

Unconventional cytoplasmic splicing of HAC1 mRNA is essential for the yeast unfolded protein response (UPR). The UPR requires translational regulation of unspliced and spliced forms of HAC1 mRNAs. Here we report that tRNA ligase, Rlg1p, which ligates HAC1 exons in its splicing, has another face as a translational regulator of HAC1 mRNA.
Abstract
The unfolded protein response (UPR) is an essential signal transduction to cope with protein-folding stress in the endoplasmic reticulum. In the yeast UPR, the unconventional splicing of HAC1 mRNA is a key step. Translation of HAC1 pre-mRNA (HAC1u mRNA) is attenuated on polysomes and restarted only after splicing upon the UPR. However, the precise mechanism of this restart remained unclear. Here we show that yeast tRNA ligase (Rlg1p/Trl1p) acting on HAC1 ligation has an unexpected role in HAC1 translation. An RLG1 homologue from Arabidopsis thaliana (AtRLG1) substitutes for yeast RLG1 in tRNA splicing but not in the UPR. Surprisingly, AtRlg1p ligates HAC1 exons, but the spliced mRNA (HAC1i mRNA) is not translated efficiently. In the AtRLG1 cells, the HAC1 intron is circularized after splicing and remains associated on polysomes, impairing relief of the translational repression of HAC1i mRNA. Furthermore, the HAC1 5′ UTR itself enables yeast Rlg1p to regulate translation of the following ORF. RNA IP revealed that yeast Rlg1p is integrated in HAC1 mRNP, before Ire1p cleaves HAC1u mRNA. These results indicate that the splicing and the release of translational attenuation of HAC1 mRNA are separable steps and that Rlg1p has pivotal roles in both of these steps.
INTRODUCTION
Protein quality control in the endoplasmic reticulum (ER) is essential for eukaryotic cells to maintain their cellular homeostasis. Because the ER faces overload of the protein-folding capacity under certain circumstances, eukaryotic cells are armed with elaborated systems to sense folding stress in the ER and to up-regulate protein folding and degradation capacities in the ER. These signal transduction systems are called the unfolded protein response (UPR; Ron and Walter, 2007; Schröder, 2007). On the UPR, ER stress signals are transferred to the cytosol by three parallel pathways, namely, the Ire1p pathway (Cox et al., 1993; Mori et al., 1993), the ATF6 pathway (Haze et al., 1999; Ye et al., 2000; Yoshida et al., 2000), and the PERK pathway (Harding et al., 1999; Scheuner et al., 2001).
Among the three systems, only the Ire1p pathway is conserved among eukaryotes, and has been studied extensively in the yeast Saccharomyces cerevisiae. The signal transducer Ire1p has a luminal domain that acts as an ER stress sensor and a cytosolic effector unit consisting of a kinase domain and an endonuclease domain (Cox et al., 1993; Mori et al., 1993; Cox and Walter, 1996). On overload of the folding capacity in the ER, Ire1p forms an oligomer to allow trans-autophosphorylation by the kinase domain. Concomitant activation of the endonuclease domain initiates unconventional cytoplasmic splicing of HAC1 pre-mRNA, HAC1u mRNA (Shamu and Walter, 1996; Kawahara et al., 1997; Bertolotti et al., 2000; Papa et al., 2003; Credle et al., 2005; Zhou et al., 2006; Oikawa et al., 2007; Lee et al., 2008). HAC1 encodes a bZIP-type transcription factor responsible for activating expression of UPR target genes (Cox and Walter, 1996; Mori et al., 1996; Travers et al., 2000; Kimata et al., 2006). The resulting exons from HAC1u mRNA are ligated to HAC1i mRNA by tRNA ligase, Rlg1p/Trl1p (Sidrauski et al., 1996). The primary role of Rlg1p is to ligate tRNA exons, which are also produced in the cytoplasm by tRNA splicing endonuclease on the mitochondria (Phizicky et al., 1986; Yoshihisa et al., 2003; Yoshihisa et al., 2007). To catalyze the tRNA and HAC1 ligation, Rlg1p has three enzymatic activities: 2′-3′ cyclic phosphodiesterase, polynucleotide kinase, and adenylate synthetase/RNA ligase (Phizicky et al., 1986; reviewed in Hopper and Phizicky, 2003). The HAC1 splicing is finished by 2′-phosphotransferase, Tpt1p, which removes a 2′-phosphate left at the splicing junction of tRNA (Culver et al., 1997; Gonzalez et al., 1999).
In the yeast UPR, there are other layers of regulation in Hac1p production than the cytoplasmic splicing. HAC1u mRNA exists in cytosolic polysomes and in an unidentified ribonucleoprotein (RNP) under nonstress conditions (Kuhn et al., 2001; Rüegsegger et al., 2001). Although a part of HAC1u mRNA is associated with polysomes, Hac1up is apparently not translated because a region of the 5′ UTR of HAC1u mRNA forms base pairs with the HAC1 intron to attenuate translation of HAC1u mRNA (Rüegsegger et al., 2001). It has been postulated that the splicing itself is sufficient to resolve base-pairing between the 5′ UTR and intron of HAC1 to restart translation. However, the precise mechanism to release translational attenuation of HAC1 mRNA has not been fully understood. Furthermore, the 3′ UTR of HAC1u mRNA contains a signal to target itself to the Ire1p cluster formed upon ER stress (Aragón et al., 2009). Interestingly, translational repression of HAC1u mRNA is a prerequisite for this ER targeting. It indicates, paradoxically, that the translational repression achieved by the 5′ UTR–intron interaction is critical for activation of Hac1p production.
In contrast to the initial step of the unconventional splicing in the Ire1p pathway, the following steps in the Ire1p pathway are divergent among species. First, substrates of Ire1p homologues are different from organisms to organisms. Only some fungi have HAC1 homologues as an Ire1p substrate, whereas metazoan Ire1p catalyzes cleavage of XBP1 mRNA, a different transcription factor for the UPR (Yoshida et al., 2001; Calfon et al., 2002; Saloheimo et al., 2003). No substrate for Ire1p has been identified in plants, whereas two Ire1p homologues are present in the Arabidopsis thaliana genome (Noh et al., 2002). Second, the ligation and finishing steps of the unconventional splicing of XBP1 mRNA in mammalian cells seem to be different from those in the yeast. For tRNA ligation, two alternative pathways were reported in mammalian cells (Filipowicz and Shatkin, 1983; Zillmann et al., 1991). One of the pathways requires the same reaction steps as yeast tRNA ligation, whereas no apparent homologue of the entire yeast ligase has been found in mammalian genomes. Indeed, a part of this pathway is mediated by a different enzyme, hClp1p kinase (Weitzer and Martinez, 2007). The yeast-type RNA ligation pathway, however, is unlikely to operate in the ligation of the XBP1 exons, because knockout of the unique 2′-phosphotransferase gene in mice abolished the completion of the yeast-type RNA ligation but did not affect the UPR (Harding et al., 2008). Recently, a plant tRNA ligase homologous to yeast Rlg1p was identified in A. thaliana. Plant tRNA ligase has the same set of enzymatic activities as yeast Rlg1p and can complement a deletion mutant of yeast RLG1 in tRNA splicing, whereas its functionality in the UPR has not been examined (Englert and Beier, 2005; Wang et al., 2006). Finally, in mammalian cells, the unspliced form of XBP1 mRNA, XBP1(U) mRNA, is not under translational repression. XBP1(U) protein is translated and supposed to act as a negative regulator of the spliced form of XBP1, pXBP1(S) (Yoshida et al., 2006). It is still obscure how and why such diversity of the later steps in the Ire1p pathway to achieve the UPR has been evolved.
To obtain more insight into the later steps of the Ire1p pathway, we analyzed the abilities of tRNA ligases from several organisms to replace functions of the yeast tRNA ligase in HAC1 mRNA splicing in vivo. Although all the Rlg1p homologues tested could complement the growth defect of yeast rlg1Δ mutant under nonstress conditions, Rlg1p from A. thaliana, AtRlg1p, failed to function in the UPR. Surprisingly, AtRlg1p could ligate HAC1 exons upon UPR, but the resulting intron was circularized and remained associated with HAC1i messenger RNP (mRNP) to prevent it from translation. Thus, the splicing step and translational restart step were unlinked in the yeast strain expressing AtRlg1p, a kind of tRNA ligase mutant. Furthermore, we showed that yeast Rlg1p is preloaded onto HAC1u mRNP under nonstress conditions and that the HAC1 5′ UTR contains a cis element(s) to regulate translation of the following open reading frame (ORF) by Rlg1p. These results collectively suggest that Rlg1p has a novel function in the yeast Ire1p pathway even after the completion of HAC1 splicing, especially in the translational regulation of Hac1p.
