

Abstract
Mutations in TNNT2, encoding cardiac troponin T, commonly shows early onset, aggressive dilated cardiomyopathy (DCM). This observation may influence the decision of whether to undertake clinical genetic testing for TNNT2 in later onset DCM. Further, the trigger for late onset DCM remains enigmatic. A 70‐year‐old woman, previously healthy with a left ventricular ejection fraction of 50%–55% at age 69, presented with DCM of unknown cause and a 4‐month history progressive heart failure requiring cardiac transplantation. Clinical genetic testing revealed a novel TNNT2 R139H mutation but no relevant variants in 18 other DCM genes. Her explanted heart showed partial fatty replacement in the right ventricle. Sequencing for five arrhythmogenic right ventricular dysplasia genes was negative. Functional studies in porcine cardiac skinned fibers reconstituted with the mutant R139H troponin T protein showed decreased Ca2+ sensitivity at pH 7, characteristic of DCM. Because fatty infiltration may acidify the myocellular environment, maximal force development examined at pH 6.5 was diminished, suggesting a possible environmental trigger. We conclude that the TNNT2 R139H mutation was likely to be disease causing. Further, later age of onset may not be relevant to exclude genetic testing for TNNT2 mutations. Clin Trans Sci 2010; Volume 3: 219–226.
Keywords: dilated cardiomyopathy, genetics
Introduction
The cardiomyopathies (dilated, DCM; hypertrophic, HCM; arrhythmogenic right ventricular dysplasia/cardiomyopathy, ARVD/C; restrictive, RCM; and left ventricular noncompaction, LVNC) are increasingly recognized as genetic disorders. 1 In support of this, the 2009 guidelines published by the Heart Failure Society of America now recommend family screening, counseling, and consideration of genetic testing as part of the genetic evaluation of cardiomyopathy. 1 For DCM, onset usually occurs 30–50 years of age, but it may also occur in children and older individuals. 2 , 3 DCM of unknown cause is also termed idiopathic dilated cardiomyopathy (IDC), which infers apparently sporadic disease. 4
Up to 50% of IDC is familial (familial dilated cardiomyopathy, FDC) and a disease‐causing mutation in any of more than 30 genes can be identified in 25%–30% of cases. 1 , 5 , 6 Emerging evidence also suggests that mutations can be present regardless of family history, 6 , 7 , 8 , 9 and in some cases multiple mutations may be at play. 6 , 7 , 9 All patterns of inheritance have been reported, however, autosomal dominant with reduced penetrance and variable expressivity is most commonly observed. 1 , 2 Genocopies caused by syndromic disease such as HFE‐related hemochromatosis are also possible, but rarer causes. 3
Few genotype–phenotype correlations exist; however, TNNT2 mutations have been associated with early onset aggressive disease, 9 , 10 , 11 , 12 , 13 and LMNA mutations have been consistently reported with conduction system disease and arrhythmia. 2 , 3 Most mutations are private, complicating results interpretation, especially in cases with a negative family history. 3 , 9 Despite these limitations, genetic testing for DCM can be informative for diagnosis confirmation, management, and identification of at‐risk family members. 3 , 14
A key question, not only for DCM genetics but of all of adult‐onset Mendelian disease, particularly disorders that show allelic heterogeneity and reduced, age‐dependent penetrance, is the enormous variation in the age of onset of disease. This question will become even more relevant with increased utilization of molecular genetic testing in DCM for the counseling of disease onset for those family members who are mutation carriers but have no evidence of disease. To this end, we present clinical, molecular genetic, pedigree, functional, and pathological data from a patient with late onset symptomatic DCM and a TNNT2 mutation, and suggest possible triggers of late onset. These data also raise the question of whether older age of DCM onset should be used to guide the decision of whether to undertake genetic testing.
Methods
Patient population
Written, informed consent was obtained from all subjects, and the Institutional Review Board of the University of Miami approved the project. The study principally included one Caucasian female proband and her family members, with clinical ascertainment methods as previously described. 7 , 15
Genetic evaluation and testing
A three‐generation cardiac family history was obtained. Genetic counseling was provided and the patient gave consent for genetic testing. Commercial genetic testing was conducted for the proband by the Laboratory for Molecular Medicine (Boston, MA) for 19 DCM genes (>30% detection rate) and by FAMILION (New Haven, CT; 40% detection rate) for five ARVD/C genes ( Table 1 ). Site‐specific genetic testing was conducted in our research laboratory, as previously described. 7 , 9 Briefly, genomic DNA was extracted from whole blood and was sequenced in our laboratory in both directions to detect and/or confirm nucleotide variants in TNNT2 in the proband and the proband’s relatives using an Applied Biosystems 3130XL capillary sequencer, as previously described. 7 , 9 Amino acid numbering was after Townsend et al. 16
Table 1.
