

Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating human neurodegenerative disease. The causes of ALS are poorly understood, although the protein TDP-43 has been suggested to play a critical role in disease pathogenesis. Here we show that Ataxin-2, a polyglutamine (polyQ) protein mutated in spinocerebellar ataxia type 2 (SCA2), is a potent modifier of TDP-43 toxicity in animal and cellular models. The proteins associate in a complex that depends on RNA. Ataxin-2 is abnormally localized in spinal cord neurons of ALS patients. Likewise, TDP-43 shows mislocalization in SCA2. To assess a role in ALS, we analyzed the Ataxin-2 gene (ATXN2) in 915 ALS patients. We found intermediate-length polyQ expansions (27–33 Qs) in ATXN2 significantly associated with ALS. These data establish ATXN2 as a relatively common ALS disease susceptibility gene. Further, these findings indicate that the TDP-43/Ataxin-2 interaction may be a promising target for therapeutic intervention in ALS and other TDP-43 proteinopathies.
Introduction
ALS, also known as Lou Gehrig’s disease, is a devastating adult onset neurodegenerative disease with no cure1. In fact, we still know little about the causes. The disease is mostly sporadic (SALS) but approximately 10% of cases have a first or second-degree relative with ALS (familial ALS (FALS)). Mutations in SOD1, encoding Cu/Zn superoxide dismutase, have been identified in ~20% of FALS cases2, for an overall incidence of ~2%. Additional ALS disease genes have been identified that are even more rare. Identifying new and potentially common genetic risk factors for ALS will accelerate understanding of the disease, aid the development of biomarkers, and spur innovative new treatments.
Recently, the 43 kDa TAR DNA binding protein (TDP-43) was identified as a major player in sporadic and familial ALS. In 2006, TDP-43 was identified as the major disease protein in ubiquitinated cytoplasmic inclusions in neurons of patients with ALS and frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-TDP)3. Subsequently, mutations in the gene encoding TDP-43 (TARDBP) were found associated with familial cases of ALS and FTLD-TDP4,5, arguing strongly for a central role of TDP-43 in disease pathogenesis. TDP-43 is normally a nuclear protein but pathological inclusions contain cytoplasmic TDP-43 aggregates, suggesting that altered subcellular localization of the protein may be critical to disease pathogenesis6. Little is known about how loss of one or more of the biological functions of TDP-43, or how a potential toxic gain-of-function, might contribute to neurodegenerative disease. Moreover, nothing is known about genetic modifiers of TDP-43 pathogenesis or how other factors that interact with TDP-43 contribute to the risk of developing ALS or the age of disease onset.
In an unbiased screen to define modifiers of TDP-43 toxicity in yeast, we identified Ataxin-2 as a potent, dose-sensitive modulator of TDP-43 toxicity across multiple model systems. We show that the two proteins associate in a complex and are mislocalized in ALS patient spinal cord neurons. Given that Ataxin-2 is a polyQ disease gene, we analyzed the length of the polyQ repeat in over 900 sporadic and familial ALS patients. This revealed a significant association of Ataxin-2 intermediate-length polyQ tract expansions with ALS (4.7% of cases). We propose Ataxin-2 is a new and potentially common ALS disease gene. Further, these findings indicate that the TDP-43/Ataxin-2 interaction may be a promising target for therapeutic intervention.
Results
Pbp1 modifies TDP-43 toxicity in yeast
To gain insight into mechanisms of TDP-43 pathogenesis, we used an unbiased genetic approach to identify genes that could suppress or enhance TDP-43 toxicity in yeast. Similar approaches have been used to discover modifiers of the Parkinson’s disease protein α–synuclein7,8. We individually transformed 5,100 yeast genes, which comprise the Yeast FLEXGene plasmid overexpresssion library7 into a yeast strain expressing TDP-43. 13 genes were identified that suppressed and 27 genes that enhanced TDP-43 toxicity when overexpressed (A.C.E. and A.D.G. unpublished). The largest functional class enriched in the screen included RNA binding proteins and proteins involved in RNA metabolism. We also identified kinases and proteases as potent modifiers of TDP-43 toxicity. Importantly, of 71 genes from this library that have been previously shown to modify α–synuclein toxicity in yeast7,8, only one also affected TDP-43 toxicity, underscoring the specificity of the screen for TDP-43 biology.
