

Abstract
Here we report that mouse embryos homozygous for a gene trap insertion in the fibulin-1 (Fbln1) gene are deficient in Fbln1 and exhibit cardiac ventricular wall thinning and ventricular septal defects with double outlet right ventricle or overriding aorta. Fbln1 nulls also display anomalies of aortic arch arteries, hypoplasia of the thymus and thyroid, underdeveloped skull bones, malformations of cranial nerves and hemorrhagic blood vessels in the head and neck. The spectrum of malformations is consistent with Fbln1 influencing neural crest cell (NCC)-dependent development of these tissues. This is supported by evidence that Fbln1 expression is associated with streams of cranial NCCs migrating adjacent to rhombomeres 2–7 and that Fbln1-deficient embryos display patterning anomalies of NCCs forming cranial nerves IX and X, which derive from rhombomeres 6 and 7. Additionally, Fbln1-deficient embryos show increased apoptosis in areas populated by NCCs derived from rhombomeres 4, 6 and 7. Based on these findings, it is concluded that Fbln1 is required for the directed migration and survival of cranial NCCs contributing to the development of pharyngeal glands, craniofacial skeleton, cranial nerves, aortic arch arteries, cardiac outflow tract and cephalic blood vessels.
Keywords: Fibulin, extracellular matrix, neural crest, anterior heart field, secondary heart field, heart development, cardiac abnormalities, outflow tract, bleeding, cranial nerves, DiGeorge syndrome, 22q11.2 deletion, motility, gene trap, knock-out
Introduction
Fibulin-1 (Fbln1) is the first described member of a seven-gene family of extracellular matrix (ECM) proteins that have a common structural signature consisting of a series of repeated EGF-like domains followed by a C-terminal element referred to as a ‘fibulin module’ (Argraves et al., 2003; Chu and Tsuda, 2004). Alternative splicing produces two Fbln1 variants in nematodes, chickens and mice and four variant polypeptides in humans. These variants differ at their C-termini and appear to have distinct biological activities based on their ability to differentially bind ECM proteins (Sasaki et al., 1995) and growth factors (Brooke et al., 2002; Perbal et al., 1999) and associate with distinct networks in the ECM (Muriel et al., 2006).
A major function ascribed to Fbln1 is regulation of cell motility and guidance. During C. elegans development, Fbln1 is required for proper guidance of migrating distal tip cells engaged in gonad morphogenesis (Kubota et al., 2004; Kubota and Nishiwaki, 2003). In Fbln1-deficient nematode embryos, an abnormal widening of sheets of gonadal cells occurs combined with a failure of distal tip cells to complete their normal migration to the midline of the animal (Hesselson et al., 2004). Based on findings from in vitro studies, Fbln1 can suppress the motility (i.e., migration velocity and persistence time) of many types of cancer cells (Hayashido et al., 1998; Lee et al., 2005; Qing et al., 1997; Twal et al., 2001). However, Fbln1 alone is apparently neither adhesive nor motility suppressive, but acts to suppress the motility promoting activity of other ECM proteins including fibronectin (FN) (Twal et al., 2001), one of its principal binding proteins (Balbona et al., 1992). Evidence that Fbln1 can inhibit motility promoting activity of other matrix proteins stems from its ability to inhibit the migration of cells through Matrigel (Qing et al., 1997), a basement membrane protein rich extract that lacks FN. This finding is consistent with the requirement for Fbln1 in regulation of distal tip cell guidance in C. elegans, a species whose genome does not encode FN and whose ECM is basement membrane-like (Kramer, 1997).
During vertebrate development, Fbln1 has been indirectly implicated as a regulator of cell motility based on the fact that it is expressed by migrating mesenchymal cells including endocardial cushion cells and neural crest cells (NCCs) (Bouchey et al., 1996; Kern et al., 2006; Spence et al., 1992; Zhang et al., 1995; Zhang et al., 1993). A number of Fbln1-binding ECM components including FN, versican, perlecan and laminin α5 have been implicated in the regulation of embryonic cell motility, particularly in the migration and guidance of cranial NCCs (Bronner-Fraser, 1993; Perris and Perissinotto, 2000). For example, in mice deficient in laminin α5 there are abnormalities in the migration of NCCs from rhombomeres 6 and 7. This results in improper condensation of the NCC primordia that give rise to cranial nerves IX and X (Coles et al., 2006). Similarly, deficiency of integrin β1, a subunit of integrins which bind both laminin α5 and FN (Kikkawa et al., 2000), leads to defective NCC migration from rhombomeres 6 and 7 and abnormal formation of cranial nerves IX and X (Pietri et al., 2004). Loss of ECM constituents that regulate NCCs can also result in cardiac defects. For example, deficiency in perlecan, a hyaluronan (HA) binding proteoglycan implicated in NCC migration, leads to increased numbers of mesenchymal cells in the cardiac outflow tract (OFT) and transposition of the great arteries (Costell et al., 2002). By contrast, increased expression of the proteoglycan versican, a known inhibitor of NCC migration (Dutt et al., 2006), correlates with a decrease in NCCs lateral to the neural tube and in the pharyngeal arches 4 and 6 of Pax 3 mutants (Henderson et al., 1997). These mutants exhibit a failure of OFT septation owing to insufficient numbers of NCCs in the OFT (Conway et al., 1997).
Here we report that mouse embryos deficient in Fbln1 display abnormalities of the OFT, cardiac septa, aortic arch arteries, pharyngeal glands, skull bones, cranial nerves and blood vessels of the head and neck. These and other findings presented support the conclusion that Fbln1 plays an important role in the process of NCC guidance and survival required for proper morphogenesis of cranial neural crest-derived structures.
Materials and Methods
Generation of Fbln1 gene trap mutant mice
To develop a Fbln1-deficient mouse strain we used an ES cell line, XST011 (parental cell line E14Tg2a.4 derived from 129P2/OlaHsd strain), which has a gene trap insertion in the Fbln1 gene (Skarnes et al., 1995; Stryke et al., 2003) (Bay Genomics, San Francisco, CA). The inserted gene trap element is comprised of a strong splice acceptor, a CD4 transmembrane domain, a β-galactosidase (β-gal) cassette and a neomycin resistance cassette (Neo). By inclusion of a CD4 transmembrane domain upstream from the β-gal insertion element, the truncated Fbln1-CD4-β-gal fusion protein is targeted for insertion into the endoplasmic reticulum (ER) in a type I configuration such that it is retained tethered to the ER membrane with the β-gal domain facing the cytosol (Skarnes et al., 1995). The XST011 ES cells were injected into C57BL/6 blastocysts that were then transferred to foster mothers to obtain chimeric mice. Two germ line competent male chimeras were generated and bred with C57BL/6 mice.
Genotyping
The genotypes of offspring were determined from tail clip genomic DNA by PCR using three primers. To detect the wild-type Fbln1 allele, PCR was performed using Fbln1 primers, 5′-AGCCCCAGCTGATTCTGAACTCCTGACC-3′ (residues 71058–71085 in GI: 15591330) and 5′-GCAACAGCAGTGTTGGGTGGAGGAAGGG-3′ (residues 71366–71339 in GI: 15591330). To detect homozygotes, the latter primer was used with a CD4 primer, 5′-CTCCTACATAGTTGGCAGTGTTTGGG-3′ (residues 743–768 from plasmid pGT2TMPFS, Bay Genomics). Cycling parameters for PCR were: 39 cycles of 95°C for 50 sec, 53°C for 30 sec and 72°C for 2 min. The expected size for the ampli con produced from the wild-type allele is 308 bp. The expected size for the amplicon produced from the targeted Fbln1 allele is 414 bp.
RT-PCR
To confirm that embryos homozygous for the gene trap insertion were deficient in each of the two mouse Fbln1 splice variants, Fbln1C and D, PCR was performed on cDNA from E9.5 embryos using a sense strand primer, 5′-CCCAATGGCCGCAACTGCCAAGACATTG-3′ (GI:13938048 Fbln1C and Fbln1D GI: 396820) and two antisense strand primers, 5′-GCCCTCCTCATTGCCAGCGGTGATGGC-3′ (residues 1959–1934 in GI: 13938048) and 5′-GGAGTCTCGAAGGTTCCCTTCTGTGATG-3′ (residues 2061–2034 in GI: 396820). Cycling parameters for PCR were: 30 cycles of 95°C for 30 sec, 60°C for 30 sec and 72°C for 1 min. The expected size for the C-specific amplicon was 332 bp and the expected size for the D-specific amplicon was 380 bp.
Mapping of the gene trap insertion element within the mouse Fbln1 gene
To map the position of the gene trap insertion element within intron 14, a series of forward primers were designed based on sequence from Fbln1 intron 14 and used in PCR together with an antisense primer 5′-GGTCCCATCACCTCACAGGTCAAAG-3′ (derived from the insertion cassette sequence in pGT2TMPFS). Amplified products were cloned and sequenced to identify the insertion site.
To determine the consequence of the gene trap insertion on Fbln1 mRNA splicing, RT-PCR analysis was performed using RNA isolated from E9.5 heterozygous embryos. A Fbln1 sense strand primer 5′-CCTCATCTGGCTACAGGCTAGCTCCC-3 (residues 1658–1683 GI:396820) and a CD4 antisense strand primer 5′-GGTCCCATCACCTCACAGGTCAAAG-3′ (residues 1167–1143 GI:7304952) were used to generate a fragment that contains the junctional region between Fbln1 and the CD4 transmembrane region. The deduced amino acid sequence of the resulting 500 bp fragment is presented as Supplementary Information (Supplementary Fig. 2).
Histology and immunohistochemistry
Embryos were harvested from pregnant females following matings of mice heterozygous for the Fbln1 gene trap insertion. A piece of the yolk sac was isolated for genotyping. Embryos were fixed in phosphate buffered saline (PBS) containing 4% paraformaldehyde and then embedded in paraffin and sectioned at 5 μm thickness. Immunohistochemical staining was performed on deparaffinized and rehydrated sections subjected to antigen unmasking using a high temperature citric acid protocol (H-3300, Vector Laboratories Burlingame, CA). Sections were incubated with rabbit anti-Fbln1 (15 μg/ml) (Argraves et al., 1990), anti-Crabp1 (1:100, Abcam, Cambridge, MA), anti-α-sarcomeric actin (1:500 ascites fluid, Sigma Chemical Corp, St. Louis, MO), anti-Pax3 (5 μg/ml, Developmental Studies Hybridoma Bank (DSHB) The University of Iowa, Department of Biological Sciences, Iowa City, IA) or anti-α-smooth muscle actin (5 μg/ml, Sigma). For mouse monoclonal anti-Fbln1 (3A11) (Tran et al., 1997) immunolabeling, a proteinase K treatment (20 μg/ml in PBS) was used in place of the citric acid procedure. Bound antibodies were detected with fluorescently conjugated secondary antibodies (Jackson Laboratories, Bar Harbor, ME). Nuclei were labeled with 1 μg/ml propidium iodide (Molecular Probes, Invitrogen Corp., Carlsbad, CA) in PBS for 5 min prior to a final series of washes in PBS. Sections were analyzed using a Leica TCS SP2 AOBS Confocal Microscope System (Leica Microsystems Inc., Exton, PA). Hematoxylin and eosin (H&E) staining was performed using standard procedures. Alizarin red/Alcian blue staining was performed on whole mounts as described (Kimmel and Trammell, 1981). TUNEL-based apoptosis analysis was performed on embryo sections using the ApopTag system (Chemicon International, Temecula, CA).
