

Abstract
Tumorigenesis is generally caused by genetic changes that activate oncogenes or inactivate tumor suppressor genes. The targeted inactivation of oncogenes can be associated with tumor regression through the phenomenon of oncogene addiction. One of the most common oncogenic events in human cancer is the activation of the MYC oncogene. The inactivation of MYC may be a general and effective therapy for human cancer. Indeed, it has been experimentally shown that the inactivation of MYC can result in dramatic and sustained tumor regression in lymphoma, leukemia, osteosarcoma, hepatocellular carcinoma, squamous carcinoma, and pancreatic carcinoma through a multitude of mechanisms, including proliferative arrest, terminal differentiation, cellular senescence, induction of apoptosis, and the shutdown of angiogenesis. Cell-autonomous and cell-dependent mechanisms have both been implicated, and recent results suggest a critical role for autocrine factors, including thrombospondin-1 and TGF-β. Hence, targeting the inactivation of MYC appears to elicit oncogene addiction and, thereby, tumor regression through both tumor cell–intrinsic and host-dependent mechanisms.
Keywords: MYC, oncogene addiction, tumorigenesis
Oncogene Addiction: A New Paradigm for the Mechanism of Targeted Therapeutics
Most cancers do not respond to conventional therapies, suggesting that new approaches are needed to treat these diseases.1 The presumption has been that by targeting the molecular underpinnings of a cancer with specific drugs, cancers may better respond to treatments. Indeed, the discovery of imatinib that inhibits several oncogenes having tyrosine kinase activity has proven to be highly effective in the treatment of chronic myelogenous leukemia and gastrointestinal stromal tumors.2,3 Many other targeted therapeutics are being developed for the treatment of cancer1; hence, there is optimism that we will be able to markedly improve the treatment of cancer by targeting specific genes with therapeutic agents.
It is not entirely clear why targeting any specific gene results in tumor regression.4 One emerging concept that attempts to explain tumor regression upon the inactivation or repair of a single mutant gene product is the idea of oncogene addiction, first described by Weinstein.5,6 In this paradigm, the survival of cancer cells becomes dependent on the continued activation of particular mutant oncogenes. However, even when tumors initially respond to targeted oncogene inactivation, most eventually recur.7 By understanding when and why oncogene inactivation elicits oncogene addiction, it should be possible to design rational therapeutic strategies to treat cancer.
The emergence of conditional transgenic mouse models that enable one to regulate oncogene activation in a tissue-specific and temporal manner has provided a unique and powerful approach to define when and why oncogene inactivation elicits oncogene addiction (Table 1). This review summarizes recent insights from conditional transgenic mouse models that have shed insight into the general mechanisms by which the inactivation of MYC elicits oncogene addiction. In particular, the review discusses how MYC inactivation induces sustained tumor regression through both tumor cell–intrinsic and host-dependent mechanisms. These mechanistic insights may be useful toward developing new therapies that target MYC for the treatment of cancer.
Table 1.
Consequences of Oncogene Inactivation in Transgenic Mouse Models
| Oncogene | Model | System | Tumor Type | Response to Inactivation | Mechanism of Tumor Regression | References |
|---|---|---|---|---|---|---|
| BCL2 | MMTV-tTATet-O-BCL-2Eµ-MYC | Tet-Off | Lymphoblastic leukemia | Regression | Apoptosis | Letai et al. 200443 |
| BCR-ABL | MMTV-tTATet-O-BCR-ABL | Tet-Off | B-cell Leukemia | Regressiona | Apoptosis | Huettner et al. 200044 |
| SCL-tTATet-O-BCR-ABL | Tet-Off | Chronic myelogenous leukemia | Regression | Not determined | Koschmieder et al. 200545 | |
| FGF-10 | CCSP-rtTA orSPC-rtTATet-O-CMV-FGF10 | Tet-On | Lung adenocarcinoma | Regression | Not determined | Clark et al. 200146 |
| HER2/NEU | MMTV-rtTATet-O-NeuNT | Tet-On | Breast adenocarcinoma | Regressiona | Decreased proliferation and apoptosis | Moody et al. 200247 |
| MET | LAP-tTATet-O-MET | Tet-On | Hepatocellular carcinoma | Regression | Decreased proliferation and apoptosis | Wang et al. 200148 |
| c-MYC | EµSRα-tTATet-O-MYC | Tet-Off | T- and B-cell lymphoma, acute myeloid leukemia | Regressiona | Cell cycle arrest, differentiation, and apoptosis | Felsher and Bishop 199949; Marinkovic et al. 200450 |
| EµSRα-tTATet-O-MYC | Tet-Off | Osteosarcoma | Regression | Differentiation | Jain et al. 200211 | |
| MMTV-rtTATet-O-MYC | Tet-On | Breast adenocarcinoma | Partial regression | Not determined | D’Cruz et al. 200118; Boxer et al. 200416 | |