MATERIALS AND METHODS
Strains and Plasmids
Yeast genetic techniques are essentially described in Guthrie and Fink (1991), and other molecular biological techniques were in Sambrook and Russell (2001). S. cerevisiae strains used in this study are summarized in Table S1. The plasmids and primers are listed in Table S2 and S3, respectively.
A DNA fragment containing the ScRLG1 gene with the 5′ and 3′ flanking sequences was amplified by PCR and cloned into low-copy vectors pRS314 and pRS316 to yield pTYSC220 and pTYSC224, respectively. Chromosomal disruption of ScRLG1 was performed with a diploid strain of W303 as described in Phizicky et al. (1992) with a YIp plasmid, pTYSC295 [rlg1Δ::kanMX4]. TMSC03 and TMSC05 harboring rlg1-4 [T180I] and rlg1-100 [H148Y], respectively, at their chromosomal RLG1 locus were constructed by the pop-in/pop-out method with W303-1A as a host after constructing these mutant rlg1 genes on plasmids based on an integration vector pRS306. TYSC335 with RLG1-protein A at its ScRLG1 locus was constructed from W303-1A by integrating a protein A::CgHIS3 gene cassette with 60-base pair tabs homologous to 3′ regions of ScRLG1 to allow in-frame fusion between ScRLG1 and protein A ORFs. ORFs of fungal RLG1 genes, ScRLG1, KlRLG1, and SpRLG1, were amplified with appropriate PCR primers from genomic DNAs of S. cerevisiae strain X2180-1A, Kluyveromyces lactis strain BY20597 and Schizosaccharomyces pombe strain 972, respectively. Resulting PCR fragments were inserted into XhoI/KpnI-digested pTYSC128, of which multicloning site was placed after the CUP1 promoter and a triple hemagglutinin (3×HA) tag, to yield pTYSC418 with HA-ScRLG1, pTYSC441 with HA-KlRLG1, and pTYSC442 with HA-SpRLG1. ScRLG1 was also subcloned to a BglII/SacII site of a FLAG-tag vector with a URA3 marker, pTYSC462 to yield pTYSC463. For SpRLG1 cloning, the original plasmid was constructed according to the S. pombe Gene DB (http://www.genedb.org/genedb/pombe/), but was found to be nonfunctional from a sequence error of the database. pTYSC442, described above, with the correct 5′-terminal region was constructed using a 5′-primer with a correct sequence (SpRLG1_5-2 in Table S3). A 3.35-kb AtRLG1 ORF from the 74th Met codon (AtRLG1[M74]) was prepared from pIVEX WG1.4-Atlig (Englert and Beier, 2005) by digestion with SacII and Cfr9I, and cloned into the SacII/Cfr9I-digested pTYSC128 to obtain pAt-R1. The same fragment was also inserted into a SacII/Cfr9I site of pTYSC461, another FLAG-tag vector with a TRP1 marker, but SacII sites of the insert and vector were blunted before ligation to allow in-frame fusion between the FLAG tag and AtRLG1 ORF (pTYSC541). A 0.66-kb fragment containing Met54-Val293 of AtRLG1 was amplified from pAt-R1 with a forward primer encoding the Met54-Lys73 sequence and an appropriate reverse primer and ligated with pAt-R1 digested with EagI and BstEII to yield pTYSC471 with HA-AtRLG1[M54].
For constructing m1–m4 HAC1 mutants, KpnI-BamHI fragments (1.98 kb) containing the HAC1 gene with no mutation, the m1 (replacing the intronic element with the 5′ UTR element), m2 (altering the intronic element complementary to the 5′ UTR element), m3 (replacing the 5′ UTR element with the intronic element), or m4 mutation (altering the 5′ UTR element complementary to the intronic element) was amplified by megaprimer PCR. These fragments were subcloned into a centromeric vector pASZ11 [CEN ADE2] to yield pTYSC445 (HAC1), pTYSC447 (hac1-m1), pTYSC446 (hac1-m2), pTYSC468 (hac1-m3), and pTYSC469 (hac1-m4). For constructing hac1Δ strains, an hac1Δ::HIS3 fragment was amplified from a YIp plasmid, pTYSC421 [hac1Δ::HIS3], by PCR, and introduced to TYSC771 [rlg1Δ/pTYSC418] and TYSC774 [rlg1Δ/pAt-R1] to yield TYSC791 and TYSC793, respectively.
For construction of HAC1-GFP (green fluorescent protein) fusion reporters, a GAL7p::GFP::SV40t vector pTYSC475 was constructed, and its 0.29-kb BamHI-AgeI region with a GAL7 promoter was replaced with a 0.55-kb BamHI-AgeI fragment containing the HAC1 promoter and its 5′ UTR to yield pTYSC476. Next, a 0.86-kb PCR fragment with the 3′ half of HAC1 (from G1121 to the end of the gene) was inserted into the SacII-KpnI sites of pTYSC476 to yield pTYSC508. Then, 24 base pairs of the BspEI-SacII region was removed, blunted, and self-ligated to obtain an in-frame fusion between GFP and HAC1u ORFs (pTYSC544).
To obtain a template for in vitro transcription of HAC1i mRNA, HAC1i cDNA was produced from yeast total RNA with SuperScript III First Strand Synthesis System (Invitrogen, Carlsbad, CA) and amplified by PCR with primers, T7-HAC1_f1211 and HAC1_r1511-491. The fragment was inserted into a KpnI/BamHI site of pUC119 to yield pTYE493. Templates for HAC1u mRNA, HAC1i mRNA, and HAC1 intron were amplified by PCR with T7-HAC1_f477 and HAC1_r1927-06 as primers and pTYSC445 as a template, T7-HAC1_f477 and HAC1_r1927-06 as primers and pTYE493 as a template, and T7-HAC1_f1211 and HAC1_r1511-491 as primers and pTYSC445 as a template, respectively. Then, RNAs were transcribed in vitro from the T7 promoter with a MEGAScript kit (Ambion, Austin, TX). After phenol/chloroform extraction and desalting with a spin column, amounts of RNAs were determined from A260.
All the PCR fragments for cloning were sequenced with BigDye Terminator ver. 1.1 and ABI Prism 3130 Genetic Analyzer (Applied BioSystems, Foster City, CA).
Northern Blotting and RT-PCR
Northern blotting was performed as essentially described in Sambrook and Russell (2001). Total RNA prepared from yeast according to Yoshihisa et al. (2003) was analyzed on 1.5% agarose gel with formaldehyde and transferred to Hybond N+ charged nylon membranes (GE Healthcare Biosciences, Piscataway, NJ) by the capillary transfer method. Antisense RNA probes of HAC1u full length, HAC1 intron, and ACT1 decorated with digoxigenin were prepared by in vitro transcription from linearized pHAC1, pHAC1-int, and pACT1, respectively, with DIG Northern Starter Kit (Roche Diagnostics, Mannheim, Germany) according to manufacturer's instructions. After hybridization in 0.2×SSC, 1.0% SDS at 68°C and immunodecoration with ALP-labeled anti-digoxigenin IgG, signals were developed by the ECF system (GE Healthcare Biosciences) and read with STORM860 Image Analyzer (Molecular Dynamics, Sunnyvale, CA), or the signals were visualized by CDP-star (Roche Diagnostics) and read with LAS-4000mini (Fujifilm, Tokyo, Japan).
For RT-PCR, first strands of cDNA were generated by Superscript III First-Strand Synthesis System for RT-PCR (Invitrogen) with an oligo-(dT)20 primer and 1.0 μg of total RNA. The cDNAs of interest were amplified from reverse transcription (RT) reactions equivalent of 0.10 μg total RNA with appropriate primer sets (see Table S3) and ExTaq DNA polymerase (Takara Bio, Otsu, Japan). The predetermined optimal cycle number was used to amplify each transcript, for example, 22 cycles for HAC1 and SRP1, 20 cycles for ERO1 and 18 cycles for ACT1.
RNase H Cleavage Assay
Oligonucleotide-directed RNase H cleavage was performed as described as in Murray and Schoenberg (2008). Briefly, 10 μg of total RNA prepared from the HA-AtRLG1 cells was hybridized with 200 pmol of anti-sense probes a (HAC1_1310-291c) or b (HAC1_1360-41c) shown in Figure 3C, and treated with 20 U of RNase H at 37°C for 30 min. RNAs were extracted by phenol/chloroform and ethanol-precipitated, and then separated with 7 M urea/5% polyacrylamide gel. The HAC1 intron and its fragments were detected by Northern blotting with anti-sense probes a or b. Complete digestion of the HAC1 intron was confirmed by the absence of the Northern signal when the same probe was used for the digestion and detection.