Neurofilament antibody labeling was performed on E10.5 whole mount embryos incubated for 12 h with PBS containing 1% fetal bovine serum (FBS, Atlantic Biologicals, Lawrenceville, GA), 1% TritonX-100 (PBS/FBS/Triton) with 0.1%H2O2. The embryos were washed with PBS/FBS/Triton for 4 h and incubated with 0.4 μg/ml neurofilament-M monoclonal antibody 2H3 in PBS/FBS/Triton. The monoclonal antibody 2H3, developed by Drs. Jessell and Dodd, was obtained from the DSHB. After washing the embryos were incubated with anti-mouse-HRP (GE Biosciences, Piscataway, NJ) and the chromogenic substrate DAB (Pierce, Rockford, IL).
Whole heart confocal imaging by cardiac endopainting
Whole heart confocal imaging was performed using a modification of the endocardial painting procedure described by Miller et al. (Miller et al., 2005). Briefly, timed pregnant mice of fibilin-1 heterozygous intercross matings were dissected and embryos harvested in PBS-containing heparin (1000 units/ml). Embryos (E17.5) were bisected in the abdomen to allow perfusion through the hepatic sinus with heparin-containing relaxation buffer (PBS, 1% lidocaine) using pulled glass needles. Hearts were then perfused with 0.05% FITC-poly-L-lysine (Sigma), which binds the endothelium of vessels and internal heart structures. The embryos were fixed in 4% paraformaldehyde for 2 h. Hearts were removed from the thoracic cavity and mounted in a precise frontal plane (Miller et al., 2005). Dehydration was performed prior to incubation in Murray’s clear. Hearts were examined by confocal microscopy using the 5X objective in 25 μm optical sections over a total depth of 800 μm.
In situ hybridization
Whole-mount in situ hybridization of E9.5-10.5 embryos was performed using antisense riboprobes for Fbln1, Sox-10 and Crabp1. cDNA inserts corresponding to each of these genes were generated using the following primer pairs: 5′-CCAAATCAGATGGCTAACCAGCACAGGGAC-3′ and 5′-ACTGGAAGCTTCCGGGGATGTTGATG-3′ for Fbln1 (GI: 6753821); 5′-CCAGGGTGTTTGGTGGTGAGGA-3′ and 5′-CAGCTCAGTCAGGGCTTGGCCT-3′ for Sox10 (GI:94399599); and 5′-AACGCAGCGTGCGACCGCTCCGCAGC-3′ and 5′-CCCCTCAAGAAGTGTCTGTGTGCAG-3′ for Crabp1 (GI:7304974). The resulting cDNA were subcloned into pCRII (Invitrogen) and used as templates to generate digoxigenin-labeled antisense riboprobes. Whole-mount in situ hybridizations were carried out essentially according to methods described in Hogan et al. (1994). DIG-labeled anti-sense RNA probes were detected by BCIP/NBT substrate (Roche, Indianapolis, IN).
Whole-mount β-galactosidase analysis
Embryos were fixed in 4% paraformaldehyde/PBS for 1 h followed by a 12 h incubation in PBS containing 0.02% sodium deoxycholate and 0.01% NP-40 at 4°C. To detect β-gal activity, embryos were incubated in PBS containing 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM MgCl2 and 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal; Pierce), for 1–4 h at 37°C. The embryos were then transferred to buffered formalin for 1 h and then stored in PBS with 0.02% sodium azide.
Fbln1 exon 1 deletion mouse
The mouse strain described by Kostka et al. (2001) carrying a targeted deletion of Fbln1 exon 1 (on a mixed C57BL/6 and 129/Sv background) was provided by Dr. Mon-Li Chu (Thomas Jefferson University). Mice were genotyped using the following primer pairs: 5′-TCACGTGACTTTCTGCCTGT-3′ and 5′-GTCCTGGGTGGGGACCTAGTG-3′ for detecting the wild-type Fbln1 allele and 5′-TCTGAGCTTTGCGCTCT-3′ and 5′-TAAAGCGCATGCTCCAGACTGC-3′ for detecting the allele containing the Fbln1 exon 1 deletion.
Results
Fbln1 gene disruption by gene trap insertion
Mapping of the gene trap insertion element within the mouse Fbln1 gene (Fig. 1A) revealed that the start of the element was at position 71145 (GI: 15591330) within intron 14 (Fig. 1B and Supplementary Fig. 1 for the sequence of the 5′ junction region of the insertion within intron 14).
Figure 1. Disruption of the Fbln1 gene by gene trap insertion.
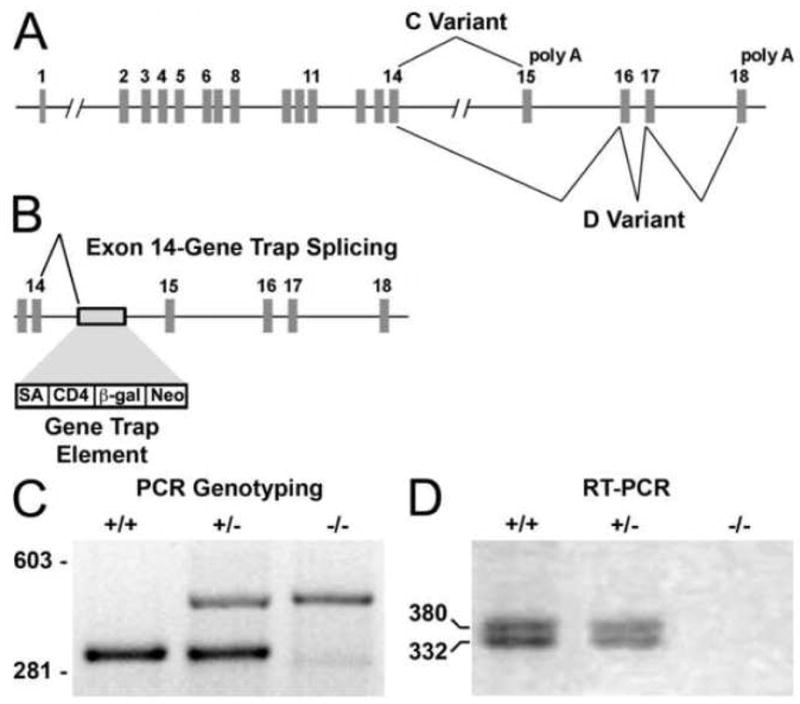
A shows the arrangement of the exons and introns in the mouse Fbln1 gene. The exons that encode the alternatively spliced portions of Fbln1C and D transcripts are connected by lines. B shows the location of the gene trap element within intron 14 and the consequential aberrant splicing of exon 14 to the splice acceptor site (SA) of the gene trap element. C, PCR genotyping of DNA isolated from a wild-type (+/+) embryo and embryos heterozygous (+/−) and homozygous (−/−) for the Fbln1 allele containing the gene trap element. D, RT-PCR analysis of RNA isolated from a wild-type embryo (+/+) and embryos heterozygous (+/−) and homozygous (−/−) for the gene trap insertion. In D, the larger product corresponds to the amplicon produced from the Fbln1D transcript and the smaller product from Fbln1C mRNA. CD4, CD4 transmembrane domain; β-gal, β-galactosidase element.
To determine the effect of the gene trap insertion on splicing of the Fbln1 transcript, RT-PCR was performed using a primer from exon 13 and a primer from the CD4 component of the gene trap element. DNA sequencing of the resulting amplicon revealed that the gene trap insertion caused an aberrant splicing of exon 14 to the splice acceptor site of the gene trap cassette. The mis-spliced mRNA encoded the first 568 amino acids of the Fbln1 protein in-frame with the CD4 transmembrane containing segment and β-gal element (Supplementary Fig. 2). Additional RT-PCR analysis showed that embryos (E9.5) homozygous for the gene trap insertion lacked transcripts encoding exons located downstream from the insertion element (i.e., exons 15–18) (Fig. 1D). These findings indicate that homozygous embryos lack Fbln1C and D transcripts.
To assess the effects of the gene trap insertion on in vivo Fbln1 protein expression, immunoblot and immunohistological analyses were performed. By immunoblot analysis using a Fbln1 polyclonal antibody there was no immunoreactive Fbln1 polypeptide in 8M urea extracts from embryos homozygous for the Fbln1 gene trap insertion (Fig. 2A). Heterozygous embryos showed approximately half the level of Fbln1 present in wild-type embryos extracts.
Figure 2. Embryos homozygous for the Fbln1 gene trap insertion have no immunologically detectable Fbln1.
