

Abstract
Ocular involvement in muscular dystrophy ranges from structural defects to abnormal electroretinograms. While the mechanisms underlying the abnormal retinal physiology in patients are not understood, it is thought that α-dystroglycan extracellular interactions are critical for normal visual function. Here we show that β-dystroglycan anchors dystrophin and the inward rectifying K+ channel Kir4.1 at glial endfeet and that disruption of dystrophin and potassium channel clustering in dystroglycan mutant mice is associated with an attenuation of the electroretinogram b-wave. Glial-specific inactivation of dystroglycan or deletion of the cytoplasmic domain of β-dystroglycan was sufficient to attenuate the electroretinogram b-wave. Unexpectedly, deletion of the β-dystroglycan cytoplasmic domain did not disrupt the laminar structure of the retina. In contrast to the role of α-dystroglycan extracellular interactions during early development of the CNS, β-dystroglycan intracellular interactions are important for visual function but not the laminar development of the retina.
Introduction
Muscular dystrophies with ocular involvement are associated with mutations that affect the expression or posttranslational processing of components of the dystrophin-glycoprotein complex (DGC) (Cohn and Campbell, 2000; Barresi and Campbell, 2006). Muscle-eye-brain disease (MEB), caused by mutations of known or putative glycosyltransferases in the biosynthetic pathway of dystroglycan (Barresi and Campbell, 2006), is characterized by brain and eye involvement, including high myopia, retinal dysplasia, and cataracts (Muntoni and Voit, 2004). In a study of 18 MEB patients, electroretinograms were isoelectric in 12 patients and the electroretinogram b-wave was attenuated in 4 patients (Santavuori et al., 1989). Prolonged electroretinogram latency was reported in one case study (Fahnehjelm et al., 2001). An attenuation of the electroretinogram b-wave is also frequently observed in Duchenne and Becker muscular dystrophies, though vision is preserved (Cibis et al., 1993; Pillers et al., 1999). Duchenne and Becker muscular dystrophies are both caused by mutations of the gene encoding dystrophin. A reduction of dystroglycan expression is also observed in the retinas of mice with mutations in the dystrophin gene (Kameya et al., 1997; Blank et al., 1999; Dalloz et al., 2001).
The components of the DGC in skeletal muscle include dystrophin, dystroglycan, the sarcoglycan/sarcospan complex, syntrophin, and dystrobrevin (Ervasti and Campbell, 1991; Ibraghimov-Beskrovnaya et al., 1992; Cohn and Campbell, 2000). Dystroglycan, several isoforms of dystrophin, and other components of the skeletal muscle DGC are broadly expressed in the CNS, including the retina (Pillers et al., 1993; D'Souza et al., 1995; Montanaro et al., 1995; Ueda et al., 2000; Dalloz et al., 2001). Dystroglycan consists of an extracellular α subunit (α-dystroglycan) and a transmembrane β subunit (β-dystroglycan), which are noncovalently associated (Ibraghimov-Beskrovnaya et al., 1993). α-Dystroglycan binds laminin, perlecan, agrin, neurexin, and pikachurin in the extracellular space (Ervasti and Campbell, 1993; Gee et al., 1994; Peng et al., 1998; Sugita et al., 2001; Sato et al., 2008), and the intracellular C-terminal tail of β-dystroglycan binds dystrophin in the cytoskeleton (Ervasti and Campbell, 1993).
In the retina, dystroglycan is concentrated in the Müller glial endfeet at the inner limiting membrane and in the glial endfeet abutting the vasculature (Montanaro et al., 1995). Dystroglycan is also expressed at ribbon synapses of rod and cone photoreceptors in the outer plexiform layer of the retina (Blank et al., 1997; Koulen et al., 1998; Blank et al., 1999; Jastrow et al., 2006). The expression of dystroglycan in the outer plexiform layer coincides with the earliest detection of synaptic markers (Blank et al., 2002), suggesting that it may have a role in synaptic physiology. Tissue-specific deletion of the dystroglycan gene in the mouse epiblast produces structural defects of both the anterior and posterior chambers of the eye (Satz et al., 2008), but the physiology of the retina has not been previously examined in dystroglycan-deficient mice.
Here we show that tissue-specific loss of dystroglycan in the neuroepithelium causes an attenuation of the electroretinogram b-wave, which resembles the defect in muscular dystrophy patients. The abnormal retinal physiology was associated with the disruption of dystrophin and the inward rectifying potassium channel Kir4.1 in glial cells. Either loss of dystroglycan from glial cells or mutation of the β-dystroglycan cytoplasmic domain was sufficient to attenuate the electroretinogram b-wave, demonstrating the importance of β-dystroglycan intracellular interactions for the physiology of the retina.
Materials and Methods
Generation of dystroglycan-deficient mice.
The generation of the floxed-dystroglycan mouse strain has been previously described (Cohn et al., 2002). Homozygous floxed dystroglycan (Dag1lox/lox) mice were bred to hemizygous mice expressing the GFAP-Cre (Zhuo et al., 2001; Moore et al., 2002) or Nestin-Cre (Tronche et al., 1999; Graus-Porta et al., 2001). Mice that were heterozygous for the floxed dystroglycan allele and hemizygous for the Cre recombinase transgene (Cre-Dag1+/lox) were bred with mice that were homozygous for the floxed dystroglycan allele (Dag1lox/lox) to generate conditional dystroglycan-deficient mice (Cre-Dag1lox/lox). The genotypes of the mice were determined by PCR analysis of tail DNA.
Generation of truncated dystroglycan mutant mice.
A 4.3 kb SalI-EcoRV fragment of mouse genomic dystroglycan gene (Williamson et al., 1997) that includes exon 2 was subcloned into pBluescript KS (+) (pBSDGSE). Two PCRs were performed using pBSDGSE as a template with primers that incorporate stop codons in all three reading frames and a HindIII site as a marker for the stop codons. The resulting PCR product was subcloned into a pPNT vector (Dr. Richard Mulligan, Harvard Medical School, Boston, MA) flanking a PGK-neomycin resistance cassette. The vector includes a thymidine kinase cassette distal to the short arm to allow for negative selection of nonrecombinants. R1 ES cells (2 × 107; from 129/Sv male mouse blastocyst) were electroporated with the NotI-linearized targeting vector. G418- and ganciclovir-resistant clones were isolated. Targeting fidelity was determined by PCR and sequence analysis. Three correctly targeted ES cell lines were aggregated with ICR out bred 8-cell stage embryos then implanted into ICR based surrogate mothers. Chimeras are pups that have an agouti coat, the percentage of which indicates acceptance of the mutated gene. To decrease screening time and to flag for germ-line transmitters, all the ICR-based chimeras were mated with ICR mice. Dark eyed pups from these matings indicate the parent chimera is able to give germ-line transmission of the genetically altered ES cells. Of the 34 chimeras, 16 were found to present germ-line transmission. The 16 chimeras were mated with C57BL/6 mice and the resulting tail DNA from the pups was screened by PCR to identify heterozygotes. Heterozygous mutant mice from the F1 generation were crossed to obtain homozygous mutant mice.
Slit lamp.
Slit-lamp images were acquired with a Topcon SL-D7 slit lamp, using a 25× objective and a Nikon D100 digital camera. Identical camera settings were used for each photo.
Electroretinogram.
All experiments were performed in accordance with protocols approved by the Animal Care and Use Committee at the University of Iowa and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All procedures were performed in total darkness with the aid of infrared night-vision goggles. All animals were tested between 9:00 A.M. and 11:00 A.M. to minimize circadian physiological variations. Mice were dark adapted overnight, anesthetized with a mixture of ketamine (70 mg/kg) and xylazine (7 mg/kg) and maintained on a heating blanket until immobile. Following dilation of the right eye with a drop of atropine (1%), both eyes were wetted using a 1.2% cellulose, 0.9% NaCl solution. Animals were placed on a heating pad inside the test chambers, a grounding electrode inserted into the tail, and DTL fiber electrodes placed on the surface of each eye. An occluding contact lens was placed on the left eye, and a transparent lens on the right. Diffuse stimuli were presented to the right eye using narrow bandwidth LEDs (λmax 520 nm) in a series of nine full-field flash stimuli with luminances ranging from 5.5 × 10−5 to 26.7 cd · s · m−2 to investigate scotopic vision, followed by a flash stimulus of 300 cd · s · m−2 against a dim adapting light of 0.4 cd · m−2 to evoke the photopic response. The scotopic threshold response was measured at 5.5 × 10−5 cd · s · m−2. Data were recorded at 1 kHz with a resolution of 0.5 μV and response averaging was used to improve the signal-to-noise ratio in the dim stimuli. All stimuli presentation and data recordings were controlled by Labview (National Instruments). Analysis of the electroretinograms (ERGs) was performed using MATLAB analysis software (The MathWorks), both systems running software kindly made available to the community by Dr. L. Pinto (Northwestern University, Chicago, IL) (www.neuromice.org). Unpaired t tests were used to determine significance in comparisons between the mutant animals and wild-type controls.