One yeast gene identified that enhanced TDP-43 toxicity, PBP1 (poly(A)-binding protein (Pab1p)-Binding Protein), was notable as an ortholog of the human Ataxin-2 gene, mutations in which cause the neurodegenerative disease spinocerebellar ataxia type 2 (SCA2). SCA2 is one of a heterogeneous group of 28 autosomal dominant hereditary ataxias9 and is caused by polyQ tract expansions in the Ataxin-2 gene (ATXN2)10–13. Interestingly, in SCA2, as in ALS, motor neurons are also known to degenerate, but these features typically occur later than the cerebellar degeneration. However, in select cases, the motor neuron features of SCA2 are sufficiently prominent to mimic an ALS presentation14,15, indicating the potential for clinicopathological overlap. Although the precise functions of yeast Pbp1 and human Ataxin-2 are not fully understood, Pbp1 interacts with Pab1 to regulate mRNA polyadenylation and is involved in stress granule assembly16. P-bodies and stress granules play important roles in regulating translation, mRNA degradation, and the subcellular localization of mRNAs17. Upregulation of Pbp1 enhanced TDP-43 toxicity in yeast (Fig. 1a), whereas Pbp1 loss-of-function suppressed toxicity (Fig. 1b), indicating that Pbp1 is a dose-sensitive modifier of TDP-43 toxicity. Up- or down-regulation of Pbp1 did not enhance the toxicity of other human neurodegenerative disease proteins, such as α–synuclein or a mutant huntingtin fragment (Fig. 1a, b), demonstrating specificity of the Pbp1 interaction for TDP-43.
Figure 1.
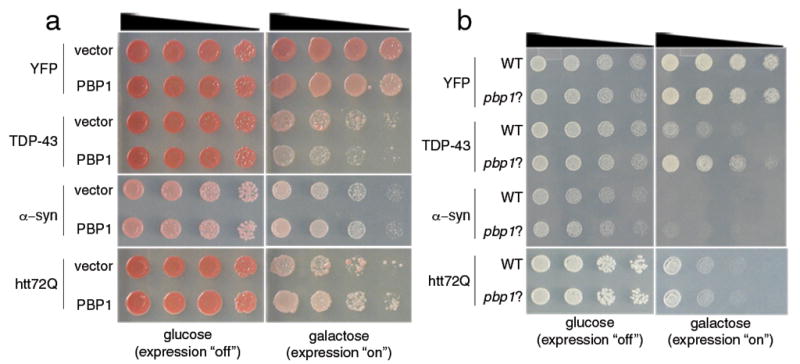
Pbp1 is a dose-sensitive modifier of TDP-43 toxicity in yeast. a) Spotting assays with yeast TDP-43 showing toxicity. Five-fold serial dilutions of yeast cells spotted onto glucose (expression repressed) or galactose (expression induced). Upregulation of PBP1 enhances TDP-43 toxicity. Whereas PBP1 has no effect on yeast viability when expressed with the control protein YFP, when co-expressed with TDP-43, it enhances toxicity. Enhancement is specific because PBP1 does not affect the toxicity of a pathogenic Huntington fragment (htt72Q) or α–synuclein. b) Spotting assays with yeast TDP-43 showing that PBP1 deletion (pbp1Δ) suppresses TDP-43 toxicity. Whereas expression of TDP-43 from a plasmid in WT yeast was toxic, this was mitigated in pbp1Δ cells. The effect was specific because α–syn or htt72Q toxicity was not suppressed by pbp1Δ
Ataxin-2 modifies TDP-43 toxicity in Drosophila
To test the relevance of the Ataxin-2/TDP-43 genetic interaction in the nervous system we used Drosophila. A series of transgenic lines were generated that expressed WT human TDP-43 or an ALS-linked mutant form (Q331K) at varying levels. This yielded a set of lines with milder or more severe effects, enabling the detection of genetic interactions in a sensitive manner (Fig. 2). Directing expression of TDP-43 to the eye of the fly caused a progressive, age-dependent degeneration of the structure (Figs. 2a and S2a). TDP-43 induced a markedly shortened lifespan when expressed in the nervous system (Fig. 2d), and led to progressive loss of motility when directed to motor neurons (Figs. 2b). Notably, expression of an ALS-linked mutant TDP-43 had a more severe effect than WT TDP-43 (Figs. 2b and S2b). Upregulation of the fly homolog of Ataxin-2, dAtx2, enhanced toxicity of TDP-43, resulting in more severe retinal degeneration (Fig. 2c) and a further reduction in lifespan (Fig. 2d). dAtx2 upregulation also enhanced toxicity of an ALS-linked mutant TDP-43 (data not shown). The effect of dAtx2 was strikingly dose-dependent, as reducing levels of dAtx2 by 50% mitigated TDP-43 toxicity in the eye (Fig. 2c) and dramatically extended lifespan (Fig. 2d), indicating that toxicity of TDP-43 is exquisitely sensitive to the levels of dAtx2. dAtx2 enhanced TDP-43 protein accumulation with upregulation, although had no effect when downregulated (Fig. 2e). The enhanced toxicity owing to dAtx2 was not attributable to simply enhanced TDP-43 protein levels, because degeneration was strikingly more severe upon upregulation of dAtx2 than with simply higher levels of TDP-43 (Fig. S3). The interaction was specific, as dAtx2 had no effect on the expression of a control protein (s-galactosidase; Fig. S4; also18,19). Moreover, upregulation of the molecular chaperone Hsp70 did not suppress TDP-43 toxicity, as it does in models of Parkinson’s disease and spinocerebellar ataxia type 3 (SCA3)20,21, nor did upregulation of Ataxin-3 (whose polyQ expansion is the basis of SCA3) have an effect (Fig. S4). These data indicate that modulation of TDP-43 toxicity by Ataxin-2 is conserved in the nervous system of the fly.