Coding sequences and near intron exon junctions of genes sequenced for variants in the proband.
| Gene | Locus | OMIM* | Protein |
|---|---|---|---|
| Dilated cardiomyopathy genes | |||
| ACTN2 | 1q42–q43 | 102573 | α‐Actinin‐2 |
| TNNT2 | 1q32 | 191045 | Cardiac troponin T |
| LMNA | 1q21.2–.3 | 150330 | Lamin A/C |
| DES | 2q35 | 125660 | Desmin |
| SGCD | 5q33–34 | 601411 | d‐Sarcoglycan |
| PLN | 6q22.1 | 172405 | Phospholamban |
| ZASP/LDB3 | 10q22.2–23.3 | 605906 | Cypher |
| VCL | 10q22.1–23 | 193065 | Metavinculin |
| MLP/CSRP3 | 11p15.1 | 600824 | Muscle LIM protein |
| MYBPC3 | 11p11.2 | 600958 | Myosin‐binding protein C |
| ABCC9 | 12p12.1 | 601439 | SUR2A |
| MYH7 | 14q12 | 160760 | b‐Myosin heavy chain |
| TPM1 | 15q22.1 | 191010 | a‐Tropomyosin |
| ACTC | 15q14 | 102540 | Cardiac actin |
| CTF1 | 16p11.2–p11.1 | 600435 | Cardiotrophin |
| Titin‐cap TCAP | 17q12 | 604488 | Titin‐cap or telethonin |
| TNNI3 | 19q13.4 | 191044 | Cardiac troponin I |
| TAZ/G4.5 | Xq28 | 300394 | Tafazzin |
| EMD | Xq28 | 300384 | Emerin |
| Arrhythmogenic right ventricular dysplasia/cardiomyopathy genes | |||
| TMEM43 | 3p25 | 612048 | Transmembrane protein 43 |
| DSP | 6p24 | 125647 | Desmoplakin |
| PKP2 | 12p11 | 602861 | Plakophilin 2 |
| DSC2 | 18q12.1 | 125645 | Desmocollin 2 |
| DSG2 | 18q12.1–q12.2 | 125671 | Desmoglein 2 |
*OMIM is online Mendelian inheritance in man, URL: http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim.
Functional studies
Briefly, the wild‐type human cardiac troponin (HCTnT‐WT) was cloned, expressed, and purified, 17 and used as a template for sequential overlapping PCR (polymerase chain reaction) with primers designed to replace the R139H mutation, as previously reported. 9 A functional assay using porcine papillary skinned fibers prepared from the left ventricles of freshly slaughtered porcine hearts was conducted. The endogenous troponin complex was displaced by an excess of exogenous HCTnT and the binary complex was reconstituted. The Ca2+ dependence of force development and maximal force were measured. The experimental results were reported as x± SEM, and analyzed for significance using unpaired Student’s t test at p < 0.05.
Results
Clinical data
The proband, a previously healthy 70‐year‐old female, presented with New York Heart Association (NYHA) Class III heart failure and DCM to an outside hospital. She had been fully active except for symptoms of progressive dyspnea on exertion and fatigue that began 2–3 months prior to her presentation.
Her past medical history was remarkable for well‐controlled, asymptomatic HFE‐associated hemochromatosis (H63D homozygote) discovered serendipitously after a routine laboratory evaluation 10 years prior to presentation. Neither clinical or biochemical findings of hemochromatosis were present, and there was no history of cardiotoxic drug exposure. One year earlier, she had an episode of atypical chest pain and dizziness after playing a tennis match. A cardiovascular evaluation at that time included an ECG that showed a normal sinus rhythm with poor R wave progression in V1–V3, with nonspecific ST‐T changes in the inferior and lateral leads. A two‐dimensional transthoracic echocardiogram showed a left ventricular (LV) end‐diastolic dimension of 5.1 cm, a LV end‐systolic dimension of 3.9 cm, with an estimated ejection fraction of 55% with no wall motion abnormalities. Her septum and posterior wall were 0.9 and 1.0 cm thick, respectively. Her left atrial dimension was 3.1 cm, and her right atrial and ventricular size and function were normal. She underwent a standard Bruce treadmill stress test where she achieved 8 minutes and 45 seconds, with a peak heart rate of 142 (100% predicted 152), with a normal blood pressure response. No ischemic ST‐T changes were observed, and only rare isolated premature ventricular contractions were observed. Nuclear images showed a fixed defect at the apex that was thought to be artifact, with a gated ejection fraction of 47% without wall motion abnormalities. A helical 64 slice gated CT angiogram showed patent coronary arteries with only minimal fibrocalcific plaquing in the proximal left anterior descending without stenosis. At that study, her LV ejection fraction was estimated at 50% with an end‐diastolic volume of 159 mL, and end‐systolic volume of 79 mL, and a stroke volume of 80 mL.