Figure 3.

HAC1 intron is circularized and retained in the AtRLG1 strain. Wild-type (RLG1), rlg1-4, rlg1-100, ire1Δ, RLG1-HA, and rlg1Δ/HA-AtRLG1[M74] strains were cultured in the presence (+ lanes) or absence (− lanes) of 2.0 μg/ml Tm for 60 min. Total RNAs were prepared and electrophoresed with 1.5% agarose/2.2 M formaldehyde gel. (A) HAC1 RNA species were detected by Northern blotting with an anti-HAC1u probe (top). ACT1 mRNA was also detected as a loading control (bottom). (B) The total RNAs from the AtRLG1[M74] cells were subjected to Northern blotting with an anti-HAC1 intron probe. Signal enhancement enables to observe the minor HAC1u mRNA in the presence of Tm. (C) Total RNAs prepared from HA-AtRLG1 cells were hybridized with probes a or b and treated with RNase H. In the lane labeled −, no oligonucleotide was added. RNAs recovered from the digestion were analyzed on a polyacrylamide gel. The HAC1 intron and its fragments were visualized by Northern blotting with either of the probe a (left) or b (right). Hybridization positions of these probes on putative linear or circular HAC1 introns are schematically described in the right.
Polysome Analysis
Polysome analysis was performed essentially as described in Inada et al. (2002). Briefly, cells cultured in appropriate conditions were treated with final 0.1 mg/ml cycloheximide and harvested. Pelleted cells were frozen and ground in the liquid N2 with a prechilled mortar and pestle. Cell powder was extracted with lysis buffer [20 mM HEPES-KOH, pH 7.4, 2.0 mM Mg(OAc)2, 100 mM KOAc, 1.0 mM DTT, 1.0 mM PMSF, 1/1000 of Protease Inhibitor Cocktail (Roche Diagnostics)] supplemented with 0.5 U/μl RNasin. After two sequential centrifugations at 9200 × g for 10 min at 4°C, supernatants were loaded onto a 10–50% sucrose gradient. The gradient was ultracentrifuged at 100,000 × g for 3 h at 4°C in a P28S swing rotor (Hitachi Koki, Tokyo, Japan), and fractions were collected from the top with Piston Gradient Fractionator (Biocomp Instruments, Fredericton, NB, Canada).
Native Immunoprecipitation
HA-fusion proteins and FLAG-fusion proteins were immunoprecipitated from the extracts prepared as above by incubating with anti-HA-agarose and with anti-FLAG M2-agarose (Sigma-Aldrich, St. Louis, MO), respectively, for 2 h at 4°C. After five-times wash with lysis buffer, protein-RNA complexes were eluted by boiling in 25 mM Tris-HCl, pH 7.5, 10 mM EDTA, and 1.0% SDS for 5 min or by incubating with 0.2 μg/μl FLAG peptide for 30 min at 4°C. After removing beads by centrifugation, a portion was set aside for Western blotting, and RNAs in the rest of the eluates were extracted with phenol/chloroform and recovered by ethanol precipitation. RNAs were also recovered from loaded and flow-through samples. For separation of polysomal and nonpolysomal fractions from the extract or the immunoprecipitate, a sample (300 μl) was loaded onto a 16/50, 18/50, or 20%/50% wt/vol discontinuous sucrose gradient (600 μl + 500 μl) and centrifuged at 160,000 × g for 1 h at 4°C with a CS100 microultra centrifuge (Hitachi Koki). A 400-μl fraction from the top and another 400-μl fraction containing the 16–20%/50% interface were recovered as nonribosomal and ribosomal fractions, respectively. These fractions were subjected to immunoprecipitation (IP) with anti-HA-agarose or protein/RNA analysis as described above.
RNA IP after Cross-Link
RNA IP after cross-link was performed as described in Gilbert et al. (2004). To cross-link between proteins and RNAs, final 1.0% formaldehyde was directly added to the appropriate cultures of yeast strains. After 30-min incubation, residual formaldehyde was quenched with final 350 mM glycine, and then cell extracts were prepared by eight 30-s bursts of glass-bead agitation at 90-s intervals on ice in FA lysis buffer (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1.0 mM EDTA, and 5.0 mM DTT) supplemented with a 1/1000 volume of Protease Inhibitor Cocktail and 100 U/ml RNasin. Then, final 1.0% Triton X-100 and 0.10% Na-deoxycholate were added, and DNA was digested by sonication and treatment with 500 U of RNase-free DNase I in the presence of 25 mM MgCl2 and 5.0 mM CaCl2. After adding final 30 mM EDTA, the extracts were centrifuged at 20,000 × g for 15 min. Resulting supernatants were subjected to IP with IgG-Sepharose in the presence of 10 U/ml RNasin. Proteins and RNAs were eluted from the IgG-Sepharose beads by heating at 65°C for 10 min in elution buffer (25 mM Tris-HCl, pH 7.5, 10 mM EDTA, 0.5% SDS) after an extensive wash with FA lysis buffer. RNA-protein adducts in the eluates were de-cross-linked by treating with proteinase K at 42°C for 1 h and further heating at 65°C for 5 h. Liberated RNAs were extracted with phenol/chloroform and recovered by ethanol precipitation. For RNase IP control, a part of the DNase I–treated samples was incubated with 20 μg/ml RNase A at 37°C for 10 min and then subjected to IP in the absence of RNasin.
Miscellaneous
Western blotting was performed with anti-ScRlg1p rabbit polyclonal antibodies (this study), anti-Hac1p affinity-purified rabbit polyclonal antibodies (gifted by Dr. K. Mori, Kyoto University), an anti-Pdi1p mouse mAb (38H8: EnCor Biotechnology, Gainesville, FL), an anti-HA mouse mAb (HA-7: Sigma-Aldrich), an anti-FLAG mouse mAb (M2: Sigma-Aldrich), and an anti-Por2p mouse mAb (16G9: Molecular Probes, Eugene, OR) as primary antibodies, and with Cy5-labeled (GE Healthcare Biosciences and Alexa488-labled (Molecular Probes) secondary antibodies. Signals were detected with STOR860 Image Analyzer (Molecular Dynamics, Sunnyvale, CA). Agarose gel images of RT-PCR analysis were captured with Gel Doc 2000 Gel Documentation System (Bio-Rad Laboratories, Hercules, CA). For quantification, RT-PCR products were analyzed with MultiNA Microchip Electrophoresis System (Shimadzu, Kyoto, Japan).
RESULTS
RLG1 Genes from Other Organisms Can Complement rlg1Δ in S. cerevisiae, But Plant Rlg1p Cannot Substitute for ScRlg1p in the UPR Function
In the yeast S. cerevisiae, Rlg1p functions both in tRNA splicing essential for cell growth and in HAC1 splicing during the UPR. The former function can be reflected in the cell growth, and the latter in sensitivity to UPR-inducing reagents, such as tunicamycin (Tm). We thus attempted to test the abilities of various Rlg1p homologues to replace these functions of the authentic yeast Rlg1p in vivo. We selected three RLG1 homologues from K. lactis, S. pombe, and A. thaliana (denoted as KlRLG1, SpRLG1, and AtRLG1, respectively). As summarized in Table 1, AtRlg1p has only weak homology to ScRlg1p (7.9% amino acid identity), whereas its overall domain organization is conserved (Englert and Beier, 2005; Wang et al., 2006). On the other hand, only K. lactis has an apparent homologue of HAC1 on its genome.
Table 1.
Comparison of various Rlg1p homologues
| Organism | Identity (%)a | Homologues on genome |
rlg1Δ complementation in S. cerevisiae |
||
|---|---|---|---|---|---|
| Ire1p | Hac1p | Growth | UPR | ||
| Saccharomyces cerevisiae | — | Yes | Yes | Yes | Yes |
| Kluyveromyces lactis | 46.6 | Yes | Yes | Yes | Yes |
| Schizosaccharomyces pombe | 27.9 | Yes | No | Yes | Yes |
| Arabidopsis thaliana | 7.9 | Yes | No | Yes | No |
a The percentage of amino acid identity to Rlg1p of S. cerevisiae was determined by Clustal W.
