

Abstract
The sympathetic nervous system modulates immune responses via the secretion of catecholamines and subsequent activation of adrenergic receptors (ARs), and systemic catecholamine levels increase markedly in the setting of endotoxemia and sepsis. Previous studies have demonstrated that stimulation of β-ARs by pharmacological agonists attenuates the inflammatory response to LPS observed in vitro, and can increase survival in animal models of endotoxemia and sepsis. However, the consequences of β-AR activation by endogenous catecholamines have not been explored in these settings. Furthermore, the relative contribution of β-ARs expressed on immune versus non-immune cells to LPS-mediated inflammation and mortality is not known. Our first goal was therefore to determine the impact of β-AR stimulation by endogenous catecholamines released during endotoxemia on LPS-mediated inflammation and mortality in vivo. To address this question, we examined the LPS response of mice lacking all three known βAR subtypes, β1-, β2- and β3-AR, and demonstrated that these β-less mice exhibited a net increase in inflammation (increased TNF-α levels and decreased IL-10 levels in serum) and a 50% decrease in survival relative to wildtype animals. The second goal of our study was to determine the relative contribution of β-ARs expressed on radiosensitive immune versus radioresistant cells to the protective action of β-ARs in the setting of endotoxemia. We therefore examined the LPS response of bone marrow chimeras generated between β-less and wildtype mice, and concluded that β-ARs expressed on radioresistant cells play the dominant role in protecting against LPS-mediated mortality and attenuating systemic TNF-α responses. Finally, we determined that β3-AR subtype does not play a significant role in regulating LPS-mediated mortality and inflammation, by evaluating mice lacking the β1- and β2-AR subtypes only.
Keywords: bone marrow chimeras, cytokines, survival, LPS, mice
INTRODUCTION
The endotoxin lipopolysaccharide (LPS) is a major component of the outer membrane of Gram-negative bacteria and a potent activator of innate immune cells via engagement of Toll-like receptor 4 (TLR-4). Macrophages are the predominant source of proinflammatory cytokines released in response to LPS-mediated activation in vivo [1]. Although such cytokine release is a critical aspect of the protective immune response aimed at clearing bacterial infection, excessive production can lead to an exaggerated inflammatory response that results in circulation collapse, multi-organ failure and death in the setting of experimental endotoxemia or septic shock. For example, administration of TNF-α, the first cytokine produced in the inflammatory cascade mediated by LPS, is sufficient to reproduce the pathological sequelae of endotoxemia or septic shock, including mortality, due to activation of downstream responses [2, 3]. In rodents, neutralization of endogenous TNF-α by passive immunization protects mice from LPS-induced death if carried out before LPS administration [4]. However, passive immunization is not protective when administered 3 hours after LPS challenge, since late mediators of lethality have already been induced [4]. LPS induces not only pro-inflammatory cytokines such as TNF-α, IL-1 and IFN-γ, but also anti-inflammatory molecules such as IL-10. IL-10 is a potent inhibitor of proinflammatory cytokine release by macrophages, and administration of IL-10 before or within 30 minutes of LPS injection prevents lethality in experimental endotoxemia [5, 6]. Consistent with these data, IL-10-deficient mice exhibit markedly increased sensitivity to LPS as shown by elevated mortality accompanied by uncontrolled TNF-α production [7].
Endotoxemia and sepsis are accompanied by activation of the sympathetic nervous system (SNS) leading to the release of high concentrations of catecholamines. The α1-, α2-, and β-adrenergic receptors (AR) comprise a class of G-protein-coupled receptors that mediate the complex physiological actions of catecholamines. Increasing evidence supports an important role for catecholamines in modulating immune responses, both systemically following release from adrenal glands, and locally following release from postganglionic sympathetic nerves innervating immune organs [8, 9]. The three known β-AR subtypes, β1-, β2-, and β3-AR, are broadly expressed on both immune and non-immune cells. β2-AR is the principal β-AR expressed by macrophages, although β1-AR may also play a role in modulating macrophage function. ®-AR stimulation is associated with down regulation of inflammatory molecules, in particular TNF-α, IL-1β, IL-6 and nitric oxide (NO), and elevated levels of anti-inflammatory IL-10 by macrophages stimulated in vitro by LPS [10-15]. Previous studies have revealed that pretreatment with pharmacological β-AR agonists results in decreased TNF-α and increased IL-10 serum levels, and increased survival in rodent models of endotoxemia [14-17]. Consistent with these agonist studies, non-specific blockade of β-AR increased lethality in a mouse model of sepsis induced by cecal ligation and puncture [18]. In addition to decreasing the net inflammatory response, β-ARs may promote survival by directly compensating for the impaired heart and vessel tone that characterizes LPS-induced shock. All three subtypes of β-AR are expressed on cardiac myocytes, and β1- and β2-AR play a major role in promoting cardiac contractility [19-23].
The first goal of the present study was to determine whether β-AR stimulation by endogenous catecholamines released during endotoxemia modifies LPS-mediated lethality and inflammation in vivo. To address this question, we first examined the LPS response of mice lacking functional β1-, β2- and β3-AR (β-less mice) [24-27]. As mentioned above, prior studies have already examined the effects of β-AR agonists or antagonists on endotoxemia and sepsis. However, interpretation of such pharmacological studies is complicated by the relative lack of specificity of many of the β-AR ligands that can be used in vivo. For example, many such agents bind to other G-protein coupled receptors such as serotonin and dopamine receptors. Likewise, the impact of pharmacological agonists on β-AR function cannot be extrapolated to that of endogenous catecholamines, since differences in activity and concentration are known to impact outcome. The second goal of our study was to determine the relative contribution of β-ARs expressed on radiosensitive immune cells versus other cells, to the mortality and inflammation seen in the setting of acute endotoxemia. In order to achieve this, we generated bone marrow chimeras between β-less and wildtype (wt) mice, and evaluated the impact of β-ARs expressed on radioresistant versus sensitive cells to survival and systemic cytokine responses to LPS in vivo. Finally, we asked whether the β1- and β2-AR subtypes played the major role in regulating LPS-mediated mortality or inflammation, by evaluating mice retaining β3-AR function, but lacking the β1- and β2-AR subtypes.