A medical evaluation was undertaken. A two‐dimensional echocardiogram showed that her LV had dilated with an end‐diastolic dimension of 6.4 cm and an end‐systolic dimension of 5.0 cm; her fractional shortening was 21.9%, and her ejection fraction was estimated at 35%–40%. Despite usual medical therapy, she had an unrelenting progression of symptoms and was hospitalized with advanced NYHA Class IV heart failure 6 weeks later. A repeat echocardiogram showed that her LV end‐diastolic dimension was unchanged, but the ejection fraction had fallen to 10%–15% with moderately severe mitral regurgitation. She was referred to us for consideration of advanced therapies. A right heart catheterization revealed a cardiac index of 1.4 L/min/m2. Inotropic therapy was started, and her clinical course stabilized. She was evaluated, accepted, and listed for cardiac transplantation, and remained stable on a continuous inotropic infusion until she was successfully transplanted.
Pathologic evaluation
The explanted heart weighed 276 g. The coronary arteries arose from their normally positioned ostia, had a right dominant circulation and had a few atherosclerotic patches with no more than 30% stenoses of the lumina. There was no bridging of the epicardial arteries. No cardiac malformation was present and the atrioventricular valves were normal except for mild myxoid degeneration of the mitral and tricuspid valves. Serial transverse sections of the ventricles showed slight dilatation of the LV and slightly more pronounced dilatation of the right ventricle (RV) (Figure 1). The LV and RV cavities measured 3.5 and 3 cm in diameter, respectively. The LV wall showed no gross abnormalities and had a uniform thickness of 1.1 cm. The heart had a moderate amount of epicardial fat and the most striking gross change was the irregular, diffuse fat infiltration of the RV, somewhat more pronounced in the apical portion with focal transmural replacement of the myocardium. There was no fat infiltration of the LV. Hematoxylin and eosin (H&E) and trichrome stains of all segments of the ventricles showed foci of slight interstitial fibrosis of the LV and pronounced fat infiltration with no fibrosis of the RV (Figure 2). There was no myocarditis or infiltrative process and no histological evidence of hemochromatosis, which was further supported by a negative iron stain.
Figure 1.
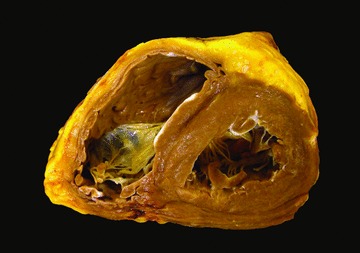
Explanted heart. A transverse section of explanted heart at about the proximal third between the atrioventricular groove and apex shows moderate amount of epicardial fat and marked fatty infiltration of the right ventricular wall. There is dilatation of the right ventricular cavity, slight dilatation of the left ventricular cavity, and the left ventricular wall has a normal uniform thickness without myocardial lesions.
Figure 2.
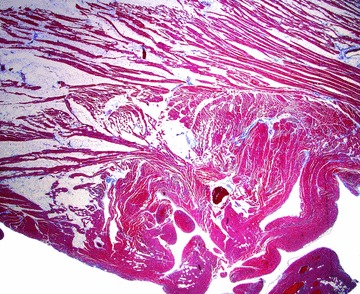
Photomicrograph of the right ventricular wall. The extensive fatty infiltration of the myocardium and absence of fibrosis is shown in this trichrome stained section.
Clinical genetic evaluation
Family history was negative for IDC or heart failure (Figure 3), although an uncle (II‐3) reportedly had an enlarged heart, and three other relatives (II‐1, II‐2, and III‐4) had cardiac related deaths confirmed by death certificate. The proband’s father (II‐1) died of myocardial failure and pulmonary edema; her mother (II‐2) died of pulmonary embolism; and her sister (III‐4) died suddenly of coronary artery disease in her 60s. Her daughter, the proband’s niece (IV‐3), had a history of palpitations and lower extremity swelling, intermittent numbness, and blanching of the fingers at age 49. Her echocardiogram was normal. Her ECG showed nonspecific ST‐T wave changes and left anterior hemiblock.
Figure 3.
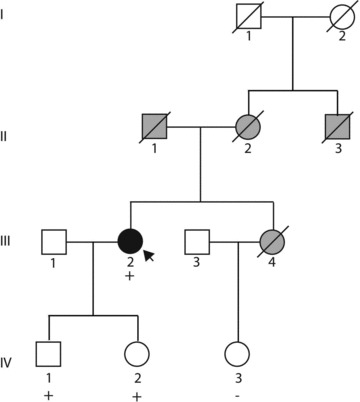
Pedigree. Squares represent males; circles represent females. The presence (+) of a troponin T mutation is indicated for those from whom DNA was available. Filled symbols indicate IDC. Gray symbols indicate another form of cardiovascular abnormality or diagnosis. See text for detailed clinical descriptions.
Oligonucleotide hybridization‐based DNA sequencing was conducted in the proband for a panel of 19 DCM genes ( Table 1 ). Only wild‐type sequence was identified in all fragments evaluated, except for two novel heterozygous variants in TNNT2: 416G>A (R139H) in exon 10 and 690–6G>A in intron 13. The intronic variant occurred within a not highly conserved location and was therefore considered a variant of unknown significance. The R139H variant was presumed pathogenic given the predicted amino acid substitution in a known DCM gene and its absence in more than 1,000 Caucasian controls.
