|
| Locus | PAR% (95% CI) | OR (95% CI) | PAR% (95% CI) | OR (95% CI) | PAR% (95% CI) | OR (95% CI) |
| 9p21 risk haplotype | 37·9% (27·7–48·1%) | 21·0 (11·2–39·1) | 9·9% (6·6–13·2%) | 5·9 (3·4–10·3) | 13·0% (9·9–16·1) | 9·0 (5·0–14·3) |
| Homozygous D90A | 25·5% (16·9–34·1%) | 202·9 (27–1518) | 2·8% (1·0–3·9%) | 21·6 (2·8–166) | 5·6% (3·8–7·4%) | 54·4 (7·4–397) |
| Both | 64·9% (54·6–75·2%) | 58·4 (30·9–110·4) | 12·8% (9·2–16·5%) | 7·3 (4·3–12·4) | 19·6% (15·8–23·4%) | 12·6 (7·6–20·8) |
Calculations were based on 27 familial ALS cases and 13 sporadic ALS cases that were homozygous carriers of the SOD1 D90A allele, and on 41 familial ALS cases and 58 sporadic ALS cases that carried the chromosome 9p21 risk haplotype and were not homozygous D90A carriers. PAR% = population attributable risk percent; OR = odds ratio; 95% CI = 95% confidence interval; 9p21 risk haplotype refers to carrier status for the 42-SNP risk haplotype identified on chromosome 9p21; Homozygous D90A refers to homozygous carrier status for the D90A allele of the SOD1 gene on chromosome 21q22; “Both” refers to the combined effects of the 9p21 risk haplotype and homozygous D90A allele carrier status.
We compared the results of the previous published studies implicating the chromosome 9p region in the pathogenesis of ALS and related neurodegenerative diseases with the results from our current study (see webappendix p 13). Data were available from five linkage studies16–21, two GWAS of ALS patients9,28, and one GWAS of FTD patients.27 The minimum overlap genomic distance defined by the linkage studies was 3.58Mb extending from 27.23Mb to 30.81Mb on the short arm of chromosome 9. In comparison, our study identified the same block of linkage disequilibrium as was reported by the other association studies.9,27,28
Discussion
Our genome-wide association study of Finnish ALS patients and controls identified loci on chromosomes 9p21 and 21q22 that represent the most significant association signals identified in a GWAS of ALS to date.2–9 The homozygous D90A allele of the SOD1 gene is a well-known cause of ALS within the Scandinavian population15, and family-based linkage studies have previously identified the chromosome 9p13.2–21.3 region as being relevant to the pathogenesis of ALS.16–21 An earlier GWAS study reported by van Es et al gave suggestive evidence of association in the same chromosomal region, though the p-value in that study did not achieve genome-wide significance (uncorrected p for rs3849942 = 1·58×10−6 compared to a threshold of 1·7×10−7 for genome-wide significance based on 293,768 SNPs analyzed in that study).9 Published contemporaneously with our study, a GWAS of 599 UK ALS cases and 4,144 controls also replicated the association signal on chromosome 9p21 (rs903603, p-value = 8.9×10−8).28 Joint analysis involving these UK samples and an additional 3,713 cases and 3,986 controls that included US, Italian and Irish samples genotyped at our laboratory2–4 yielded a p-value of 6.64×10−10 for rs3849942.28 While this cannot be considered to be an independent replication because of the sample overlap with the van Es et al GWAS9, these data support the notion that the 9p21 locus is important across multiple populations.