MATERIALS AND METHODS
Mice
Mice lacking all three known β-ARs (β-less) and wild-type (wt) mice, both derived from the same original strain [24], were generously provided by Dr. Brian Kobilka. Age- (6-8 weeks) and sex-matched animals were employed in all experiments. Genotyping was performed on tail DNA by PCR with the following primer sets:
β1-AR Forward 5′-CCGCTGCTACCACGACCCCAAG-3′,
β1-AR Reverse 5′-AGCCAGTTGAAGAGCAAGAGGCG-3′,
β2-AR Forward 5′-GGTTATCGTCCTGGCCATCGTGTTTG-3′,
β2-AR Reverse 5′-TGGTTCGTGAAGTCACAGCAAGTCTC-3′,
β3-AR Forward 5′-AGTGGACGTGCTCTGTGTAACTGTAACTGCTAG-3′
β3-AR Reverse 5′-CTCCAACATGCCCTATGCGCTGCT-3′
Animals were housed in pathogen free facilities, receiving standard chow and water ad libitum, and lighting was maintained on a 12-hour cycle. Animal experiments were conducted in accordance with federal regulations and guidelines, and approved by the Institutional Animal Care and Use Committee of the University of Virginia.
Survival studies
Mice were injected intraperitoneally (i.p.) with 50 mg/kg of LPS (E. coli serotype O55:B5) purchased from Sigma Aldrich (St Louis, MO) suspended in sterile PBS, and were subsequently observed every 12 hours for movement, grooming, conjunctivitis and survival for a period of 72 hours.
Measurements of serum cytokine levels
Mice were injected with LPS as described above, and blood was removed by cardiac puncture at 0, 1, and 3 hours after injection. Serum samples were stored at −20°C. Serum levels of TNF-α IL-10, and IL-12 p40 were measured by ELISA employing DuoSet® reagents purchased from R&D Systems (Minneapolis, MN).
Bone marrow chimeras
Bone marrow cells were isolated from the femurs and tibias of ®-less and wt mice, and resuspended in McCoy’s 5A medium with 5% penicillin/streptomycin. Chimeras were generated by injecting 5 ×106 bone marrow cells in a volume of 200 ul into the tail vein of recipient mice that had undergone total body irradiation (2 × 550 rads, with an interval of 3 hours). Animals were allowed to reconstitute for two months post-transplant. The efficiency of reconstitution for β-less→wt and wt→β-less chimeras was determined by quantitative real-time PCR analysis of genomic DNA from peripheral blood leukocytes to determine the relative frequency of wt versus β-AR alleles, and averaged 84%. The following primer sets were employed:
Neomycin Forward 5′-AGACAATCGGCTCTGAT-3′
Neomycin Reverse 5′-CTCGTCCTGCAGTTCCATTCA-3′
®1-AR Forward 5′-GCTGATCTGGTCATGGGATT-3′
®1-AR Reverse 5′-CACACAGGGTCTCAATGCTG-3′
®2-AR Forward 5′-AAGAATAAGGCCCGAGTGGT-3′
®2-AR Reverse 5′-GTAGGCCTGGTTCGTGAAGA-3′
®3-AR Forward 5′-ACAGGAATGCCACTCCAATC-3′
®3-AR Reverse 5′-TTAGCCACAACGAACACTCG-3′
Statistics
Survival and cytokine data were analyzed using GraphPad Prism version 4 for Macintosh. Specifically, survival curves were generated using the product limit method of Kaplan and Meier, and compared by the Log-rank test. Cytokine data are presented as the mean +/− the standard error (s.e.m.). P-values of 0.05 or less were considered significant for both survival and cytokine data.
RESULTS
β-ARs decrease LPS-mediated mortality
The contribution of β-AR function to survival was examined in a standard model of murine endotoxemia. ®-less and wt mice were given a single dose of LPS (50 mg/kg, i.p.) and survival was then monitored over a 72-hour period. Mice lacking β-ARs had a 50% mortality rate after 3 days, whereas all the wt mice survived (Fig. 1). LPS-mediated mortality in the β-less group was relatively rapid, with the majority of deaths occurring within the first 24 hours following LPS administration. Lack of β-AR also exacerbated the development of clinical manifestations of endotoxin morbidity, including ruffled fur, loss of mobility, and conjunctivitis (data not shown).
Fig. 1. β-ARs protect against LPS-mediated mortality.
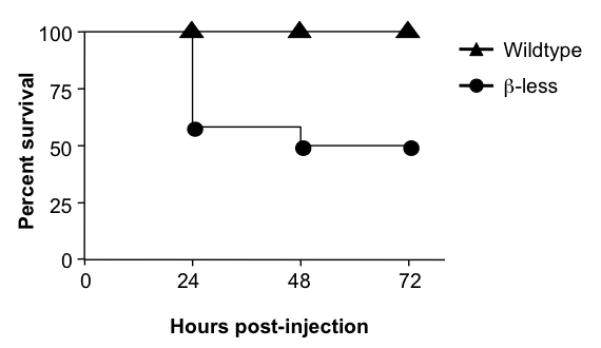
Wildtype (n=12) and β-less (11) mice were injected i.p. with 50 mg/kg of LPS and then monitored for survival over a 72 hour period. Survival curves were generated using the product limit method of Kaplan and Meier, and compared by the Log-rank test. Statistically significant differences in survival were observed between the two groups (p<0.01). (Similar results were obtained with the Gehan-Breslow-Wilcoxon test).