We expressed these Rlg1p homologues N-terminally fused with a 3×HA tag from the CUP1 promoter under uninduced conditions. These RLG1 genes could complement growth defects of chromosomal deletion of ScRLG1 (for clarity, we hereafter refer RLG1 of S. cerevisiae as ScRLG1; Figure 1A). In the case of SpRlg1p, one nucleotide deletion was found in the database sequence, resulting in N-terminal extension of the ORF. Only this revised SpRLG1 ORF was functional in S. cerevisiae (Figure 1A, bottom left). For AtRLG1, similar complementation test was already reported with a short form of AtRlg1p, AtRlg1p[M74], starting from the 3rd Met codon in the longest ORF (Wang et al., 2006). Because the N-terminal ∼50 residues encoded by the longest AtRLG1 ORF turned out to be a chloroplastic targeting signal (Englert et al., 2007), we also expressed AtRlg1p from the 2nd Met (AtRlg1p[M54]) and found that this was also functional. All the four Rlg1 proteins from the three species were expressed as proteins with expected molecular weights (Figure S1A). They were localized in the yeast cytoplasm and excluded from the nucleus (Figure S1B). We also confirmed that both the authentic ScRlg1p and protein A–tagged ScRlg1p were mainly localized in the cytosolic dots in the wild-type yeast cells (Figure S1C). Therefore, all the Rlg1p homologues can act as an RNA ligase in the cytoplasmic splicing of pre-tRNA in S. cerevisiae.
Figure 1.

RLG1 genes from various organisms complement lethality of rlg1Δ in S. cerevisiae, but only plant RLG1 fails to complement UPR defects. (A) RLG1 genes from K. lactis, S. pombe, and A. thaliana were expressed as N-terminal 3×HA fusion proteins in a S. cerevisiae strain TYSC474 (rlg1Δ/pTYSC224 [URA3 ScRLG1]), and whether these genes complement rlg1Δ was tested using 5′ fluoroorotic acid selection. W303-1A was also streaked as a wild-type control. None, vector plasmid; HA-ScRLG1, RLG1 of S. cerevisiae; HA-KlRLG1, RLG1 of K. lactis; HA-SpRLG1, RLG1 of S. pombe with a corrected ORF; HA-SpRLG1*, RLG1 of S. pombe with an ORF predicted from the genome database; HA-AtRLG1[M74], RLG1 of A. thaliana from the 3rd Met ([M74]); HA-AtRLG1[M54], that from the 2nd Met ([M54]). (B) The yeast cells expressing heterologous RLG1 genes were tested for ability of the UPR by streaking these strains on a 0.25 μg/ml Tm plate. Strains tested are shown on the right.
Next, we asked whether the RLG1 homologues can substitute for ScRLG1 in the UPR. The yeast strains with KlRLG1 and SpRLG as the sole RLG1 gene, as well as the ScRLG1 strain, grew in the presence of 0.25 μg/ml Tm, which caused accumulation of unfolded proteins in the ER through inhibition of N-glycosylation of newly synthesized secretory proteins (Figure 1B; see also Figure 2A). On the other hand, the AtRLG1[M54] and AtRLG1[M74] strains showed growth defects on the Tm plate, whereas the former grew better than the latter. Tm sensitivity of AtRLG1-expressing cells was quantified with the disk assay. An average diameter of the inhibition zone caused by a 6-mm filter disk absorbing 0.25 μg of Tm was 14.3 ± 0.3 mm for the wild-type yeast, 17.5 ± 0.3 mm for the HA-AtRLG1[M54] strain, and 18.5 ± 0.4 mm for the HA-AtRLG1[M74] strain, which confirms the higher sensitivity of HA-AtRLG1[M74] to Tm. These results suggest that the fungal homologues but not the plant homologue of Rlg1p can replace ScRLG1 in the UPR in vivo.
Figure 2.

AtRlg1 proteins can splice HAC1 mRNA, whereas AtRLG1 strains are defective in the UPR. (A) Lysates were prepared from cultures of indicated strains in the absence (−) or presence (+) of 2.0 μg/ml Tm and then analyzed by Western blotting with anti-Hac1p antibodies (top). Appearance of unglycosylated Pdi1p (◁, ug-Pdi1p) was monitored to confirm effects of Tm (middle), and Por2p (mitochondrial porin) was used as a loading control (bottom). In the left part, yeast strains harboring ScRLG1 genes with no mutation (wt), rlg1-4 mutation (defective only in tRNA ligation), or rlg1-100 mutation (defective only in HAC1 mRNA ligation) on their chromosome were analyzed as control. Right, lysates from rlg1Δ strains complemented with various RLG1 genes were analyzed. In the bottom square, twofold more of lysates and the fivefold higher concentration of the anti-Hac1p antibodies were used to detect small amounts of Hac1p produced in the AtRLG1[M74] and AtRLG1[M54] cells. (B) Total RNAs (0.10 μg) prepared from the same set of strains in A was subjected to RT-PCR of HAC1 with a set of primers whose target was represented in the schematic drawing (arrows). ERO1 RT-PCR was also carried out to monitor the UPR, and ACT1 RT-PCR was done as a loading control. The top part represents RT-PCR products from yeast cells with ScRLG1 alleles on their chromosome. In the bottom panel, RT-PCR products from rlg1Δ strains complemented with various RLG1 genes on plasmids were analyzed. RT, reverse-transcription. ▶, RT-PCR fragment derived from HAC1u mRNA; ◁, that from HAC1i mRNA; gray triangles, RT-PCR fragments of ERO1 and ACT1 mRNAs. (C) A typical electrogram of sequencing reactions of the RT-PCR fragment amplified from the AtRLG1[M74] cells treated with Tm. Corresponding sequence is shown beneath the electrogram. The exon–exon junction is indicated by a vertical arrow.
The AtRLG1 Strain Can Splice HAC1u mRNA Normally
The above results suggested that Rlg1p from A. thaliana is defective in HAC1u mRNA splicing in S. cerevisiae. Inability to activate the UPR though the Ire1p-Hac1p system was further tested by Western blotting of Hac1p. The UPR was induced by treatment with 2.0 μg/ml Tm for 60 min, and the effects of Tm was confirmed by appearance of the unglycosylated form of Pdi1p in Western blotting (Figure 2A, middle, ug-Pdi1p). Yeast strains with the authentic ScRLG1 (Figure 2A, lanes marked with wt) and fungal HA-RLG1 genes on plasmids (Figure 2A, right gel set, lanes marked with Sc, Kl, and Sp) and the rlg1-4 mutant only defective in tRNA splicing (lanes rlg1-4) expressed Hac1ip when exposed to 2.0 μg/ml Tm. A particular rlg1 mutant sensitive to Tm, rlg1-100 (Sidrauski et al., 1996), could not produce Hac1ip (lanes rlg1-100). As expected, AtRLG1 strains produced only small amounts of Hac1ip when exposed to Tm. The AtRLG1[M54] strain produced a larger amount of Hac1ip than the AtRLG1[M74] strain (Figure 2A, bottom panel). These results correlate well to the sensitivity of these strains to Tm and indicate that the AtRLG1 strains are defective in the Ire1p–Hac1p system to activate the UPR.
Then, we tested whether AtRlg1p joins the HAC1 exons to the HAC1i mRNA in vivo by RT-PCR. In the wild-type and ScRLG1 strains, considerable amounts of HAC1i mRNA were detected in the presence of 2.0 μg/ml Tm, whereas only HAC1u mRNA was detected in the absence of Tm (Figure 2B). The rlg1-100 cells lost HAC1u mRNA but did not accumulate HAC1i mRNA in the presence of Tm, whereas rlg1-4 cells normally converted the HAC1u mRNA to the HAC1i mRNA (Figure 2B, top left). Surprisingly, the AtRLG1 strains did splice HAC1u mRNA to HAC1i mRNA. As shown in Figure 2B, lanes with At[M54] and At[M74], AtRLG1 cells produced HAC1i mRNA in the presence of Tm. The splicing efficiency was also comparable to the wild-type strain, indicating that AtRlg1p is functional as HAC1 ligase in the yeast. On the other hand, transcriptional activation of ERO1, one of the UPR-regulated genes (Travers et al., 2000), was not obvious in the Tm-treated AtRLG1 strains and was correlated to their Tm sensitivity shown in Figure 1B. We noticed that the AtRLG1 strains also produced a small but detectable amount of HAC1i mRNA even in the absence of Tm. Deficiencies in the UPR in these strains may perturb maintenance of protein folding environments in the ER even under nonstress conditions, resulting in partial activation of Ire1p in the absence of Tm.