Morris water maze.
The protocol for the visible platform test of the Morris water maze was based on the design by Richard Morris (Morris, 1981; Wemmie et al., 2002; Lim et al., 2004). The maze consisted of a circular pool 105 cm in diameter, 60 cm deep, with a water level of 40 cm. The water was made opaque with nontoxic white paint. A black platform, 12 cm in diameter, was raised 1 cm above the water surface level. The mice were placed along the edge of the pool and allowed a maximum time of 60 s to locate the platform. The time to reach the platform was recorded. Each mouse was tested on two successive trials.
Visual cliff test.
The visual cliff test protocol was based on the design by M. W. Fox (Fox, 1965; Crawley, 2000). A test chamber was constructed with a Plexiglas floor and positioned 0.5 m above the ground. One half of the Plexiglas floor was covered from the outside with black-and-white checkered contact paper and the other half of the floor remained uncovered, creating the illusion of a cliff. Checkered contact paper extended from the platform and covered the ground below. Mice were placed on a metal ridge at the interface of the checkered (“safe”) or uncovered (“unsafe”) sides of the test chamber and allowed to move freely to either side. The movement of the mouse to the “safe” or “unsafe” side of the test chamber was recorded. Each mouse was tested in 10 successive trials. The whiskers of the mice were shaved before testing to minimize tactile cues.
Biochemistry.
For immunoblot, retinas were solubilized in TBS containing 1% Triton X-100 and protease inhibitors. The solubilized fraction was incubated with WGA-agarose beads (Vector Laboratories) for 24 h. The beads were washed three times in TBS containing 0.1% Triton X-100 and protease inhibitors. The bound proteins were eluted with TBS containing 0.1% Triton X-100, protease inhibitors, and 300 mm N-acetyl-glucosamine. Proteins were separated by 3–15% SDS-PAGE and transferred to polyvinylidene fluoride membranes.
Histology and immunofluorescence.
For histology, adult mice 10–30 weeks of age were deeply anesthetized with ketamine and killed by intracardiac perfusion with 4% paraformaldehyde. The eyes were processed and embedded in paraffin or acrylamide. Sections were stained with hematoxylin and eosin and imaged on a Leica DMRXA microscope. For immunofluorescence, the eyes were removed and snap frozen in Tissue-Tek O.C.T. compound or fixed for 4 h in 4% paraformaldehyde and frozen in an acrylamide embedding solution (Johnson and Blanks, 1984; Thompson et al., 2008). Sections (8 μm thick) were cut on a cryostat (Leica RM2135) and stored at −80°C before use. Sections were fixed for 10 min in 2% paraformaldehyde and blocked for 30 min with 4% BSA and 0.3% Triton X-100 in PBS. After overnight incubation of the primary antibody at 4°C and washes with PBS, sections were incubated with CY3- or FITC-conjugated secondary antibodies (Jackson Laboratories), AlexaFluor 488- or AlexaFluor 594-labeled secondary antibodies (Invitrogen), or biotinylated secondary antibodies (Vector Laboratories) and streptavidin conjugated to FITC or CY3 (Jackson Laboratories). Images were acquired on DMRXA (Leica), BX41 (Olympus), M1 (Zeiss), and MRC600 (Biorad) microscopes.
Antibodies.
The following antibodies were used: anti-α-dystroglycan, IIH6 (Ervasti and Campbell, 1991) and goat 20 (Ervasti and Campbell, 1991); anti-β-dystroglycan, AP83 (Duclos et al., 1998); anti-laminin (Sigma L9393); anti-aquaporin 4 (Millipore); anti-dystrophin, rabbit 31 (Ohlendieck and Campbell, 1991; Duclos et al., 1998) and MANDRA1 (Developmental Studies Hybridoma Bank, The University of Iowa); anti-Kir4.1 (Alomone Labs); anti-mGluR6 (Neuromics). IIH6 is a monoclonal antibody to the fully glycosylated species of α-dystroglycan (Ervasti and Campbell, 1991). Goat 20 recognizes the core protein of α-dystroglycan (Ibraghimov-Beskrovnaya et al., 1992).
Quantification of axon density.
Optic nerves were dissected from nonperfused mice, fixed by immersion in 2.5% glutaraldehyde in cacodylate buffer, and processed into epon blocks. Semithin epon cross sections of optic nerve were cut at 2 μm, counterstained with toluidine blue, and photographed with a light microscope (Zeiss M1) at 60× under oil immersion. Quantifications of axon density, optic nerve cross-sectional area, and optic nerve perimeter were done on Adobe Photoshop. A square grid of 150 μm per side was applied to the image and the number of complete myelinated axons was counted in every third square (see supplemental Fig. 7A, available at www.jneurosci.org as supplemental material). Axon density was calculated as the number of axons per 100 μm2. Optic nerve cross-sectional area and perimeter were measured by tracing the perimeter of the optic nerve.
Results
Generation of Nestin-Cre/DG-null mice
The dystroglycan-null mutation causes embryonic lethality in mice (Williamson et al., 1997), and mice with conditional deletion of dystroglycan in all epiblast-derived tissues (MORE-DG null) die before the maturation of the eye (Satz et al., 2008), presenting a barrier to the study of visual function. To generate mice with conditional deletion of dystroglycan in the CNS, floxed-dystroglycan mice (Cohn et al., 2002) were crossed with mice that express Cre recombinase transgene under the control of the rat Nestin enhancer (Tronche et al., 1999; Graus-Porta et al., 2001; MacPherson et al., 2004). Nestin, a type VI intermediate filament protein, is widely expressed in neuroepithelial precursor cells. In the retina, the rat Nestin-Cre transgene is expressed as early as embryonic day 9.5 (MacPherson et al., 2004) and the Nestin-Cre transgene has been previously demonstrated to be effective for gene deletion in the retina (MacPherson et al.; Honjo et al., 2007; Martins et al., 2008).
The Nestin-Cre allele was maintained as a hemizygous locus on a background that was heterozygous for the floxed-dystroglycan allele. The compound heterozygous mice were bred with mice homozygous for the floxed-dystroglycan allele. Crosses of floxed-dystroglycan and Nestin-Cre mice generated dystroglycan-deficient (Nestin-Cre/DG-null) mice according to Mendelian ratio. A spectrum of ocular malformations was observed in the dystroglycan-deficient mice, including synechia of the iris and cornea, corneal opacities, microphthalmia, and buphthalmos (supplemental Figs. 1–3, available at www.jneurosci.org as supplemental material). The pupils of the Nestin-Cre/DG-null mice were dilated but responsive to light (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
Dystroglycan is normally expressed in the corneal endothelium, the lens epithelium, the iris, the ciliary epithelia, and the retina (Ueda et al., 2000). To confirm that the Nestin-Cre transgene abolished dystroglycan expression in eye, vertical sections through Nestin-Cre/DG-null and wild-type littermate control eyes were labeled with antibodies to dystroglycan. Nestin-Cre transgene activity has been reported in the iris, lens, and the ciliary body of the eye (Calera et al., 2006) and Nestin-Cre/DG-null eyes showed no detectable dystroglycan in these structures (supplemental Fig. 5, available at www.jneurosci.org as supplemental material, and data not shown). Dystroglycan expression was also notably absent in the cornea of Nestin-Cre/DG-null mice (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Although expression of the nestin-Cre transgene has not been previously demonstrated in the cornea, nestin is expressed in the cornea during early embryonic development at E12.5 (Yang et al., 2000). In the retina, dystroglycan is normally concentrated at Müller glia endfeet abutting the inner limiting membrane, perivascular glial endfeet, and in the outer plexiform layer. In Nestin-Cre/DG-null mice, dystroglycan was completely absent from glial endfeet (Fig. 1D). Some residual dystroglycan expression was detected in the outer plexiform layer in the Nestin-Cre/DG-null mice (Fig. 1D); however, the amount of expression was greatly reduced compared with wild-type littermate controls, and dystroglycan expression was not detected on immunoblots of Nestin-Cre/DG-null retinas (Fig. 1E).
Figure 1.

Loss of dystroglycan expression in Nestin-CRE/DG-null retina. A, B, Sections of adult wild-type (A) and Nestin-CRE/DG-null (B) retina stained with hematoxylin and eosin. C, D, Sections of adult wild-type (C) and Nestin-Cre/DG-null (D) retinas labeled with the AP83 antibody to β-dystroglycan. E, Immunoblot of WGA-enriched retina homogenates from wild-type littermate (control) and Nestin-Cre/DG-null (Nestin) retinas labeled with the IIH6 and AP83 antibodies to α- and β-dystroglycan. The sections were counterstained with propidium iodide. GCL, Ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer.
Previously we have shown that retinal nonattachment and dysgenesis occur in mice with MOX2-Cre-mediated deletion of dystroglycan in the mouse epiblast (Satz et al., 2008). Unexpectedly, the laminar organization of the Nestin-CRE/DG-null retina appeared normal by light microscopy of hematoxylin and eosin-stained sections (Fig. 1B; supplemental Fig. 6, available at www.jneurosci.org as supplemental material). The Nestin-Cre transgene is activated at embryonic day 9.5 (MacPherson et al., 2004) in the retina whereas MOX2-Cre is activated earlier in development at embryonic day 5 and affects all epiblast-derived tissues (Tallquist and Soriano, 2000).