β-ARs differentially regulate LPS-induced TNF-α and IL-10 production
In order to determine the impact of β-ARs on inflammatory responses initiated by LPS, β-less and wt mice were injected with a single dose of LPS (50 mg/kg, i.p.), and serum analyzed at 0, 1, and 3 hour post-injection (Fig. 2). As previously reported for wt mice, serum levels of TNF-α increased rapidly in response to LPS, peaking at 1 hour and returning close to baseline by 3 hours. Although the kinetics of cytokine production was similar in both strains, serum levels of TNF-α were significantly higher (1.6-fold) at 1 hour post-injection in the β-less group (Fig. 2A). As expected, wt mice displayed a steady increase in IL-10 serum levels over the 3-hour time period following LPS injection. The β-less mice group also exhibited a gradual increase in serum IL-10, but by 1 hour post-injection, IL-10 levels were significantly lower (1.7-fold) than those of wt mice (Fig. 2B). The ratio between the pro-inflammatory cytokine TNF-α and the anti-inflammatory cytokine IL-10 in the systemic circulation has been used as an estimate of net inflammatory activity in experimental murine and human endotoxemia [28, 29]. The TNF-α:IL-10 ratios for the wt and β-less mice were 3.2 and 8.7 respectively, demonstrating that lack of β-AR function increased this “inflammatory” index by 2.7-fold. In contrast to TNF-α and IL-10, the levels of LPS-induced IL-12p40, IL-6 and IL-1β did not differ significantly between the two groups of mice (Fig. 2).
Fig. 2. β-ARs differently regulate LPS-induced TNF-α and IL-10 production.
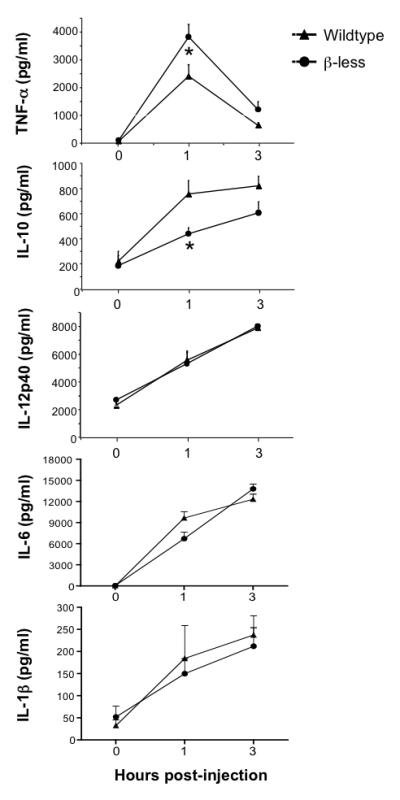
Wildtype and β-less mice were injected i.p. with 50 mg/kg LPS. At 0, 1 and 3 hours post-injection, serum was collected and analyzed for TNF-α, IL-10 and IL-12p40 concentration by ELISA. Data are expressed as mean ± s.e.m. of 7 to 10 animals per group per time point. TNF-α and IL-10 plasma levels were significantly different between the two groups at 1 hr post-injection as determined by ANOVA with Bonferroni post tests (*, p<0.01).
Relative contribution of β-ARs expressed on radiosensitive versus resistant cells to protection against LPS-mediated mortality
The above studies demonstrate that β-ARs have a protective role against LPS-mediated lethality. In order to determine the relative contribution of β-ARs expressed on hematopoietic versus radioresistant cells to survival in the setting of endotoxemia, we generated bone marrow chimeras between wt and β-less mice. Lethally irradiated ®-less or wt recipients were reconstituted with bone marrow from either wt or β-less donors, to generate 4 groups of mice. After reconstitution, each group was injected with LPS and survival monitored over a 72-hour period (Fig. 3). β-less mice reconstituted with β-less marrow (BB) exhibited a marked increase in LPS-mediated mortality relative to wt mice reconstituted with wt marrow (WW), consistent with, and even more dramatic than the differential mortality previously observed for unmanipulated β-less and wt mice. Strikingly, β-less mice reconstituted with wt bone marrow (WB) exhibited a frequency and kinetics of mortality that was very similar that of the BB controls, whereas wt mice reconstituted with β-less bone marrow (BW) exhibited a mortality frequency closer to that of the WW controls. These data demonstrate that although β-ARs expressed on immune cells contribute to protection against LPS-mediated lethality, those expressed on radioresistant cells clearly play the dominant role.
Fig. 3. Relative contribution of β-AR expressed on radio-resistant versus -sensitive cells to protection against LPS-mediated mortality.
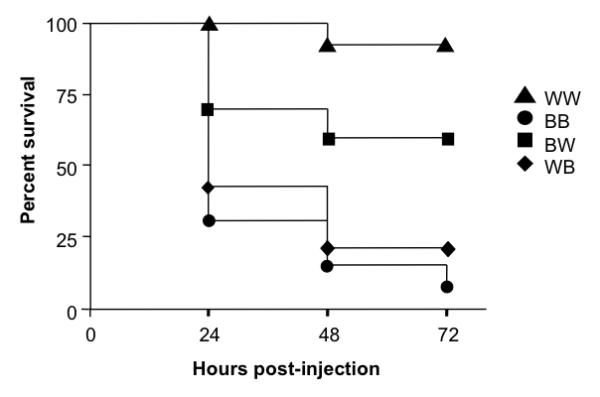
Chimeric mice expressing β-AR on radiation-resistant non-bone marrow-derived cells only were generated by reconstituting wildtype (W) mice with bone marrow from β-less (B) mice (BW mice, n=10). Conversely, chimeric mice expressing β-AR on radiation-sensitive bone marrow-derived cells only were generated by reconstituting β-less (B) mice with bone marrow from wildtype (W) mice (WB mice, n=13). In parallel, wildtype (W) and β-less (B) mice were reconstituted with wildtype (W) and β-less (B) marrow respectively, to generate WW (n=13) and BB (n=13) controls. The four groups of reconstituted mice were injected i.p. with 50 mg/kg of LPS and monitored for survival over a 72 hour period. Survival curves were generated using the product limit method of Kaplan and Meier, and compared by the Log-rank test. Statistically significant differences in survival were observed for the following groups: WW versus BW p=0.05; BW versus BB, p <0.01; WW versus WB, p<0.0001; WW versus BB, p<0.0001.