The proband’s offspring (IV‐1, IV‐2), in their 5th decade, had normal physical exams, echocardiograms, and ECGs. Both were found to carry R139H and 690–6G>A, proving that the variants are in cis.
Because of the fatty infiltration noted in the right ventricle of the proband, genetic testing for five ARVD/C genes was undertaken with DNA from the proband ( Table 1 ). Only wild‐type sequence was identified.
Functional characterization of the TNNT2 R139H mutation
The R139H mutation significantly decreased Ca2+ sensitivity of force but did not exert a significant change in maximal force recovery at pH 7.0 (Figure 4). The pCa50 (pCa needed to reach 50% of the maximal force) for skinned fibers exchanged with the HCTnT WT or R139H was 5.66 ± 0.01 and 5.54 ± 0.01, respectively. At pH 6.5 the mutation also desensitized the myofilament to Ca2+ (−0.1 pCa units) and further decreased the maximal force recovery of approximately 20% (Figure 4).
Figure 4.

Normalized pCa‐force relationship in skinned cardiac muscle fibers. The Ca2+ dependence of force development was measured in each preparation after whole troponin (Tn) displacement and reconstitution at pH 7.0 and pH 6.5. The maximal force recovery was also evaluated and no significant difference was observed at pH 7.0 between fibers containing wild‐type human cardiac troponin T (HCTnT‐WT) and HCTnT‐R139H (57.4 ± 1.8 vs. 58.9 ± 4.7), respectively. However, a difference in maximal force recovery was observed at pH 6.5 between HCTnT‐WT and HCTnT‐R139H (53.9 ± 2.6 vs. 43.8 ± 2.7), respectively. Data in each experiment are the average of 5 experiments and expressed as mean ± S.E. *=p < 0.05 compared to the HCTnT‐WT.
Age of detection of clinical disease of previously published TNNT2 mutation carriers
The mean age of detection of DCM in patients with TNNT2 mutations reported from our group 9 and others 10 , 11 , 13 from 55 subjects was 29.7 ± 18.1 years; the median age was 26 years, and ranged from 0.1 to 84 years ( Table 2 ). The median age of death, implantation of a left ventricular device (LVAD) or heart transplant was 21 years (range 0.1–74 years) ( Table 2 ).
Table 2.
Age of detection of disease and age of outcome (death, LVAD or transplant), TNNT2 mutation carriers, from four prior publications.*
| Subject | Age of detection, years | Age at death, LVAD or transplant, years | Heart failure (Y/N) or NYHA Class (I–IV) | Phenotypic features |
|---|---|---|---|---|
| Kamisago et al. 10 | ||||
| Family C: Lys 210 deletion | ||||
| II‐3 | 26 | 26 | I | Sudden death |
| II‐4 | 27 | 27 | I | Sudden death |
| III‐3 | 53 | III | Alive, age 59 | |
| III‐4 | 26 | 26 | I | |
| III‐6 | 48 | III | Alive, age 51 | |
| III‐7 | 0.1 | 0.1 | I | Sudden death |
| III‐8 | 0.7 | 0.7 | I | Sudden death |
| IV‐1 | 23 | II | ||
| Family D: Lys 210 deletion | ||||
| I‐1 | 49 | 68 | IV | Cerebrovascular accident |
| II‐1 | 17 | 17 | IV | HF |
| II‐2 | 19 | 19 | IV | Postpartum HF, sudden death |
| II‐4 | 23 | II | Alive, age 44 | |
| III‐1 | 14 | 15 | IV | HF |
| Li et al. 11 | ||||
| Pedigree 1: Arg‐141‐Trp | ||||
| II‐1 | 84 | III | EF 25%, bradycardia | |
| III‐2 | 40 | IV | EF<20%, tachycardia, IRBBB | |
| III‐4 | 42 | II | EF 40%, inferior MI | |
| III‐6 | 46 | I | EF 45%, bradycardia | |
| IV‐5 | 27 | I | EF 38%, IRBBB | |
| IV‐17 | 15 | II | EF 32% | |
| IV‐19 | 17 | 17 | IV | Heart transplant |
| IV‐20 | 9 | I | EF 40%, prolonged QT | |
| IV‐21 | 6 | I | EF 45%, prolonged QT | |
| IV‐27 | 37 | II | EF 40%, prolonged QT | |
| IV‐38 | 21 | 21 | IV | Heart transplant |
| IV‐39 | 22 | II | EF 49% | |
| IV‐43 | 60 | II | EF 38% | |
| Mogensen et al. 13 | ||||
| Family B: Arg131Trp | ||||
| II‐3 | 23 | III | ||
| II‐2 | 16 | 16 | I | Sudden death |
| I‐2 | 34 | 34 | III | Died, HF |
| Family C: Arg205Leu | ||||
| III‐1 | 16 | 16 | III | Heart transplant, age 16 |
| III‐2 | 20 | 20 | IV | LVAD |
| II‐2 | 45 | II | ||
| I‐2 | 24 | 24 | I | Sudden death |
| Family D: Lys210del | ||||
| III‐3 | 25 | 26 | III | Died, HF |
| III‐4 | 21 | 22 | IV | Heart transplant, age 22 |
| III‐1 | 35 | I | ||
| II‐1 | 36 | 36 | I | Sudden death |
| Family E: Asp270Asn | ||||
| II‐1 | 38 | 38 | IV | Heart transplant, age 38 |
| III‐2 | 19 | I | ||
| Hershberger et al. 9 | ||||
| Pedigree A: ARG‐134‐GLY | ||||
| A.1 | 63 | No | ||
| A.4 | 34 | No | ICD | |
| A.6 | 34 | 36 | No | Sudden death |
| A.8 | 12 | No | 1AVB, asymptomatic | |
| A.10 | 6 | 10 | No | Heart transplant, age 10 |
| Pedigree B: ARG‐151‐CYS | ||||