| LAP-tTATet-O-MYC | Tet-Off | Hepatocellular carcinoma | Regression | Apoptosis and differentiation | Beer et al. 200451; Shachaf et al. 200412 | |
| Plns- MycERTam | Tamoxifen | Pancreatic islet cell | Regression | Growth arrest, differentiation, cellular adhesion, vascular collapse | Pelengaris et al. 200214 | |
| Involucrin-MycERTam | Tamoxifen | Papilloma | Regression | Growth arrest and differentiation | Pelengaris et al. 199952; Flores et al. 200453 | |
| RAS | Tyr-rtTAH-Ras(V12G) Ink4a−/− | Tet-On | Melanoma | Regressiona | Apoptosis, epidermal growth factor receptor expression required | Chin et al. 199954; Wong and Chin 200055 |
| SP-r-rtTTARtTA-KiRas(G12C) | Tet-On | Lung adenocarcinoma | Regression | Not determined | Floyd et al. 200556 | |
| CCSP-rtTATet-O-KiRas(G12C) | Tet-On | Lung adenocarcionma | Regression | Not determined | Floyd et al. 200556 | |
| CCSP-rtTATet-op-K-Ras4B(G12D) | Tet-On | Lung adencarcinoma | Regression | Apoptosis | Fisher et al. 200157 | |
| Nestin-TVARCAS-tTARCAS-AktRCAS-Tet-O-KRas | RCAS | Glioblastoma | Regression | Apoptosis | Holmen and Williams 200558 | |
| WNT | MMTV-rtTATet-O-WNT1P53−/− | Tet | Mammary adenocarcinoma | Regressiona | Not determined | Gunther et al. 200359 |
Most tumors regress upon initial oncogene inactivation, but some tumors reoccur.
MYC: A Potential Ultimate Target for Cancer Treatment
The MYC oncogene has protean effects on many biological processes, including gene transcription, protein translation, and DNA replication. These processes in turn coordinate many cellular functions, including proliferation, apoptosis, differentiation, self-renewal/senescence, and angiogenesis, as reviewed in this issue. The overexpression of MYC is one of the most common events associated with tumorigenesis, providing us with many reasons to consider that targeting this gene may be an effective treatment for human cancer.
Transgenic mouse models have been employed as a tractable approach for investigating the mechanism by which MYC and other oncogenes contribute to the initiation and maintenance of tumorigenesis.8–10 In particular, the development of conditional strategies for the regulation of gene expression in transgenic mouse models has been useful to define the circumstances under which oncogene inactivation will result in tumor regression (Table 1). It has become clear from these experiments in transgenic mouse models that the consequences of MYC inactivation are often dramatic and so result in sustained tumor regression. However, the specific mechanisms are contextual. Moreover, it has emerged that both tumor cell–intrinsic and host-dependent mechanisms are involved in oncogene addiction.1
Two major approaches have been used to generate conditional transgenic mouse models to dissect the role of MYC in the initiation and maintenance of tumorigenesis. The tetracycline system can be used to generate conditional transgenic models.13 In this model, a gene of interest is placed upstream of the tetracycline response element, and the tetracycline transactivating gene is placed upstream of a tissue-specific promoter. Transgenes of both constructs are generated, and mice that contain both transgenes express the gene of interest in a tissue-specific and temporally controlled manner. A similar strategy has been developed by utilizing a chimeric gene product between a gene of interest and the estradiol receptor.14 Here, the fusion gene exhibits conditional activity in the presence of tamoxifen. Both approaches have been used to demonstrate that the inactivation of a single oncogene can have profound effects on a tumor and in many cases result in a complete reversal of tumorigenesis in vivo.
Conditional transgenic mouse models have demonstrated that MYC inactivation in tumors is associated with proliferative arrest, differentiation, senescence, and/or apoptosis (Figure 1). Interestingly, the consequences of oncogene inactivation depend on the cellular and genetic context (Figure 2). Most notably, the inactivation of the MYC oncogene has been shown to be potent in inducing tumor regression.14 MYC inactivation in lymphoma, leukemia, pancreatic islet cell tumors, and skin squamous carcinomas results in rapid tumor cell elimination through apoptosis.15 Many mouse models in which the MYC gene has been disrupted exhibit sustained tumor regression. In other tumor types, MYC inactivation appears to more generally induce terminal differentiation. For example, MYC inactivation in osteogenic sarcoma results in the terminal differentiation of tumor cells into mature bone cells.11 Note that these cells are now terminally differentiated and generally incapable of becoming tumor cells. Thus, even brief MYC inactivation can induce the sustained loss of a neoplastic phenotype.