Disruption of the inner limiting membrane has been shown to cause reduced viability of retinal ganglion cells during development (Halfter et al., 2005) and optic nerve hypoplasia is a phenotype that has been reported in some congenital muscular dystrophy patients (Muntoni and Voit, 2004). For this reason, we quantified the axon density, cross-sectional area, and perimeter from control and Nestin-Cre/DG-null mice. A reduction in the number of ganglion cell axons in the optic nerve was observed in one Nestin-Cre/DG-null mouse that was affected by severe hydrocephalus (data not shown). Ganglion cell loss has been previously associated with hydrocephalus (Williamson et al., 1992). This example of optic nerve atrophy was not representative, as no significant difference was observed in the optic nerve cross-sectional area, perimeter, or myelinated axon density of three Nestin-Cre/DG-null mice that were not affected by hydrocephalus (supplemental Fig. 7B–E, available at www.jneurosci.org as supplemental material).
Selective disruption of DGC components in the dystroglycan-deficient eye
In skeletal muscle (Cohn et al., 2002) and brain (Moore et al., 2002; Satz et al., 2008), the loss of dystroglycan is associated with a disruption of the expression of other components of the DGC. The expression of DGC components in the retina was examined by immunofluorescence to determine whether dystroglycan is necessary for their localization. Retinas from Nestin-Cre/DG-null and littermate control mice were labeled with antibodies to laminin, dystrophin, and aquaporin 4. Immunofluorescence labeling of laminin in the Nestin-CRE/DG-null eye showed intact expression at the inner limiting membrane (Fig. 2B). In contrast, we had previously observed discontinuous localization of laminin at the inner limiting membrane in mice with MOX2-Cre-mediated excision of dystroglycan (Satz et al., 2008). Mice with MOX2-Cre-mediated deletion of dystroglycan also exhibited retinal detachment and dysgenesis, whereas the laminar histology of the retina is preserved in the Nestin-Cre/DG-null mice. Comparison of the MORE-DG-null mice and the Nestin-Cre/DG-null mice suggests that dystroglycan is necessary for the integrity of the inner limiting membrane early in development but that it is dispensable for its later maintenance.
Figure 2.
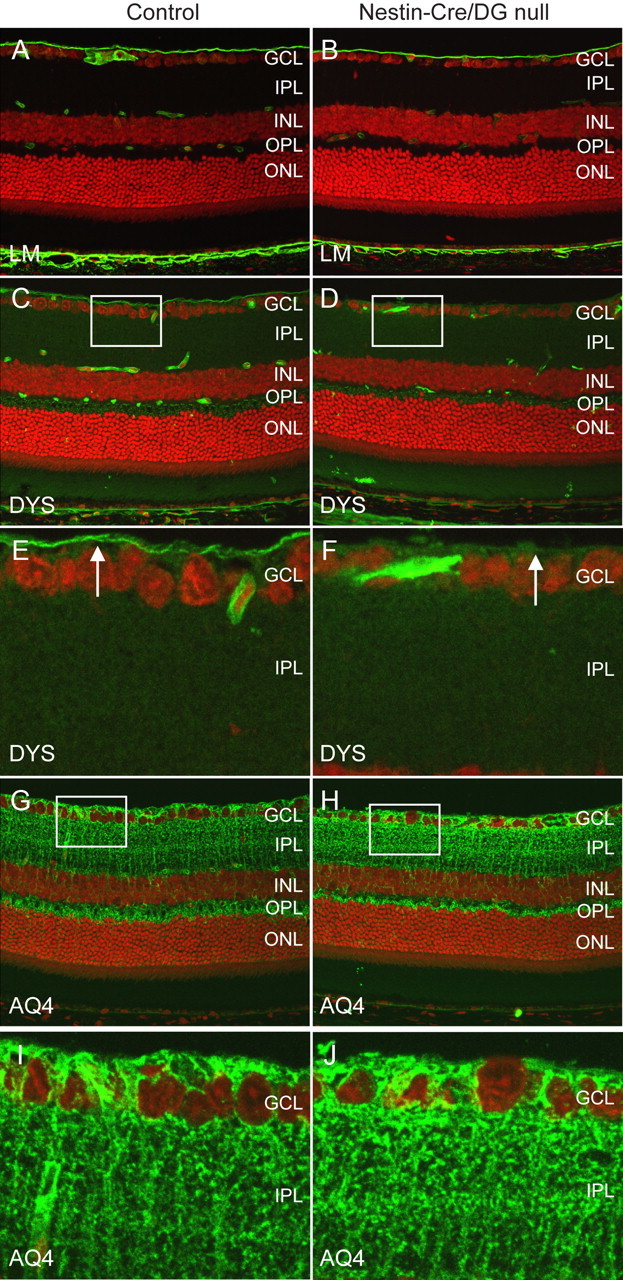
Selective loss of components of the dystrophin–glycoprotein complex. A–J, Sections of adult wild-type littermate (left column) and Nestin-CRE/DG-null (right column) retina labeled with antibodies to laminin (A, B), dystrophin (C–F), and aquaporin 4 (G–J). The sections were counterstained with propidium iodide. Note the selective loss of staining with the dystrophin antibody at the inner limiting membrane (arrows in E, F) of the Nestin-Cre/DG-null mouse. E, F and I, J are high-magnification views of the boxed regions in C, D and G, H, respectively. GCL, Ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer.
Dystrophin is normally concentrated at Müller glial endfeet at the inner limiting membrane, perivascular glial endfeet, and photoreceptor ribbon synapses in the outer plexiform layer of the retina. The predominant dystrophin isoform in Müller glia and astrocytes is DP71 (Howard et al., 1998). A retina specific isoform of dystrophin, DP260, is concentrated in photoreceptor ribbon synapses (D'Souza et al., 1995; Howard et al., 1998). In the Nestin-Cre/DG-null eye dystrophin expression was selectively lost from the inner limiting membrane (Fig. 2D,F). The expression of dystrophin in the outer plexiform layer was comparable to the expression in wild-type littermate control retinas (Fig. 2D).
Aquaporin 4 is normally expressed in Müller glial cells in retina. The expression of Aquaporin 4 (Fig. 2G,H) was similar in Nestin-Cre/DG-null and control mice but a reduction of expression in the perivascular glial end feet was observed (Fig. 2H).
Impaired performance on visual tasks
Visual function in wild-type and Nestin-CRE/DG-null mice was assessed on the visible platform test of the Morris water maze (Fig. 3A) and the visual cliff test (Fig. 3B). Nestin-CRE/DG-null mice that were affected by hydrocephalus or ocular aberrations that were visible by slit-lamp photography (e.g., microphthalmia and corneal opacities) were excluded from the studies.
Figure 3.
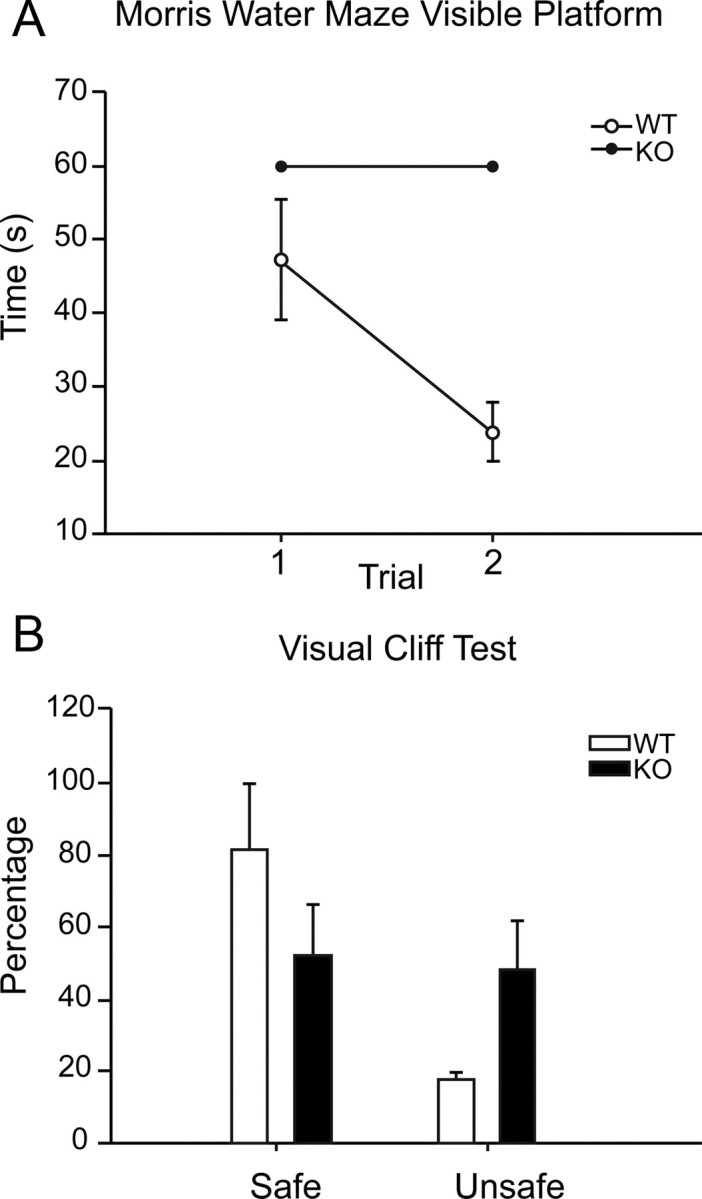
Nestin-CRE/DG-null mice show poor performance on visual tasks. A, Morris water maze visible platform test. B, Visual cliff test.