Relative contribution of β-ARs expressed on radiosensitive versus resistant cells to LPS-induced cytokine production
Our data show that β-ARs exert an overall anti-inflammatory effect in the setting of endotoxemia, as shown by an increase in the TNF-α:IL10 ratio in β-less versus wt mice. We next generated bone marrow chimeras as described above, in order to determine the relative contribution of β-ARs expressed on radioresistant versus radiosensitive immune cells to this anti-inflammatory effect. Mice were injected with LPS and serum levels of TNF-α, IL-10 and IL-12p40 were determined at 0, 1 and 3 hours post-injection (Fig. 4). Consistent with the data obtained from unmanipulated β-less and wt mice, BB mice exhibited significantly higher serum levels of TNF-α (Fig. 4A) and lower levels of IL-10 (Fig. 4B) relative to WW mice, at 1 hour post-injection. The magnitude of the TNF-α and IL-10 responses of reconstituted wildtype and beta-less controls was greater than that observed for their unmanipulated counterparts (Fig. 2), although that of IL-12p40 was very similar. The reason for this is unclear, but may be related to the bone marrow transplant setting. The TNF-α:IL-10 ratios for WW and BB mice were 2.0 and 12 revealing a six-fold difference in the inflammatory index between these two groups of mice. This result is consistent with that observed for unmanipulated wt and β-less mice, but reveals an even greater anti-inflammatory impact (six-fold) of β-AR. As expected, IL-20p40 serum levels were similar for all four groups of mice (Fig. 4C). The level of TNF-α produced by the WB chimeras was intermediate between that of WW and BB mice (Fig. 4A). However, only the difference between WB and WW mice was statistically different. Consistent with a role for βAR on radioresistant cells in mediating TNF-α suppression, serum TNF-α levels of BW and WW mice were very similar, and the WB chimeras produced higher levels of TNF-α than the BW chimeras (p=0.07). Although not statistically significant, the trend for IL-10 showed BW mice with cytokine levels intermediate between those of WW and BB at 1 hour post-injection (Fig. 4B).
Fig. 4. Relative contribution of β-AR on radio-resistant versus -sensitive cells to LPS-induced cytokine production.
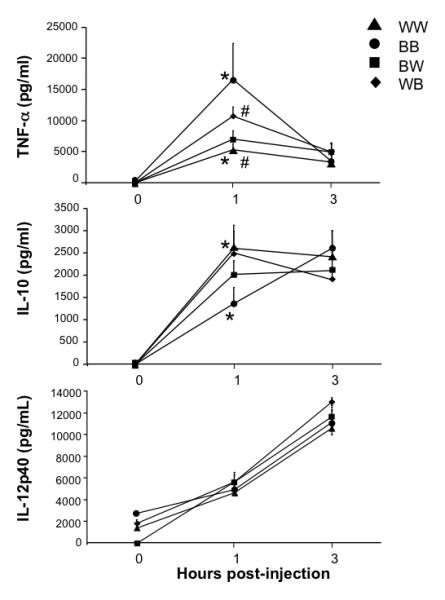
Chimeric mice expressing β-AR on radiation-resistant non-bone marrow-derived cells only (BW mice) or on radiation-sensitive bone marrow-derived cells only (WB) were generated as described in the legend to Figure 3. As before, wildtype mice reconstituted with wildtype bone marrow (WW mice) and β-less mice reconstituted with β-less bone marrow (BB) served as radiation controls. Following reconstitution, WW, BB, WB and WB mice were injected i.p. with 50 mg/kg LPS. At 0, 1 and 3 hours post-injection, serum was collected and analyzed for TNF-α, IL-10 and IL-12p40 concentration by ELISA. Data are expressed as mean ± s.e.m. of 6 to 15 animals per group. Analysis by ANOVA with Bonferroni post tests revealed statistically significant differences between WW and BB groups at the 1 hour time point for both TNF-α and IL-10 serum levels (*, p<0.05), and between WW and WB groups at the 1 hour time point for TNF-α serum levels only (#, p< 0.05).
Role of β1- and β2-ARs in LPS-mediated lethality or inflammation
To highlight the role of the β1 and β2-AR subtypes per se in survival and inflammation following LPS challenge, we employed mice lacking β1- and β2-ARs, but retaining functional β3-ARs in our final set of experiments. The double knockout mice (β1/2−/−) and their wt controls were injected with LPS, and mortality followed over a 72-hour period as before (Fig. 5). Although wt mice died between 24 and 48 hours after injection, β1/2−/− mice exhibited a significant increase in mortality relative to wt mice at 24 hours post-injection. Finally, β1/2−/− and wt mice were injected with LPS, and serum TNF-α, IL-10 and IL-12p40 levels evaluated at 0, 1 and 3 hours (Fig. 6). Similar to the response of animals lacking all three β-AR subtypes, β1/2−/− mice produced significantly higher levels of TNF-α (2.5-fold) and lower levels of IL-10 (1.9-fold) relative to wt controls. The TNF-α:IL-10 ratios of the wt and β1/2−/− mice were 1.1 and 5.2 respectively, demonstrating that lack of β1/2-AR function increased the inflammatory index by 4.7-fold.
Fig. 5. β1- and β2-AR protect against LPS-mediated mortality.
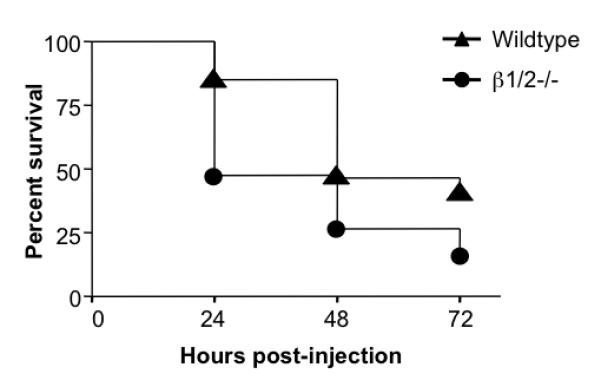
Wildtype (n=26) and β1/2−/− (n=19) mice were injected i.p. with 50 mg/kg of LPS and then monitored for survival over a 72 hour period. Survival curves were generated using the product limit method of Kaplan and Meier, and compared by the Log-rank test. Statistically significant differences in survival were observed between the two groups (p<0.05). (Similar results were obtained with the Gehan-Breslow-Wilcoxon test).