Figure 1.
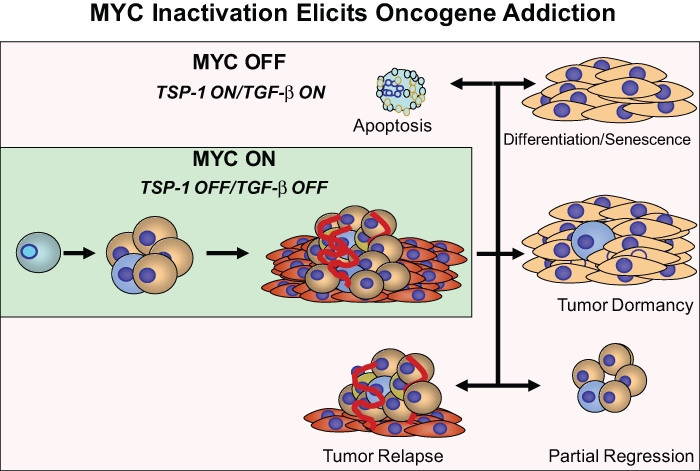
MYC inactivation has different outcomes in different types of tumors, including proliferative arrest, differentiation, apoptosis, and/or cellular senescence. Although the consequences are different for each type of tumor, proliferative arrest, apoptosis, and differentiation/senescence appear to be common mechanisms. Tumors can also become dormant, partially regress, and reoccur.
Figure 2.
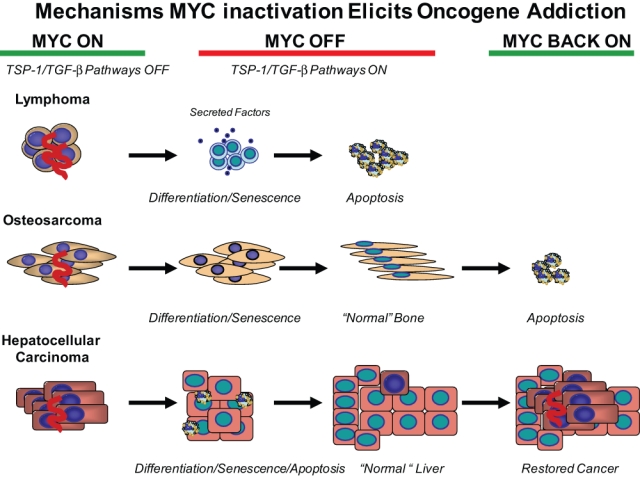
MYC inactivation elicits oncogene addiction by multiple mechanisms that differ depending on tumor type. MYC inactivation in lymphoma induces proliferative arrest, differentiation/senescence, and widespread apoptosis. Both tumor cell–intrinsic and host-dependent mechanisms (angiogenesis) have been shown to be important. Secreted factors, such as TSP-1, are important for the regulation of angiogenesis, and TGF-β is important for the regulation of cellular senescence. MYC inactivation in osteosarcoma induces proliferative arrest and differentiation/senescence but not apoptosis. MYC reactivation does not restore tumorigenesis. MYC inactivation in liver adenocarcinoma induces proliferative arrest, differentiation/senescence, and apoptosis. MYC reactivation can result in restoration of the tumor.
In contrast, brief inactivation of MYC fails to reverse tumorigenesis in epithelial tumors, such as hepatocellular carcinoma and breast cancer. In these contexts, some of these otherwise normal-appearing tumor cells can rapidly regain their neoplastic properties upon MYC reactivation.12,16 Hence, in this case, MYC inactivation is associated with the terminal differentiation of many of the tumor cells; in the case of hepatocellular carcinoma, the tumor cells can give rise to what appear to be normal liver cells, including hepatocytes and biliary cells. Yet, some of these cells retain the latent capacity to become tumor cells upon MYC reactivation. Thus, some tumor cells can, upon oncogene inactivation, appear normal and behave normally but actually be in a state of dormancy.17 In essence, sustained MYC inactivation results in tumor regression, but the reactivation of MYC can restore their neoplastic properties.
Genetic context has been shown to be critical in defining the consequence of the inactivation of MYC.7 Breast adenocarcinomas that have acquired a mutation in K-ras fail to undergo sustained regression upon MYC inactivation.18 Similarly, loss of p53 function markedly impedes the ability of MYC inactivation to induce sustained tumor regression.19 Snail has been identified as one gene product that appears to facilitate the escape from oncogene dependence.19 Loss of p53 function has also been shown to impede the sustained regression of lymphoma and leukemia through disruption of the ability of oncogene inactivation to induce the shutdown of angiogenesis.20 Finally, the escape from MYC inactivation induces the regression of hematopoietic tumors and appears to be associated with the acquisition of specific chromosomal translocations.21 Hence, there appear to be multiple genetic mechanisms that can predictably impede the ability of MYC inactivation to induce sustained tumor regression. One could speculate that these genetic events block MYC inactivation from inducing sustained regression solely because they prevent the triggering of oncogene addiction.