On the visible platform test of the Morris water maze, the mice were subjected to two successive trials and the escape latencies were recorded. The escape latencies of wild-type littermate control mice were between 40 and 50 s on the first trial and between 20 and 30 s on the second trial (n = 9). Nestin-CRE/DG-null mice failed to find the visible platform in both trials and were removed from the maze after 60 s (n = 6). The visible platform test is dependent on motivation and motor function as well as vision, but the mice showed no difficulty swimming or balancing on a rotarod (data not shown).
As an additional evaluation of vision, the mice were also tested in 10 successive trials on the visual cliff test. The littermate control mice chose the “safe” side 80% of the time (n = 10) whereas Nestin-CRE/DG-null mice chose the “safe” and “unsafe” side with approximately equal frequency (n = 10), corresponding to chance.
Loss of dystroglycan expression in the GFAP-Cre/DG-null retina
GFAP-CRE/DG-null mice have previously been characterized (Moore et al., 2002) but the eye was not examined. The human GFAP-Cre transgene drives expression of Cre recombinase in radial glia and astrocytes, silencing dystroglycan in these cells and a subset of neurons that are the progeny of radial glia (Zhuo et al., 2001; Moore et al., 2002). Immunofluorescence detection of dystroglycan (Fig. 4A,B) and dystrophin (Fig. 4C,D) in the GFAP-CRE/DG-null eye showed discontinuous expression at the inner limiting membrane but preserved expression in the outer plexiform layer. As had been observed in Nestin-Cre/DG-null mice, the expression of laminin was preserved in the GFAP-Cre/DG-null retina (Fig. 4E,F). Dystroglycan expression was also preserved in the cornea and the lens, which appeared normal on histological examination (data not shown).
Figure 4.
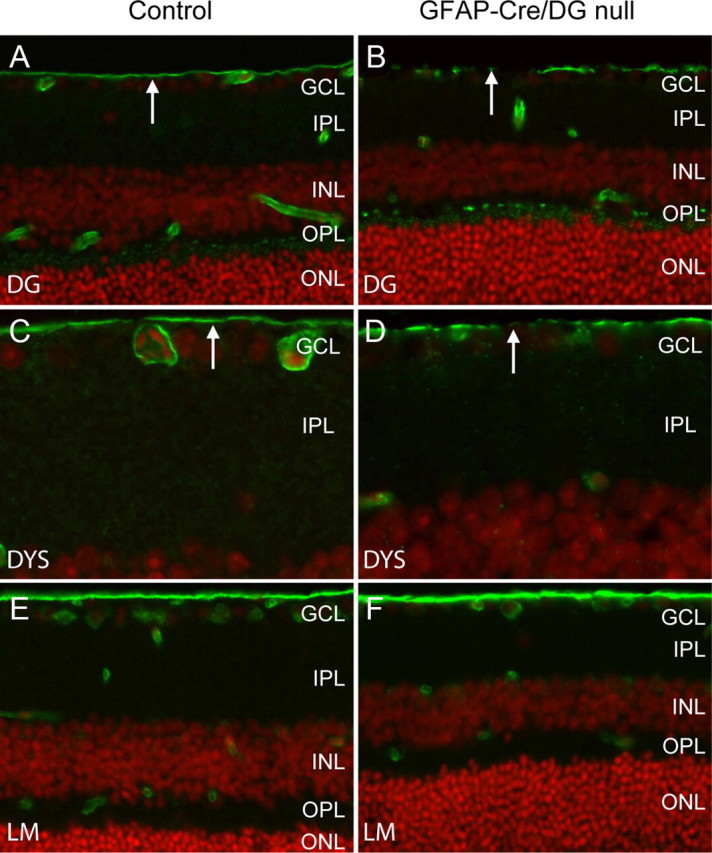
Loss of dystroglycan expression in GFAP-Cre/DG-null retina. A–F, Sections of wild-type littermate (left column) and GFAP-CRE/DG-null (right column) retina labeled with the AP83 antibody to β-dystroglycan (A, B) and MANDRA1 antibody to dystrophin (C, D) and laminin (E, F). The sections were counterstained with propidium iodide. GCL, Ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer.
Attenuation of the scotopic threshold response and electroretinogram b-wave
The affect of dystroglycan deletion on the physiology of the Nestin-Cre/DG-null and GFAP-Cre/DG-null retinas was examined by electroretinogram (ERG). Mice with indications of hydrocephalus or ocular aberrations, which were detected by slit lamp, were excluded from the ERG studies.
The scotopic threshold response (STR) of the electroretinogram (ERG) is elicited by very dim flashes of light that selectively stimulate the rod photoreceptors. The STR has positive (pSTR) and negative (nSTR) components. In mice, the pSTR is mediated through a cone off ganglion cell pathway while the nSTR is mediated in part through an AII-type amacrine cell (Frishman, 2006). A significantly reduced pSTR was observed in both the Nestin-CRE/DG-null (p = 0.0014) and GFAP-CRE/DG-null (p = 0.0249) mice (Fig. 5). The nSTR was statistically normal in the Nestin-CRE/DG-null mice, while significantly greater than normal (p = 0.0241) in the GFAP-CRE/DG-null mice (Fig. 5).
Figure 5.

Scotopic threshold responses (STR). A, ERG scotopic threshold responses of wild-type, Nestin-CRE/DG-null, and GFAP-CRE/DG-null mice elicited at 5.5 × 10–5 cd · s · m−2. B, Negative scotopic threshold responses. C, Positive scotopic threshold responses. Note the attenuation of the positive scotopic threshold responses in the Nestin-CRE/DG-null and GFAP-CRE/DG-null mice.
The bright flash scotopic ERG is composed of a-, b-, and c-waves. The ERG a-wave reflects the electrical activity of the rod and cone photoreceptor cells. (Peachey et al., 1995; Goto et al., 1996; Lyubarsky and Pugh, 1996). The ERG a-waves in the Nestin and GFAP mice were statistically indistinguishable from the wild-type controls (Fig. 6), suggesting that the function of the photoreceptors is normal. The representative response in Figure 6 is from a high-luminance stimulus, which reflects both rod and cone photoreceptor activity. In this figure, the wild-type animal a-wave is terminated rapidly by the onset of the b-wave. In the Nestin-Cre/DG-null and GFAP-Cre/DG-null animals, the reduced amplitude b-wave (see below) is associated with a prolongation of the a-wave.
Figure 6.

Full intensity response scotopic electroretinograms. A, Scotopic electroretinograms at varying stimulus intensities from wild-type, Nestin-CRE/DG-null, and GFAP-CRE/DG-null mice (B) Electroretinogram a-wave response at 26.7 cd · s · m−2. C, Maximum a-wave values. D, Electroretinogram b-wave responses at 4.02 cd · s · m−2. E, Normalized b-waves. Note that the Nestin-CRE/DG-null and GFAP-CRE/DG-null mice exhibited robust a-waves but attenuated b-waves.
The b-wave responses are generated predominantly by activity of ON-bipolar cells (Robson and Frishman, 1995; Hood and Birch, 1996; Robson and Frishman, 1996). In both the Nestin-CRE/DG-null and GFAP-CRE/DG-null mice, the b-wave responses were severely attenuated (p = 0.0123 and p = 0.0223 respectively) (Fig. 6). The log of the luminance at half maximal amplitude of the b-wave (sigmaB) was statistically indistinguishable for Nestin-Cre/DG-null and GFAP-Cre/DG-null animals. This suggests that while the maximum response is severely attenuated, the response itself is still graded with respect to the luminance, i.e., a brighter stimulus elicits a stronger response. There was a significant amount of variation in the results within the Nestin-CRE/DG-null and GFAP-CRE/DG-null mice (p < 0.0001 and p = 0.0004, respectively) indicating variation in the phenotypic severity. The c-wave response originates in the retinal pigment epithelium (Pinto and Enroth-Cugell, 2000). We did not observe any statistical differences between the c-wave responses measured in the wild-type and mutant animals (data not shown).