| B.9 | 19 | 19 | Yes | Heart transplant, age 19 |
| Pedigree C: ARG‐159‐GLN | ||||
| C.3 | 20 | Yes | Peripartum cardiomyopathy | |
| Pedigree D: ARG‐205‐TRP | ||||
| D.3 | 0.5 | 12 | Yes | Heart transplant, age 12 |
| Pedigree E: Lys 210 deletion | ||||
| E.2 | 31 | 31 | Yes | Heart transplant, age 31 |
| Pedigree F: Lys 210 deletion | ||||
| F.4 | 37 | No | Asymptomatic, age 50 | |
| F.6 | 26 | Yes | ||
| Pedigree H: Lys 210 deletion | ||||
| H.6 | 74 | 74 | Yes | Died, HF |
| H.11 | 59 | Yes | ||
| H.15 | 52 | Yes | ||
| H.16 | 47 | No | Asymptomatic at age 47 | |
| Pedigree I: GLU‐244‐ASP | ||||
| I.3 | 13 | 13 | Yes | LVAD, heart transplant, age 13 |
| Number of subjects | 55 | 27 | ||
| Average age, years | 29.7 | 24.6 | ||
| Std. dev. | 18.1 | 16.5 | ||
| Median | 26 | 21 | ||
| Minimum | 0.1 | 0.1 | ||
| Maximum | 84 | 74 | ||
*LVAD, left ventricular assist device; NYHA, New York Heart Association; HF, heart failure; ICD, implantable cardiac defibrillator.
The proband of this current report, with almost normal cardiovascular screening at age 69 but with DCM and frank heart failure at age 70, becomes the third oldest reported subject ( Table 2 ). Both of the two older subjects (84 years, Li et al. 11 and 74 years, Hershberger et al. 9 ) belong to FDC kindreds, carried their family TNNT2 mutations and had DCM and heart failure. The age of onset in both of these pedigrees varied widely, and some family members had advanced disease in adolescence or early adulthood.
Discussion
This novel report of late onset, sporadic IDC at age 70 caused by a TNNT2 mutation, accompanied by life‐threatening progressive heart failure requiring cardiac transplantation, raises clinical and mechanistic questions regarding the timing of onset of adult‐onset partially penetrant Mendelian rare variant disease. The age of onset of 70 years presented here contrasts distinctly with the median age of onset of 26 years in the 55 previously reported TNNT2 mutation carriers, or the median age of 21 at which heart transplantation, LVAD or death occurred ( Table 2 ). We 7 , 9 , 12 and others 10 , 11 , 13 have previously pointed out that the usual age of onset of DCM‐causing TNNT2 mutations is within the first 3 decades of life.
The clinical evidence that this patient had advanced DCM and heart failure is incontrovertible: her cardiac function was markedly reduced (cardiac index of 1.4 L/min/m2, ejection fraction 10%–20%), and she required intravenous inotropic support until undergoing cardiac transplantation. Because of the recent guideline suggestions that molecular genetic testing should be considered even in apparently sporadic DCM, usually termed IDC, 1 the proband underwent molecular genetic testing for 19 genes associated with DCM in a commercial laboratory; a rare TNNT2 nonsynonymous variant was identified. The molecular genetic evidence is also clear: a nonsynonymous mutation not found in >1,000 control DNAs and identified in a gene known to harbor DCM‐causing variants 7 , 9 , 10 , 11 , 12 , 13 indicates that it is possibly disease‐causing, as we have attributed to such cases in our prior studies. 6 , 7 When combined with the functional evidence presented above and based on extensive prior reports from us 18 , 19 , 20 and others 18 , 21 , 22 , 23 , 24 using this approach, the collective evidence is compelling that the identified TNNT2 mutation was highly likely relevant, if not the principal causative factor for her DCM.
Despite the incontrovertible echocardiographic and hemodynamic evidence of life‐threatening DCM, this patient’s heart did not present the usual structural abnormalities of DCM, consistent with the short duration of disease. The heart was not heavy from hypertrophy, the left ventricle was only slightly dilated, the LV wall had normal thickness and only minute foci of interstitial fibrosis were present on microscopic examination. This presentation might also have been precipitated by acute myocarditis, but there was no clinical or pathological evidence to support that possibility. The most pronounced changes were those of the right ventricle with extensive fat infiltration that raised the possibility of the lipomatous form of arrhythmogenic right ventricular cardiomyopathy, a consideration that was not sustained by the age of onset, molecular studies and the nature of the fat displacing the myocardium in a manner indistinguishable from the more commonly observed fat infiltration of the right ventricle.