MYC Inactivation and the Induction of Senescence and the Shutdown of Angiogenesis
At least 2 general mechanisms appear to explain how MYC inactivation induces tumor regression. First, intrinsic tumor cell mechanisms are responsible, including proliferative arrest and apoptosis. In addition, one important mechanism by which the inactivation of MYC and other oncogenes induces tumor regression is through the induction of cellular senescence.22 MYC inactivation of osteosarcoma and lymphomas essentially induces all the tumor cells to undergo cellular senescence. However, if these tumors have acquired defects—or are engineered to have defects—in gene products known to be required to induce senescence (e.g., p16, RB, p53), then tumors do not undergo senescence and do not exhibit sustained tumor regression upon MYC inactivation.22 MYC inactivation also induces cellular senescence in hepatocellular carcinoma; however, not all the cells undergo senescence, suggesting that these cells can regain their tumorigenic properties upon MYC reactivation. Thus, MYC inactivation has been shown to induce cellular senescence in tumors in several contexts.
Second, host-dependent mechanisms of tumor regression are important. Upon MYC inactivation, tumor cells not only arrest, differentiate, and undergo senescence and apoptosis through autonomous mechanisms but do so through the shutdown of the host microenvironment that promotes cellular proliferation. These pathways are in turn associated with the shutdown of angiogenesis,20 which is likely an important mechanism of oncogene addiction and could even contribute to tumor dormancy that is observed upon oncogene inactivation.23
The mechanism by which MYC inactivation induces the shutoff of the angiogenic switch appears to involve a p53-dependent induction of thrombospondin-1 (TSP-1).20 There are likely to be multiple mechanisms by which angiogenesis and the general host microenvironment are restored upon inactivation of an oncogene within the tumor. In general, the results suggest that by targeting the MYC oncogene within a tumor, it is possible to not only reverse the neoplastic properties of the tumor but also restore the host microenvironment. The important implication of these results is that oncogene addiction may be a consequence of cell-autonomous effects with the tumor and through cell-dependent effects on the host environment.
These results also suggest that inactivation of MYC can reverse the limitless proliferation or self-renewal properties of cancer cells and induce cellular senescence. The mechanism of cellular senescence could be related to many possible mechanisms.22 One possibility is a tumor cell’s intrinsic response to DNA damage.24 In addition, the restoration of autocrine growth regulatory mechanisms could be important, as described in the next section. Regardless, these results could be highly relevant to the notion of cancer stem cells.25,26 Turning off MYC appears to elicit oncogene addiction in tumor cells by blocking self-renewal of cancer cells.27 Therefore, oncogene addiction may be a manifestation of the restoration of limited life span or mortality of tumors. MYC inactivation has been also shown to induce senescence in normal cells28 as well as in human melanoma tumors.29
MYC Inactivation Induces Tumor Regression through Autocrine Mechanisms
The notion that secreted factors such as TSP-1 may be important to the mechanism of tumor regression upon MYC inactivation suggests the possibility that other soluble factors may be involved. Indeed, recent evidence suggests that a TGF-β-dependent autocrine pathway is essential to the mechanism by which MYC inactivation induces cellular senescence and impedes tumorigenesis (van Riggelen et al., in press, Genes and Development).
Furthermore, MYC-induced lymphomas express high amounts of TGF-β, TGF-2, and TGF-3 and exhibit activation of SMADs. TGF-β signaling may be important for restraining MYC from inducing tumorigenesis via Miz-1-induction of cyclin-dependent kinase inhibitors (CKIs), which in turn trigger cellular senescence. However, ectopic MYC bypasses these programs and stimulates tumor formation. Upon MYC inactivation, the effects of TGF-β signaling are restored, resulting in CKI expression and thereby inducing cellular senescence. The introduction of a mutant TGF-β receptor that blocks signaling prevents MYC inactivation from inducing cellular senescence and sustained tumor regression. TGF-β is therefore a key autocrine factor that contributes to MYC-associated oncogene addiction of lymphomas.