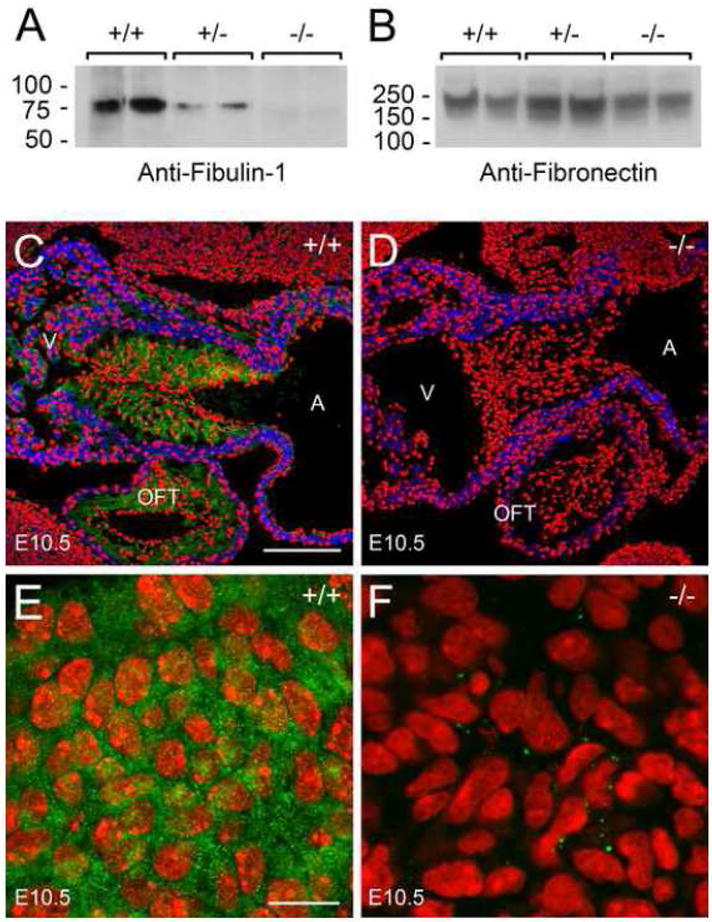
A and B, 8M urea extracts from wild-type embryos (+/+), embryos heterozygous (+/−) and homozygous (−/−) for the Fbln1 gene trap insertion (E13.5) were subjected to immunoblot analysis using polyclonal antibodies to Fbln1 (A) and FN (B). C and D, anti-Fbln1 (green) and anti-α-sarcomeric actin (blue) immunolabeling of tissue sections containing embryonic hearts from wild-type (+/+) embryos (C) and embryos homozygous for the Fbln1 gene trap insertion (−/−) (D) (E10.5). Nuclei were stained using propidium iodide (red). A, atrium; V, ventricle; OFT, outflow tract; AVC, AV cushion. E and F, monoclonal Fbln1 antibody 3A11 immunolabeling of tissue sections containing pharyngeal arch of wild-type E10.5 embryo (E) and an E10.5 embryo homozygous for the Fbln1 gene trap insertion (−/−) (F). The epitope for monoclonal antibody 3A11, which has been mapped to amino acid residues 30–153 of Fbln1, is contained within the Fbln1-CD4-β-gal fusion protein expressed in Fbln1 gene trap mutants since the fusion protein contains the first 568 amino acid residues of the Fbln1 polypeptide. Nuclei were stained using propidium iodide (red). Bar in C = 150 μm and also applies to panel D. Bar in E = 10μm and applies to F.
Embryonic heart tissue sections from wild-type embryos and embryos homozygous for the Fbln1 gene trap insertion (E10.5) were next evaluated by immunohistochemistry. In wild-type embryos, Fbln1 staining is prominent in the ECM surrounding mesenchymal cells of the AV and OFT cushions (Fig. 2C). By contrast, no Fbln1 staining was evident in these tissues (Fig. 2D) or any other tissues examined from Fbln1 gene trap mutants (data not shown). Using a recombinant amino terminal fragment of mouse Fbln1 (amino acid residues 1–568), we confirmed that the polyclonal antibody used in immunohistochemistry and immunoblotting was reactive with the truncated portion of Fbln1 in the Fbln1-CD4-β-gal fusion protein (data not shown). We performed additional immunohistochemical analysis using a Fbln1 monoclonal antibody directed to an amino terminal epitope contained in the Fbln1-CD4-β-gal fusion protein (Tran et al., 1997). Pronounced Fbln1 staining was evident in the wild-type tissues (E10.5) (Fig. 2E), but extremely low levels were detectable in tissues of embryos homozygous for the gene trap (Fig. 2F). Furthermore, the low levels of immunoreactive material appeared as discrete intracellular foci. Based on these findings, it can be concluded that the Fbln1-CD4-β-gal fusion protein becomes quantitatively degraded similar to what has been described in mice homozygous for other genes containing secretory gene trap insertions (Schymeinsky et al., 2002; Zhou et al., 2004). Taken together, the findings indicate that embryos homozygous for the Fbln1 gene trap insertion are effectively Fbln1-deficient.
Lethality occurs in embryos homozygous for the Fbln1 gene trap insertion
Genotypic analysis was performed on 117 offspring from heterozygous matings. As a result, 37 wild-type, 80 heterozygous and 0 homozygous offspring were detected (Supplementary Table 1). This non-Mendelian ratio indicated that lethality was occurring in offspring homozygous for the gene trap insertion. To determine the stage of lethality, retrograde genotypic analysis was performed on embryos from timed heterozygous matings. From E9.5-18.5, homozygous embryos were detected at a frequency in accordance with Mendelian expectations (Supplementary Table 1). These findings indicated that homozygous embryos were dying at birth. When litters were examined immediately after birth, null embryos were observed that were hemorrhagic and either dead or experiencing labored breathing.
Fbln1−/− embryos have blood vessel and lung anomalies
Morphological analysis revealed that homozygous embryos exhibit bleeding within the eyes and petechial bleeding in the head and neck (Fig. 3). In addition, fewer superficial blood vessels are apparent in the heads of mutant embryos (Fig. 3A). These abnormalities, which were 100% penetrant, could be seen as early as E14.5 and the severity increased with gestational age. In later stage null embryos (E18.5), blood cells could be found in extravascular spaces around the spinal cord. Several homozygous embryos also displayed significant edema on the back of the head overlying the 4th ventricle. Upon histological examination of the eyes of E15.5 Fbln1-deficient embryos, the hyaloid blood vessels, which extend through the hyaloid cavity to the caudal surface of the lens capsule, appear distended and filled with blood cells (Fig. 3D and E).
Figure 3. Mice homozygous for the Fbln1 gene trap insertion display a hemorrhagic phenotype.
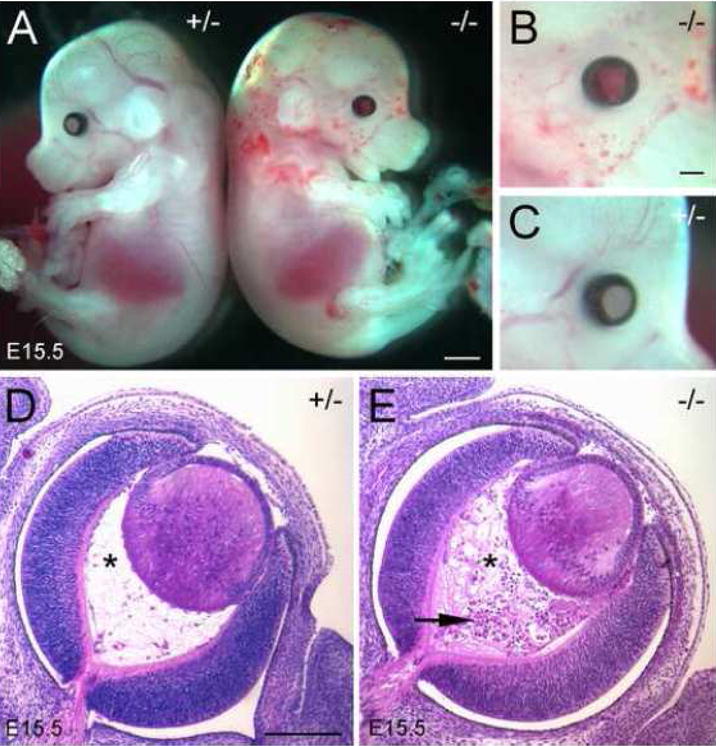
A shows E15.5 embryos heterozygous (+/−) and homozygous (−/−) for the Fbln1 gene disruption. B and C, high magnification views of the facial regions of the homozygous and heterozygous embryos shown in A. B, blood filled eye and petechial bleeding in head, neck and proximal portion of the forelimb and fewer superficial blood vessels are apparent in the mutant embryo. D and E, H&E stained sections of eyes from E15.5 embryos heterozygous (D) and homozygous (E) for the Fbln1 gene trap insertion. Asterisks indicate the hyaloid cavity. Arrow indicates blood cell accumulation in the hyaloid cavity. Bar in A = 1 mm. Bar in B = 700μm and also applies to C. Bar in D = 200μm and also applies to E.
The lungs of the Fbln1 null embryos (E17.5) appeared smaller than normal. Histological analysis reveals that the lungs of homozygous embryos have a greater cell density as compared to lungs of wild-type or heterozygote embryos of the same gestational age (Supplementary Fig. 3). Most notable is that lungs of Fbln1 nulls have few saccules.
Outflow tract abnormalities in Fbln1−/− embryos
Abnormalities of the remodeling of the OFT were evidenced by the high incidence of double outlet right ventricle (DORV) (Fig. 4B and Supplementary Fig. 4A–F) and overriding aorta in homozygotes (Table 1). Persistent truncus arteriosus (PTA), resulting from a failure of OFT septation, was not observed in Fbln1−/− embryos. Together, these observations indicate that Fbln1 is not required for the septation of the aorta and pulmonary artery, but that it is required for the proper rotation of the aorta and pulmonary arteries during remodeling of the OFT.
Figure 4. Fbln1−/− hearts have DORV, VSD and aortic arch artery abnormalities.