Our data highlight the utility of performing GWA studies in population isolates with a high incidence of a particular disease. Compared to outbred European ancestry cohorts, the genetic homogeneity of the Finnish population increased our power to detect associated loci by decreasing both genetic variability (i.e. the number of genes in a population that cause disease), and allelic heterogeneity (i.e. the number of mutant alleles within a causative gene).10 Indeed, of the 36 monogenic diseases that are highly enriched in the Finnish population (known as the Finnish Disease Heritage), a single founder mutation accounts for 70–100% of these disease alleles.13,14,29
A comparison of epidemiological studies of ALS found the incidence of the disease in Finland to be 8·2 per 100,000 of the population among the 50 to 79 age-group, representing the highest occurrence of motor neuron degeneration outside of the Pacific Rim.11,12 Our study demonstrates that a sizeable proportion of this excess occurrence stems from an overrepresentation of the chromosome 9p21 risk haplotype and homozygous D90A SOD1 mutations within the Finnish population. In particular, the chromosome 9p21 risk haplotype was found in 44 out of 93 familial cases (47·3%), representing a larger proportion than was explained by the known D90A SOD1 allele (27 out of 93 cases, 29·0%). This scenario of a small number of loci underlying a disease arises from the unique history of Finland with a small founder population limited to several thousand individuals, and multiple population bottlenecks over the last few millennia, where a significant percentage of a population is killed or prevented from reproducing.13,14 Although homozygous D90A SOD1 allele is relatively uncommon outside of Scandinavia and Russia, a recent study has shown that the chromosome 9p21 locus accounts for a sizeable proportion of French ALS cases.20 However, because referral bias is known to occur in clinic-based cohorts of ALS patients30, additional longitudinal, population-based genetic studies are required to determine if our results are generalizable to the entire Finnish ALS population. Furthermore, the lack of age- and gender-matching between the cases and controls included in this study may have lead to overestimation of the contribution of the identified loci in the pathogenesis of ALS in Finland, though this impact was likely mitigated by the close matching of genetic ancestry (see webappendix p 2).31
Our data dramatically narrow the region of interest within the chromosome 9p21 ALS locus from the known 3.58Mb minimum overlap region based on published linkage studies down to a 232Kb block of linkage disequilibrium (LD). Within this LD block, we identified a 42-SNP haplotype that was significantly associated with ALS, and which serves as a marker for the underlying founder mutation. The chromosome 9p21 ALS locus most likely represents a monogenic form of disease, as the association signal was strongest in an analysis restricted to familial ALS cases, and the original identification of the region was based on linkage studies of ALS families.16–21 The high odds ratio associated with carrying the 42-SNP risk haplotype is also consistent with monogenic, high penetrance inheritance. Although this 232Kb LD block encodes only four genes, mutation screening of these coding regions in our Finnish ALS cases failed to identify a pathogenic amino acid altering variant. This is consistent with reports from other groups who have similarly failed to identify a coding variant in the larger region, and suggests that the genetic factor underlying chromosome 9p21-related ALS may lie in an intronic or intergenic region, and may give rise to disease either by abnormally altering splicing, or by influencing gene expression.
A recently published GWAS of patients diagnosed with FTD detected a tentative association signal in the same LD block on chromosome 9p21 as found in our study of ALS patients.27 Furthermore, the partial risk haplotype reported in the FTD study was identical to that identified in our cohort of Finnish ALS patients.27 This observation is consistent with growing clinical, epidemiological, and genetic evidence indicating that ALS and FTD form a spectrum of disease, characterized neuropathologically by TDP-43 staining ubiquitin neuronal inclusions.32 In our study, ~4% of the patients linked to the chromosome 9p21 locus presenting with motor neuron dysfunction ultimately developed cognitive changes consistent with frontal lobe degeneration. Taken together, these data strongly suggest that the chromosome 9p21 locus represents a single founder mutation that underlies any combination of motor neuron and frontal cortex degeneration (i.e. pure ALS, pure FTD, ALS-FTD, and FTD with motor neuron disease). It is noteworthy that the prevalence rate of FTD in Scandinavia is among the highest in the world33, suggesting an underlying genetic predisposition within this geographical region. Further studies will be required to confirm that the increased occurrence of FTD in Finland is also due to the chromosome 9p21 mutation.
In summary, our genome-wide study of the genetically homogeneous Finnish population identified the strongest associations signals of any ALS GWAS reported to date. Together, the chromosome 9p21 locus and the D90A allele of the SOD1 gene account for a large proportion of the excess incidence of ALS cases in Finland, and our observations further demonstrate the utility of performing GWAS in isolated populations. Furthermore, our data define a 42-SNP haplotype on chromosome 9p21 that significantly increased the risk of developing ALS, and which was identical to the risk haplotype recently tentatively suggested in FTD cases. The high frequency of this risk haplotype in the Finnish ALS population, as well as its identification in a recent FTD GWAS, suggests that chromosome 9p21-related disease arose from a single founder mutation with the highest frequency in Northern Europe, and which has subsequently disseminated throughout other European populations. Despite the inability to identify the specific genetic variant underlying this locus, our data make it feasible to identify chromosome 9p21-linked ALS cases by sequencing samples for this risk haplotype.20 Our future studies will continue to exploit the unique genetic structure of the Finnish population to understand the specific genetic factor underlying the chromosome 9p21 locus and to identify additional loci important in the pathogenesis of this fatal neurodegenerative disorder.