Abnormal Kir4.1 clustering but normal expression of mGluR6 in the Nestin-Cre/DG-null retina
The attenuation of the ERG b-wave suggested that the localization of metabotropic glutamate receptor 6 (mGluR6) in the outer plexiform layer may be disrupted by the loss of dystroglycan expression in Nestin-Cre/DG-null mice. mGluR6 mediates the response of ON bipolar cells and deletion of mGluR6 in mice causes an attenuation of the electroretinogram b-wave (Masu et al., 1995). Immunofluorescence labeling showed that the expression of mGluR6 was indistinguishable between Nestin-CRE/DG-null mice and littermate controls (Fig. 7A,B). Furthermore, the electroretinogram b-wave was also attenuated in GFAP-Cre/DG-null mice, which have preserved expression of dystroglycan in the outer plexiform layer, suggesting an alternative mechanism for the abnormal physiology.
Figure 7.
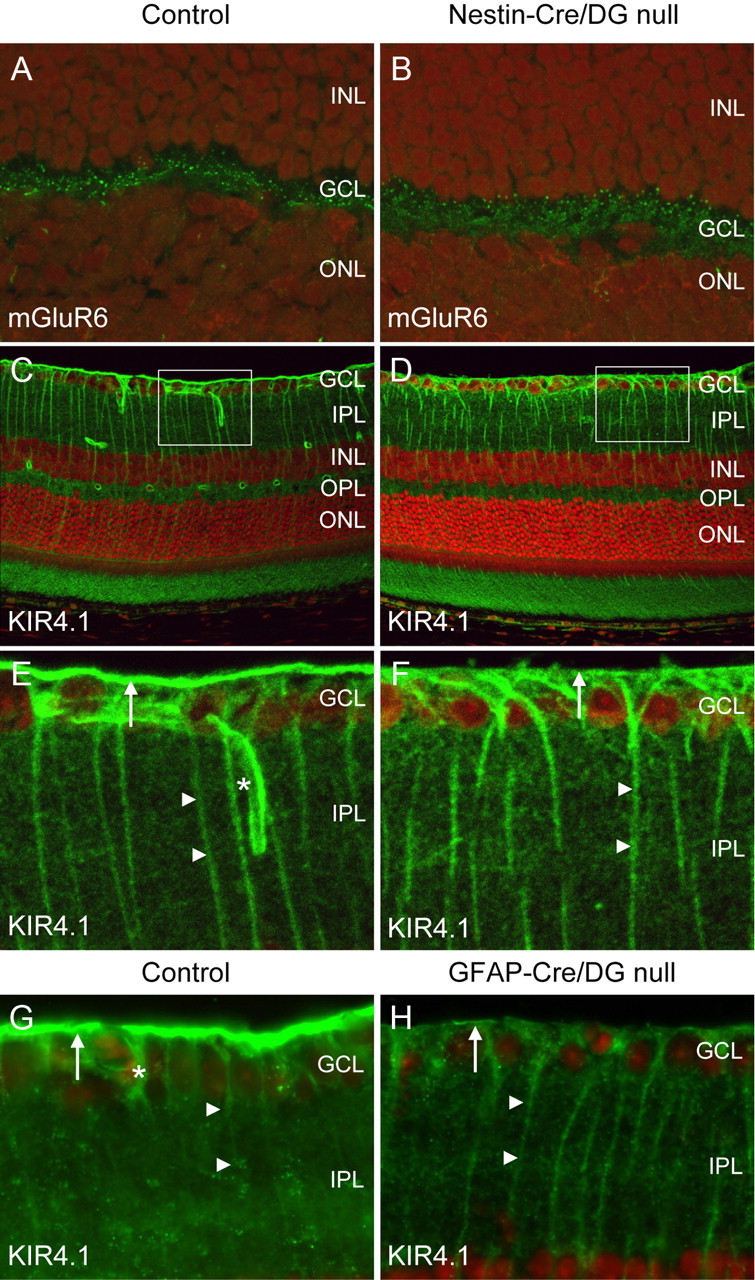
Localization of mGluR6 and Kir4.1. A–F, Sections of adult wild-type littermate (left column) and Nestin-CRE/DG-null (right column) retina labeled with antibodies to mGluR6 (A, B) and Kir4.1 (C–F). G, H, Sections of wild-type littermate (left column) and GFAP-CRE/DG-null (right column) retina labeled with an antibody to Kir4.1. Note the loss of Kir4.1 localization in perivascular glial endfeet (normal marked by asterisks in E, G) and the loss of Kir4.1 clustering in Müller glial endfeet at the inner limiting membrane (arrows). Müller glial processes (arrow heads) retained Kir4.1 expression. The sections were counterstained with propidium iodide. E, F, High-magnification views of the boxed regions in C and D, respectively. GCL, Ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer.
Defects in the clustering of Kir4.1, an inward rectifying potassium channel in Müller glia, have been demonstrated in mice with mutations of dystrophin (Connors and Kofuji, 2002) and the selective loss of dystrophin at the inner limiting membrane in the Nestin-Cre/DG-null retina suggested that Kir 4.1 clustering may also be impaired in the mice. In the wild-type littermate control mouse retina, Kir4.1 expression was detected in perivascular glial endfeet and in Müller glial processes. Expression of Kir 4.1 was enriched in Müller glia endfeet abutting the inner limiting membrane. In Nestin-Cre/DG-null and GFAP-Cre/DG-null mice, the expression of Kir4.1 was retained in the processes of Müller glial cells however Kir4.1 was not enriched in Müller glial endfeet or perivascular glial endfeet (Fig. 7D,F,H).
Deletion of the cytoplasmic tail of β-dystroglycan is sufficient to disrupt potassium channel clustering and the physiology of the retina
In Nestin-Cre/DG-null mice, deletion of dystroglycan caused a selective loss of dystrophin and Kir4.1 expression at the inner limiting membrane. In contrast, the localization of dystrophin was preserved in the outer plexiform layer despite reduced dystroglycan expression. We have previously shown that a peptide corresponding to the C-terminal last 15 aa of β-dystroglycan (KNMTPYRSPPPYVPP) binds to dystrophin in vitro (Jung et al., 1995). To test whether the cytoplasmic tail is required for dystrophin membrane-anchoring in vivo and to examine whether disruption of β-dystroglycan intracellular interactions is sufficient to attenuate the electroretinogram b-wave, we generated transgenic mice lacking the entire cytoplasmic region of β-dystroglycan (DGβcyt/βcyt) (supplemental Fig. 8, available at www.jneurosci.org as supplemental material).
Premature stop codons were introduced in all three reading frames of the Dag1 gene in murine 129J embryonic stem (ES) cells (supplemental Fig. 8B, available at www.jneurosci.org as supplemental material). The targeted truncation ends at Lys-778, including only 4 aa in the predicted cytoplasmic part of β-dystroglycan (Ibraghimov-Beskrovnaya et al., 1992). The chimera mice derived from three independent heterozygous ES clones were separately backcrossed to C57BL/6J. Heterozygous mice were crossed to generated homozygous mutant offspring. The targeted truncation of the cytoplasmic regions of β-dystroglycan was confirmed by tail DNA genotyping (supplemental Fig. 8C, available at www.jneurosci.org as supplemental material). The number of DGβcyt/βcyt mice from heterozygous intercrosses was smaller than the Mendelian ratio (35:55:9, +/+:+/βcyt:βcyt/βcyt), suggesting that some of the homozygous animals died in utero.
To confirm that the βcyt mutant dystroglycan was expressed, sections of the retina were labeled with antibodies to dystroglycan. The AP83 antibody, which recognizes the C terminus of β-dystroglycan (Duclos et al., 1998), showed no signal in the βcyt/βcyt mouse confirming the deletion of the C-terminal tail of β-dystroglycan (Fig. 8B). The G20 antibody, which recognizes α-dystroglycan (Michele et al., 2002), showed that dystroglycan was correctly localized in the βcyt/βcyt mice (Fig. 8D).
Figure 8.
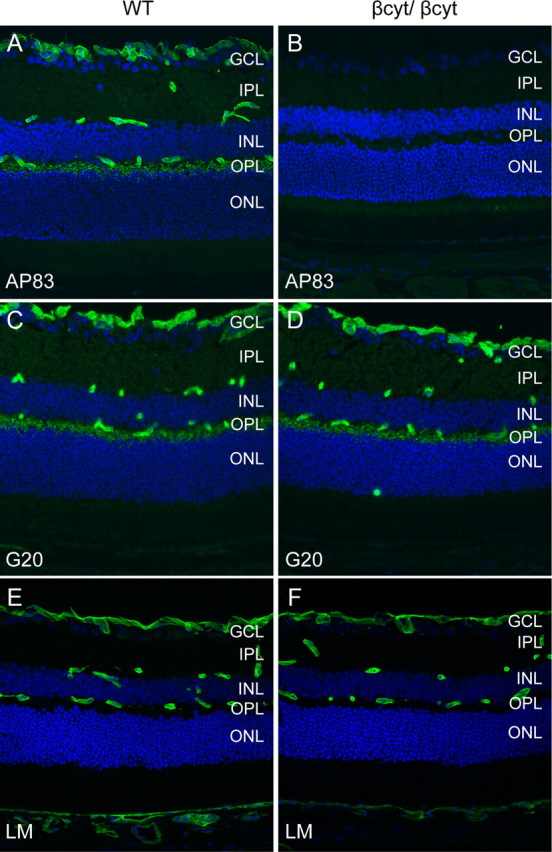
Deletion of the β-dystroglycan cytoplasmic domain. A–F, Sections of adult wild-type littermate (left column) and βcyt/βcyt (right column) retina labeled with antibodies to the C terminus of β-dystroglycan, AP83 (A, B), α-dystroglycan, G20 (C, D), and laminin (E, F). The sections were counterstained with 4′, 6-diamidino-2-phenylindoledihydrochloride.