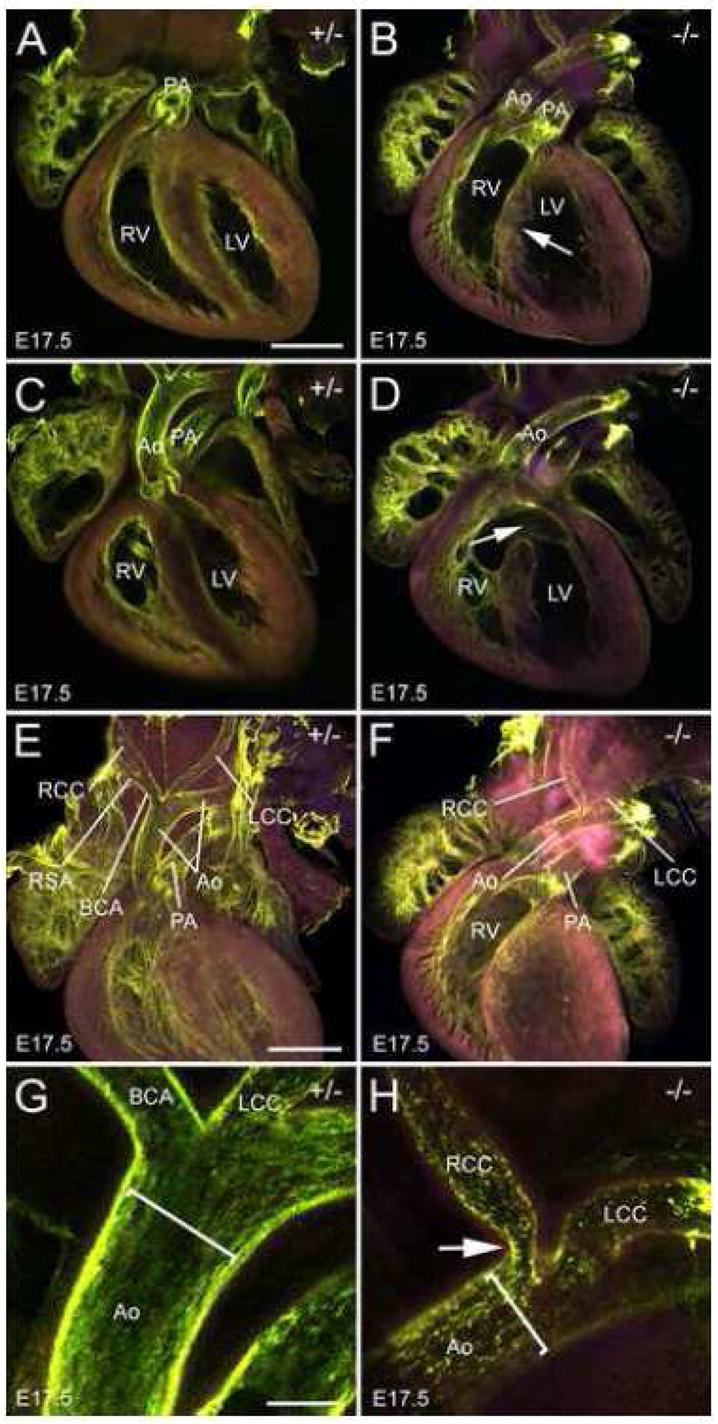
Shown are confocal images of whole mount E17.5 hearts perfused with FITC-poly-L-lysine (green). A and C are single optical sections from hearts of embryos heterozygous for the Fbln1 gene trap insertion (+/−). B and D are single optical sections from embryos homozygous for the Fbln1 gene trap insertion (−/−). A DORV and VSD (arrow) are apparent in B. A VSD (arrow) is also apparent in D. In the heterozygous heart (A), the pulmonary leaflets are located dorsal to the aortic leaflets (175μm apart) whereas in the homozygous heart (B), the aortic and pulmonary leaflets appear near the same focal plane. The later configuration is consistent with a failure in the rotation of the aorta and pulmonary valve sites during OFT remodeling. E and G, optical sections from hearts of embryos heterozygous for the Fbln1 gene trap insertion. F and H, optical sections from embryos homozygous for the Fbln1 gene trap insertion. In F, the right subclavian artery (RSA) is absent from the brachiocephalic artery (BCA). E is derived from a collapsed z-series of multiple optical sections spanning 950μm. F-H are single optical sections. Arrow in H points to a constriction of the right common carotid (RCC). Brackets in G and H indicate the width of the aorta (Ao) lumen. The purple color is the result of the merger of the autofluorescence from 543nm and 633nm excitation. LV, left ventricle, RV, right ventricle; PA, pulmonary artery; LCC, left common carotid. Bars in A and E = 600μm and apply to A-F. Bar in G = 150μm and also applies to H.
Table 1.
Penetrance of cardiac defects in embryos homozygous for gene trap disruption of the fibulin-1 gene.
| Cardiac/OFT Anomaly | No. of Embryos Displaying the Anomaly | Penetrance (%) |
|---|---|---|
| ASD | 6 (n = 15)1 | 40 |
| VSD | 5 (n = 15)1 | 334 |
| VSD with DORV | 7 (n = 15)1 | 47 |
| VSD with OA | 1 (n = 15)1 | 7 |
| ASD with DORV or OA | 4 (n = 15)1 | 27 |
| Aortic Arch Defect2 | 3 (n = 15)3 | 20 |
| Thin Myocardium | 6 (n = 15)3 | 40 |
Information was compiled from analysis of E15.5-18.5 null embryos. The background of embryos homozygous for the Fbln1 gene trap insertion was 129P2/OlaHsd:C57BL/6 backcrossed to C57BL/6 for 2 generations.
The principal aortic arch defects observed in fibulin-1 nulls were an absence of the right subclavian artery from its normal branch point from the brachiocephalic artery.
Information was compiled from analysis of 16.5–18.5 dpc null embryos. Abbreviations: ASD, atrial septal defects; AVSD, atrial ventricular septal defect (i.e., common AV valve/canal); VSD, ventricular septal defects; DORV, double-outlet right ventricle; OA, overriding aorta.
Eleven of 13 were membranous VSDs; 2 of 13 were muscular VSDs; and 2 of 13 had both membranous muscular VSDs.
Cardiac septation defects in Fbln1−/− embryos
Ventricular septal defects (VSD) (both muscular and membranous) were seen in 86% of the hearts from Fbln1−/− embryos (Fig. 4D, Supplementary Fig. 4E and F and Table 1). The majority of these VSDs (53%) occurred in combination with either DORV or overriding aorta. Atrial septal defects (ASDs) were also observed in 40% (6 out of 15) of Fbln1−/− hearts and included septum primum and septum secundum anomalies Supplementary Fig. 4G. The prevalence of VSD and ASD in Fbln1−/− hearts emphasizes the important role of Fbln1 in proper cardiac septation.
Aortic arch abnormalities in Fbln1−/− embryos
Abnormalities of morphology of the aortic arch arteries were apparent in Fbln1 null embryos. The right subclavian artery (RSA), which is derived from the right 4th-pharyngeal arch artery and branches off the brachiocephalic artery was missing from its normal position in 20% of the Fbln1−/− mutants (Fig. 4F). Moreover, in several of these mutants, there was an anomalous blood vessel that emerged from the aorta, caudal to the left subclavian, that passed behind the esophagus (i.e., retroesophageal RSA). In addition, constrictions of the brachiocephalic artery were also apparent in one Fbln1 null (Fig. 4H). These anomalies are consistent with defective remodeling of the pharyngeal arch arteries, a process that requires proper NCC investment into the arch artery sheath layers (Bockman et al., 1989).
Myocardial wall defects in Fbln1−/− embryos
In 40% of Fbln1−/− hearts (6 of 15 at E17.5-18.5), the compact layer of the ventricular wall myocardium of both ventricles was thinner compared to that of wild-type hearts (Supplementary Fig. 5).
Pharyngeal gland abnormalities in Fbln1−/− embryos
Examination of the pharyngeal glands of Fbln1-deficient embryos revealed a number of morphological abnormalities. In all Fbln1 null embryos examined (E16.5-18.5, n = 15), the thymus, which is derived from the third pharyngeal pouch, was hypoplastic (Fig. 5A). Thymic aplasia or uni-lobe thymus was not observed in null embryos. Fbln1−/− embryos also displayed hypoplasia of the thyroid, which is derived from the 3rd and 4th pharyngeal arches and pouches (Fig. 5A).
Figure 5. Hypoplastic thymus and thyroid and under mineralized and reduced sized skull bones in Fbln1−/− embryos.
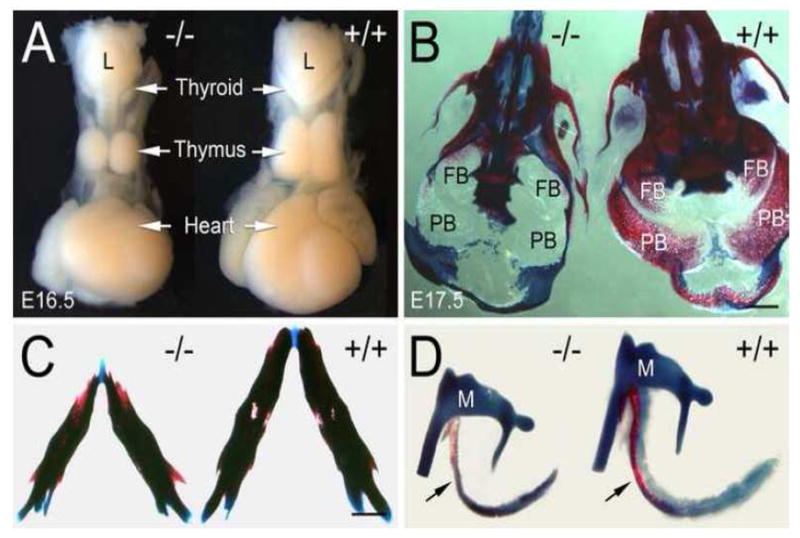
A, hypoplastic thymus and thyroid in a Fbln1−/− embryo (E16.5) as compared with normal size thymus and thyroid glands in a wild-type embryo (+/+). B, ventral view of Alizarin red/Alcian blue stained skulls from E17.5 embryos homozygous (−/−) and heterozygous (+/−) for the Fbln1 gene trap insertion. Note that the frontal (FB) and parietal bones (PB) of the null are membranous and show little Alizarin red staining as compared to the same bones in the +/− skull, which are extensively mineralized. C, lower jaws from Fbln1−/− and Fbln1+/− E17.5 embryos. Note that the mandibles of the Fbln1 null embryos are smaller (micrognathia) than those of the heterozygote. D, ear bones from Fbln1−/− and Fbln1+/− E17.5 embryos. Arrows in D point to the tympanic rings. Note that the tympanic ring in the mutant is under mineralized and smaller than the control. L, larynx; M, malleus. Bars in B and C = 1 mm.
Craniofacial skeletal abnormalities in Fbln1−/− embryos
Analysis of the ossified and cartilaginous tissues of the craniofacial skeleton of Fbln1 mutant embryos (E17.5) revealed decreased ossification of most cranial bones, particularly the frontal, parietal, tympanic ring, nasal and premaxillary bones (Fig. 5B–D). There is also reduction in the size of many skull bones in the nulls including the mandible (micrognathia) (Fig. 5C) and tympanic ringbones (Fig. 5D) as well as an overall reduced cranial size as compared to littermate controls. No cleft palate was observed in the Fbln1 nulls. The neural crest-derived elements of the cartilages of the throat (i.e., hyoid and thyroid) also appear reduced in the nulls as compared to wild-type embryos (data not shown).
Fbln1 expression in the neural crest, somites, pharyngeal arches and distal OFT/secondary heart field
As a result of the gene trapping, β-gal is placed under the control of the Fbln1 promoter. X-gal staining was performed to establish the pattern of Fbln1 expression in heterozygous embryos. X-gal staining was apparent in presomitic mesoderm from E8.75 to E9.0 (Fig. 6A–D). X-gal staining was also prominent in somites from E8.75 to E10.5 (Fig. 6A–E). X-gal staining was also apparent along the margins of neural folds in caudal regions of the embryo and dorsal regions of the neural tube (Fig. 6A and C, arrowheads). At E10.5, the dorsal neural tube region was relatively free of X-gal stained cells, however, pronounced X-gal staining was apparent in populations of cells lateral to the hindbrain, around the otic vesicle, in pharyngeal arches 1–3, in anterior and posterior regions of the somites, in the limb buds and in the region cardiac inflow tract (Fig. 6E).
Figure 6. Fbln1 expression in the neural crest, somites, secondary heart field and pharyngeal arches.