Fig.6. β1/2-AR differentially regulate LPS-induced TNF-α and IL-10 production.
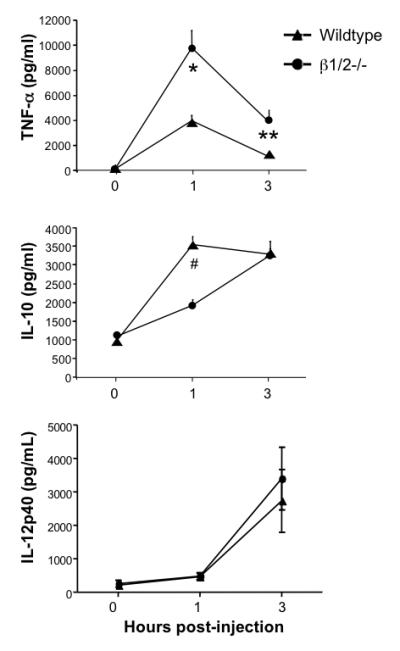
Wildtype (n=12) and β1/2−/− (n=18) mice were injected I.p. with 50 mg/kg LPS. At 0, 1 and 3 hours post-injection, serum was collected and analyzed for TNF-α, IL-10 and IL-12p40 concentration by ELISA. Data are expressed as mean ± s.e.m. per group at each time point. Analysis by ANOVA with Bonferroni post test revealed that significant difference between the two groups for serum TNF-α levels at 1 hour (*p<0.0001) and 3 hours (**p<0.05) post-injection, and for serum IL-10 levels at 1 hour post-injection (# p<0.00003).
DISCUSSION
β-ARs stimulated by pharmacological agonists in vivo (presumably in concert with circulating catecholamines) promote survival and ameliorate inflammation in rodent models of endotoxemia. However, the extent to which such findings are relevant to the native actions of β-ARs in the setting of endotoxemia/septic shock is unclear, since many of the β-AR agonists employed exhibit a relative lack of specificity, and likely differ from endogenous circulating catecholamines (whose levels are increased in response to LPS) in their temporal and spatial activity in vivo. We therefore extended these studies to evaluate the impact of β-ARs modulated by their native ligands, the catecholamines, on LPS-mediated inflammation and mortality. Specifically, we have utilized mice lacking β-AR function to demonstrate for the first time that β-AR stimulation by endogenously released catecholamines increases survival and reduces inflammation in the setting of endotoxemia.
A brief comparison of the results of the present endotoxemia study evaluating the role of endogenous catecholamines with those of previous studies evaluating the impact of pharmacological β-AR agonists, yields some interesting similarities and differences. Pre-treatment of LPS-challenged rodents with the β2-AR agonist, terbutaline, resulted in inhibition of TNF-α and augmentation of IL-10 production by similar magnitudes to those observed in our study, and also increased survival [16]. Similarly, pre-treatment of LPS-challenged mice with the broad-spectrum β-AR agonist isoproterenol resulted in decreased TNF-α serum levels [15]. In contrast to our study however, LPS-challenged rodents pre-treated with isoproterenol or the β2-AR-selective agonist clenbuterol exhibited reduced levels of serum IL-12p40 [30] or IL-1β and IL-6 [17] respectively. We observed that the LPS-induced serum levels of these three cytokines did not differ between β-less and wildtype mice. The differential cytokine responses observed may reflect important differences in the actions of endogenous catecholamines versus pharmacological agonists on β-AR function not just in terms of their receptor specificity, but also their relative activity or tissue accessibility. With respect to activity, the pharmacological agonists employed in these studies are presumably stimulating β-ARs beyond the level already achieved by endogenous catecholamines. One example of differential tissue accessibility is that catecholamines have access to β-AR expressed in the CNS, whereas the β-AR agonists are inefficient at crossing the blood-brain barrier. An alternative explanation is that disruption of β-AR gene function may have activated secondary changes during development that compensate for lack of β-AR regulation of these particular cytokines, but not TNF-α and IL-10. In summary, our data demonstrate that β-ARs respond to endogenous catecholamines released in response to LPS challenge by decreasing the inflammatory response and increasing survival.
Having established that β-ARs serve a protective role in endotoxemia in this genetic knockout model, we were then able to determine the extent to which β-ARs expressed on bone marrow-derived versus radioresistant cells contribute to survival and inflammation in the setting of endotoxemia. Analysis of reciprocal bone marrow chimeras revealed that β-ARs expressed on radioresistant cells played the predominant role in promoting survival, since the survival rate of β-less mice reconstituted with wt bone marrow was about as poor as that of mice reconstituted with β-less marrow. Nonetheless, β-ARs expressed on hematopoietic cells must also contribute to some extent to survival, since wt mice reconstituted with β-less marrow exhibited increased mortality relative to those reconstituted with wt marrow. The data suggest that β-ARs expressed on radioresistant cells play an essential role in protecting from LPS-induced death regardless of whether or not β-ARs are expressed on bone marrow-derived cells, whereas the protective role of β-ARs present on the latter compartment only becomes apparent when β-AR function on radioresistant cells is intact. In a subsequent set of experiments, we compared the cytokine responses of LPS-challenged bone marrow chimeras in order to determine whether β-AR location played an important role in inflammatory responses. We observed that the TNF-α response of mice lacking β-ARs only on radioresistant cells was significantly higher than that of mice that retained β-AR function on all cells, demonstrating that β-ARs expressed in this compartment not only play an important role in survival, but also in inhibition of LPS-induced TNF-α responses in vivo. Consistent with this observation, the TNF-α response of mice lacking β-ARs on radiosensitive hematopoietic cells alone was only marginally increased relative to (and not statistically significant from) those animals with wt cells. Although we did observe differences in the IL-10 responses of reciprocal chimeras compared to WW or BB controls, they did not reach statistical significance. For simplicity’s sake, the relative mortality exhibited by the 4 experimental groups can be represented as BB≥WB<<BW>WW, and the relative TNF-α response as BB≥WB<<BW≥WW. Thus, the relative order of the 4 groups is similar with respect to mortality and TNF-α responses. Although these data do not demonstrate a direct correlation between mortality and TNF-α levels, they do reveal a dominant role for β-AR expressed on radioresistant cells in promoting survival and decreasing pro-inflammatory TNF-α responses in vivo. Our data do not preclude a role for β-ARs expressed on immune cells to dampening of pro-inflammatory TNF-α responses, but suggest that it is subordinate to those expressed on radioresistant cells.