Thus, MYC inactivation elicits oncogene addiction through cell-intrinsic and autocrine regulatory pathways (Figure 3), which may be a vital part of the mechanism of oncogene addiction. In general, secreted factors could provide a link between cell-intrinsic and host-dependent microenvironment changes associated with tumorigenesis and tumor regression. TSP-1 and TGF-β have emerged as 2 possible critical factors in this process, but other cytokines are likely to be involved as well. The identification of these factors and their source and role in oncogene addiction are not well defined. The role of such factors in the induction and regulation of the relevant signaling pathways may be an important consideration for the effective treatment of cancer through the targeted inactivation of oncogenes.
Figure 3.
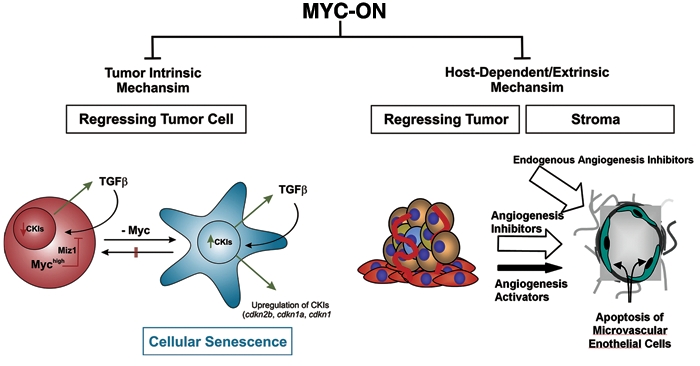
MYC inactivation elicits oncogene addiction through tumor cell–intrinsic and hostdependent mechanisms. Cancers exhibit autonomous behavior resulting in immortality, self-renewal, and proliferation. Cancers also influence the microenvironment to support tumorigenesis. MYC inactivation restores physiological safety switches and results in proliferative arrest, differentiation, apoptosis, cellular senescence, and the restoration of a physiologic microenvironment, including the suppression of angiogenesis. Tumor cell–intrinsic, autocrine, and host-dependent mechanisms are involved in oncogene addiction. The TSP-1 and TGF-β cytokine pathways are 2 examples of secreted factors that may play a critical role in the mechanism by which MYC inactivation elicits oncogene addiction.
MYC Inactivation, Genetic/Epigenetic “Amnesia,” and Cancer Stem Cells
In many ways oncogene addiction may be conceived as a consequence of the fact that cancer is in a state of genetic and epigenetic “amnesia.” The “awareness” of the cancer and the host is restored with the physiologic restoration of the normal level of activity of a specific oncogene.27 In this model, cancers can grow autonomously and independently of their host and the cellular regulatory mechanisms that normally restrain proliferation. Oncogenes such as MYC could interfere with host mechanisms that restrain growth and proliferation and regulate processes such as angiogenesis, immortalization, cell cycle, DNA repair, and apoptosis, among many physiological programs. It is perhaps less surprising that the inactivation of oncogenes would result in the restoration of these processes. In a sense, an awareness of the tumor to its state of genomic disruption is associated with a cancerous state and so results in proliferative arrest, senescence, and apoptosis.22,24,27
To explain differences in the precise consequences of MYC inactivation, one could speculate that the biology of each type of cancer is strongly influenced by epigenetic features particular to the specific cell type initially transformed. Epithelial tumors, such as hepatocellular carcinomas and breast adenocarcinomas, may necessarily involve the initiation of malignant transformation of cells that have or will acquire stem cell features. Then, as stem cells, they could necessarily give rise to malignant progenitor cells. Upon MYC inactivation, these stem cell features could result in the differentiation into cells that are normal in appearance. Some of these cells may retain their latent stem cell properties and upon MYC reactivation rapidly regain their neoplastic properties as well. This notion is consistent with the proposed cancer stem cell hypothesis,25,26 although this hypothesis does not presume that cancer cells necessarily have any features of stem cells other than their self-renewal properties. An alternative and nonmutually exclusive explanation would be that MYC, as an iPS gene, may directly elicit stem cell features in cells.
Other Possible Mechanisms by Which MYC Inactivation May Elicit Oncogene Addiction
Our current models of oncogene addiction or oncogenic amnesia still do not explicitly take into account several other possible nonmutually exclusive mechanisms that could play a role in the mechanism by which MYC inactivation elicits oncogene addiction. Several recent findings have suggested that MYC may play a critical role in cellular metabolism, protein synthesis, DNA replication, and cell cycle machinery. All these processes may contribute to oncogene addiction upon cessation of MYC activity.
First, MYC has been shown to play a central role in regulation of many aspects of cellular metabolism.30–34 MYC inactivation could induce tumor regression because tumor cells are no longer capable of coordinating glucose metabolism, producing essential metabolites, regulating hypoxia, and/or generating sufficient energy. Hence, MYC inactivation could result in direct activation of apoptotic programs or indirectly result in a metabolic catastrophe that induces tumor regression.