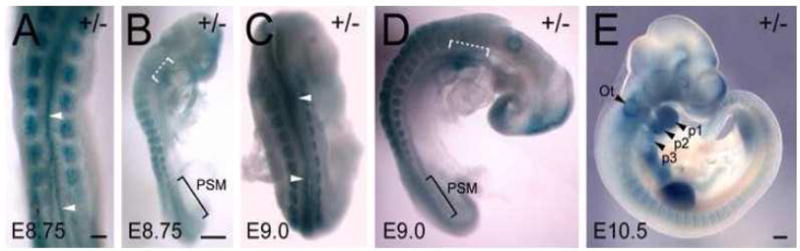
A–E are dorsal and lateral views of X-gal stained embryos heterozygous for the Fbln1 gene trap insertion at E8.75 (A and B), E9.0 (C and D) and E10.5 (E). Arrowheads in A and C indicate X-gal-positive cells in neural folds and dorsal neural tube regions. White brackets in B and D indicate X-gal staining in presumptive secondary heart field regions. PSM, presomitic mesoderm; Ot, otic vesicle; p1, p2 and p3 indicate pharyngeal arches 1, 2 and 3. Bars in A and B = 250μm. Bar in E = 300μm.
Consistent with the X-gal staining, immunohistological analysis showed pronounced Fbln1 immunolabeling in the ECM of the mesenchyme lateral to the neural tube at the level of the pharyngeal arches in E9.5-10.5 embryos (Fig. 7A and B). By contrast, relatively low levels of Fbln1 immunolabeling were detected in the arch mesenchyme directly adjacent to the aortic sac/distal OFT (Fig. 7A and B, brackets). At E9.5, this area was rich in Pax3-expressing NCCs (Fig. 7C, brackets). Prominent Fbln1 labeling was evident in the ECM of the distal OFT, in the region of the SHF (Fig. 7C and D, arrowheads). Pharyngeal arch mesenchyme underlying the neural tube had high levels of Fbln1 and few Pax3-positive cells (Fig. 7C, asterisk). Little or no Fbln1 labeling was apparent in pharyngeal arch ectoderm or endoderm at E9.5-10.5.
Figure 7. Fbln1 expression in the head mesenchyme and secondary heart field.
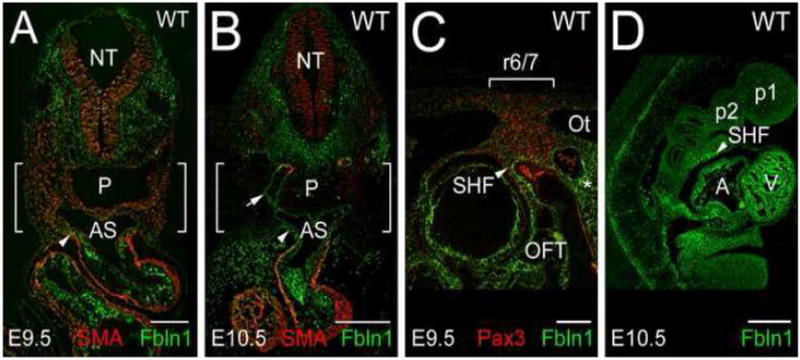
A and B show cross sections of a wild-type E9.5 and E10.5 embryo immunolabeled with antibodies to Fbln1 and αSM actin (SMA). C, sagittal section of a wild-type E9.5 embryo immunolabeled with antibodies to Fbln1 and Pax3, a NCC marker. D, sagittal section of a wild-type E10.5 embryo immunolabeled with antibodies to Fbln1. Arrowheads point to Fbln1 staining in the secondary heart field (SHF) region. Arrow in B points to Fbln1 staining in the subendothelial ECM of the 4th aortic arch artery. Bracketed areas in A–B contain pharyngeal arch regions displaying relatively low levels of Fbln1 immunolabeling. Bracketed area in C indicates the region of pharyngeal arch 3/4 that contains streams of Pax3-positive cells emanating from rhombomere 6 and 7 (r6/7). NT, neural tube; P, pharynx; AS, aortic sac; Ot, otic vesicle; A, atrium; V, ventricle; OFT, outflow tract; p1 and p2 indicate pharyngeal arches 1 and 2. Bars in A and C = 150μm. Bars in B and D = 300μm.
Whole-mount in situ RNA hybridization analysis of wild-type E8.3-9.5 embryos showed that Fbln1 mRNA is expressed in presomitic mesoderm and somites, similar to what was observed in X-gal stained embryos heterozygous for the gene trap insertion (Fig. 8). Fbln1 RNA was also detected in rhombomeres 2, 4, 6/7 and in pharyngeal arches 1–4 (Fig. 8C and D). The rhombomere expression of Fbln1 is similar to that of the NCC marker Crabp1 (Fig. 8E). Together, the findings from Fbln1 expression analysis indicate that Fbln1 expression is associated with migratory pathways of NCCs derived from rhombomeres 2–7, which contribute to morphogenesis of the OFT and pharyngeal glands. However, it is not known whether Fbln1 is expressed by migrating NCCs or by non-neural crest derived-mesenchymal cells located along the path of migrating NCCs.
Figure 8. Fbln1 in situ RNA hybridization analysis.
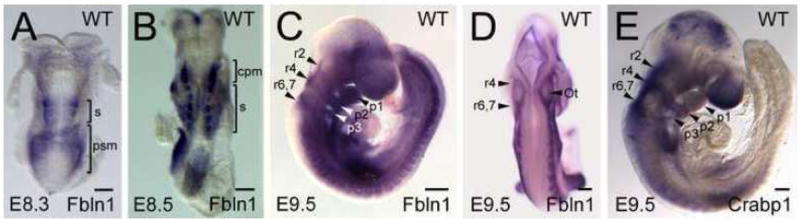
Shown are wild-type embryos (E8.3-9.5) after whole mount in situ hybridization with antisense riboprobes for Fbln1 (A–D) and Crabp1 (E), a marker of migrating NCCs. S, somite; psm, presomitic mesoderm; cpm, cranial paraxial mesoderm; r2, r4, r6 indicate rhombomeres 2, 4 and 6; p1, p2 and p3 indicate pharyngeal arches 1, 2 and 3. Bars in A–E = 200μm.
Fbln1 gene trap nulls have cranial nerve patterning abnormalities
NCCs derived from rhombomeres 2, 4, 6 and 7 also contribute to the formation of the cranial nerves. In situ hybridization analysis using a Sox-10 probe revealed abnormalities of the patterning of cranial nerve NCCs in Fbln1 null embryos (Fig. 9A–C). In particular, interruptions and inappropriately directed streams of Sox-10-positive NCCs that comprise the primordia for cranial nerves IX (glossopharyngeal) and X (vagus) were observed (Fig. 9B and C). The apparent misguidance of NCCs resulted in abnormal mixing of the distal regions of the glossopharyngeal and vagal streams (Fig. 9B and C, brackets), which may be interpreted to have resulted from a caudal misdirection of the glossopharyngeal stream. Abnormal branching of the proximal region of the vagal stream was also observed in some embryos (data not shown).
Figure 9. Fbln1 deficiency leads to anomalies of cranial NCC patterning and cranial nerve morphogenesis as well as increased apoptosis in hindbrain regions.
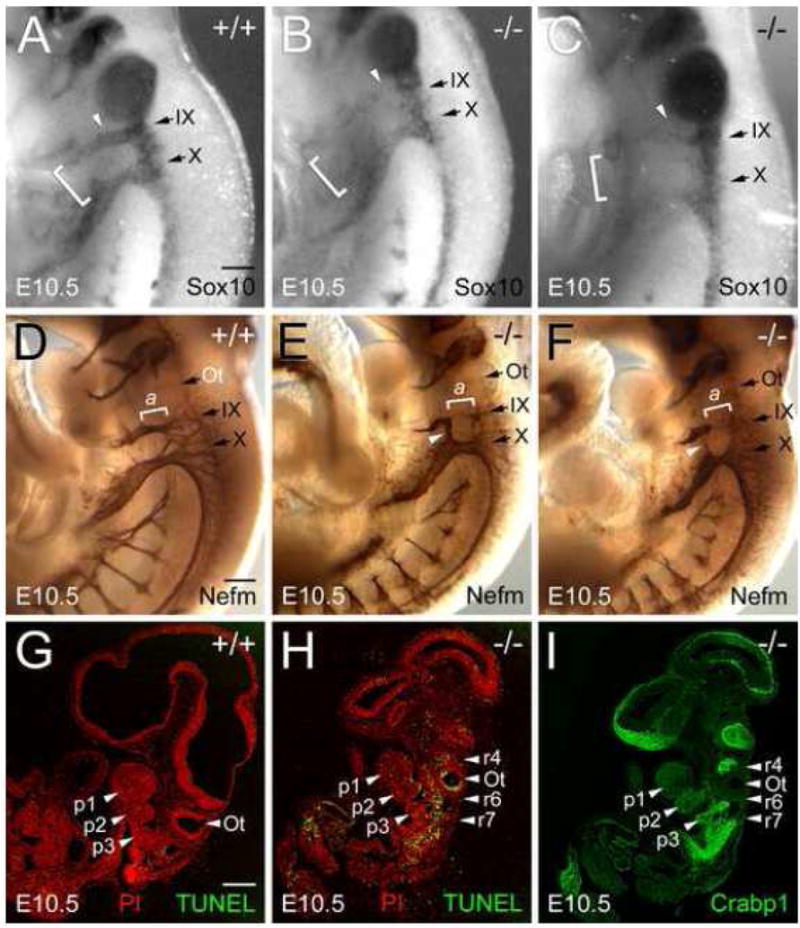
A–C, lateral views of E10.5 Fbln1+/+ (A) and Fbln1−/− embryos (B and C) after whole mount in situ hybridization with antisense riboprobe for Sox-10, a transcription factor expressed by migratory NCCs that form the cranial nerves. Arrows in A–C indicate the forming glossopharyngeal and vagus nerves (cranial nerves IX and X). Note the decreased level of Sox-10 expression in the forming glossopharyngeal nerves (white arrowheads in B and C). Abnormal fusion of glossopharyngeal ganglia with the distal ganglia of cranial nerve X is apparent in the nulls (brackets in B and C). In the embryo shown in C, cranial nerve X has a decreased level of Sox-10 expression relative to the control. The results depicted in A–C are representative of in situ hybridizations from 3 sets of embryos. D–F, lateral views of wild-type (D) and Fbln1−/− (E and F) E10.5 embryos with cranial nerves immunolabeled with monoclonal neurofilament-M antibody. Brackets labeled a in D–F indicate the proximal portion of the cranial nerve IX, which is hypoplastic and absent in the Fbln1 null embryos shown in panels E and F, respectively. White arrowheads in E and F indicate bridging in the epibranchial placode-derived regions of cranial nerves IX and X. G and H, confocal images of sagittal sections from E10.5 Fbln1+/+ and Fbln1−/− embryos assayed for apoptosis (TUNEL, green). Nuclei were stained with propidium iodide (red). I shows an anti-Crabp1 immunolabeled (green) adjacent section of the Fbln1 null embryo shown in H. p1, p2 and p3 indicate pharyngeal arches 1, 2 and 3; r4, r6 and r7 indicate rhombomeres 4, 6 and 7. Ot, otic vesicle. Bar in A = 300μm and applies to A–C. Bar in D = 300μm and applies to D–F. Bar in G = 100μm and applies to G–I.