Figure 2.

Ataxin-2 is a dose-sensitive modifier of TDP-43 toxicity in Drosophila. a) Expression of TDP-43 caused a dose-dependent disruption of retinal structure. Genotypes: gmr-GAL4(YH3) in trans to UAS-YFP (control), UAS-TDP-43(M) or UAS-TDP-43-YFP(S). (M) and (S) are moderate and strong TDP-43 expression, respectively (see Methods and Fig. S2). b) TDP-43 caused motility deficits when expressed in motor neurons. TDP-43.Q331K causes a more severe loss of motility than the WT protein at the same level of expression (see Fig. S2). Genotypes: D42-GAL4 in trans to +, UAS-TDP-43 or UAS-TDP-43.Q331K. c) dAtx2 modulates TDP-43 toxicity. Flies expressing TDP-43 or dAtx2 alone (dAtx2EP) have a mild effect on retinal structure. TDP-43 toxicity is more severe upon upregulation of dAtx2 (dAtx2EP3145). TDP-43 toxicity is markedly mitigated upon reduction of dAtx2 (flies in trans to null allele dAtx2X1). Genotypes: control: gmr-GAL4/UAS-YFP. TDP-43: UAS-TDP-43(M)/+, gmr-GAL4(YH3)/+. dAtx2EP: gmr-GAL4(YH3)/dAtx2EP3145. TDP43+dAtx2EP: UAS-TDP-43(M)/+, gmr-GAL4(YH3)/dAtx2EP3145. TDP-43;dAtx2X1/+: UAS-TDP-43(M)/+, gmr-GAL4(YH3)/dAtx2X1. dAtx2X1/+ has wild type retinal structure. d) dAtx2 modulates reduced lifespan conferred by TDP-43. Expression of TDP-43 in the nervous system reduces lifespan (black, compared to normal in blue). Upregulation of dAtx2 causes more rapid death (red, compared to TDP-43 in black). Upregulation of dAtx2 on its own has no effect (purple). Reduction of dAtx2 significantly extends lifespan (green, compared to TDP-43 in black). Heterozygous loss of dAtx2 on its own has no effect (not shown). quantitative-RT-PCR showed that TDP-43 expression has no effect on levels of dAtx2 transcript in dAtx2EP flies. Genotypes. elav/+: elavC155/Y. elav/dAtx2EP: elavC155/Y; +/dAtx2EP3145. elav, TDP-43/+: elavC155/Y; UAS-TDP-43(M)/+. elav, TDP-43/dAtx2EP: elavC155/Y; UAS-TDP-43(M)/+; +/dAtx2EP3145. elav, TDP-43; dAtx2X1/+: elavC155/Y; UAS-TDP-43(M)/+; +/dAtx2X1. e) TDP-43 immunoblot upon dAtx2 modulation. Reduction of dAtx2 has no effect on TDP-43 levels, whereas upregulation of dAtx2 enhances TDP-43 protein levels. dAtx2 has no effect on the transgene expression system and 2x TDP-43 does not produce the same phenotype as TDP-43+dAtx2EP (see Figs. S2 and S3). Genotypes: —, control lane. TDP-43 lanes, —: UAS-TDP-43(M)/+, gmr-GAL4(YH3)/+. dAtx2X1:UAS-TDP-43(M)/+, gmr-GAL4(YH3)/dAtx2X1. dAtx2EP: UAS-TDP-43(M)/+, gmr-GAL4(YH3)/dAtx2EP3145. Bottom, quantitation of immunoblots from independent experiments; normalized to tubulin.
TDP-43 and Ataxin-2 interactions
Given the striking effects of Ataxin-2 on TDP-43 toxicity in yeast and flies, we determined whether the two proteins could physically interact in yeast and human cells. Yeast cells were transformed with YFP-tagged TDP-43 and either CFP-tagged Pbp1 or CFP alone, and the localization of the proteins visualized by fluorescence microscopy. Upregulation of Pbp1 increased the number of TDP-43-YFP foci present in yeast cells compared to situations with TDP-43-YFP (23 and A.C.E. and A.D.G. unpublished observations). These studies showed that Pbp1 localized to TDP-43 cytoplasmic accumulations (Fig. S5). To determine whether Pbp1 and TDP-43 could associate in the same protein complex, we performed immunoprecipitation assays with an antibody directed against the Pbp1 epitope tag followed by immunoblotting to detect TDP-43. These studies confirmed the ability of TDP-43 to interact with Pbp1 in the same complex (Fig. S5). To determine whether this interaction was conserved in human cells, HEK293T cells were transfected with YFP-tagged TDP-43 or YFP alone. TDP-43-YFP, but not YFP alone, immunoprecipitated endogenous human Ataxin-2 (Fig. 3a). This interaction was specific to Ataxin-2 because Ataxin-3 did not co-immunoprecipitate with TDP-43 (Fig. S6). Finally, an ALS-linked TDP-43 mutant (Q331K) also interacted with Ataxin-2 (Fig. S6). These data indicate that Ataxin-2 and TDP-43 can physically interact in both yeast and human cells.