Second, MYC globally coordinates protein synthesis through many different mechanisms.35,36 In particular, MYC regulates ribosomal biogenesis. MYC inactivation in cancer can permanently suppress ribosomal biogenesis expression, thereby globally suppressing protein synthesis.37 Thus, MYC inactivation could induce tumor regression because tumor cells are not generally capable of making critical proteins required for proliferation, the suppression of apoptosis, the regulation of self-renewal, or alternatively, the expression of specific critical protein products.38
Third, MYC directly regulates DNA replication.39 MYC overexpression in tumors could enforce replication in a manner that necessarily impedes DNA repair, leading to widespread genomic damage. The abrupt suppression of MYC in a tumor could then result in incomplete DNA replication, resulting in widespread DNA damage. Consequently, MYC inactivation may result in oncogene addiction through the ability of the tumor cell to now be aware of this damage and have the appropriate response in the induction of apoptosis and/or cellular senescence.24
Fourth, MYC appears to regulate the function of many cell cycle regulatory proteins critical to the regulation of self-renewal and cellular senescence. Notably, MYC has been shown to directly regulate CDK2 function, and it may play an important role in suppressing cellular senescence programs.40–42 The suppression of MYC could restore these regulatory programs and result in tumor regression.
Thus, many mechanisms could work in concert to mediate the antineoplastic consequences of MYC suppression. Different mechanisms could operate in different types of cancer that depend on differences both epigenetic and genetic. Most likely, it is precisely because MYC plays such a central role in the coordination of so many critical cellular programs that the abrupt cessation of MYC expression in a cancer cell has such profound effects. Indeed, one of the major current challenges is to explain why the consequences of MYC inactivation differ depending on cellular and genetic context. These insights could better predict when the targeting of MYC and other oncogenes will be successful as a therapy for cancer.
General Implications for MYC as a Target for the Treatment of Human Cancer
MYC appears to be a potentially useful target for the treatment of cancer. Experimentally, MYC inactivation has been shown to induce sustained regression of many tumor types. However, results in experimental models suggest several important mechanisms to consider in the analysis of agents designed to target MYC as a treatment for cancer. First, evidence suggests that MYC inactivation may be able to convert cancer cells into what appear to be biologically normal cells. Thus, liver cancer cells seem capable of giving rise to what appears to be normal liver cells.12 Similarly, osteosarcoma cells appear to be capable of giving rise to normal bone.11 This means that a therapy that targets MYC does not necessarily need to eliminate tumor cells to have clinical efficacy.
Second, these results illustrate that the consequences of targeted MYC inactivation is not necessarily the death of the tumor cells but rather a change in the biology of the cells, including cellular differentiation and cellular senescence. Thus, simply looking for cell death may not be the only criteria used to identify agents that can be used to target MYC for the treatment of cancer. This is important because most screens for drugs that target oncogene presume that the response is tumor cell death. In particular, MYC inactivation can result in cancer cells appearing and apparently functioning as normal cells.
Third, the results imply that cancer can be caused by oncogenic events that usurp control of an epigenetically susceptible state. Inactivating MYC can revert the physiologic mechanisms of a tumor cell and cause it to behave normally. This implies that genetic events that impede these epigenetic mechanisms block the clinical efficacy of agents that target MYC to treat cancer.
Fourth, MYC’s ability to regulate host mechanisms such as angiogenesis are critical to understanding the mechanism by which MYC inactivation induces tumor regression. These results illustrate that host mechanisms play a key role in the mechanism by which MYC inactivation elicits oncogene addiction; they also suggest that the ability of MYC inactivation to elicit the expression of key regulators of angiogenesis, such as TSP-1, may be important for tumor regression. Finally, these results suggest that it will be important to look in vivo in an intact host to measure the true effects of a drug that targets MYC.
Fifth, the ability of MYC to abrogate autocrine mechanisms—such as TGF-β-mediated regulation of cell cycle gene products important for proliferation and senescence—may be critical to the therapeutic mechanism of MYC inactivation. The integrity of this and other autocrine pathways could be important toward anticipating the efficacy of agents that target MYC for the treatment of cancer. Hence, the precise understanding of the mechanisms by which MYC inactivation elicits oncogene addiction and induces tumor regression is likely critical for effectively developing, identifying, and testing agents that target MYC and, for that matter, other oncogenes as treatments for human cancer.
In the era of targeted therapeutics, it is humbling that no explanation has been provided for why existing agents that target specific oncogenes or tumor suppressor genes result in the regression of a tumor. Indeed, a priori, there would be many alternative possible outcomes. Insight into the mechanism by which the inactivation of MYC and other oncogenes induces tumor regression may guide the development of new treatments for cancer.