The laminar organization of the retinas from βcyt/βcyt mice was indistinguishable from wild-type littermate controls by light microscopy of hematoxylin and eosin-stained sections (data not shown), and laminin was normally expressed at basement membrane interfaces formed by glial endfeet at the inner limiting membrane and vasculature (Fig. 8F). Dystrophin was selectively lost from Müller glial endfeet at the inner limiting membrane and perivascular glial endfeet, but expression in the outer plexiform layer was preserved (Fig. 9B). The deletion of the cytoplasmic tail of β-dystroglycan was also sufficient to disrupt the clustering of Kir 4.1 in Müller glial endfeet (Fig. 9D).
Figure 9.

Selective loss of dystrophin and Kir4.1 in βcyt/βcyt retina. Sections of 5-week-old wild-type littermate (left column) and βcyt/βcyt (right column) retina labeled with antibodies to dystrophin (A, B) and Kir4.1 (C, D). The sections were counterstained with propidium iodide. Note the selective loss of staining with the dystrophin antibody at perivascular sites and the inner limiting membrane (arrows in C, D) of the βcyt/βcyt mouse. GCL, Ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer.
Electroretinograms were recorded from the βcyt/βcyt to examine the effect of disruption of β-dystroglycan intracellular interactions on the physiology of the retina. The ERG a-waves in βcyt/βcyt mice were indistinguishable from those of the wild-type controls (Fig. 10A), suggesting that the function of the photoreceptors is normal in the mutant mice, but the b-wave responses were attenuated (Fig. 10B).
Figure 10.

Scotopic electroretinograms of βcyt/βcyt mice. A, Electroretinogram a-wave response at 26.7 cd · s · m−2. B, Electroretinogram b-wave responses at 4.02 cd · s · m−2. Note that the βcyt/βcyt mice exhibited robust a-waves but attenuated b-waves.
Discussion
An attenuation of the electroretinogram b-wave is characteristic of Duchenne and Becker muscular dystrophies and muscle-eye-brain disease, but the mechanisms underlying the abnormal retinal physiology in patients are not understood. Here we show that deletion of dystroglycan in the CNS causes an attenuation of the electroretinogram b-wave similar to what is observed in patients. The abnormal retinal physiology was associated with a selective loss of dystrophin and Kir4.1 clustering in glial endfeet, suggesting a critical role for dystroglycan intracellular interactions for the physiology of the retina.
In skeletal muscle, loss of dystroglycan results in the disruption of dystrophin and other components of the DGC. In the retina, β-dystroglycan anchors dystrophin in Müller glial endfeet and perivascular glial endfeet, but it is not necessary for the localization of dystrophin in the outer plexiform layer, suggesting that another protein anchors dystrophin in the absence of dystroglycan. Although dystrophin does not require dystroglycan for its localization in the outer plexiform layer, the two proteins are closely associated at the synapse. DP260, a retina-specific isoform of dystrophin, is localized at photoreceptor synapses (D'Souza et al., 1995) and its disruption results in a selective loss of dystroglycan in the outer plexiform layer (Kameya et al., 1997). Mice with impaired expression of DP260 have an electroretinogram with a prolonged b-wave implicit time but no change in b-wave amplitude (Kameya et al., 1997). Interestingly, we observed a delay in the implicit time of electroretinograms from Nestin-Cre/DG-null mice although the expression of dystrophin was preserved, suggesting that dystroglycan is important for the physiology of the ribbon synapse.
Recently, it has been shown that a novel α-dystroglycan-binding protein, pikachurin, is necessary for the apposition of presynaptic and postsynaptic termini in the photoreceptor ribbon synapse (Sato et al., 2008). Deletion of pikachurin produced an abnormal electroretinogram (Sato et al., 2008) similar to that observed in Nestin-Cre/DG-null mice. Similar defects are also present in mice with mutations of laminin β2 chain (Libby et al., 1999) but it is not known if dystroglycan binds to the laminin β2 chain. Patients with muscle-eye-brain disease (Santavuori et al., 1989) and mouse models with mutations of large or fukutin show an attenuation of the electroretinogram b-wave (Holzfeind et al., 2002; Takeda et al., 2003; Lee et al., 2005), suggesting that a disruption of α-dystroglycan glycosylation and extracellular ligand binding is sufficient to disrupt its function. Hypoglycosylation of dystroglycan may also affect the intracellular interactions of dystroglycan by disrupting dystroglycan localization.
Our results suggest that dystroglycan contributes to the physiology of the retina by more than one mechanism. GFAP-Cre inactivation of dystroglycan in glial cells was sufficient to attenuate the b-wave even though dystroglycan was present in the outer plexiform layer. Although it remains controversial, there is evidence that potassium channels in Müller glia may contribute to the b-wave (Wen and Oakley, 1990; Connors and Kofuji, 2002). In the GFAP-Cre/DG-null retina the localization of dystrophin and Kir4.1 was disrupted in Müller glial endfeet, supporting the hypothesis that abnormal glial potassium currents may contribute to the abnormal retinal physiology. Mutation of the cytoplasmic domain of β-dystroglycan was sufficient to disrupt the localization of dystrophin and Kir4.1 in glial endfeet and attenuate the electroretinogram b-wave. Prior research shows that deficiency in Kir4.1 causes a defect in the amplitude of the c-wave but the b-wave is preserved (Wu et al., 2004). One of the important roles of Müller glial potassium channels is spatial buffering (Connors et al., 2004). In contrast to the Kir4.1-deficient mice, dystroglycan-deficient mice lose Kir4.1 expression selectively at glial endfeet and expression is preserved on the Müller glial processes. Furthermore, a selective disruption of Kir4.1 expression in glial endfeet is also associated with an attenuation of electroretinogram b-wave in mice with mutations of dystrophin (Pillers et al., 1995; Connors and Kofuji, 2002), providing support for this hypothesis. Alternatively, other yet to be identified β-dystroglycan intracellular interactions in Müller glia may also contribute to the b-wave amplitude.
Loss of dystroglycan expression in the neuroepithelium in Nestin-Cre/DG-null mice causes an attenuation of the scotopic threshold response as well as the b-wave of the bright flash scotopic electroretinogram. Based on the electrophysiology, we would predict that the animals would have some degree of useful scotopic and photopic vision but that it would be attenuated with respect to the wild-type mice. The Nestin-Cre/DG-null mice demonstrated severe visual impairment on two visual paradigms, which is likely attributed to defects in the CNS in addition to the abnormal retinal physiology. Structural defects, which may have been missed on examination by slit-lamp photography, may have also contributed to the poor performance of the mice on visual tasks. A combination of brain and eye defects is also likely to contribute to the loss of vision in congenital muscular dystrophy patients.
Preservation of the scotopic a-wave but attenuation of the b-wave observed in both Nestin-Cre/DG-null and GFAP-Cre/DG-null animals is also similar to that observed in “typical” juvenile X-linked retinoschisis (XLRS) (Pawar et al., 1996; Heckenlively and Arden, 2006). Exceptions have been observed and are termed “atypical” (Sieving et al., 1999). Sieving et al. (1999) assess the cause of the reduced b-wave as “defective signaling by depolarizing bipolar cells in the rod pathway” (Heckenlively and Arden, 2006). There is also a similarity to two different types of congenital stationary night blindness (CSNB) (Heckenlively and Arden, 2006 MIT press 2006). Incomplete CSNB has the same b-wave attenuation observed in the Nestin-Cre/DG-null and GFAP-Cre/DG-null animals but unlike the mutant animals, incomplete CSNB typically has a severely attenuated cone response. Complete CSNB has an intact cone response, as observed in the Nestin-Cre/DG-null and GFAP-Cre/DG-null animals, but has an absent scotopic b-wave. Thus the absent pSTR observed in these animals is a notable difference from the responses of human patients affected with XLRS or CSNB. This facet of the Nestin-Cre/DG-null and GFAP-Cre/DG-null mutant animals may be a valuable tool for the further dissection of retinal physiology.
Footnotes
This work was supported in part by Paul D. Wellstone Muscular Dystrophy Cooperative Research Center Grant NS053672 and National Institutes of Health Grant NS041407. K.P.C. and E.M.S. are investigators of the Howard Hughes Medical Institute. We thank members of the Campbell laboratory for comments on this work. We thank Mandy Pattersen, Jason Flanagan, Baoli Yang, Keith Garringer, and Melissa Hassebrock for technical support and John A. Wemmie for the use of the Morris water maze.