Figure 3.

Ataxin-2 and TDP-43 interact in a manner dependent on RNA. a) TDP-43 and Ataxin-2 associate in mammalian cells in a manner dependent on the RRMs. HEK293T cells were transfected with plasmids encoding YFP, TDP-43-YFP, TDP-43 ΔNLS-YFP (NLS mutant localizes to cytoplasm), TDP-43 ΔNLS,5F→L-YFP (NLS mutant + RNA-binding mutant), or TDP-435F→L-YFP (RNA-binding mutant). Protein was immunoprecipitated with anti-GFP antibody, and then subjected to immunoblotting with anti-Ataxin-2 to detect endogenous Ataxin-2. Whereas TDP-43 and TDP-43 ΔNLS both interact with Ataxin-2, RNA-binding mutant versions do not. b) Co-IP in HEK293T cells as in (a), but now with lysates treated with RNase. The interaction between Ataxin-2 and TDP-43 seen normally was abolished upon RNase treatment. c) HEK293T cells transfected with YFP-tagged WT and mutant TDP-43 constructs then immunostained for endogenous Ataxin-2. Normally, Ataxin-2 is localized to the cytoplasm forming occasional cytoplasmic accumulations. TDP-43 localized to the nucleus in a diffuse pattern. TDP-43 ΔNLS localized to the cytoplasm where it occasionally formed cytoplasmic aggregates; these aggregates always co-localized with Ataxin-2 cytoplasmic accumulations (arrow). Abolishing the ability of TDP-43 to interact with RNA (TDP-43 ΔNLS, 5F→L or TDP-435F→L) eliminated Ataxin-2 colocalization (arrowheads). TDP-435F→L-YFP was restricted to the nucleus where it formed multiple foci. Scale bar is 2.5 μm for merge panels and 0.5 μm for high mag. merge panels. d) Yeast spotting assays for TDP-43 toxicity. Whereas WT and TDP-43 ΔNLS constructs are toxic, mutations of TDP-43 that prevent RNA binding abolish toxicity.
Because both TDP-43 and Ataxin-2 are involved in RNA metabolism22,23, we considered that RNA binding may be important for the TDP-43/Ataxin-2 interaction. TDP-43 is an RRM-containing protein with highly conserved RNP-1 and RNP-2 consensus motifs in each RRM. Within these motifs, specific aromatic residues have been shown to be necessary for RNA base stacking interactions and mutation of these residues (Phe to Leu) reduces the ability of TDP-43 to bind RNA in vitro22. To address the significance of RNA binding by TDP-43, we mutated all 5 of these residues to generate a TDP-43(5F→L)-YFP protein, and determined the effect on the interaction with Ataxin-2. Mutation of the RRM domain abolished the ability of TDP-43 to interact with Ataxin-2 (Fig. 3a), suggesting that RNA likely serves as a bridge between the two proteins. To rule out potential effects of these RRM mutations on protein-protein interactions or TDP-43 stability, we performed several additional control experiments. We tested single, double, and triple mutants within the RRMs of TDP-43 and found that even single mutations markedly diminished the interaction with Ataxin-2 (Fig. S6), with double and triple mutations abolishing the interaction completely (Fig. S6). To further demonstrate a role for RNA in mediating the TDP-43/Ataxin-2 interaction and exclude potential deleterious effects of point mutations on TDP-43 folding or stability, we performed the immunoprecipitation with full-length WT protein in the presence of RNase. RNase treatment abolished the interaction between TDP-43 and Ataxin-2 (Fig. 3b). Finally, HEK293T cells were transfected with YFP-tagged WT and RRM-domain mutant TDP-43 constructs and immunostained for endogenous Ataxin-2 (Fig. 3c). Consistent with previous reports, Ataxin-2 was predominantly localized to the cytosol and occasionally formed punctate cytoplasmic accumulations24. TDP-43-YFP appeared mostly in the nucleus, though has been reported to shuttle back and forth to the cytoplasm6, However, a form of TDP-43 with a mutated nuclear localization signal (NLS; ΔNLS-TDP-43-YFP) to restrict TDP-43 to the cytosol, which is its presumed pathogenic localization in disease6, remained in the cytosol where it occasionally formed aggregates; these aggregates always co-localized with Ataxin-2 (Fig. 3c). Strikingly, mutating the RRM domains of TDP-43 in the context of the ΔNLS construct, despite also resulting in TDP-43 aggregation in the cytosol, never led to co-localization with Ataxin-2 (Fig. 3c). In addition to blocking the interaction between TDP-43 and Ataxin-2, mutating the RRMs of TDP-43 also completely eliminated TDP-43 toxicity (Fig. 3d). Taken together, these data indicate that TDP-43 and Ataxin-2 can, though perhaps transiently, interact in a complex in the cytoplasm—the site of toxic function of TDP-43 in disease—and that this interaction likely depends on RNA binding. In a broader sense, these results also highlight that cytoplasmic aggregation per se is not sufficient for toxicity but that an RNA binding component is also involved, which provides some mechanistic insight into TDP-43 pathogenesis.