Acknowledgments
I would like to thank the current and former members of the Felsher laboratory for their contribution to the results described in this review. In particular, I would like to kindly thank Dr. Jan van Riggelen for help in generating the figures.
Footnotes
Our work has been generously supported by grants from the National Institutes of Health (2R01CA89305, R01CA105102, P50CA114747, P01CA034233, U56CA112973), Lymphoma and Leukemia Society, Burroughs Wellcome Fund, Damon Runyon Foundation, and the Lymphoma Research Foundation.
The author declares no conflicts of interest with respect to the publication of this review.
References
- 1. Arvanitis C, Bendapudi PK, Bachireddy P, Felsher DW. Identifying critical signaling molecules for the treatment of cancer. Recent Results Cancer Res 2007;172:5-24 [DOI] [PubMed] [Google Scholar]
- 2. Gorre ME, Sawyers CL. Molecular mechanisms of resistance to STI571 in chronic myeloid leukemia. Curr Opin Hematol 2002;9:303-7 [DOI] [PubMed] [Google Scholar]
- 3. Sherbenou DW, Druker BJ. Applying the discovery of the Philadelphia chromosome. J Clin Invest 2007;117:2067-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Felsher DW. Cancer revoked: oncogenes as therapeutic targets. Nat Rev Cancer 2003;3:375-80 [DOI] [PubMed] [Google Scholar]
- 5. Weinstein IB. Cancer: addiction to oncogenes. The Achilles heal of cancer. Science 2002;297:63-4 [DOI] [PubMed] [Google Scholar]
- 6. Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction. A rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol 2006;3:448-57 [DOI] [PubMed] [Google Scholar]
- 7. Giuriato S, Felsher DW. How cancers escape their oncogene habit. Cell Cycle 2003;2:329-32 [PubMed] [Google Scholar]
- 8. Van Dyke T, Jacks T. Cancer modeling in the modern era: progress and challenges. Cell 2002;108:135-44 [DOI] [PubMed] [Google Scholar]
- 9. Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov 2006;5:741-54 [DOI] [PubMed] [Google Scholar]
- 10. Gutmann DH, Hunter-Schaedle K, Shannon KM. Harnessing preclinical mouse models to inform human clinical cancer trials. J Clin Invest 2006;116:847-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jain M, Arvanitis C, Chu K, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002;297:102-4 [DOI] [PubMed] [Google Scholar]
- 12. Shachaf CM, Kopelman AM, Arvanitis C, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004;431:1112-7 [DOI] [PubMed] [Google Scholar]
- 13. Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol 2006;16:313-7 [DOI] [PubMed] [Google Scholar]
- 14. Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer 2002;2:764-76 [DOI] [PubMed] [Google Scholar]
- 15. Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002;109:321-34 [DOI] [PubMed] [Google Scholar]
- 16. Boxer RB, Jang JW, Sintasath L, Chodosh LA. Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell 2004;6:577-86 [DOI] [PubMed] [Google Scholar]
- 17. Shachaf CM, Felsher DW. Rehabilitation of cancer through oncogene inactivation. Trends Mol Med 2005;11:316-21 [DOI] [PubMed] [Google Scholar]
- 18. D’Cruz CM, Gunther EJ, Boxer RB, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med 2001;7:235-9 [DOI] [PubMed] [Google Scholar]
- 19. Moody SE, Perez D, Pan TC, et al. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 2005;8:197-209 [DOI] [PubMed] [Google Scholar]
- 20. Giuriato S, Ryeom S, Fan AC, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc Natl Acad Sci U S A 2006;103:16266-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karlsson A, Giuriato S, Tang F, Fung-Weier J, Levan G, Felsher DW. Genomically complex lymphomas undergo sustained tumor regression upon MYC inactivation unless they acquire novel chromosomal translocations. Blood 2003;101:2797-803 [DOI] [PubMed] [Google Scholar]
- 22. Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A 2007;104:13028-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Folkman J, Ryeom S. Is oncogene addiction angiogenesis-dependent? Cold Spring Harb Symp Quant Biol 2005;70:389-97 [DOI] [PubMed] [Google Scholar]
- 24. Karlsson A, Deb-Basu D, Cherry A, Turner S, Ford J, Felsher DW. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc Natl Acad Sci U S A 2003;100:9974-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weissman IL. Normal and neoplastic stem cells. Novartis Found Symp 2005;265:35-50 [PubMed] [Google Scholar]
- 26. Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med 2007;58:267-84 [DOI] [PubMed] [Google Scholar]
- 27. Felsher DW. Oncogene addiction versus oncogene amnesia: perhaps more than just a bad habit? Cancer Res 2008;68:3081-6 [DOI] [PubMed] [Google Scholar]