Although AtRlg1p can join the HAC1 exons, it might be possible that formation of the splice junction is not correct and a frame shift is introduced to disrupt synthesis of normal Hac1ip. In fact, production of aberrantly spliced HAC1 mRNA was reported when ScRLG1 deletion is complemented by a combination of genes encoding T4 RNA ligase 1 and T4 polynucleotide kinase/3′ phosphatase (Schwer et al., 2004). To examine this possibility, we sequenced the RT-PCR fragment of HAC1i mRNA amplified from the AtRLG1[M74] cells. The sequence around the splice junction of HAC1i mRNA was the same as that from the wild-type yeast, and no minor peaks of other nucleotides were detected in the electrogram (Figure 2C). Therefore, we concluded that AtRlg1p can join the HAC1 exons correctly like the authentic ScRlg1p in vivo even though the Arabidopsis genome does not have an apparent homologue of HAC1. Furthermore, these results indicate that the posttranscriptional defect of the AtRLG1 strains in production of Hac1ip does not come from the splicing but from the translation. Because the UPR defect is more prominent in the AtRLG1[M74] strain than in the AtRLG1[M54] strain, we decided to analyze the former strain further.
HAC1 Intron Is Circularized and Remains Associated with HAC1i mRNA in AtRLG1 Cells
We examined RNA species accumulating in the AtRLG1[M74] cells in more detail by Northern blotting. As shown in Figure 3A, AtRLG1 cells contained both HAC1u and HAC1i mRNA even in the absence of ER stress, and HAC1i mRNA increased in amount under the ER stress conditions. This is in marked contrast to rlg1-100 cells in which HAC1u mRNA was lost under ER stress conditions. In addition, a short RNA that was hybridized with an anti-HAC1u probe was detected in the AtRLG1 cells. This RNA species was absent in the wild-type and other ScRLG1 mutant strains. Northern blotting with an intron specific probe revealed that this is the HAC1 intron (Figure 3B).
One possible reason to explain accumulation of the HAC1 intron in the HA-AtRLG1 strain is that the intron is circularized by the action of AtRlg1p and escapes from exonucleolytic degradation. Indeed, it is reported that AtRlg1p but not ScRlg1p can circularize an intron cleaved from a pre-tRNA by splicing endonuclease in vitro (Phizicky et al., 1986; Englert and Beier, 2005). To examine whether the HAC1 intron is circularized in the HA-AtRLG1 cells in vivo, we performed oligonucleotide-directed RNase H cleavage (Murray and Schoenberg, 2008). As shown in the right panel of Figure 3C, RNase H treatment of the HAC1 intron hybridized with an antisense oligonucleotide will produce two linear fragments if the intron is linear, but only one fragment slightly shorter than the intron will be produced if the intron is circular. We hybridized a 20mer antisense probe a (corresponding to 88th-107th of the intron) or probe b (138th-157th) to total RNA prepared from the HA-AtRLG1 cells and then treated with RNase H. When no oligonucleotide was added, we detected two HAC1 intron-related bands: one migrated slower than a 500-nt DNA marker and the other migrated near the 250-nt marker on 7 M urea/5% polyacrylamide gel. These bands were converted to single bands appearing near the 250-nt marker when treated with either of the oligonucleotides. No smaller fragments corresponding to possible derivatives from the linear intron were detected, indicating that most, if not all, of the HAC1 intron is circularized in the HA-AtRLG1 strain. We still do not know why there are two forms of the circular HAC1 intron. It might be possible that the longer form is a dimer or concatemer of the HAC1 intron.
The existence of the stable intron in the AtRLG1 strain suggests that base-pairing between the 5′ UTR of the HAC1i mRNA and the cleaved intron is still maintained in this strain to attenuate Hac1p translation. To examine this possibility, we performed sucrose density gradient centrifugation to compare distribution of HAC1 RNA species and polysomes. HAC1u and HAC1i mRNAs both in ScRLG1 and AtRLG1 extracts appeared in two peaks in the sucrose density gradient as reported (Rüegsegger et al., 2001; Kuhn et al., 2001): one peak sedimented near 40S and 60S ribosomal subunits, and the other cosedimented with polysomes (Figure 4). More HAC1i was associated with polysomes than HAC1u. In the AtRLG1 extracts, some HAC1 intron was detected in the lighter (nonpolysomal) fraction, but its significant portion was cosedimented with polysomes (Figure 4, B and D). No intron was detected in the fractions of ScRLG1 extracts (Figure 4, A and C).
Figure 4.

HAC1 intron is associated with polysomes in the HA-AtRLG1 strain. Distribution of HAC1 RNA species was examined by polysome analysis. Extracts from HA-ScRLG1 cells (A and C) and HA-AtRLG1[M74] cells (B and D) cultured in the absence (A and B, −Tm) or the presence (C and D, +Tm) of 2.0 μg/ml Tm were subjected to 10–50% sucrose density gradient ultracentrifugation. Fractions were collected from the top (left) to the bottom (right). In each set of three panels, the top, middle, and bottom panels represent A260 trace, HAC1 RNAs, and ACT1 RNA, respectively. In the top panel of A, peak assignments are shown. In the middle panel, positions of HAC1 RNA species were indicated in the right. Amounts of HAC1-related RNAs in the sucrose gradient fractions of HA-AtRLG1 extracts from control (E) or Tm-treated (F) cells were compared with standard RNAs transcribed in vitro. In the bottom panel, Northern signals of HAC1u mRNA (□), HCA1i mRNA (▩) and HAC1 intron (■) were quantified and expressed in arbitrary units. L, cell extract; Std, 0.22 fmol each of HAC1u mRNA, HAC1i mRNA, and HAC1 intron.
Next, we examined whether the HAC1 intron on polysomes forms a one-to-one complex with the HAC1i mRNA to secure its association with the polysomes by measuring amounts of HAC1 RNAs in the sucrose gradient fractions. The HAC1u and HAC1i mRNAs without poly-(A) and the HAC1 intron were transcribed in vitro to obtain RNA standards, and sucrose gradient fractions from HA-AtRLG1 cell extracts were analyzed by Northern blotting with defined amounts of these standard RNAs. As shown in Figure 4, E and F, molar amounts of the HAC1i mRNA detected in the polysomal fractions were much lower than those of the HAC1 intron when compared with a mixture of 0.22 fmol each RNA standards (lane Std). This was more prominent in the case of Tm-untreated extracts (Figure 4E). These results suggest that, in the AtRLG1 cells, some HAC1 intron is retained on the HAC1i mRNA even after its splicing and remains on the polysomes after completion of translation of the mRNA and that this is one of the reasons why the AtRLG1 strain cannot produce Hac1ip upon ER stress. Furthermore, the existence of the HAC1 intron in the nonribosomal fractions indicates that the free intron is a poor substrate for cytosolic RNA degradation machinery because of its circular nature.
The 5′ UTR of HAC1 Possesses a cis Element to Regulate Translation of the Following ORF, and This Regulation Depends on Rlg1p
To confirm that the base-paring between the 5′ UTR and intron of HAC1 mRNA is responsible for the translational defect of HAC1i mRNA in the AtRLG1 strain, we constructed a series of hac1 mutants. As summarized in Figure 5A, the 5′ UTR element from −38 to −20 of HAC1u mRNA is almost complementary to the intronic element from 764 to 782 (Rüegsegger et al., 2001). We disrupted this complementarity by replacing the intronic element with the 5′ UTR element or vice versa (m1 and m3). We also changed either of the elements to the sequence perfectly complementary to the other (m2 and m4). These mutant hac1 genes on a centromeric plasmid were introduced to an ScRLG1 hac1Δ strain or an AtRLG1 hac1Δ strain. First, we monitored the splicing efficiency of HAC1u mRNA by RT-PCR. AtRLG1 cells harboring mutant HAC1 genes with the same sequences in the 5′ UTR, and the intron could not splice HAC1u mRNA even in the presence of Tm (Figure 5B, m1 and m3; see below). On the other hand, when the complementarity was strengthened, the efficiency of HAC1u splicing increased even in the absence of Tm (Figure 5B, m2 and m4). Similar results were obtained with the ScRLG1 strains. Then, translation of Hac1p was checked by Western blotting. The AtRLG1 cells harboring m1 and m3 hac1 mutants expressed Hac1up from the unspliced mRNA even in the absence of ER stress. A considerable increase of Hac1up in amount was observed in the m1 mutant upon ER stress (Figure 5C). These results indicate that disruption of the base-paring releases translational attenuation of HAC1 mRNA even in the AtRLG1 strain. On the other hand, m2 and m4 mutant cells produced amounts of Hac1ip similar to those of the wild-type HAC1 cells despite the fact that the mutants produced more HAC1i mRNA in the absence of Tm. The effects of hac1 mutations in the ScRLG1 background were also similar to those seen in the AtRLG1 strains, but the effect of m1 mutation is modest in the ScRLG1 strain when compared with that in the AtRLG1 strain. A recent report demonstrated that the splicing of HAC1u is dependent on translational attenuation under nonstress conditions (Aragón et al., 2009). Our results are consistent with this observation. The defects of HAC1u splicing in the m1 and m3 mutants expressed in the AtRLG1 background may be due to translation of HAC1u mRNA in the absence of Tm. These results also support the idea that the defects of AtRLG1 on the UPR are linked to the impaired intron release from HAC1i mRNP. As described above, Hac1up production from the m1 mutant is up-regulated by ER stress, whereas that from m3 is not. Therefore, the 5′ UTR element on HAC1 mRNA may be an important cis-element that is directly recognized by the translational machinery for regulation.