Increased survival may be due to β-AR actions that reduce TNF-α production per se, and to those that modulate other protective mechanisms. With regard to the former, an attenuated TNF-α response could result in decreased expression of lethality mediators that are induced much later in the cytokine cascade. Recent studies have demonstrated that the major source of LPS-induced TNF-α in vivo is the macrophage population, and mice with a macrophage-specific defect in TLR expression are relatively resistant to LPS-induced death [1]. Mice reconstituted with bone marrow lacking TLR-4 exhibit a markedly decreased TNF-α response to LPS challenge, highlighting radiosensitive macrophages as a major source of this cytokine in the setting of endotoxemia [31]. The TNF-α response of these chimeric mice is still readily detectable however, suggesting that radioresistant cells do contribute to this inflammatory response [31]. Our observation that radioresistant cells play a significant role in β-AR-mediated inhibition of TNF-α responses is intriguing. One possible explanation is that this radioresistant population may comprise resident macrophages that fail to be efficiently replaced by donor cells in the transplant setting. For example, the lung and liver contain relatively radioresistant macrophage populations [32-34]. An alternative explanation for the involvement of the radioresistant compartment in β-AR-modulation of TNF-α production is that β-ARs expressed in the CNS act in an indirect manner to suppress TNF-α production by radiosensitive macrophages in the periphery. Catecholamines are released within the CNS in response to systemic LPS administration, and β-ARs are expressed on microglia, astrocytes and neurons [35-37]. Central β-AR stimulation has the potential to modulate peripheral immune responses via regulation of sympathetic tone and activation of the hypothalamic-pituitary-adrenal axis for example. Indeed, depletion of central catecholamines by chemical lesioning of dopaminergic neurons results in impaired peripheral host immunity [38, 39].
An alternative explanation for the protective role of β-ARs expressed on radioresistant cells is that they can directly protect against LPS-induced mediators of lethality. For example, β1-, β2-, and to a lesser extent β3-AR, are expressed on cardiac myocytes where they regulate cardiac contractile activity. Specifically, β1- and β2-ARs mediate the inotropic and chronotropic effects of catecholamines, whereas the effect of β3-AR is less clear. However, sepsis/endotoxemia is associated with a net decrease in cardiac contractile activity despite elevated sympathetic tone, and available evidence suggests that this is due, at least in part, to inhibition of the positive inotropic action of β1/β2-AR [19, 23]. It is therefore possible that the increased mortality of β-less relative to wt mice is due, at least in part, to markedly deficient cardiac contractile activity secondary to loss of positive inotropic β1/2-AR action on myocytes. The elevated TNF-α levels present in LPS-challenged mice lacking β-ARs could contribute to this effect. Similarly, lack of β-AR function on endothelial cells may increase the severity of microvessel hyperpermeability associated with mortality in the setting of endotoxemia. β-ARs expressed in the CNS may also play a role in promoting survival, by acting indirectly to promote cardiac output and protect against tissue injury. For example, systemic administration of LPS has been reported to stimulate the synthesis and/or projection of catecholamines in several areas of the CNS [35-37], and activation of central β-AR has been reported to increase sympathetic outflow to the heart [40].
In order to examine the role of β1- and β2-ARs per se to the increased mortality and inflammation we observed in the β-less mice, we evaluated these parameters in mice with intact β3-AR function, but lacking functional β1- and β2-ARs. β3-ARs are primarily expressed in adipose tissue where they mediate lipolytic and thermogenic responses. β3-AR expression in myocardium is markedly elevated in the setting of sepsis [19], but our current understanding of β3-AR function in inflammation or vascular/cardiac function is relatively limited. As observed for mice lacking all three β-AR subtypes, the double knockout mice exhibited increased mortality relative to wt controls. The relative increase in mortality between β1/2−/− and control mice approached that seen for the β-less study at 24 hours post-LPS injection. In addition, as observed for β-less mice, β1/β2−/− mice exhibited increased TNF-α and decreased IL-10 serum levels in response to LPS challenge relative to wt animals. Caution should be taken in making a direct comparison between the β-less and double knockout mice in this study, because the control mice in each case exhibited differential sensitivity to LPS, presumably because of differences in their respective genetic backgrounds. Nevertheless our data demonstrate that β3-ARs do not play a major role in promoting survival and decreasing inflammation in the setting of endotoxemia.
In summary, our data demonstrate that β1- and β2-ARs play the dominant protective role in the setting of endotoxemia, and respond to endogenous catecholamines by increasing survival and dampening inflammatory responses initiated by LPS. Although our data suggest that β-ARs expressed on immune cells can contribute, it appears that β1- and β2-ARs expressed on radioresistant cells play the major role in inhibiting LPS-induced lethality and TNF-α responses. It is likely that the pro-survival actions of β-AR are mediated both by dampening the acute TNF-α response to LPS and by promoting cardiac/vascular function. It will be of great interest to identify the radioresistant tissues whose β-AR function plays such a critical role in protecting against LPS-induced mortality.
ACKNOWLEDGEMENTS
The authors are very grateful to Dr. Patrice Guyenet for his critical reading of this manuscript.
Sponsored in part by funding from the National Institutes of Health (NS41213).