Supplementary Material
Acknowledgements
This work was supported in part by the Intramural Research Program of the NIH, and the National Institute on Aging (Z01-AG000949-02). The work was also funded by Microsoft Research, the ALS Association, the Helsinki University Central Hospital, the Finnish Academy, the Finnish Medical Society Duodecim, and Kuopio University. We thank the DNA extraction and storage facility of the National Institute for Health and Welfare/FIMM, Helsinki, Finland, and Dr Tuomo Polvikoski, Institute for Ageing and Health, Campus for Ageing and Vitality, Newcastle University, Newcastle upon Tyne, UK for their help in DNA extraction of the patients diagnosed with ALS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributors
HL, TP, and JCS all contributed equally to this study. The authors wish it to be known that PJT and BJT should be considered as joint last authors of this study. HL was responsible for the collection and characterisation of patients, was involved in the design of the study, and participated in critical revision of the manuscript. TP, JCS, SWS, S-LL, LJ, and DGH participated in the laboratory-based experiments and data analysis, and in critical revision of the manuscript. LM and RS were responsible for the collection and characterisation of the control subjects, were involved in the design of the study, and participated in critical revision of the manuscript. JRG and MAN did the data handling and analysis, and participated in critical revision of the manuscript. DH and PJT were involved in the design of the study, and undertook critical revision of the manuscript. BJT participated in the laboratory-based experiments and data analysis, drafted the manuscript, and designed and supervised the study. All authors had full access to all of the data in the study. All authors have seen and approved the final version of this manuscript and held final responsibility for the decision to submit for publication.
Conflicts of interest
David Heckerman is Senior Director of the eScience Research Group at Microsoft Research. Hannu Laaksovirta has received payment from Sanofi-Aventis and Rhone-Poulenc Rorer for development of educational presentations including services on speakers’ bureaus, as well as travel and accommodation expenses from Rhone-Poulenc-Rorer. None of the other authors have any conflicts of interest.
References
- 1.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 2.Chiò A, Schymick JC, Restagno G, et al. A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18:1524–1532. doi: 10.1093/hmg/ddp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schymick JC, Scholz SW, Fung HC, et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2007;6:322–328. doi: 10.1016/S1474-4422(07)70037-6. [DOI] [PubMed] [Google Scholar]
- 4.Cronin S, Berger S, Ding J, et al. A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum Mol Genet. 2008;17:768–774. doi: 10.1093/hmg/ddm361. [DOI] [PubMed] [Google Scholar]
- 5.Dunckley T, Huentelman MJ, Craig DW, et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N Engl J Med. 2007;357:775–788. doi: 10.1056/NEJMoa070174. [DOI] [PubMed] [Google Scholar]
- 6.van Es MA, Van Vught PW, Blauw HM, et al. ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol. 2007;6:869–877. doi: 10.1016/S1474-4422(07)70222-3. [DOI] [PubMed] [Google Scholar]
- 7.van Es MA, van Vught PW, Blauw HM, et al. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat Genet. 2008;40:29–31. doi: 10.1038/ng.2007.52. [DOI] [PubMed] [Google Scholar]
- 8.Landers JE, Melki J, Meininger V, et al. Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2009;106:9004–9009. doi: 10.1073/pnas.0812937106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Es MA, Veldink JH, Saris CG, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;40:29–31. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- 10.Shifman S, Darvasi A. The value of isolated populations. Nat Genet. 2001;28:309–310. doi: 10.1038/91060. [DOI] [PubMed] [Google Scholar]