From a clinical genetic testing viewpoint, this case raises the issue of using age of onset to influence whether genetic testing should be undertaken, or the selection of genes of interest. The latter point is rapidly becoming irrelevant, as many of the known DCM genes are now included in testing panels. This case would suggest that even for late onset, even apparently sporadic disease, clinical genetic testing may well be appropriate.
What remains unknown is why, at 70 years, this patient would have onset of life‐threatening DCM and heart failure, when after 69 years she had essentially no detectable clinically relevant cardiovascular abnormalities. This illustrates one of the most vexing questions of adult‐onset rare variant Mendelian clinical genetics: what underlies the variation in the age of onset of disease in subjects who carry disease‐causing mutations?
In a classic dichotomous view of genetic disease, a Mendelian condition is caused by a single gene mutation of large effect, while a polygenic disorder is caused by small, combined effects of several common variants. 25 It is more likely, however, that all genetic conditions fall somewhere in the middle of the spectrum, 26 and perhaps particularly so for DCM because of its many genes implicated. 25 Modifier genes are now known to influence Mendelian disorders 26 (which may explain in part penetrance and variable expressivity) and rare variants are now argued to play a role in common (usually conceived as polygenic) disease. 27
Penetrance and variable expressivity are “catch all” terms describing the factors that modulate these phenotypes lying in the middle of the genetic disease spectrum. In this light, most genetic conditions are complex and thus, the challenge to the field is determining where in the spectrum genetic DCM best fits, and to what extent environmental and stochastic effects influence a genetically determined phenotype.
We can only speculate about the underlying causes of the proband’s late onset phenotype, which may be due to interactions between the mutant TNNT2 and one or more, yet to be identified genetic variants acting as modifiers of TNNT2 or the cardiac troponin T protein, or other unknown or undetected environmental influences. While HFE was known to carry a variably penetrant iron disease‐associated variant, we could find no evidence of iron‐related myocyte injury. Other possible genetic modifiers could be a second rare nonsynonymous variant in one or more DCM‐related genes, which we have identified in 3% of a cohort that has had resequencing of 15 DCM‐related genes. 6 In this regard, we noted fatty infiltration predominantly in the RV of the patient’s explanted heart, prompting molecular genetic testing for ARVD, as ARVD has been shown at times to involve the LV. 28 However, no rare nonsynonymous variants were identified in the five genes encoding desmosomal proteins known to most commonly cause ARVD ( Table 1 ). We note, however, that the ARVD genetic testing in this case was not fully sensitive, as eight autosomal genes have been identified (the five in Table 1 and rarely JUP, RYR2, and TGFB3), 3 raising the possibility that a mutation in a known or unknown ARVD‐related gene variant may have been missed. We also note an HCM case caused by a MYH7 mutation was found to have fibrofatty replacement of the right ventricle at pathology examination, 29 suggesting a more complex relationship of fibrofatty replacement with genetic cardiomyopathy. In that case, genetic testing for PKP2, DSP, DSG2, and DSC2 was also negative. 29
The late onset in this case may in part also be explained by the novel observation that at pH 6.5, the TNNT2 R139H mutation significantly decreased both the Ca2+ sensitivity and maximal force development compared to the wild‐type. At pH 7.0, only the Ca2+ sensitivity was shown to be affected by the R139H mutation. Therefore, the late onset DCM in this case may have been related in part to a progressively acidic environment brought on by fatty infiltration of the right ventricle (even though the lack of LV fat infiltration does not explain the observed prominent LV dysfunction). We have previously shown that skinned cardiac fibers exchanged with the TNNT2 mutations R92L, R92W, and R94L, which cause hypertrophic cardiomyopathy, to have a blunted Ca2+ sensitivity of force development response with an acidic pH. 20 A positive charge is present in the side chain of arginine residues (that is encoded at position 139 in the wild‐type sequence), and changes in the pH can alter the pK a of such charged amino acids, affecting protein conformation and function. A substitution with histidine (R139H), another positively charged amino acid, as occurred for this individual, may have lead to other pH‐dependent functional perturbations (due to pK a value that is similar to that of imidazole at physiological pH), as one histidine residue in fast skeletal TnI was responsible for the difference in acidic pH resistance of skeletal versus cardiac muscle. 30
We also note possible allelic interactions between the identified in cis TNNT2 variants, as the intronic variant may have had an effect on RNA production given its −6 position within the polypyrimydine tract that is known to promote spliceosome assembly. 31 If the intronic variant was functional, mutant protein levels may have been reduced, possibly delaying disease onset. Phenotype amelioration by in cis allelic interactions has been reported in a case of neonatal diabetes, developmental delay, and epilepsy 32 and discussed as a possibility in familial HCM. 33 An in cis phenotypic effect has also been reported in the CFTR gene associated with cystic fibrosis 34 resulting from interactions between an intronic repeat variant and a coding mutation. 35
The proband’s offspring, as carriers of the R139H TNNT2 mutation, are now known to be at risk of developing DCM, and thus cardiac screening has been recommended. 1 Notably they remain with normal cardiovascular testing in their 5th decade.