Ataxin-2 localization perturbed in ALS
The genetic interactions between TDP-43 and Ataxin-2 in yeast and Drosophila, and the physical association in yeast and mammalian cells, suggested that Ataxin-2 might show abnormal localization in human disease. To address this, we examined Ataxin-2 localization in spinal cord neurons from six ALS patients and three neurologically normal controls (Fig. 4). Normally, Ataxin-2 is localized in a diffuse or fine-granular pattern throughout the cytoplasm of spinal cord neurons (Fig. 4a, b). However, in ALS spinal cord neurons, Ataxin-2 localization was altered, showing more distinct cytoplasmic accumulations (see arrows, Fig. 4c, d; 27% of ALS spinal cord neurons vs. 5% of control neurons). Interestingly, we did not observe a significant difference in Ataxin-2 localization in spinal cord neurons from ALS patients with normal or expanded Ataxin-2 polyQ lengths (Fig. 4e, cases 1–3 vs. cases 4–6; see below). Importantly, mislocalization of Ataxin-2 in ALS appeared specific because Ataxin-3 localization was unaffected (Fig. S7). These studies indicate that Ataxin-2 localization is altered in spinal cord neurons of ALS patients (see Supplemental Results for further analysis of Ataxin-2 localization in FTLD-TDP and TDP-43 analysis in SCA2).
Figure 4.

Ataxin-2 localization is perturbed in ALS patient neurons. a–d, immunostaining for Ataxin-2 in spinal cord. a–b) In control spinal cord neurons, Ataxin-2 is localized throughout the cytoplasm in a diffuse pattern (c–f and Fig. S10). In ALS spinal cord neurons, Ataxin-2 was present in distinct cytoplasmic accumulations (arrows). In some cases, Ataxin-2-positive accumulations were adjacent to clearings indicative of TDP-43 aggregates (* in (b)). e, f) Quantitation of Ataxin-2 large accumulations in control (normal) vs. ALS spinal cord neurons. 27.2 +/− 12.3% of spinal cord neurons in ALS patients had large accumulations of Ataxin-2 compared to 4.7+/− 2.6% of control neurons. ALS patients with normal (ALS 1–3) and intermediate-length (ALS 4–6) Ataxin-2 polyQ repeats (See Fig. 5) were included and the Ataxin-2 pathology was not significantly different. An ALS case with both an SOD1 mutation and Ataxin-2 polyQ expansion (27Q) was included (case ALS-4). Scale bar, 1.25 μm for a; 5 μm for b–d.
Ataxin-2 polyQ expansions in ALS
These genetic, biochemical, and neuropathological interactions between Ataxin-2 and TDP-43 raised the possibility that mutations in Ataxin-2 could play a causative role in ALS. The Ataxin-2 polyQ tract length, though variable, is most frequently 22–23, with expansions of > 34 causing SCA210–13. However, the variable nature of the Ataxin-2 repeat suggested a mechanism by which such mutations in Ataxin-2 could be linked to ALS: we hypothesized that intermediate-length repeat expansions greater than 23 but below the threshold for SCA2 (e.g. 24–34) may be associated with ALS (Fig. 5a and Fig. S1). To test this, the Ataxin-2 polyQ repeat length was defined in genomic DNA from 915 individuals diagnosed with ALS and 980 neurologically normal controls (Fig. 5b). We found that 24 of 980 control cases (2.4%) were found to harbor a single intermediate-length Ataxin-2 allele, while 50 of 915 ALS cases (5.5%) possessed one allele with an intermediate-length Ataxin-2 repeat (mean repeat length 28, range 24–33, p=8.0×10−4, odds ratio 2.3 with 95% confidence interval of 1.41–3.76). Ataxin-2 polyQ repeat lengths of 22 and 23 are the most common alleles, however our findings and other studies have reported slightly longer repeat lengths in some control individuals10,11,13. Receiver operating characteristic (ROC) analysis of our dataset revealed that a cutoff of ≥27 polyQ repeats in Ataxin-2 provided the greatest sensitivity and specificity for discriminating normal vs. ALS. By using this cutoff (≥27 Qs), the association with ALS is even stronger (Table 1, p=3.6×10−5, odds ratio 2.8 with 95% confidence interval of 1.54–5.12). Notably, in 980 neurologically normal controls, we identified only 3 individuals with expansions >28 (all 3 of these were under age 60), whereas 24 ALS cases fell in this range (Fig. 5c). Moreover, we never observed an Ataxin-2 polyQ repeat length greater than 31 in controls, whereas 10 ALS patients harbored repeat lengths of 32 or 33 (Fig. 5c). Thus, intermediate-length Ataxin-2 polyQ repeat expansions are significantly associated with ALS.