To evaluate the impact of Fbln1 deficiency on cranial nerves, E10.5 embryos were immunostained to detect neurofilament-M. The results reveal abnormalities in cranial nerves IX and X (Fig. 9D–F). Null embryos display an interruption in the proximal portion of cranial nerve IX and a less than normal degree of branching in the distal segment (Fig. 9E and F). Furthermore, there is an abnormal fusion of cranial nerves IX and X in the epibranchial placode region (Fig. 9E). Cranial nerve X in Fbln1 null embryos also appeared more compact and less reticulated than in wild-type embryos (Fig. 9E and F).
Fbln1 gene trap nulls have increased apoptosis of NCCs in the hindbrain
TUNEL analysis was performed to assess the consequence of Fbln1 deficiency on NCC survival. Null E10.5 embryos displayed increased levels of TUNEL-positive cells in the hindbrain region as compared to wild-type embryos (Fig. 9G and H). In particular, relatively high levels of TUNEL-positive cells were apparent in the otic vesicle epithelium and around rhombomeres 4, 6 and 7 as compared to wild-type embryos. To determine if the apoptotic cells observed in Fbln1 nulls might correspond to NCCs, immunolabeling was performed using antibodies to the NCC marker, Crabp1. Crabp1-positive cells were observed in null embryos in streams extending from rhombomeres 4, 6 and 7, which corresponded to areas having high levels of TUNEL-positive cells (Fig. 9I).
Genetic background influences penetrance of cardiac anomalies in Fbln1 nulls
While the bleeding, lung abnormalities and perinatal lethality in Fbln1 gene trap homozygotes are in agreement with an earlier analysis of mouse Fbln1 deficiency (Kostka et al., 2001), abnormalities of the OFT, arch arteries, pharyngeal glands, cranial nerves and cephalic skeleton were not previously reported. Differences in genetic background could account for the phenotypic disparities given that the Fbln1 gene trap homozygotes were a mixed C57BL/6 and 129P2/OlaHsd background whereas embryos homozygous for Fbln1 exon 1 deletion were a mixed C57BL/6 and 129/Sv background (Kostka et al., 2001). When we examined embryos homozygous for the Fbln1 exon 1 deletion we found hypoplastic thymus (100% penetrance) as well as cardiac defects corresponding to those observed in embryos homozygous for the Fbln1 gene trap mutation (e.g., VSD associated with DORV) (Supplementary Fig. 6). However, the penetrance of cardiac defects in the Fbln1 exon 1 mutants was lower than that of the Fbln1 gene trap mutants (Table 2). We subsequently found that penetrance of cardiac defects in the Fbln1 gene trap could be affected by the genetic background. When the Fbln1 gene trap line was backcrossed further onto a C57BL6 background the penetrance of cardiac defects was reduced to levels comparable with the Fbln1 exon 1 deletion on a mixed C57BL/6 and 129/Sv background. Fbln1-deficient embryos were also generated by mating the Fbln1 gene trap strain (mixed C57BL/6 and 129P2/OlaHsd background) to the Fbln1 exon 1 deletion strain (C57BL/6 and 129/Sv background). As shown in Table 2, the hybrids displayed a level of penetrance higher than the Fbln1 exon 1 deletion homozygotes having the mixed C57BL/6 and 129/Sv background. These findings suggest that the severity of cardiac defects in embryos deficient in Fbln1 is increased by genetic modifiers found within the129P2/OlaHsd background.
Table 2.
Influence of background on penetrance of cardiac defects in embryos homozygous for the Fbln1 gene trap insertion or homozygous for Fbln1 exon 1 deletion.
| Cardiac/OFT Anomaly | Anomaly Penetrance (%) | |||
|---|---|---|---|---|
| Fbln1 gene trap homozygotes(129P2/OlaHsd: C57BL6)1 | Fbln1 gene trap homozygotes(C57BL6)2 | Fbln1 Δ exon 1 homozygotes(C57BL6)3 | Fbln1 gene trap(129P2/OlaHsd: C57BL/6)/Fbln1 Δ exon 1(C57BL6)4 | |
| ASD | 40 | 23 | 14 | 44 |
| VSD | 33 | 38 | 0 | 22 |
| VSD with DORV | 47 | 0 | 7 | 11 |
| VSD with Overriding Aorta | 7 | 8 | 0 | 22 |
The background of embryos homozygous for the Fbln1 gene trap insertion was 129P2/OlaHsd:C57BL/6 backcrossed to C57BL/6 for 2 generations. Percentages indicated are based on evaluation of 15 embryos homozygous for the Fbln1 gene trap insertion.
The background of embryos homozygous for the Fbln1 gene trap insertion was 129P2/OlaHsd:C57BL/6 backcrossed to C57BL/6 for 5 generations. Percentages indicated are based on evaluation of 13 embryos (E15.5-18.5) homozygous for the Fbln1 gene trap insertion.
The background of the mouse containing the Fbln1 exon 1 deletion was 129Sv:C57BL/6 backcrossed to C57BL/6 for indeterminate number of generations. We subsequently backcrossed it to C57BL/6 for 2 generations. Percentages indicated are based on evaluation of 14 embryos (E15.5-18.5) homozygous for the Fbln1 exon 1 deletion.
Percentages indicated are based on evaluation of 9 embryos (E15.5-18.5) each having one Fbln1 allele carrying the Fbln1 gene trap insertion (129P2/OlaHsd:C57BL/6 background as described in column 1) and the other Fbln1 allele carrying the Fbln1 exon 1 deletion (C57BL/6 background as described in column 3). Note that all embryos of this genotype display thymic hypoplasia.
Discussion
Collectively, the newly revealed malformations of Fbln1-deficient embryos are similar to congenital defects known as neurocristopathies, which result from defective NCC development. Indeed, with the exception of an absence of OFT septation defects in Fbln1 nulls, the spectrum of malformations observed in the nulls are similar to those that result from ablation of the cephalic premigratory neural crest (Bockman and Kirby, 1984; Bockman et al., 1987; Hutson and Kirby, 2003). For example, neural crest ablation is associated with aplasticity or hypoplasticity of the thymus, parathyroid and thyroid glands and a failure of aortic arch arteries three, four and six to develop (Bockman and Kirby, 1984; Bockman et al., 1987). Fbln1 mutants display hypoplasia of the thymus and thyroid and anomalies of aortic arch arteries. Thus, the anomalies observed in the Fbln1 nulls are consistent with an insufficiency in numbers of specific populations of NCCs. Our finding that Fbln1 nulls have increased apoptosis in hindbrain regions that contain NCC populations migrating into the pharyngeal arches suggests that reduced NCC survival may be a contributing factor to NCC insufficiency. The bleeding that occurs primarily in the head and neck regions of Fbln1 deficient embryos may also represent a neurocristopathy since cephalic NCCs contribute pericytes and vascular smooth muscle cells to all of the blood vessels of the head including pericytes of the hyaloid vasculature (Etchevers et al., 2001; Gage et al., 2005; Korn et al., 2002). Even the lung abnormalities observed in the Fbln1 nulls might be neural crest related given the fact that vagal NCCs colonize the developing lung buds (Burns et al., 2008).
NCCs also influence the contribution of cells to the heart from the SHF, a region of pharyngeal arch mesenchyme that underlies the caudal pharynx and surrounds the aortic sac (Mjaatvedt et al., 2001; Waldo et al., 2001; Yelbuz et al., 2002). When premigratory neural crest is ablated, insufficient addition of myocardial progentitors from the SHF is believed to be the basis for the incomplete rotation of the aorticopulmonary septum and incorrect ventriculoarterial alignment (i.e., overriding aorta and DORV) (Yelbuz et al., 2002). Fbln1 nulls display both DORV and overriding aorta. The SHF also contributes cells to the right ventricular myocardium and interventricular septum (Verzi et al., 2005). Fbln1 mutant hearts show thinning of the right ventricular myocardium as well as interventricular septal defects (e.g., muscular VSD). The DORV, overriding aorta, thin myocardium and VSD anomalies together with the finding that Fbln1 is expressed in the SHF region suggest that Fbln1 influences the indirect roles of NCCs in promoting addition of myocardium from the SHF to the OFT, right ventricle and interventricular septum. However, it cannot be ruled out that the thin ventricular myocardium and membranous VSDs observed in Fbln1 nulls might be related to anomalies of non-NCC mesenchymalization (Kirby, 2002) given that Fbln1 expression is associated with epithelial-mesenchymal transition of the epicardium (WSA, unpublished observation) and AV cushions (Kern et al., 2006).
Cardiac neural crest ablation is also associated with an absence or poor development of cranial nerves IX and X (Kuratani et al., 1991). The IXth cranial nerve, the glossopharyngeal nerve, derives from NCCs of rhombomere 6 and innervates the third pharyngeal arch (D’Amico-Martel and Noden, 1983). The Xth cranial nerve, the vagus nerve, derives from NCCs of rhombomere 7 and innervates the 4th pharyngeal arch. In Fbln1 nulls, hypoplasticity and patterning anomalies of the NCC precursors for these cranial nerves is observed in E10.5 embryos as revealed by Sox-10 in situ hybridizations. The patterning anomalies include interruptions and apparent misguidance in the streams of Sox-10-positive NCCs that comprise each nerve primordia. The apparent misguidance is manifested by abnormal mixing of the distal regions of the glossopharyngeal and vagal streams of Sox-10-positive NCCs. These abnormalities in Sox-10-expressing NCCs are mirrored by abnormalities in the cranial nerves of Fbln1 nulls as revealed by neurofilament-M immunolabeling. Specifically, the proximal portion of cranial nerve IX in nulls was missing and the distal epibranchial placode-derived region was often fused with the vagus nerve. Given the evidence that post otic rhombomere-derived NCCs (i.e., rhombomere 6-7 Sox-10-positive cells) contribute to the heart (Montero et al., 2002), the observed abnormalities in rhombomere 6-7 NCC patterning might also be a contributing factor to the OFT defects in Fbln1 nulls.