In our experience, genetic testing in older individuals with DCM is not common practice, although IDC is commonly diagnosed in patients in their 7th, 8th, and even 9th decades. Ongoing genetic testing in sporadic late onset cases will help to clarify this question. If proven to be true, genetic DCM may turn out to have a wider phenotypic presentation. Similar observations have been made for MYBPC3 mutations in late onset HCM. 36 , 37
Limitations
We were unable to functionally assess the significance, if any, of the 690–6G>A intronic variant of unknown significance that is in cis with the R139H TNNT2 mutation.
Conclusion
In conclusion, DCM mutations may be present in late onset sporadic IDC cases, and research studies designed to identify and characterize genetic cause in sporadic DCM is warranted. Clinical genetic testing may be considered in patients with late onset IDC for diagnosis confirmation and family risk management. Research addressing underlying genetic factors including penetrance, variable expressivity, and mixed cardiomyopathy phenotypes is also warranted, as this information could greatly benefit mutation carriers and the physicians advising them. Ultimately, high throughput exome or whole genome sequencing in large cohorts will be required to evaluate the extent to which genetic susceptibility contributes to late onset sporadic IDC.
Conflict of Interest
There is no conflict of interest of any kind and no relationships with industry.
Acknowledgments
We thank the proband and members of this family for their participation in the Familial Dilated Cardiomyopathy Research Project, without whom these studies would not have been possible. We also thank Michelle Jones and Jingsheng Liang for their valuable assistance with the recombinant troponin T production and skinned fiber assays.
This work was supported by NIH awards RO1‐HL58626 (Dr. Hershberger), and R01‐HL42325 (Dr. Potter), and a postdoctoral fellowship from the American Heart Association AHA‐0825368E (Dr. Pinto).
References
- 1. Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J Card Fail. 2009; 15: 83–97. [DOI] [PubMed] [Google Scholar]
- 2. Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005; 45: 969–981. [DOI] [PubMed] [Google Scholar]
- 3. Hershberger RE, Cowan J, Morales A, Siegfried JD. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail. 2009; 2: 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hershberger RE, Kushner JK, Parks SP. Dilated Cardiomyopathy Overview. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Available at: http://www.genetests.org. Accessed July 10, 2008. [Google Scholar]
- 5. Judge DP, Johnson NM. Genetic evaluation of familial cardiomyopathy. J Cardiovasc Trans Res. 2008; 1: 144–154. [DOI] [PubMed] [Google Scholar]
- 6. Hershberger R, Norton N, Morales A, Li D, Siegfried J, Gonzalez‐Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1 And TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010; 3: 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J, Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Translat Sci. 2008; 1: 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008; 156:161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hershberger R, Pinto J, Parks S, Kushner J, Li D, Ludwigsen S, Cowan J, Morales A, Parvatiyar M, Potter J. Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ Genet. 2009; 2: 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG, Seidman CE. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000; 343:1688–1696. [DOI] [PubMed] [Google Scholar]
- 11. Li D, Czernuszewicz GZ, Gonzalez O, Tapscott T, Karibe A, Durand JB, Brugada R, Hill R, Gregoritch JM, Anderson JL, Quinones M, Bachinski LL, Roberts R. Novel cardiac troponin T mutation as a cause of familial dilated cardiomyopathy. Circulation. 2001; 104:2188–93. [DOI] [PubMed] [Google Scholar]
- 12. Hanson E, Jakobs P, Keegan H, Coates K, Bousman S, Dienel N, Litt M, Hershberger R. Cardiac troponin T lysine‐210 deletion in a family with dilated cardiomyopathy. J Card Fail. 2002; 8: 28–32. [DOI] [PubMed] [Google Scholar]
- 13. Mogensen J, Murphy RT, Shaw T, Bahl A, Redwood C, Watkins H, Burke M, Elliott PM, McKenna WJ. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2004; 44: 2033–2040. [DOI] [PubMed] [Google Scholar]
- 14. Judge DP. Use of genetics in the clinical evaluation of cardiomyopathy. JAMA. 2009; 302: 2471–2476. [DOI] [PubMed] [Google Scholar]
- 15. Kushner JD, Nauman D, Burgess D, Ludwigsen S, Parks S, Pantely G, Burkett EL, Hershberger R. Clinical characteristics of 304 kindreds evaluated for familial dilated cardiomyopathy. J Card Fail. 2006; 12: 422–429. [DOI] [PubMed] [Google Scholar]