Figure 5.

Intermediate-length Ataxin-2 polyQ expansions linked to ALS. a) The ATXN2 gene contains a trinucleotide repeat encoding polyQ. The repeat length is normally 22–23Q. Expansions of > 34 cause SCA212. We hypothesized that intermediate-length polyQ expansions (e.g. 24–34) could be linked to ALS. The Ataxin-2 polyQ length was defined by Genescan analysis of ALS cases and neurologically normal controls (for details, see Table 1 and Methods). b) Representative examples of Genescan analysis of polyQ lengths from control and ALS cases. c) The distribution of Ataxin-2 polyQ repeat lengths in ALS and control cases. PolyQ lengths ≥27 are significantly enriched in ALS vs. controls. Additionally, polyQ lengths >31 were never observed in our controls but we found 10 ALS patients above this threshold. d) In a selected cohort of ALS patients (n=65), those with Ataxin-2 polyQ expansions showed significantly lower age of onset (compared by survival analysis; also see Table 2).
Table 1.
Increased frequency of intermediate-length Ataxin-2 polyQ repeat expansions in ALS
| Total | ≤ 26 Repeats | 27–33 Repeats | % 27–33 Repeats | P-value | OR (95% CI) | |
|---|---|---|---|---|---|---|
| ALS | 915 | 872 | 43 | 4.7% | 3.6×10−5 | 2.80 (1.54–5.12) |
| Neurologically normal | 980 | 966 | 14 | 1.4% |
OR, odds ratio; CI, confidence interval
For a subset of the ALS cases (n=65; screened negative for mutations in SOD1, TARDBP, and FUS/TLS), extensive clinical details have been assessed. We interrogated the clinical characteristics in this cohort and compared the ALS cases with (n=8) and without (n=57) intermediate-length Ataxin-2 repeats (≥24 Qs). Strikingly, this analysis revealed that the age of onset was significantly earlier in ALS patients in this cohort with intermediate-length Ataxin-2 repeats (Fig. 5d, mean age 47.8 yrs vs. 59.4 yrs in ALS without intermediate-length Ataxin-2 repeat expansion, p=0.01), though it will be important to extend this analysis to a larger ALS population. Taken together, these data suggest a strong link between intermediate-length polyQ expansions in Ataxin-2 and ALS. In addition, Ataxin-2 associated ALS cases may have distinct characteristics, such as early age of onset (see also clinical anecdote in Supplemental Text).
Effects of Ataxin-2 polyQ expansions
To provide insight into how an intermediate-length Ataxin2 polyQ repeat could enhance pathogenesis we considered whether the expanded polyQ repeat might increase Ataxin-2 stability and/or affect its degradation, as is the case for other polyQ proteins25. Therefore, we analyzed Ataxin-2 protein levels in patient-derived lymphoblastoid cells from ALS cases harboring intermediate-length polyQ expansions, ALS cases with normal range repeat lengths, and controls. These studies showed that whereas the steady-state levels of Ataxin-2 were comparable, cycloheximide treatment, which blocks new protein synthesis, revealed an increase in stability (or decreased degradation) of Ataxin-2 in cells with intermediate-length polyQ repeats (Fig. S8). Thus, intermediate-length repeats could increase Ataxin-2 stability or inhibit its degradation, which could result in an increase in the effective concentration of Ataxin-2. This may further promote TDP-43 pathology beyond the interactions of Ataxin-2 harboring normal repeat lengths.
We next examined the effect of polyQ repeat expansions in Ataxin-2 on its interaction with TDP-43. We transfected HEK293T cells with YFP-tagged TDP-43 or YFP alone along with Ataxin-2 containing 22Q, 31Q, or 39Q (Figs. S8 and S9). As before, TDP-43-YFP, but not YFP alone, immunoprecipitated endogenous Ataxin-2 (Fig. 6c, long exposure). Interestingly, this interaction was enhanced by longer polyQ lengths; Ataxin-2 with 39Qs immunoprecipitated with TDP-43-YFP more robustly than Ataxin-2 with 22Qs (Fig. S8). These data suggest that polyQ expansions in Ataxin-2 might enhance its interaction with TDP-43.