REFERENCES
- 1.Yang Y, Liu B, Dai J, Srivastava PK, Zammit DJ, Lefrancois L, Li Z. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26:215–26. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, Hariri RJ, Fahey TJ, 3rd, Zentella A, Albert JD, Shires GT, Cerami A. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–4. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- 3.Tracey KJ, Lowry SF, Fahey TJ, 3rd, Albert JD, Fong Y, Hesse D, Beutler B, Manogue KR, Calvano S, Wei H, Cerami A, Shires GT. Cachectin/tumor necrosis factor induces lethal shock and stress hormone responses in the dog. Surg Gynecol Obstet. 1987;164:415–22. [PubMed] [Google Scholar]
- 4.Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869–71. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- 5.Gerard C, Bruyns C, Marchant A, Abramowicz D, Vandenabeele P, Delvaux A, Fiers W, Goldman M, Velu T. Interleukin 10 reduces the release of tumor necrosis factor and prevents lethality in experimental endotoxemia. J Exp Med. 1993;177:547–50. doi: 10.1084/jem.177.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howard M, Muchamuel T, Andrade S, Menon S. Interleukin 10 protects mice from lethal endotoxemia. J Exp Med. 1993;177:1205–8. doi: 10.1084/jem.177.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berg DJ, Kuhn R, Rajewsky K, Muller W, Menon S, Davidson N, Grunig G, Rennick D. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J Clin Invest. 1995;96:2339–47. doi: 10.1172/JCI118290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987-2007) Brain Behav Immun. 2007;21:736–45. doi: 10.1016/j.bbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molina PE. Neurobiology of the stress response: contribution of the sympathetic nervous system to the neuroimmune axis in traumatic injury. Shock. 2005;24:3–10. doi: 10.1097/01.shk.0000167112.18871.5c. [DOI] [PubMed] [Google Scholar]
- 10.Spengler RN, Chensue SW, Giacherio DA, Blenk N, Kunkel SL. Endogenous norepinephrine regulates tumor necrosis factor-alpha production from macrophages in vitro. J Immunol. 1994;152:3024–31. [PubMed] [Google Scholar]
- 11.Ignatowski TA, Spengler RN. Regulation of macrophage-derived tumor necrosis factor production by modification of adrenergic receptor sensitivity. J Neuroimmunol. 1995;61:61–70. doi: 10.1016/0165-5728(95)00074-c. [DOI] [PubMed] [Google Scholar]
- 12.Hasko G, Nemeth ZH, Szabo C, Zsilla G, Salzman AL, Vizi ES. Isoproterenol inhibits Il-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages. Brain Res Bull. 1998;45:183–7. doi: 10.1016/s0361-9230(97)00337-7. [DOI] [PubMed] [Google Scholar]
- 13.Boomershine CS, Lafuse WP, Zwilling BS. Beta2-adrenergic receptor stimulation inhibits nitric oxide generation by Mycobacterium avium infected macrophages. J Neuroimmunol. 1999;101:68–75. doi: 10.1016/s0165-5728(99)00134-4. [DOI] [PubMed] [Google Scholar]
- 14.Szelenyi J, Kiss JP, Puskas E, Szelenyi M, Vizi ES. Contribution of differently localized alpha 2- and beta-adrenoceptors in the modulation of TNF-alpha and IL-10 production in endotoxemic mice. Ann N Y Acad Sci. 2000;917:145–53. doi: 10.1111/j.1749-6632.2000.tb05378.x. [DOI] [PubMed] [Google Scholar]
- 15.Szelenyi J, Kiss JP, Vizi ES. Differential involvement of sympathetic nervous system and immune system in the modulation of TNF-alpha production by alpha2- and beta-adrenoceptors in mice. J Neuroimmunol. 2000;103:34–40. doi: 10.1016/s0165-5728(99)00234-9. [DOI] [PubMed] [Google Scholar]
- 16.Wu CC, Liao MH, Chen SJ, Chou TC, Chen A, Yen MH. Terbutaline prevents circulatory failure and mitigates mortality in rodents with endotoxemia. Shock. 2000;14:60–7. doi: 10.1097/00024382-200014010-00011. [DOI] [PubMed] [Google Scholar]
- 17.Izeboud CA, Hoebe KH, Grootendorst AF, Nijmeijer SM, van Miert AS, Witkamp RR, Rodenburg RJ. Endotoxin-induced liver damage in rats is minimized by beta 2-adrenoceptor stimulation. Inflamm Res. 2004;53:93–9. doi: 10.1007/s00011-003-1228-y. [DOI] [PubMed] [Google Scholar]
- 18.Schmitz D, Wilsenack K, Lendemanns S, Schedlowski M, Oberbeck R. beta-Adrenergic blockade during systemic inflammation: impact on cellular immune functions and survival in a murine model of sepsis. Resuscitation. 2007;72:286–94. doi: 10.1016/j.resuscitation.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 19.Moniotte S, Belge C, Sekkali B, Massion PB, Rozec B, Dessy C, Balligand JL. Sepsis is associated with an upregulation of functional beta3 adrenoceptors in the myocardium. Eur J Heart Fail. 2007;9:1163–71. doi: 10.1016/j.ejheart.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Moniotte S, Kobzik L, Feron O, Trochu JN, Gauthier C, Balligand JL. Upregulation of beta(3)-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation. 2001;103:1649–55. doi: 10.1161/01.cir.103.12.1649. [DOI] [PubMed] [Google Scholar]
- 21.Rohrer DK, Schauble EH, Desai KH, Kobilka BK, Bernstein D. Alterations in dynamic heart rate control in the beta 1-adrenergic receptor knockout mouse. Am J Physiol. 1998;274:H1184–93. doi: 10.1152/ajpheart.1998.274.4.H1184. [DOI] [PubMed] [Google Scholar]