- 28. Guney I, Wu S, Sedivy JM. Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16(INK4a). Proc Natl Acad Sci U S A 2006;103:3645-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhuang D, Mannava S, Grachtchouk V, et al. C-MYC overexpression is required for continuous suppression of oncogene-induced senescence in melanoma cells. Oncogene 2008;27:6623-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shim H, Chun YS, Lewis BC, Dang CV. A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc Natl Acad Sci U S A 1998;95:1511-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang H, Gao P, Fukuda R, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007;11:407-20 [DOI] [PubMed] [Google Scholar]
- 32. Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008;105:18782-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007;12:108-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res 2009;15:6479-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol 2005;7:295-302 [DOI] [PubMed] [Google Scholar]
- 36. Boon K, Caron HN, van Asperen R, et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J 2001;20:1383-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu CH, Sahoo D, Arvanitis C, Bradon N, Dill DL, Felsher DW. Combined analysis of murine and human microarrays and ChIP analysis reveals genes associated with the ability of MYC to maintain tumorigenesis. PLoS Genet 2008;4:e1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer 2010;10:301-9 [DOI] [PubMed] [Google Scholar]
- 39. Dominguez-Sola D, Ying CY, Grandori C, et al. Non-transcriptional control of DNA replication by c-Myc. Nature 2007;448:445-51 [DOI] [PubMed] [Google Scholar]
- 40. Hydbring P, Bahram F, Su Y, et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc Natl Acad Sci U S A 2010;107:58-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Campaner S, Doni M, Hydbring P, et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat Cell Biol 2010;12:54-9 [DOI] [PubMed] [Google Scholar]
- 42. van Riggelen J, Felsher DW. Myc and a Cdk2 senescence switch. Nat Cell Biol 2010;12:7-9 [DOI] [PubMed] [Google Scholar]
- 43. Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell 2004;6:241-9 [DOI] [PubMed] [Google Scholar]
- 44. Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet 2000;24:57-60 [DOI] [PubMed] [Google Scholar]
- 45. Koschmieder S, Göttgens B, Zhang P, et al. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood 2005;105:324-34 [DOI] [PubMed] [Google Scholar]
- 46. Clark JC, Tichelaar JW, Wert SE, et al. FGF-10 disrupts lung morphogenesis and causes pulmonary adenomas in vivo. Am J Physiol Lung Cell Mol Physiol 2001;280:L705-15 [DOI] [PubMed] [Google Scholar]
- 47. Moody SE, Sarkisian CJ, Hahn KT, et al. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell 2002;2:451-61 [DOI] [PubMed] [Google Scholar]
- 48. Wang R, Ferrell LD, Faouzi S, Maher JJ, Bishop JM. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J Cell Biol 2001;153:1023-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 1999;4:199-207 [DOI] [PubMed] [Google Scholar]
- 50. Marinkovic D, Marinkovic T, Kokai E, Barth T, Möller P, Wirth T. Identification of novel Myc target genes with a potential role in lymphomagenesis. Nucleic Acids Res 2004;32:5368-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Beer S, Zetterberg A, Ihrie RA, et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol 2004:e332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pelengaris S, Littlewood T, Khan M, Elia G, Evan G. Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol Cell 1999;3:565-77 [DOI] [PubMed] [Google Scholar]
- 53. Flores I, Murphy DJ, Swigart LB, Knies U, Evan GI. Defining the temporal requirements for Myc in the progression and maintenance of skin neoplasia. Oncogene 2004;23(35):5923-30 [DOI] [PubMed] [Google Scholar]
- 54. Chin L, Tam A, Pomerantz J, et al. Essential role for oncogenic Ras in tumour maintenance. Nature 1999;400:468-72 [DOI] [PubMed] [Google Scholar]
- 55. Wong AK, Chin L. An inducible melanoma model implicates a role for RAS in tumor maintenance and angiogenesis. Cancer Metastasis Rev 2000;19:121-9 [DOI] [PubMed] [Google Scholar]
- 56. Floyd HS, Farnsworth CL, Kock ND, et al. Conditional expression of the mutant Ki-rasG12C allele results in formation of benign lung adenomas: development of a novel mouse lung tumor model. Carcinogenesis 2005;26:2196-206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fisher GH, Wellen SL, Klimstra D, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev 2001;15:3249-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Holmen SL, Williams BO. Essential role for Ras signaling in glioblastoma maintenance. Cancer Res 2005;65:8250-5 [DOI] [PubMed] [Google Scholar]
- 59. Gunther EJ, Moody SE, Belka GK, et al. Impact of p53 loss on reversal and recurrence of conditional Wnt-induced tumorigenesis. Genes Dev 2003;17:488-501 [DOI] [PMC free article] [PubMed] [Google Scholar]