Figure 5.
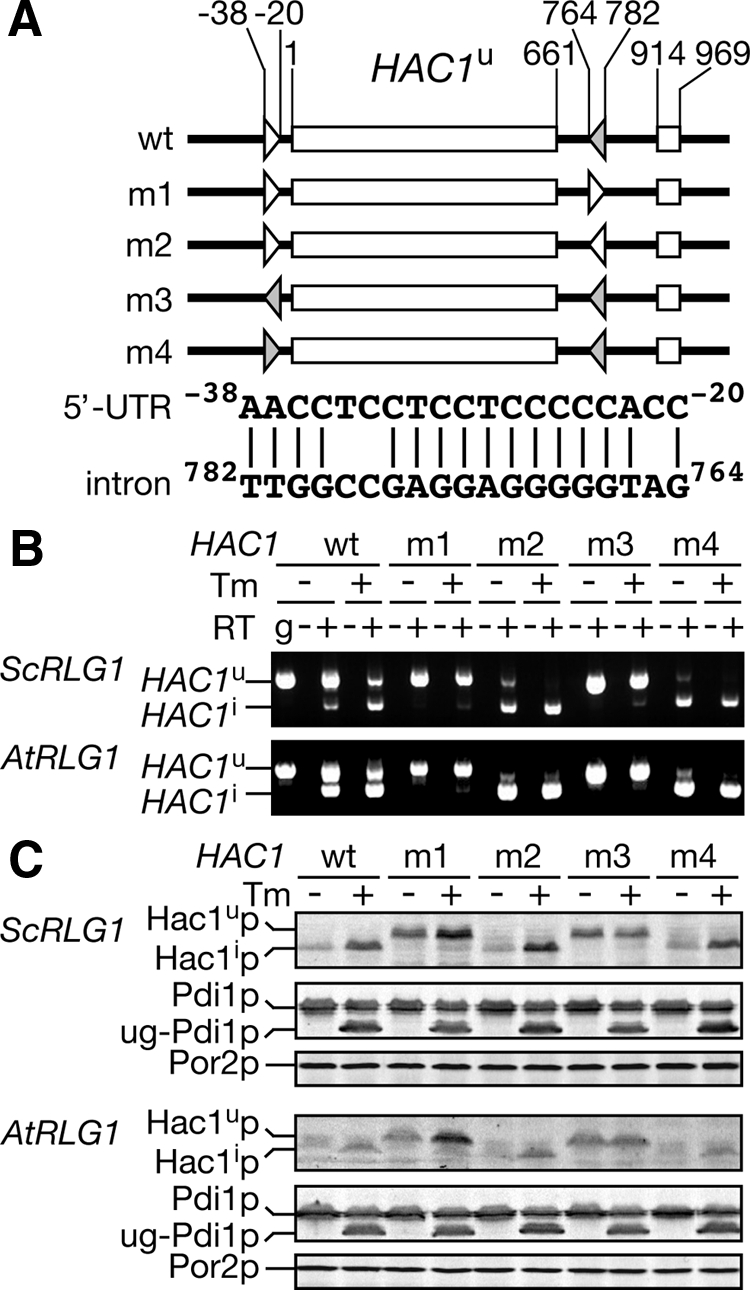
Disruption of the base-paring between the 5′ UTR and intron of HAC1 releases translational attenuation even in the AtRLG1 strain. (A) Schematic drawing of hac1 mutants and comparison of the 5′ UTR (open triangles) and intronic (gray triangles) elements of the wild-type HAC1. (B) RT-PCR analysis of hac1 mutant mRNAs produced in the ScRLG1 and AtRLG1 strains. The ScRLG1 hac1Δ (top) and AtRLG1 hac1Δ (bottom) strains with either one of the hac1 mutants shown in A or the wild-type HAC1 were cultured in the absence (Tm, −) or presence (Tm, +) of 2.0 μg/m Tm, and total RNA fractions prepared from these cultures were subjected to RT-PCR as in Figure 2B. (C) The protein samples were prepared from the cultures in B and analyzed by Western blotting with antibodies shown in the left as in Figure 2A.
The above results that AtRLG1-expressing cells accumulate the circular intron and that prolonged interaction between the HAC1 5′ UTR and intron causes translational attenuation in these cells may suggest that the accumulation of the intron itself fully explains the reason for repressed translation of the spliced HAC1i mRNA. This further suggests that the HAC1 intron may repress the translation of unrelated ORFs following the HAC1 5′ UTR in trans. To test this hypothesis, we constructed HAC1-GFP fusion reporters described in Figure 6A. In an HGH construct, a large part of the 1st exon of HAC1 was replaced with a GFP ORF in frame to allow unconventional splicing upon ER stress. On the other hand, an HG construct has only the HAC1 5′ UTR in front of the GFP ORF. Both of them are expressed from the HAC1 promoter. We introduced these reporter genes into HA-ScRLG1 or HA-AtRLG1 strains with wild-type HAC1 or hac1Δ background and then analyzed how much of GFP proteins are translated. As shown in Western blotting of Figure 6B, GFP production from both HGH and HG constructs was not affected by HAC1 deletion in the HA-AtRLG1 cells (see At/Wt lanes vs. At/Δ lanes), indicating that existence of the HAC1 intron on ribosomes does not affect translation of the reporter genes with the 5′ UTR of HAC1 in trans. Interestingly, we noticed that about twofold more GFP was translated from the HG construct in the HA-ScRLG1 cells than that in the HA-AtRLG1 cells. This is not an overall effect of the “HA-AtRLG1 mutation,” because translational efficiency of the control GG construct, which is under the control of the galactose-inducible promoter and does not contain any HAC1 sequence, was not affected by RLG1 genes (Figure 6B, right). These results imply that a cis element in the HAC1 5′ UTR can act to repress translation of the following ORF even in the absence of the HAC1 intron in cis. This negative regulation is compensated by ScRlg1p but not by AtRlg1p. On the basis of these observations, we conclude that yeast Rlg1p has a pivotal role in translational reactivation of HAC1i mRNA in addition to the unconventional splicing.
Figure 6.

Rlg1p affects translation of HAC1 5′ UTR-containing mRNA not through splicing of the mRNA. (A) A schematic diagram of GFP reporter genes with or without HAC1 5′ UTR. Annealing sites of RT-PCR primers are shown by short arrows. The pair a (black arrows) was used to amplify a GFP portion of the reporter mRNAs, whereas the pair b (gray arrows) was used to monitor unconventional splicing of the HGH reporter. (B) HA-ScRLG1 (Sc) or HA-AtRLG1 (At) strains with the HAC1 (Wt) or hac1Δ (Δ) background were transformed with a plasmid expressing either HG, HGH, or GG type GFP reporter gene. Total proteins and RNAs were prepared from the cells treated with (+) or without (−) Tm, and subjected to Western blotting and RT-PCR, respectively, to detect the gene products shown in the left. Open arrowhead, GFP-Hac1ip; closed arrowhead, GFP. The SRP1 gene products were monitored as loading controls. Amounts of GFPs and corresponding RT-PCR products were quantified, and their ratios are shown in the bottom with an arbitrary unit.