Finally, although we were unable to detect a physical interaction by co-immunoprecipitation between endogenous TDP-43 and Ataxin-2, with or without polyQ expansions, in ALS patient-derived lymphoblastoid cells (data not shown), we did detect a functional interaction. Both Ataxin-2 and TDP-43 have been shown to re-localize to stress granules, sites of RNA processing, under various stress situations (heat shock, oxidative stress)26–28. Under normal conditions, TDP-43 was localized to the nucleus and Ataxin-2 to the cytoplasm, in both control cells and cells harboring Ataxin-2 polyQ repeat expansions (Fig. S8). However, following 1-hr heat shock at 44°C, Ataxin-2 coalesced into multiple discreet cytoplasmic foci (Fig S8). By blinded analysis, in the same situation, there was a significant increase in the number of cells with TDP-43 mislocalization to the cytoplasm in polyQ-expanded Ataxin-2 cases vs. controls (53% vs. 23%, p=0.02, n=4 Ataxin-2 expanded cases, n=3 control cases, Fig. S8). While further analysis is required in disease-relevant cells, these data suggest a mechanism by which intermediate-length Ataxin-2 polyQ repeats might confer genetic risk for ALS: by making TDP-43 more prone to mislocalize from the nucleus to cytoplasm under situations of stress.
Discussion
We present evidence for intermediate-length polyQ expansions in the Ataxin-2 gene as a potentially very common genetic contributor to ALS. This finding extends from a simple modifier screen in yeast for genes whose activity affects TDP-43 toxicity. Confirmation of these studies in the fly and human cells, followed by biochemical analysis in yeast and human cells revealed that Ataxin-2 and TDP-43 can associate in a complex, and that the interaction depends on RNA. Further, Ataxin-2 is abnormally localized in ALS patient motor neurons, and TDP-43 pathology characterizes SCA2. Whereas long polyQ expansions in Ataxin-2 are the cause of SCA2, our studies reveal that intermediate-length polyQ expansions of 27–33 are associated with ALS, with a frequency of 4.7% in cases unselected for family history. These findings indicate that intermediate-length polyQ expansions in Ataxin-2 may be the most common genetic risk factor for ALS defined to date (Fig. S1, see also Supplemental Discussion).
The identification of a novel and potentially common ALS disease gene from a simple yeast screen, leveraged by the more complex model organism Drosophila, underscores the extraordinary power of yeast and fly as model systems for gaining insight into human disease pathogenesis. There is no cure for ALS and currently the only treatment is riluzole, which slows disease progression by only 3 months29. The identification of pathological interactions between Ataxin-2 and TDP-43, together with the strong genetic association of Ataxin-2 intermediate-length polyQ expansions and ALS, will empower the development of new therapies for this devastating disease.
Methods Summary
The yeast TDP-43 modifier screen was performed similar to previous screens7,8. Transgenic flies expressing human TDP-43 were generated by standard techniques using the pUAST vector. Co-immunoprecipitation in yeast and mammalian cells (HEK293T) were performed using standard techniques. Lymphoblastoid cell lines were obtained from patients with ALS or unaffected normal controls (Coriell) and cultured according to instructions from Coriell. Detailed immunohistochemistry protocols are available at http://www.uphs.upenn.edu/mcrc. For Ataxin-2 polyQ repeat size determination, we amplified Ataxin-2 CAG repeats from individual samples by polymerase chain reaction (PCR). The 5′ primer was SCA2-Anew: 5′ - CCCCGCCCGGCGTGCGAGCCGGTGTATG -3′. The 3′ primer was SCA2-B: 5′ - CGGGCTTGCGGACATTGG - 3′. PCR cycles were as follows: 2 min 94°C, 35 cycles (1 min 94°C, 1 min 60°C, 1 min 72°C), and 5 min 72°C. We determined Ataxin-2 CAG repeat lengths by capillary electrophoresis, incorporating 6FAM fluorophor into PCR products in 5′ SCA2-Anew primer. PCR products were mixed with Liz-500 size standard (Applied Biosystems) and processed for size determination on an ABI3730 sequencer. Repeat sizes were determined with GeneMapper 4.0 software (Applied Biosystems).
Supplementary Material
Acknowledgments
This work was supported in part by a Pilot grant from the University of Pennsylvania Institute on Aging (A.D.G.), an NIH Director’s New Innovator Award 1DP2OD004417-01 (A.D.G.), 1R01NS065317-01 (A.D.G.), P01 AG-09215 (N.B.), AG-10124 (J.Q.T.) and AG-17586, (V.M.-Y.L.). A.D.G. is a Pew Scholar in the Biomedical Sciences, supported by The Pew Charitable Trusts. A.C.P. is supported by a Burroughs Wellcome Fund Career Award and NIH K08 AG-033101-01. N.M.B. is an Investigator of the Howard Hughes Medical Institute. U.R. has support from the DHAG and ADCA Vereniging Nederland, G.A. from EuroSCA and the DFG (AU96/11-1). We acknowledge Wilfred den Dunnen and Ewout Brunt for autopsy tissue and Mariette Babl for excellent technical assistance. We thank Jon Epstein, Jim Shorter, Tony Cashmore and members of the Gitler laboratory for comments on the manuscript and useful discussions. We are truly grateful for the dedication of the patients and their families and for their invaluable contributions to this research.