- 11.Cronin S, Hardiman O, Traynor BJ. Ethnic variation in the incidence of ALS: a systematic review. Neurology. 2007;68:1002–1007. doi: 10.1212/01.wnl.0000258551.96893.6f. [DOI] [PubMed] [Google Scholar]
- 12.Murros K, Fogelholm R. Amyotrophic lateral sclerosis in Middle-Finland: an epidemiological study. Acta Neurol Scand. 1983;67:41–47. doi: 10.1111/j.1600-0404.1983.tb04543.x. [DOI] [PubMed] [Google Scholar]
- 13.Kere J. Human population genetics: lessons from Finland. Annu Rev Genomics Hum Genet. 2001;2:103–128. doi: 10.1146/annurev.genom.2.1.103. [DOI] [PubMed] [Google Scholar]
- 14.Peltonen L, Jalanko A, Varilo T. Molecular genetics of the Finnish disease heritage. Hum Mol Genet. 1999;8:1913–1923. doi: 10.1093/hmg/8.10.1913. [DOI] [PubMed] [Google Scholar]
- 15.Andersen PM, Nilsson P, Ala-Hurula V, et al. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat Genet. 1995;10:61–66. doi: 10.1038/ng0595-61. [DOI] [PubMed] [Google Scholar]
- 16.Luty AA, Kwok JB, Thompson EM, et al. Pedigree with frontotemporal lobar degeneration--motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol. 2008;8:32. doi: 10.1186/1471-2377-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morita M, Al-Chalabi A, Andersen PM, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66:839–844. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- 18.Valdmanis PN, Dupre N, Bouchard JP, et al. Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol. 2007;64:240–245. doi: 10.1001/archneur.64.2.240. [DOI] [PubMed] [Google Scholar]
- 19.Vance C, Al-Chalabi A, Ruddy D, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain. 2006;129:868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 20.Le Ber I, Camuzat A, Berger E, et al. Chromosome 9p–linked families with frontotemporal dementia associated with motor neuron disease. Neurology. 2009;72:1669–1676. doi: 10.1212/WNL.0b013e3181a55f1c. [DOI] [PubMed] [Google Scholar]
- 21.Boxer AL, Mackenzie IR, Boeve BF, et al. Clinical, neuroimaging and neuropathological features of a new chromosome 9p–linked FTD-ALS family. J Neurol Neurosurg Psychiatry. 2010 doi: 10.1136/jnnp.2009.204081. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial "Clinical limits of amyotrophic lateral sclerosis" workshop contributors. J Neurol Sci. 1994;124 Suppl:96–107. doi: 10.1016/0022-510x(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 23.Polvikoski T, Sulkava R, Myllykangas L, et al. Prevalence of Alzheimer's disease in very elderly people: a prospective neuropathological study. Neurology. 2001;56:1690–1696. doi: 10.1212/wnl.56.12.1690. [DOI] [PubMed] [Google Scholar]
- 24.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 26.Walter SD. Calculation of attributable risks from epidemiological data. Int J Epidemiol. 1978;7:175–182. doi: 10.1093/ije/7.2.175. [DOI] [PubMed] [Google Scholar]
- 27.Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shatunov A, Mok K, Newhouse S, et al. Chromosome 9p21.2 is the only replicable susceptibility locus for sporadic ALS. Lancet Neurol. 2010 [Google Scholar]
- 29.Anttonen AK, Metzidis A, Avela K, Aula P, Peltonen L. [accessed 07/06/2010];Finnish Disease Database. Available from: www.findis.org.
- 30.Sorenson EJ, Mandrekar J, Crum B, Stevens JC. Effect of referral bias on assessing survival in ALS. Neurology. 2007;68:600–602. doi: 10.1212/01.wnl.0000254501.58158.e7. [DOI] [PubMed] [Google Scholar]
- 31.Luca D, Ringquist S, Klei L, et al. On the Use of General Control Samples for Genome-wide Association Studies: Genetic Matching Highlights Causal Variants. Am J Hum Genet. 2008;82:453–463. doi: 10.1016/j.ajhg.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ince PG, Lowe J, Shaw PJ. Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology. Neuropathol Appl Neurobiol. 1998;24:104–117. doi: 10.1046/j.1365-2990.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- 33.Gislason TB, Sjogren M, Larsson L, Skoog I. The prevalence of frontal variant frontotemporal dementia and the frontal lobe syndrome in a population based sample of 85 year olds. J Neurol Neurosurg Psychiatry. 2003;74:867–871. doi: 10.1136/jnnp.74.7.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.