- 22.Tavernier G, Toumaniantz G, Erfanian M, Heymann MF, Laurent K, Langin D, Gauthier C. beta3-Adrenergic stimulation produces a decrease of cardiac contractility ex vivo in mice overexpressing the human beta3-adrenergic receptor. Cardiovasc Res. 2003;59:288–96. doi: 10.1016/s0008-6363(03)00359-6. [DOI] [PubMed] [Google Scholar]
- 23.Yasuda S, Lew WY. Lipopolysaccharide depresses cardiac contractility and beta-adrenergic contractile response by decreasing myofilament response to Ca2+ in cardiac myocytes. Circ Res. 1997;81:1011–20. doi: 10.1161/01.res.81.6.1011. [DOI] [PubMed] [Google Scholar]
- 24.Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC, Kobilka BK, Lowell BB. betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science. 2002;297:843–5. doi: 10.1126/science.1073160. [DOI] [PubMed] [Google Scholar]
- 25.Rohrer DK, Desai KH, Jasper JR, Stevens ME, Regula DP, Jr., Barsh GS, Bernstein D, Kobilka BK. Targeted disruption of the mouse beta1-adrenergic receptor gene: developmental and cardiovascular effects. Proc Natl Acad Sci U S A. 1996;93:7375–80. doi: 10.1073/pnas.93.14.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chruscinski A, Brede ME, Meinel L, Lohse MJ, Kobilka BK, Hein L. Differential distribution of beta-adrenergic receptor subtypes in blood vessels of knockout mice lacking beta(1)- or beta(2)-adrenergic receptors. Mol Pharmacol. 2001;60:955–62. doi: 10.1124/mol.60.5.955. [DOI] [PubMed] [Google Scholar]
- 27.Bachman ES, Hampton TG, Dhillon H, Amende I, Wang J, Morgan JP, Hollenberg AN. The metabolic and cardiovascular effects of hyperthyroidism are largely independent of beta-adrenergic stimulation. Endocrinology. 2004;145:2767–74. doi: 10.1210/en.2003-1670. [DOI] [PubMed] [Google Scholar]
- 28.Coimbra R, Melbostad H, Loomis W, Tobar M, Hoyt DB. Phosphodiesterase inhibition decreases nuclear factor-kappaB activation and shifts the cytokine response toward anti-inflammatory activity in acute endotoxemia. J Trauma. 2005;59:575–82. [PubMed] [Google Scholar]
- 29.Fijen JW, Kobold AC, de Boer P, Jones CR, van der Werf TS, Tervaert JW, Zijlstra JG, Tulleken JE. Leukocyte activation and cytokine production during experimental human endotoxemia. Eur J Intern Med. 2000;11:89–95. doi: 10.1016/s0953-6205(00)00068-6. [DOI] [PubMed] [Google Scholar]
- 30.Hasko G, Szabo C, Nemeth ZH, Salzman AL, Vizi ES. Stimulation of beta-adrenoceptors inhibits endotoxin-induced IL-12 production in normal and IL-10 deficient mice. J Neuroimmunol. 1998;88:57–61. doi: 10.1016/s0165-5728(98)00073-3. [DOI] [PubMed] [Google Scholar]
- 31.Nolte MA, Leibundgut-Landmann S, Joffre O, e Sousa C Reis. Dendritic cell quiescence during systemic inflammation driven by LPS stimulation of radioresistant cells in vivo. J Exp Med. 2007;204:1487–501. doi: 10.1084/jem.20070325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matute-Bello G, Lee JS, Frevert CW, Liles WC, Sutlief S, Ballman K, Wong V, Selk A, Martin TR. Optimal timing to repopulation of resident alveolar macrophages with donor cells following total body irradiation and bone marrow transplantation in mice. J Immunol Methods. 2004;292:25–34. doi: 10.1016/j.jim.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 33.Noulin N, Quesniaux VF, Schnyder-Candrian S, Schnyder B, Maillet I, Robert T, Vargaftig BB, Ryffel B, Couillin I. Both hemopoietic and resident cells are required for MyD88-dependent pulmonary inflammatory response to inhaled endotoxin. J Immunol. 2005;175:6861–9. doi: 10.4049/jimmunol.175.10.6861. [DOI] [PubMed] [Google Scholar]
- 34.Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, Pierce RH, Crispe IN. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. 2007;110:4077–85. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavicky J, Dunn AJ. Endotoxin administration stimulates cerebral catecholamine release in freely moving rats as assessed by microdialysis. J Neurosci Res. 1995;40:407–13. doi: 10.1002/jnr.490400316. [DOI] [PubMed] [Google Scholar]
- 36.Linthorst AC, Flachskamm C, Holsboer F, Reul JM. Intraperitoneal administration of bacterial endotoxin enhances noradrenergic neurotransmission in the rat preoptic area: relationship with body temperature and hypothalamic--pituitary--adrenocortical axis activity. Eur J Neurosci. 1995;7:2418–30. doi: 10.1111/j.1460-9568.1995.tb01040.x. [DOI] [PubMed] [Google Scholar]
- 37.Lacosta S, Merali Z, Anisman H. Behavioral and neurochemical consequences of lipopolysaccharide in mice: anxiogenic-like effects. Brain Res. 1999;818:291–303. doi: 10.1016/s0006-8993(98)01288-8. [DOI] [PubMed] [Google Scholar]
- 38.Filipov NM, Cao L, Seegal RF, Lawrence DA. Compromised peripheral immunity of mice injected intrastriatally with six-hydroxydopamine. J Neuroimmunol. 2002;132:129–39. doi: 10.1016/s0165-5728(02)00321-1. [DOI] [PubMed] [Google Scholar]
- 39.Pacheco-Lopez G, Niemi MB, Kou W, Bildhauser A, Gross CM, Goebel MU, del Rey A, Besedovsky HO, Schedlowski M. Central catecholamine depletion inhibits peripheral lymphocyte responsiveness in spleen and blood. J Neurochem. 2003;86:1024–31. doi: 10.1046/j.1471-4159.2003.01914.x. [DOI] [PubMed] [Google Scholar]
- 40.Gourine A, Bondar SI, Spyer KM, Gourine AV. Beneficial effect of the central nervous system beta-adrenoceptor blockade on the failing heart. Circ Res. 2008;102:633–6. doi: 10.1161/CIRCRESAHA.107.165183. [DOI] [PubMed] [Google Scholar]