Abnormalities in Fbln1 nulls, including patterning defects of cranial nerves IX and X, are also seen in Hoxa3 null mice (Chisaka and Capecchi, 1991). Like the Fbln1 nulls, mice deficient in Hoxa3 die at or shortly after birth and display hypoplastic thymus and thyroid (Chisaka and Capecchi, 1991). Hoxa3 nulls also display arch artery and heart abnormalities resembling those seen in Fbln1 nulls (Chisaka and Capecchi, 1991). Hoxa3 is expressed in rhombomeres 5–8, which contribute NCCs to the formation of cranial nerves IX–XII. In Hoxa3 nulls, the formation of cranial nerves IX and X is affected in a manner similar to that seen in Fbln1 nulls, with a fusion of the epibranchial placode regions of the two nerves and a gap in the proximal portion of IX (Watari et al., 2001). Evidence that cranial nerve anomalies in Hoxa3 nulls are a consequence of NCCs defects comes from the finding of reduced Sox-10-positive cells in streams emanating from rhombomeres 6/7 (Watari et al., 2001). Again, these findings are consistent with our results showing an apparent reduction in the level of Sox-10 expression in the stream of NCCs that comprise the primordia for cranial nerve IX as well as a mixing of Sox-10 positive cells in distal regions of the glossopharyngeal and vagal streams.
An underlying basis for the mixing and interruptions of NCC streams emanating from rhombomeres 6 and 7 seen in Fbln1 nulls may be defects in NCC motility and guidance. Defective migration of Sox-10-positive NCCs emanating from rhombomeres 6 and 7 has been implicated in cranial nerve IX interruptions observed in mice deficient in Hoxa3 (Watari et al., 2001). Migration defects of rhombomere 6-7-derived, Sox-10-positive NCCs have also been speculated to account for the gap in glossopharyngeal nerves observed in mice doubly deficient for the Nk-related homeodomain transcription factors Msx1 and Msx2 (Ishii et al., 2005). Additional evidence for NCCs of Msx1/Msx2 double mutants having migration problems comes from the finding of Sox-10-positive NCCs in the normally NCC-free zone of rhombomere 3 (Ishii et al., 2005). As a result of this apparent misguidance/unrestricted migration, there is a fusion of the trigeminal and facial cranial nerves. Indeed, in Fbln1 nulls we observed Sox-10-positve cells in the normally NCC-free zone between the glossopharyngeal and vagal NCCs streams and a subsequent fusion of the two nerves. These findings are consistent with the known roles of Fbln1 as a suppressor of motility (Hayashido et al., 1998; Lee et al., 2005; Qing et al., 1997; Twal et al., 2001) and a regulator of directed cell migration (Kubota et al., 2004).
The abnormalities of Fbln1 nulls are similar to the constellation of congenital malformations of the aortic arch, heart, thymus, thyroid, parathyroid glands and bones of the head that are associated with DiGeorge syndrome (DGS) (Baldini, 2004; Liao et al., 2004). The majority of DGS individuals are heterozygous for a microdeletion of human chromosome 22q11.2; however, the remaining individuals do not possess a 22q11.2 deletion indicating that mutation of genes outside of the region may cause the disorder. The Fbln1 gene is located outside of the 22q11.2 deletion region at 22q13.3 (Korenberg et al., 1995). Since heterozygous deletion of Fbln1 in mice did not lead to OFT, aortic arch, cardiac, pharyngeal gland or craniofacial anomalies, Fbln1 haploinsufficiency does not lead to the same outcome as occurs with haploinsufficiency of genes located within the 22q11.2 deletion region. The variable penetrance and severity of DGS also indicate that genes from outside of the microdeletion region may act as modifiers of the DGS pathogenesis pathway. Several candidate DGS modifier genes have been identified through gene deletion studies in mice. For example, Hoxa3 deficiency in mice leads to a DGS-like phenotype (Chisaka and Capecchi, 1991). In addition, Fgf8, and Gbx2, an Fgf8-inducible transcription factor (Liu and Joyner, 2001), when deleted individually or in combination in mice leads to cardiac, aortic arch artery and craniofacial anomalies consistent with features of 22q11 deletion in humans (Abu-Issa et al., 2002; Byrd and Meyers, 2005; Frank et al., 2002).
The similarities that exist between the DGS phenotype and that of Fbln1-deficient embryos raises the possibility that Fbln1 has links to the mechanism by which 22q11 deletion causes DGS pathogenesis. Interaction with the Fgf8 signaling pathway represents one plausible mechanism. Fgf8 signaling is mediated by Fgfr1 and 2 interaction with the adaptor protein, Crkl (Moon et al., 2006), whose deficiency in mice causes many of the anomalies seen in DGS (Guris et al., 2001) and in Fbln1 nulls. Mechanistically, Crkl participates in activation of the Raf-1/MEK/Erk signaling pathway (Arai et al., 2006) and mediates FN-integrin-induced cell migration (Li et al., 2003) and cell survival. Indeed, Crkl deficiency in mice leads to decreased survival of NCCs that contribute to the pharyngeal arches and OFT (Moon et al., 2006), an outcome similar to what is observed in the Fbln1 nulls. Although Fbln1 is not known to interact with Fgf8, Fbln1 does regulate Erk activation and FN-induced cell migration (Twal et al., 2001). Future studies will need to test this and other possible interactions between Fbln1 and the repertoire of genes associated with DGS.
Acknowledgments
This work was supported by NIH grants HL52813 (WSA), HL080168 (CJD) and American Heart Association (AHA) Grant-in-Aid 0755346U (WSA), AHA Beginning Grant-in-Aid 0765236U (CBK) and AHA Postdoctoral Fellowship 0120537U (MAC). We also acknowledge Dr. Demetri Spyropoulos and the MUSC Gene Targeting and Knockout Mouse Core (Supported by NIH grant RR16434) for assistance with the development of the Fbln1 gene trap knockout mouse. We thank Joyce Edmonds, Aimee Phelps and Sally Fairey for performing tissue embedding and sectioning. We thank Dr. Mon-Li Chu (Thomas Jefferson University) for providing us with the Fbln1 exon 1 knockout line described by Kostka et al., (2001).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could a3ect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abu-Issa R, et al. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129:4613–25. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
- Arai A, et al. CrkL plays a role in SDF-1-induced activation of the Raf-1/MEK/Erk pathway through Ras and Rac to mediate chemotactic signaling in hematopoietic cells. Cell Signal. 2006 doi: 10.1016/j.cellsig.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Argraves WS, et al. Fibulins: physiological and disease perspectives. EMBO Rep. 2003;4:1127–31. doi: 10.1038/sj.embor.7400033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argraves WS, et al. Fibulin is an extracellular matrix and plasma glycoprotein with repeated domain structure. J Cell Biol. 1990;111:3155–64. doi: 10.1083/jcb.111.6.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbona K, et al. Fibulin binds to itself and to the carboxyl-terminal heparin-binding region of fibronectin. J Biol Chem. 1992;267:20120–5. [PubMed] [Google Scholar]
- Baldini A. DiGeorge syndrome: an update. Curr Opin Cardiol. 2004;19:201–4. doi: 10.1097/00001573-200405000-00002. [DOI] [PubMed] [Google Scholar]
- Bockman DE, Kirby ML. Dependence of thymus development on derivatives of the neural crest. Science. 1984;223:498–500. doi: 10.1126/science.6606851. [DOI] [PubMed] [Google Scholar]
- Bockman DE, et al. Alteration of early vascular development after ablation of cranial neural crest. Anat Rec. 1989;225:209–17. doi: 10.1002/ar.1092250306. [DOI] [PubMed] [Google Scholar]
- Bockman DE, et al. Effect of neural crest ablation on development of the heart and arch arteries in the chick. Am J Anat. 1987;180:332–41. doi: 10.1002/aja.1001800403. [DOI] [PubMed] [Google Scholar]
- Bouchey D, et al. Fibulin-1, vitronectin, and fibronectin expression during avian cardiac valve and septa development. Anat Rec. 1996;244:540–51. doi: 10.1002/(SICI)1097-0185(199604)244:4<540::AID-AR12>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Bronner-Fraser M. Mechanisms of neural crest cell migration. Bioessays. 1993;15:221–30. doi: 10.1002/bies.950150402. [DOI] [PubMed] [Google Scholar]
- Brooke JS, et al. Latent transforming growth factor beta-binding protein-3 and fibulin-1C interact with the extracellular domain of the heparin-binding EGF-like growth factor precursor. BMC Cell Biol. 2002;3:2. doi: 10.1186/1471-2121-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns AJ, et al. Development of the neural crest-derived intrinsic innervation of the human lung. Am J Respir Cell Mol Biol. 2008;38:269–75. doi: 10.1165/rcmb.2007-0246OC. [DOI] [PubMed] [Google Scholar]
- Byrd NA, Meyers EN. Loss of Gbx2 results in neural crest cell patterning and pharyngeal arch artery defects in the mouse embryo. Dev Biol. 2005;284:233–45. doi: 10.1016/j.ydbio.2005.05.023. [DOI] [PubMed] [Google Scholar]
- Chisaka O, Capecchi MR. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nature. 1991;350:473–9. doi: 10.1038/350473a0. [DOI] [PubMed] [Google Scholar]
- Chu ML, Tsuda T. Fibulins in development and heritable disease. Birth Defects Res C Embryo Today. 2004;72:25–36. doi: 10.1002/bdrc.20003. [DOI] [PubMed] [Google Scholar]
- Coles EG, et al. Abnormalities in neural crest cell migration in laminin alpha5 mutant mice. Dev Biol. 2006;289:218–28. doi: 10.1016/j.ydbio.2005.10.031. [DOI] [PubMed] [Google Scholar]
- Conway SJ, et al. Development of a lethal congenital heart defect in the splotch (Pax3) mutant mouse. Cardiovasc Res. 1997;36:163–73. doi: 10.1016/s0008-6363(97)00172-7. [DOI] [PubMed] [Google Scholar]
- Costell M, et al. Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ Res. 2002;91:158–64. doi: 10.1161/01.res.0000026056.81424.da. [DOI] [PubMed] [Google Scholar]
- D’Amico-Martel A, Noden DM. Contributions of placodal and neural crest cells to avian cranial peripheral ganglia. Am J Anat. 1983;166:445–68. doi: 10.1002/aja.1001660406. [DOI] [PubMed] [Google Scholar]