Footnotes
Author Contributions: M.P.H., H.J.K., A.C.P., F.G., U.R., G.A., J.Q.T., V.M.Y.L., V.M.V.D., N.M.B., A.D.G. designed experiments. A.C.E., H.J.K., M.P.H., B.S.J., X.F., M.A., R.G., M.M.L., U.R., A.D.G. performed research. D.J. and P.J.G. provided reagents. D.C. and V.M.V.D. collected clinical data. A.C.P., L.E., L.M. assessed clinical characteristics. M.P.H., H.J.K., A.C.P., F.G., A.P., L.E., L.M., U.R., G.A., J.Q.T., V.M.Y.L., V.M.V.D., N.M.B., A.D.G. analyzed and interpreted data. N.M.B. and A.D.G. wrote the paper with contributions from all authors.
References
- 1.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–19. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 2.Valentine JS, Hart PJ. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2003;100:3617–22. doi: 10.1073/pnas.0730423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 4.Pesiridis GS, Lee VM, Trojanowski JQ. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18:R156–62. doi: 10.1093/hmg/ddp303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–4. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winton MJ, et al. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283:13302–9. doi: 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper AA, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–8. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gitler AD, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41:308–15. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 10.Imbert G, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet. 1996;14:285–91. doi: 10.1038/ng1196-285. [DOI] [PubMed] [Google Scholar]
- 11.Lorenzetti D, Bohlega S, Zoghbi HY. The expansion of the CAG repeat in ataxin-2 is a frequent cause of autosomal dominant spinocerebellar ataxia. Neurology. 1997;49:1009–13. doi: 10.1212/wnl.49.4.1009. [DOI] [PubMed] [Google Scholar]
- 12.Pulst SM, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet. 1996;14:269–76. doi: 10.1038/ng1196-269. [DOI] [PubMed] [Google Scholar]
- 13.Sanpei K, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996;14:277–84. doi: 10.1038/ng1196-277. [DOI] [PubMed] [Google Scholar]
- 14.Infante J, et al. Spinocerebellar ataxia type 2 with Levodopa-responsive parkinsonism culminating in motor neuron disease. Mov Disord. 2004;19:848–52. doi: 10.1002/mds.20090. [DOI] [PubMed] [Google Scholar]
- 15.Nanetti L, et al. Rare association of motor neuron disease and spinocerebellar ataxia type 2 (SCA2): a new case and review of the literature. J Neurol. 2009 doi: 10.1007/s00415-009-5237-9. [DOI] [PubMed] [Google Scholar]
- 16.Mangus DA, Amrani N, Jacobson A. Pbp1p, a factor interacting with Saccharomyces cerevisiae poly(A)-binding protein, regulates polyadenylation. Mol Cell Biol. 1998;18:7383–96. doi: 10.1128/mcb.18.12.7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buchan JR, Muhlrad D, Parker R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J Cell Biol. 2008;183:441–55. doi: 10.1083/jcb.200807043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Ramahi I, et al. dAtaxin-2 mediates expanded Ataxin-1-induced neurodegeneration in a Drosophila model of SCA1. PLoS Genet. 2007;3:e234. doi: 10.1371/journal.pgen.0030234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lessing D, Bonini NM. Polyglutamine genes interact to modulate the severity and progression of neurodegeneration in Drosophila. PLoS Biol. 2008;6:e29. doi: 10.1371/journal.pbio.0060029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295:865–8. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 21.Warrick JM, et al. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–8. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 22.Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276:36337–43. doi: 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- 23.Lastres-Becker I, Rub U, Auburger G. Spinocerebellar ataxia 2 (SCA2) Cerebellum. 2008;7:115–24. doi: 10.1007/s12311-008-0019-y. [DOI] [PubMed] [Google Scholar]
- 24.Huynh DP, Yang HT, Vakharia H, Nguyen D, Pulst SM. Expansion of the polyQ repeat in ataxin-2 alters its Golgi localization, disrupts the Golgi complex and causes cell death. Hum Mol Genet. 2003;12:1485–96. doi: 10.1093/hmg/ddg175. [DOI] [PubMed] [Google Scholar]
- 25.Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell. 2004;14:95–104. doi: 10.1016/s1097-2765(04)00151-0. [DOI] [PubMed] [Google Scholar]
- 26.Colombrita C, et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111:1051–61. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- 27.Nonhoff U, et al. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell. 2007;18:1385–96. doi: 10.1091/mbc.E06-12-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freibaum BD, Chitta RK, High AA, Taylor JP. Global Analysis of TDP-43 Interacting Proteins Reveals Strong Association with RNA Splicing and Translation Machinery. J Proteome Res. 9:1104–20. doi: 10.1021/pr901076y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson M, Llado J, Rothstein JD. Therapeutic developments in the treatment of amyotrophic lateral sclerosis. Expert Opin Investig Drugs. 2002;11:1343–64. doi: 10.1517/13543784.11.10.1343. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.