Rlg1 Proteins Interact with HAC1u mRNA, HAC1i mRNA, and HAC1 Intron
If Rlg1p is involved in the translational control of the HAC1 mRNAs, Rlg1p should interact with HAC1 RNA species, including the HAC1u and/or HAC1i mRNA that are not true substrates for the ligation reaction. At least in the case of tRNA ligation, Rlg1p is known to interact with pre-tRNAs in vitro even before they are cleaved by the tRNA splicing endonuclease (Tanner et al., 1988). To examine interaction between Rlg1p and HAC1 RNA species in vivo, we immunoprecipitated Rlg1 proteins through their N-terminally attached 3×HA tag under nondenaturing conditions and analyzed associated RNA species by Northern blotting. As shown in Figure 7A, both the AtRlg1p and ScRlg1p fractions immunoprecipitated from Tm-treated extracts contained both the HAC1u and HAC1i mRNAs, indicating that both Rlg1 proteins are associated with the HAC1u mRNA before it is cleaved by Ire1p. In the same fraction, we also detected the HAC1 intron in the case of AtRlg1p immunoprecipitate. All the HAC1 RNA species were recovered when 10 mM EDTA was added to the extracts to disrupt polysomes, suggesting that interactions between Rlg1 proteins and the HAC1 RNAs is not mediated by active translation. Because the HAC1 intron can form base-pairing with the 5′ UTR of the HAC1 mRNAs, the HAC1 intron may be coprecipitated with AtRlg1p through an indirect interaction via HAC1 mRNAs. To test this possibility, we fractionated the AtRLG1 extracts into polysomal and nonpolysomal fractions using a two-step sucrose gradient and analyzed the fractions by IP. Preliminary experiments revealed that polysomes can sediment though a 16–20% sucrose layer by ultracentrifugation at 150,000 × g, whereas other proteins and RNAs do not go into the sucrose layer. As shown in Figure 7B, a large portion of tRNAs but only minute amounts of 5S and 5.8S rRNAs were recovered in the upper fraction. rRNAs were mainly recovered in the lower fraction above the 50% sucrose cushion (lanes R). When AtRlg1p was immunoprecipitated from the upper and lower fractions of the 16/50% sucrose density gradient, the intron was recovered in immunoprecipitates both from the upper (nonribosomal) and lower (ribosomal) fractions. IP after the sucrose gradient reproducibly enhanced recovery of HAC1 RNP for unknown reason. These results indicate that AtRlg1p forms an RNP(s) with the HAC1 intron irrespective of the presence of ribosome.
Figure 7.

Rlg1 proteins bind both HAC1 mRNAs and intron. (A) HA-ScRlg1p and HA-AtRlg1p[M74] were immunoisolated in the presence (Mg2+) or absence (EDTA) of Mg2+ from Tm-treated cell extracts with anti-HA-agarose. RNAs in each fraction were subjected to Northern blotting with the anti-HAC1u probe. T, one-tenth of total; U, one-third of unbound fraction; B, all of bound fraction. (B) HA-AtRLG1[M74] extracts were subjected to discontinuous sucrose density gradients (16/50, 18/50, or 20%/50%) to separate the nonribosomal (N) and the ribosomal (R) fraction described schematically in the upper region. Left, a urea-PAGE image of low-molecular-weight RNAs in the N, R, and T (total) fractions of sucrose density gradients. Right, the extract from Tm-treated AtRLG1 cells was separated to nonribosomal and ribosomal fractions with the 16/50% sucrose density gradient. RNA samples from these fractions were subjected to IP with anti-HA-agarose. Coimmunoprecipitated RNAs were analyzed as in A. Recoveries of HAC1 RNA species in the bound fractions were shown in the bottom of the Northern images.
Then we devised a setup to examine whether ScRlg1p binds the HAC1 intron because the intron accumulates only in the AtRLG1 strains. When we constructed an rlg1Δ strain harboring both HA-AtRLG1[M74] and FLAG-ScRLG1 on plasmids, it showed intermediate Tm sensitivity (Figure 8A). Then, we immunoprecipitated FLAG-ScRlg1p from Tm-treated cell extracts of this strain and found that HAC1u mRNA, HAC1i mRNA, and HAC1 intron were coprecipitated as in the case of AtRlg1p, whereas no such RNA was detected in the immunoprecipitate from the control strain that did not express FLAG-ScRlg1p (Figure 8B, top). When we analyzed protein compositions of the immunoprecipitates, only negligible amounts of HA-AtRlg1p were found in anti-FLAG immunoprecipitates and vice versa (Figure 8B, bottom). These results indicate that ScRlg1p can bind the HAC1 intron produced by splicing reaction with HA-AtRlg1p, as well.
Figure 8.
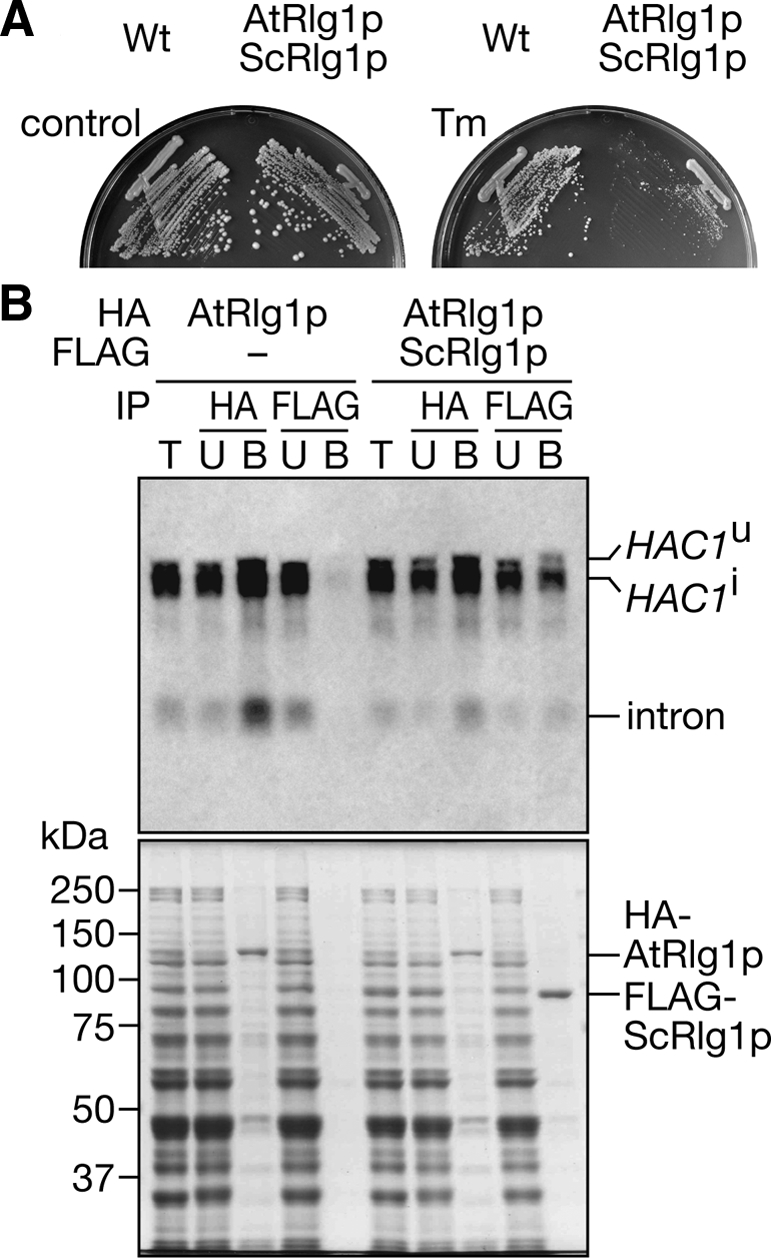
ScRlg1p also interacts with the HAC1 intron spliced by AtRlg1p. (A) HA-AtRlg1[M74]p and FLAG-ScRlg1p were coexpressed under the rlg1Δ background, and their ability to compliment UPR defects was tested on a 0.25 μg/ml Tm-containing plate (right). (B) Cell extracts were prepared from the HA-AtRLG1[M74] strain (left set) and the HA-AtRLG1[M74]/FLAG-ScRLG1 strain (right set), and tagged proteins were immunoprecipitated with immobilized antibody-resins indicated in the row IP. Top, Northern blotting with the anti-HAC1u probe. T, 1/10 of total sample; U, 1/10 of unbound fraction; B, 3/4 of bound fraction. Bottom, protein staining of the SDS-gel analyzing the same samples.