- Dutt S, et al. Versican V0 and V1 guide migratory neural crest cells. J Biol Chem. 2006;281:12123–31. doi: 10.1074/jbc.M510834200. [DOI] [PubMed] [Google Scholar]
- Etchevers HC, et al. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development. 2001;128:1059–68. doi: 10.1242/dev.128.7.1059. [DOI] [PubMed] [Google Scholar]
- Frank DU, et al. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage PJ, et al. Fate maps of neural crest and mesoderm in the mammalian eye. Invest Ophthalmol Vis Sci. 2005;46:4200–8. doi: 10.1167/iovs.05-0691. [DOI] [PubMed] [Google Scholar]
- Guris DL, et al. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet. 2001;27:293–8. doi: 10.1038/85855. [DOI] [PubMed] [Google Scholar]
- Hayashido Y, et al. Estradiol and fibulin-1 inhibit motility of human ovarian- and breast-cancer cells induced by fibronectin. Int J Cancer. 1998;75:654–8. doi: 10.1002/(sici)1097-0215(19980209)75:4<654::aid-ijc26>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Henderson DJ, et al. Over-expression of the chondroitin sulphate proteoglycan versican is associated with defective neural crest migration in the Pax3 mutant mouse (splotch) Mech Dev. 1997;69:39–51. doi: 10.1016/s0925-4773(97)00151-2. [DOI] [PubMed] [Google Scholar]
- Hesselson D, et al. GON-1 and Fibulin Have Antagonistic Roles in Control of Organ Shape. Curr Biol. 2004;14:2005–10. doi: 10.1016/j.cub.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Hogan B, et al. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Press; New York: 1994. [Google Scholar]
- Hutson MR, Kirby ML. Neural crest and cardiovascular development: a 20-year perspective. Birth Defects Res C Embryo Today. 2003;69:2–13. doi: 10.1002/bdrc.10002. [DOI] [PubMed] [Google Scholar]
- Ishii M, et al. Combined deficiencies of Msx1 and Msx2 cause impaired patterning and survival of the cranial neural crest. Development. 2005;132:4937–50. doi: 10.1242/dev.02072. [DOI] [PubMed] [Google Scholar]
- Kern CB, et al. Proteolytic cleavage of versican during cardiac cushion morphogenesis. Dev Dyn. 2006 doi: 10.1002/dvdy.20838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkawa Y, et al. Integrin binding specificity of laminin-10/11: laminin-10/11 are recognized by alpha 3 beta 1, alpha 6 beta 1 and alpha 6 beta 4 integrins. J Cell Sci. 2000;113(Pt 5):869–76. doi: 10.1242/jcs.113.5.869. [DOI] [PubMed] [Google Scholar]
- Kimmel CA, Trammell C. A rapid procedure for routine double staining of cartilage and bone in fetal and adult animals. Stain Technol. 1981;56:271–3. doi: 10.3109/10520298109067325. [DOI] [PubMed] [Google Scholar]
- Kirby ML. Molecular embryogenesis of the heart. Pediatr Dev Pathol. 2002;5:516–43. doi: 10.1007/s10024-002-0004-2. [DOI] [PubMed] [Google Scholar]
- Korenberg JR, et al. Localization of the human gene for fibulin-1 (FBLN1) to chromosome band 22q13.3. Cytogenet Cell Genet. 1995;68:192–3. doi: 10.1159/000133911. [DOI] [PubMed] [Google Scholar]
- Korn J, et al. Neuroectodermal origin of brain pericytes and vascular smooth muscle cells. J Comp Neurol. 2002;442:78–88. doi: 10.1002/cne.1423. [DOI] [PubMed] [Google Scholar]
- Kostka G, et al. Perinatal lethality and endothelial cell abnormalities in several vessel compartments of fibulin-1-deficient mice. Mol Cell Biol. 2001;21:7025–34. doi: 10.1128/MCB.21.20.7025-7034.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JM. Extracellular matrix. C. elegans II. In: Riddle DL, et al., editors. Extracellular Matrix. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1997. pp. 471–500. [PubMed] [Google Scholar]
- Kubota Y, et al. A Fibulin-1 Homolog Interacts with an ADAM Protease that Controls Cell Migration in C. elegans. Curr Biol. 2004;14:2011–8. doi: 10.1016/j.cub.2004.10.047. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Nishiwaki K. Mutations in a fibulin-1 homolog bypass the requirement of the MIG-17 ADAM protease for cell migration in C. elegans. Mol Biol Cell. 2003;14:261a. doi: 10.1016/j.cub.2004.10.047. [DOI] [PubMed] [Google Scholar]
- Kuratani SC, et al. Development of cranial nerves in the chick embryo with special reference to the alterations of cardiac branches after ablation of the cardiac neural crest. Anat Embryol (Berl) 1991;183:501–14. doi: 10.1007/BF00186439. [DOI] [PubMed] [Google Scholar]
- Lee NV, et al. Fibulin-1 acts as a cofactor for the matrix metalloprotease ADAMTS-1. J Biol Chem. 2005;280:34796–804. doi: 10.1074/jbc.M506980200. [DOI] [PubMed] [Google Scholar]
- Li L, et al. Translocation of CrkL to focal adhesions mediates integrin-induced migration downstream of Src family kinases. Mol Cell Biol. 2003;23:2883–92. doi: 10.1128/MCB.23.8.2883-2892.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, et al. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum Mol Genet. 2004;13:1577–85. doi: 10.1093/hmg/ddh176. [DOI] [PubMed] [Google Scholar]
- Liu A, Joyner AL. EN and GBX2 play essential roles downstream of FGF8 in patterning the mouse mid/hindbrain region. Development. 2001;128:181–91. doi: 10.1242/dev.128.2.181. [DOI] [PubMed] [Google Scholar]
- Miller CE, et al. Confocal imaging of the embryonic heart: how deep? Microsc Microanal. 2005;11:216–23. doi: 10.1017/S1431927605050464. [DOI] [PubMed] [Google Scholar]
- Mjaatvedt CH, et al. The outflow tract of the heart is recruited from a novel heart-forming field. Dev Biol. 2001;238:97–109. doi: 10.1006/dbio.2001.0409. [DOI] [PubMed] [Google Scholar]
- Montero JA, et al. Expression of Sox8, Sox9 and Sox10 in the developing valves and autonomic nerves of the embryonic heart. Mech Dev. 2002;118:199–202. doi: 10.1016/s0925-4773(02)00249-6. [DOI] [PubMed] [Google Scholar]
- Moon AM, et al. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev Cell. 2006;10:71–80. doi: 10.1016/j.devcel.2005.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriel JM, et al. Selective assembly of fibulin-1 splice variants reveals distinct extracellular matrix networks and novel functions for perlecan/UNC-52 splice variants. Dev Dyn. 2006;235:2632–40. doi: 10.1002/dvdy.20888. [DOI] [PubMed] [Google Scholar]
- Perbal B, et al. The C-terminal domain of the regulatory protein NOVH is sufficient to promote interaction with fibulin 1C: a clue for a role of NOVH in cell-adhesion signaling. Proc Natl Acad Sci U S A. 1999;96:869–74. doi: 10.1073/pnas.96.3.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perris R, Perissinotto D. Role of the extracellular matrix during neural crest cell migration. Mech Dev. 2000;95:3–21. doi: 10.1016/s0925-4773(00)00365-8. [DOI] [PubMed] [Google Scholar]
- Pietri T, et al. Conditional beta1-integrin gene deletion in neural crest cells causes severe developmental alterations of the peripheral nervous system. Development. 2004;131:3871–83. doi: 10.1242/dev.01264. [DOI] [PubMed] [Google Scholar]
- Qing J, et al. Suppression of anchorage-independent growth and matrigel invasion and delayed tumor formation by elevated expression of fibulin-1D in human fibrosarcoma-derived cell lines. Oncogene. 1997;15:2159–2168. doi: 10.1038/sj.onc.1201385. [DOI] [PubMed] [Google Scholar]
- Sasaki T, et al. Structural characterization of two variants of fibulin-1 that differ in nidogen affinity. J Mol Biol. 1995;245:241–50. doi: 10.1006/jmbi.1994.0020. [DOI] [PubMed] [Google Scholar]
- Schymeinsky J, et al. Gene structure and functional analysis of the mouse nidogen-2 gene: nidogen-2 is not essential for basement membrane formation in mice. Mol Cell Biol. 2002;22:6820–30. doi: 10.1128/MCB.22.19.6820-6830.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, et al. Capturing genes encoding membrane and secreted proteins important for mouse development. Proc Natl Acad Sci U S A. 1995;92:6592–6. doi: 10.1073/pnas.92.14.6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence SG, et al. Fibulin is localized at sites of epithelial-mesenchymal transitions in the early avian embryo. Dev Biol. 1992;151:473–84. doi: 10.1016/0012-1606(92)90186-k. [DOI] [PubMed] [Google Scholar]
- Stryke D, et al. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003;31:278–81. doi: 10.1093/nar/gkg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H, et al. Human fibulin-1D: Molecular cloning, expression and similarity with S1-5 protein, a new member of the fibulin gene family. Matrix Biol. 1997;15:479–93. doi: 10.1016/s0945-053x(97)90021-4. [DOI] [PubMed] [Google Scholar]
- Twal WO, et al. Fibulin-1 suppression of fibronectin-regulated cell adhesion and motility. J Cell Sci. 2001;114:4587–98. doi: 10.1242/jcs.114.24.4587. [DOI] [PubMed] [Google Scholar]
- Verzi MP, et al. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. 2005;287:134–45. doi: 10.1016/j.ydbio.2005.08.041. [DOI] [PubMed] [Google Scholar]
- Waldo KL, et al. Conotruncal myocardium arises from a secondary heart field. Development. 2001;128:3179–88. doi: 10.1242/dev.128.16.3179. [DOI] [PubMed] [Google Scholar]
- Watari N, et al. Hoxa3 regulates integration of glossopharyngeal nerve precursor cells. Dev Biol. 2001;240:15–31. doi: 10.1006/dbio.2001.0447. [DOI] [PubMed] [Google Scholar]
- Yelbuz TM, et al. Shortened outflow tract leads to altered cardiac looping after neural crest ablation. Circulation. 2002;106:504–10. doi: 10.1161/01.cir.0000023044.44974.8a. [DOI] [PubMed] [Google Scholar]
- Zhang HY, et al. Extracellular matrix protein fibulin-2 is expressed in the embryonic endocardial cushion tissue and is a prominent component of valves in adult heart. Dev Biol. 1995;167:18–26. doi: 10.1006/dbio.1995.1003. [DOI] [PubMed] [Google Scholar]
- Zhang HY, et al. The extracellular matrix glycoproteins BM-90 and tenascin are expressed in the mesenchyme at sites of endothelial-mesenchymal conversion in the embryonic mouse heart. Differentiation. 1993;52:211–20. doi: 10.1111/j.1432-0436.1993.tb00633.x. [DOI] [PubMed] [Google Scholar]
- Zhou HM, et al. Essential role for ADAM19 in cardiovascular morphogenesis. Mol Cell Biol. 2004;24:96–104. doi: 10.1128/MCB.24.1.96-104.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]