The authors declare no competing financial interests.
References
- Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- Blank M, Koulen P, Kröger S. Subcellular concentration of beta-dystroglycan in photoreceptors and glial cells of the chick retina. J Comp Neurol. 1997;389:668–678. [PubMed] [Google Scholar]
- Blank M, Koulen P, Blake DJ, Kröger S. Dystrophin and beta-dystroglycan in photoreceptor terminals from normal and mdx3Cv mouse retinae. Eur J Neurosci. 1999;11:2121–2133. doi: 10.1046/j.1460-9568.1999.00636.x. [DOI] [PubMed] [Google Scholar]
- Blank M, Blake DJ, Kröger S. Molecular diversity of the dystrophin-like protein complex in the developing and adult avian retina. Neuroscience. 2002;111:259–273. doi: 10.1016/s0306-4522(02)00032-5. [DOI] [PubMed] [Google Scholar]
- Calera MR, Topley HL, Liao Y, Duling BR, Paul DL, Goodenough DA. Connexin43 is required for production of the aqueous humor in the murine eye. J Cell Sci. 2006;119:4510–4519. doi: 10.1242/jcs.03202. [DOI] [PubMed] [Google Scholar]
- Cibis GW, Fitzgerald KM, Harris DJ, Rothberg PG, Rupani M. The effects of dystrophin gene mutations on the ERG in mice and humans. Invest Ophthalmol Vis Sci. 1993;34:3646–3652. [PubMed] [Google Scholar]
- Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–1471. doi: 10.1002/1097-4598(200010)23:10<1456::aid-mus2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Cohn RD, Henry MD, Michele DE, Barresi R, Saito F, Moore SA, Flanagan JD, Skwarchuk MW, Robbins ME, Mendell JR, Williamson RA, Campbell KP. Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell. 2002;110:639–648. doi: 10.1016/s0092-8674(02)00907-8. [DOI] [PubMed] [Google Scholar]
- Connors NC, Kofuji P. Dystrophin Dp71 is critical for the clustered localization of potassium channels in retinal glial cells. J Neurosci. 2002;22:4321–4327. doi: 10.1523/JNEUROSCI.22-11-04321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors NC, Adams ME, Froehner SC, Kofuji P. The potassium channel Kir4.1 associates with the dystrophin-glycoprotein complex via alpha-syntrophin in glia. J Biol Chem. 2004;279:28387–28392. doi: 10.1074/jbc.M402604200. [DOI] [PubMed] [Google Scholar]
- Crawley JN. New York: Wiley; 2000. What's wrong with my mouse?: behavioral phenotyping of transgenic and knockout mice. [Google Scholar]
- Dalloz C, Claudepierre T, Rodius F, Mornet D, Sahel J, Rendon A. Differential distribution of the members of the dystrophin glycoprotein complex in mouse retina: effect of the mdx(3Cv) mutation. Mol Cell Neurosci. 2001;17:908–920. doi: 10.1006/mcne.2001.0978. [DOI] [PubMed] [Google Scholar]
- D'Souza VN, Nguyen TM, Morris GE, Karges W, Pillers DA, Ray PN. A novel dystrophin isoform is required for normal retinal electrophysiology. Hum Mol Genet. 1995;4:837–842. doi: 10.1093/hmg/4.5.837. [DOI] [PubMed] [Google Scholar]
- Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, Durbeej M, Lebakken CS, Ettinger AJ, van der Meulen J, Holt KH, Lim LE, Sanes JR, Davidson BL, Faulkner JA, Williamson R, Campbell KP. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahnehjelm KT, Ygge J, Engman ML, Mosskin M, Santavuori P, Malm G. A child with muscle-eye-brain disease. Ophthalmological and neurological characteristics. Acta Ophthalmol Scand. 2001;79:72–75. doi: 10.1034/j.1600-0420.2001.079001072.x. [DOI] [PubMed] [Google Scholar]
- Fox MW. The visual cliff test for the study of visual depth perception in the mouse. Anim Behav. 1965;13:232–233. doi: 10.1016/0003-3472(65)90040-0. [DOI] [PubMed] [Google Scholar]
- Frishman LJ. Rockville, MD: ARVO; 2006. Towards guidelines for mouse electroretinography. [Google Scholar]
- Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-alpha, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Goto Y, Peachey NS, Ziroli NE, Seiple WH, Gryczan C, Pepperberg DR, Naash MI. Rod phototransduction in transgenic mice expressing a mutant opsin gene. J Opt Soc Am A Opt Image Sci Vis. 1996;13:577–585. doi: 10.1364/josaa.13.000577. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Müller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Halfter W, Willem M, Mayer U. Basement membrane-dependent survival of retinal ganglion cells. Invest Ophthalmol Vis Sci. 2005;46:1000–1009. doi: 10.1167/iovs.04-1185. [DOI] [PubMed] [Google Scholar]
- Heckenlively JR, Arden GB. Ed 2. Cambridge, MA: MIT; 2006. Principles and practice of clinical electrophysiology of vision. [Google Scholar]
- Holzfeind PJ, Grewal PK, Reitsamer HA, Kechvar J, Lassmann H, Hoeger H, Hewitt JE, Bittner RE. Skeletal, cardiac and tongue muscle pathology, defective retinal transmission, and neuronal migration defects in the Large(myd) mouse defines a natural model for glycosylation-deficient muscle-eye-brain disorders. Hum Mol Genet. 2002;11:2673–2687. doi: 10.1093/hmg/11.21.2673. [DOI] [PubMed] [Google Scholar]
- Honjo Y, Nagineni CN, Larsson J, Nandula SR, Hooks JJ, Chan CC, Karlsson S, Kulkarni AB. Neuron-specific TGF-beta signaling deficiency results in retinal detachment and cataracts in mice. Biochem Biophys Res Commun. 2007;352:418–422. doi: 10.1016/j.bbrc.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood DC, Birch DG. Beta wave of the scotopic (rod) electroretinogram as a measure of the activity of human on-bipolar cells. J Opt Soc Am A Opt Image Sci Vis. 1996;13:623–633. doi: 10.1364/josaa.13.000623. [DOI] [PubMed] [Google Scholar]
- Howard PL, Dally GY, Wong MH, Ho A, Weleber RG, Pillers DA, Ray PN. Localization of dystrophin isoform Dp71 to the inner limiting membrane of the retina suggests a unique functional contribution of Dp71 in the retina. Hum Mol Genet. 1998;7:1385–1391. doi: 10.1093/hmg/7.9.1385. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Milatovich A, Ozcelik T, Yang B, Koepnick K, Francke U, Campbell KP. Human dystroglycan: skeletal muscle cDNA, genomic structure, origin of tissue specific isoforms and chromosomal localization. Hum Mol Genet. 1993;2:1651–1657. doi: 10.1093/hmg/2.10.1651. [DOI] [PubMed] [Google Scholar]
- Jastrow H, Koulen P, Altrock WD, Kröger S. Identification of a beta-dystroglycan immunoreactive subcompartment in photoreceptor terminals. Invest Ophthalmol Vis Sci. 2006;47:17–24. doi: 10.1167/iovs.05-0597. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Blanks JC. Application of acrylamide as an embedding medium in studies of lectin and antibody binding in the vertebrate retina. Curr Eye Res. 1984;3:969–974. doi: 10.3109/02713688409167215. [DOI] [PubMed] [Google Scholar]
- Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP. Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J Biol Chem. 1995;270:27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- Kameya S, Araki E, Katsuki M, Mizota A, Adachi E, Nakahara K, Nonaka I, Sakuragi S, Takeda S, Nabeshima Y. Dp260 disrupted mice revealed prolonged implicit time of the b-wave in ERG and loss of accumulation of beta-dystroglycan in the outer plexiform layer of the retina. Hum Mol Genet. 1997;6:2195–2203. doi: 10.1093/hmg/6.13.2195. [DOI] [PubMed] [Google Scholar]