- 16. Townsend PJ, Farza H, MacGeoch C, Spurr NK, Wade R, Gahlmann R, Yacoub MH, Barton PJ. Human cardiac troponin T: identification of fetal isoforms and assignment of the TNNT2 locus to chromosome 1q. Genomics. 1994; 21: 311–316. [DOI] [PubMed] [Google Scholar]
- 17. Gomes AV, Guzman G, Zhao J, Potter JD. Cardiac troponin T isoforms affect the Ca2+ sensitivity and inhibition of force development. Insights into the role of troponin T isoforms in the heart. J Biol Chem. 2002; 277: 35341–35349. [DOI] [PubMed] [Google Scholar]
- 18. Venkatraman G, Harada K, Gomes AV, Kerrick WG, Potter JD. Different functional properties of troponin T mutants that cause dilated cardiomyopathy. J Biol Chem. 2003; 278: 41670–41676. [DOI] [PubMed] [Google Scholar]
- 19. Venkatraman G, Gomes AV, Kerrick WG, Potter JD. Characterization of troponin T dilated cardiomyopathy mutations in the fetal troponin isoform. J Biol Chem. 2005; 280: 17584–17592. [DOI] [PubMed] [Google Scholar]
- 20. Harada K, Potter JD. Familial hypertrophic cardiomyopathy mutations from different functional regions of troponin T result in different effects on the pH and Ca2+ sensitivity of cardiac muscle contraction. J Biol Chem. 2004; 279: 14488–14495. [DOI] [PubMed] [Google Scholar]
- 21. Morimoto S, Lu QW, Harada K, Takahashi‐Yanaga F, Minakami R, Ohta M, Sasaguri T, Ohtsuki I. Ca(2+)‐desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proc Natl Acad Sci USA. 2002; 99: 913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu QW, Morimoto S, Harada K, Du CK, Takahashi‐Yanaga F, Miwa Y, Sasaguri T, Ohtsuki I. Cardiac troponin T mutation R141W found in dilated cardiomyopathy stabilizes the troponin T‐tropomyosin interaction and causes a Ca2+ desensitization. J Mol Cell Cardiol. 2003; 35: 1421–1427. [DOI] [PubMed] [Google Scholar]
- 23. Mirza M, Marston S, Willott R, Ashley C, Mogensen J, McKenna W, Robinson P, Redwood C, Watkins H. Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J Biol Chem. 2005; 280: 28498–28506. [DOI] [PubMed] [Google Scholar]
- 24. Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha‐tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. 2007; 101: 1266–1273. [DOI] [PubMed] [Google Scholar]
- 25. Hershberger RE. A glimpse into multigene rare variant genetics: triple mutations in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010; 55: 1454–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Badano JL, Katsanis N. Beyond Mendel: an evolving view of human genetic disease transmission. Nat Rev Genet. 2002; 3: 779–789. [DOI] [PubMed] [Google Scholar]
- 27. Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008; 40: 695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kies P, Bootsma M, Bax J, Schalij MJ, Van Der Wall EE. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: screening, diagnosis, and treatment. Heart Rhythm. 2006; 3: 225–234. [DOI] [PubMed] [Google Scholar]
- 29. Roberts JD, Veinot JP, Rutberg J, Gollob MH. Inherited cardiomyopathies mimicking ARVC. Cardiovasc Pathol. 2009. [DOI] [PubMed] [Google Scholar]
- 30. Dargis R, Pearlstone JR, Barrette‐Ng I, Edwards H, Smillie LB. Single mutation (A162H) in human cardiac troponin I corrects acid pH sensitivity of Ca2+‐regulated actomyosin S1 ATPase. J Biol Chem. 2002; 277: 34662–34665. [DOI] [PubMed] [Google Scholar]
- 31. Lomelin D, Jorgenson E, Risch N. Human genetic variation recognizes functional elements in noncoding sequence. Genome Res. 2010; 20: 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mannikko R, Jefferies C, Flanagan SE, Hattersley A, Ellard S, Ashcroft FM. Interaction between mutations in the slide helix of Kir6. 2 associated with neonatal diabetes and neurological symptoms. Hum Mol Genet. 19: 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blair E, Price SJ, Baty CJ, Ostman‐Smith I, Watkins H. Mutations in cis can confound genotype‐phenotype correlations in hypertrophic cardiomyopathy. J Med Genet. 2001; 38: 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. GeneReviews at GeneTests: Medical Genetics Information Resource. GeneTests/GeneClinics. Available at: http://www.genetests.org. Accessed September 12, 2008. [Google Scholar]
- 35. Mickle JE, Cutting GR. Clinical implications of cystic fibrosis transmembrane conductance regulator mutations. Clin Chest Med. 1998; 19: 443–458. [DOI] [PubMed] [Google Scholar]
- 36. Niimura H, Patton KK, McKenna WJ, Soults J, Maron BJ, Seidman JG, Seidman CE. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. 2002; 105: 446–51. [DOI] [PubMed] [Google Scholar]
- 37. Niimura H, Patton KK, McKenna WJ, Soults J, Maron BJ, Seidman JG, Seidman CE. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. 2002; 105: 446–451. [DOI] [PubMed] [Google Scholar]