- Koulen P, Blank M, Kröger S. Differential distribution of beta-dystroglycan in rabbit and rat retina. J Neurosci Res. 1998;51:735–747. doi: 10.1002/(SICI)1097-4547(19980315)51:6<735::AID-JNR7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Lee Y, Kameya S, Cox GA, Hsu J, Hicks W, Maddatu TP, Smith RS, Naggert JK, Peachey NS, Nishina PM. Ocular abnormalities in Large(myd) and Large(vls) mice, spontaneous models for muscle, eye, and brain diseases. Mol Cell Neurosci. 2005;30:160–172. doi: 10.1016/j.mcn.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Libby RT, Lavallee CR, Balkema GW, Brunken WJ, Hunter DD. Disruption of laminin β2 chain production causes alterations in morphology and function in the CNS. J Neurosci. 1999;19:9399–9411. doi: 10.1523/JNEUROSCI.19-21-09399.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R, Zaheer A, Khosravi H, Freeman JH, Jr, Halverson HE, Wemmie JA, Yang B. Impaired motor performance and learning in glia maturation factor-knockout mice. Brain Res. 2004;1024:225–232. doi: 10.1016/j.brainres.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Lyubarsky AL, Pugh EN., Jr Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recordings. J Neurosci. 1996;16:563–571. doi: 10.1523/JNEUROSCI.16-02-00563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson D, Sage J, Kim T, Ho D, McLaughlin ME, Jacks T. Cell type-specific effects of Rb deletion in the murine retina. Genes Dev. 2004;18:1681–1694. doi: 10.1101/gad.1203304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins RA, Zindy F, Donovan S, Zhang J, Pounds S, Wey A, Knoepfler PS, Eisenman RN, Roussel MF, Dyer MA. N-myc coordinates retinal growth with eye size during mouse development. Genes Dev. 2008;22:179–193. doi: 10.1101/gad.1608008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masu M, Iwakabe H, Tagawa Y, Miyoshi T, Yamashita M, Fukuda Y, Sasaki H, Hiroi K, Nakamura Y, Shigemoto R, Takada M, Nakamura K, Nakao K, Katsuki M, Nakanishi S. Specific deficit of the ON response in visual transmission by targeted disruption of the mGluR6 gene. Cell. 1995;80:757–765. doi: 10.1016/0092-8674(95)90354-2. [DOI] [PubMed] [Google Scholar]
- Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- Montanaro F, Carbonetto S, Campbell KP, Lindenbaum M. Dystroglycan expression in the wild type and mdx mouse neural retina: synaptic colocalization with dystrophin, dystrophin-related protein but not laminin. J Neurosci Res. 1995;42:528–538. doi: 10.1002/jnr.490420411. [DOI] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- Morris RG. Spatial localization does not require the presence of local cues. Learn Motiv. 1981;12:239–260. [Google Scholar]
- Muntoni F, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord. 2004;14:635–649. doi: 10.1016/j.nmd.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K, Campbell KP. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar H, Bingham EL, Hiriyanna K, Segal M, Richards JE, Sieving PA. X-linked juvenile retinoschisis: localization between (DXS1195, DXS418) and AFM291wf5 on a single YAC. Hum Hered. 1996;46:329–335. doi: 10.1159/000154373. [DOI] [PubMed] [Google Scholar]
- Peachey NS, Goto Y, Quiambao AB, al-Ubaidi MR. Functional consequences of oncogene-induced photoreceptor degeneration in transgenic mice. Vis Neurosci. 1995;12:513–522. doi: 10.1017/s0952523800008427. [DOI] [PubMed] [Google Scholar]
- Peng HB, Ali AA, Daggett DF, Rauvala H, Hassell JR, Smalheiser NR. The relationship between perlecan and dystroglycan and its implication in the formation of the neuromuscular junction. Cell Adhes Commun. 1998;5:475–489. doi: 10.3109/15419069809005605. [DOI] [PubMed] [Google Scholar]
- Pillers DA, Bulman DE, Weleber RG, Sigesmund DA, Musarella MA, Powell BR, Murphey WH, Westall C, Panton C, Becker LE, Worton RG, Ray PN. Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nat Genet. 1993;4:82–86. doi: 10.1038/ng0593-82. [DOI] [PubMed] [Google Scholar]
- Pillers DA, Weleber RG, Woodward WR, Green DG, Chapman VM, Ray PN. mdxCv3 mouse is a model for electroretinography of Duchenne/Becker muscular dystrophy. Invest Ophthalmol Vis Sci. 1995;36:462–466. [PubMed] [Google Scholar]
- Pillers DA, Fitzgerald KM, Duncan NM, Rash SM, White RA, Dwinnell SJ, Powell BR, Schnur RE, Ray PN, Cibis GW, Weleber RG. Duchenne/Becker muscular dystrophy: correlation of phenotype by electroretinography with sites of dystrophin mutations. Hum Genet. 1999;105:2–9. doi: 10.1007/s004399900111. [DOI] [PubMed] [Google Scholar]
- Pinto LH, Enroth-Cugell C. Tests of the mouse visual system. Mamm Genome. 2000;11:531–536. doi: 10.1007/s003350010102. [DOI] [PubMed] [Google Scholar]
- Robson JG, Frishman LJ. Response linearity and kinetics of the cat retina: the bipolar cell component of the dark-adapted electroretinogram. Vis Neurosci. 1995;12:837–850. doi: 10.1017/s0952523800009408. [DOI] [PubMed] [Google Scholar]
- Robson JG, Frishman LJ. Photoreceptor and bipolar cell contributions to the cat electroretinogram: a kinetic model for the early part of the flash response. J Opt Soc Am A Opt Image Sci Vis. 1996;13:613–622. doi: 10.1364/josaa.13.000613. [DOI] [PubMed] [Google Scholar]
- Santavuori P, Somer H, Sainio K, Rapola J, Kruus S, Nikitin T, Ketonen L, Leisti J. Muscle-eye-brain disease (MEB) Brain Dev. 1989;11:147–153. doi: 10.1016/s0387-7604(89)80088-9. [DOI] [PubMed] [Google Scholar]
- Sato S, Omori Y, Katoh K, Kondo M, Kanagawa M, Miyata K, Funabiki K, Koyasu T, Kajimura N, Miyoshi T, Sawai H, Kobayashi K, Tani A, Toda T, Usukura J, Tano Y, Fujikado T, Furukawa T. Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat Neurosci. 2008;11:923–931. doi: 10.1038/nn.2160. [DOI] [PubMed] [Google Scholar]
- Satz JS, Barresi R, Durbeej M, Willer T, Turner A, Moore SA, Campbell KP. Brain and eye malformations resembling Walker-Warburg syndrome are recapitulated in mice by dystroglycan deletion in the epiblast. J Neurosci. 2008;28:10567–10575. doi: 10.1523/JNEUROSCI.2457-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieving PA, Bingham EL, Kemp J, Richards J, Hiriyanna K. Juvenile X-linked retinoschisis from XLRS1 Arg213Trp mutation with preservation of the electroretinogram scotopic b-wave. Am J Ophthalmol. 1999;128:179–184. doi: 10.1016/s0002-9394(99)00144-0. [DOI] [PubMed] [Google Scholar]
- Sugita S, Saito F, Tang J, Satz J, Campbell K, Südhof TC. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol. 2001;154:435–445. doi: 10.1083/jcb.200105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Kondo M, Sasaki J, Kurahashi H, Kano H, Arai K, Misaki K, Fukui T, Kobayashi K, Tachikawa M, Imamura M, Nakamura Y, Shimizu T, Murakami T, Sunada Y, Fujikado T, Matsumura K, Terashima T, Toda T. Fukutin is required for maintenance of muscle integrity, cortical histiogenesis and normal eye development. Hum Mol Genet. 2003;12:1449–1459. doi: 10.1093/hmg/ddg153. [DOI] [PubMed] [Google Scholar]
- Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 2000;26:113–115. doi: 10.1002/(sici)1526-968x(200002)26:2<113::aid-gene3>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Thompson S, Mullins RF, Philp AR, Stone EM, Mrosovsky N. Divergent phenotypes of vision and accessory visual function in mice with visual cycle dysfunction (Rpe65 rd12) or retinal degeneration (rd/rd) Invest Ophthalmol Vis Sci. 2008;49:2737–2742. doi: 10.1167/iovs.07-1546. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Ueda H, Baba T, Kashiwagi K, Iijima H, Ohno S. Dystrobrevin localization in photoreceptor axon terminals and at blood-ocular barrier sites. Invest Ophthalmol Vis Sci. 2000;41:3908–3914. [PubMed] [Google Scholar]
- Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman JH, Jr, Welsh MJ. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34:463–477. doi: 10.1016/s0896-6273(02)00661-x. [DOI] [PubMed] [Google Scholar]
- Wen R, Oakley B., 2nd K(+)-evoked Muller cell depolarization generates b-wave of electroretinogram in toad retina. Proc Natl Acad Sci U S A. 1990;87:2117–2121. doi: 10.1073/pnas.87.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson EC, Pearson HE, McAllister JP., 2nd Gliosis and ganglion cell death in the developing cat retina during hydrocephalus and after decompression. Brain Res Dev Brain Res. 1992;70:47–52. doi: 10.1016/0165-3806(92)90102-3. [DOI] [PubMed] [Google Scholar]
- Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y, Ibraghimov-Beskrovnaya O, Campbell KP. Dystroglycan is essential for early embryonic development: disruption of Reichert's membrane in Dag1-null mice. Hum Mol Genet. 1997;6:831–841. doi: 10.1093/hmg/6.6.831. [DOI] [PubMed] [Google Scholar]
- Wu J, Marmorstein AD, Kofuji P, Peachey NS. Contribution of Kir4.1 to the mouse electroretinogram. Mol Vis. 2004;10:650–654. [PMC free article] [PubMed] [Google Scholar]
- Yang J, Bian W, Gao X, Chen L, Jing N. Nestin expression during mouse eye and lens development. Mech Dev. 2000;94:287–291. doi: 10.1016/s0925-4773(00)00301-4. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]