Next, we asked whether the interaction between Rlg1 proteins and HAC1 mRNAs is direct or through ribosomes. As shown above, EDTA treatment does not inhibit coimmunoprecipitation between Rlg1 proteins and HAC1 mRNAs. However, it may be possible that Rlg1 proteins have affinity to either ribosomes or translation factors whose affinity allows Rlg1 proteins to associate with HAC1 mRNPs existing in the absence of Mg2+. To test this possibility, we performed two experiments. First, immunoprecipitates of Rlg1 proteins purified in the presence of EDTA were fractionated by the two-step sucrose density gradient to see whether HAC1 mRNA is recovered in the nonribosomal fraction. As shown in Figure 9A, large parts of HAC1u and HAC1i mRNAs were sedimented through the 16% sucrose layer both in the Mg2+ and EDTA buffers. This suggests that HAC1 mRNAs are still associated with some large structure(s) even in the presence of 10 mM EDTA, which can disassemble polysomes. Second, we examined whether association of Rlg1 proteins with polysomes depends on HAC1 mRNAs. We prepared extracts from the HA-AtRLG1 and HA-ScRLG1 strains with the HAC1 or hac1Δ background and applied them to polysome analysis. Rlg1 proteins were mainly found in top fractions, as expected from their primary function as tRNA ligase. On the other hand, small but detectable portions of both Rlg1 proteins were recovered in polysomal fractions (Figure 9, B and D). The sedimentation patterns were not affected by deletion of the HAC1 gene from the chromosome (Figure 9, C and E). These results imply that portions of both AtRlg1p and ScRlg1p are recruited to polysomes not through the HAC1 mRNA but through direct interaction with ribosomal proteins or translation factors. Similar results were obtained when the wild-type yeast strain expressing authentic Rlg1p was analyzed (Figure 9, F and G). Indeed, similar Rlg1p distribution on the sucrose gradient was seen in HAC1 and hac1Δ strains. These and other results suggest that both ScRlg1p and AtRlg1p form the HAC1 mRNP in part through their affinity to the translational machinery.
Figure 9.

Rlg1 proteins interact with polysomes irrespective of existence of HAC1 mRNAs. (A) FLAG-AtRlg1p and FLAG-ScRlg1p were immunoprecipitated with anti-FLAG M2 agarose and eluted from the resin by incubating with a 3×FLAG peptide. The eluates were loaded onto a 16/50% step sucrose density gradient as in Figure 7B. (B–E) Extracts were prepared from HA-ScRLG1 HAC1 (B), HA-ScRLG1 hac1Δ (C), HA-AtRLG1[M74] HAC1 (D), and HA-AtRLG1[M74] hac1Δ (E) and sedimented through a 10–50% sucrose density gradient as in Figure 4. The fractions were subjected to Western blotting with an anti-HA antibody. In F and G, wild-type (W303-1A) and hac1Δ (TMSC09) strains with the authentic RLG1 on their chromosome were analyzed as above, and ScRlg1p in the fractions was detected with anti-Rlg1p antibodies. L, cell extract.
Finally, we examined whether ScRlg1p interacts with HAC1 mRNAs under more physiological expression conditions because ScRlg1p was overproduced in the HA-ScRLG1 strain (Figure S1). We also adopted cross-link and immunoprecipitation (CLIP) procedures where more extensive wash can be applied (Gilbert et al., 2004). This would help us to confirm the interaction between ScRlg1p and HAC1 mRNA by a method different from native IP where mild conditions are required to retain protein–RNA complexes. First, we constructed a yeast strain harboring an RLG1-protein A fusion gene that was integrated in the chromosomal RLG1 locus under the control of its own promoter. Then, we treated the yeast cells with formaldehyde, prepared lysates by glass-bead lysis, and performed IP with IgG-Sepharose. RNAs recovered in the immunoprecipitates were detected by RT-PCR. Under nonstress conditions, a PCR fragment corresponding to HAC1u mRNA was amplified from the immunoprecipitates of the RLG1-protein A strain, whereas essentially negligible amounts of the PCR fragment were detected when immunoprecipitates were prepared from wild-type or unrelated KAP95-TAP strains (Figure 10A, −Tm panel; quantification graph). The HAC1u fragment was not detected when extracts were pretreated with RNase A (lanes “RNase IP”). Amplification of HAC1u mRNA was specific because more abundant ACT1 mRNA was not amplified from the Rlg1-protein A immunoprecipitate (Figure 10B). This suggests that the ScRlg1p interaction with translational machinery as seen in Figure 9 has some specificity to translating mRNAs. When yeast cells were treated with Tm to induce the UPR, both HAC1u and HAC1i mRNAs were recovered in the immunoprecipitate (Figure 10A, +Tm panel). These results indicate that ScRlg1p interacts with the HAC1 mRNAs irrespective to the action of Ire1p. The results also support the idea that ScRlg1p acts on translational control of Hac1p after splicing as a component of HAC1 mRNP.
Figure 10.

Authentic ScRlg1p interacts with both HAC1u and HAC1i mRNAs. Logarithmically growing yeast cells with no (wt), KAP95-TAP (KAP95-TAP), or ScRLG1-protein A (ScRLG1-prot. A) gene integrated into the chromosome were treated with (+Tm) or without (−Tm) Tm for 30 min and then were analyzed by CLIP. After cross-linking with 1.0% formaldehyde for 30 min, extracts were prepared and subjected to IP with IgG-Sepharose. Immunoprecipitated HAC1 mRNAs (A) and ACT1 mRNA (B) were amplified by RT-PCR with primers represented as thick arrows in schematic drawings of the genes. T, total extract; FT, flow through; RNase IP, immunoprecipitate from the extract treated with RNase A; IP, immunoprecipitate. In A, recovery of HAC1 mRNAs (HAC1u in the absence of Tm and HAC1i in the presence of Tm) in the immunoprecipitates was quantified by semiquantitative RT-PCR with a microchip electrophoresis system. The values were obtained from three independent IPs.
DISCUSSION
In this study, we compared tRNA ligases originated from different phylogenetic positions on their functionality in the unfolded protein response in S. cerevisiae, where the unconventional cytoplasmic splicing and intramolecular base-pairing of HAC1 mRNA regulate the UPR. First, we have demonstrated that Rlg1 proteins from various organisms have robust substrate recognition for ligation in the cytoplasmic splicing of HAC1u mRNA. SpRlg1p and AtRlg1p do ligate HAC1 exons despite the fact that both organisms do not possess any apparent homologue of HAC1. This is in a great contrast to substrate specificity of splicing endonuclease Ire1p. The A. thaliana genome has two Ire1p homologues, whereas heterologous expression of a yeast HAC1 mini-gene in A. thaliana protoplasts failed to yield the spliced form of HAC1 mRNA (Noh et al., 2002). Similar results were obtained in HeLa cells (Bowring and Llewellyn, 2001). These facts ostensibly suggest that substrate selection relies on Ire1p and that Rlg1p just joins any pairs of RNAs with a 5′-OH group and a 2′-3′ cyclic phosphodiester bond in an appropriate distance. However, our data discussed below in detail indicate that Rlg1p recognizes certain RNA structures, and the property of Rlg1p on substrate recognition seems to be conserved.
Although both plant and fungal Rlg1 proteins can ligate yeast HAC1 exons produced by yeast Ire1p in vivo, only the fungal Rlg1 proteins can substitute for ScRlg1p in full activity of the UPR. Indeed, we found that AtRlg1p has some problem on releasing translational attenuation of the HAC1i mRNA conferred by base-pairing between the 5′ UTR and intron of the HAC1 mRNA and that the HAC1 intron remains associated with the HAC1i mRNA on polysomes. This phenotype of the AtRLG1-expressing cells is in great contrast to that of rlg1-100 cells. In rlg-100 cells, in which the UPR is also impaired, the first endonucleolytic step of the cytoplasmic splicing seems to be normal, but no HAC1i mRNA is detected (Sidrauski et al., 1996; see also Figure 3), indicating that the mutant suffers from a HAC1 exon-specific ligation defect. On the other hand, AtRLG1 is a kind of “mutant RLG1” that can ligate the HAC1 exons but lacks functions for translational restart of the HAC1i mRNA. This is partly due to accumulation of the circularized HAC1 intron. AtRlg1p has ability to circularize an intron cleaved from a pre-tRNA in vitro. Similar intramolecular ligation by tRNA ligase was reported in other organisms from archaea to mammal (Perkins et al., 1985; Salgia et al., 2003), and yeast Rlg1p might have evolved to fit requirement for the fungal UPR. Because well-known degradation systems for cytoplasmic mRNAs rely on cytoplasmic exosome and Xrn1p, both of which are exonucleases, the circular intron may well be a poor substrate for cytoplasmic RNA degradation systems. AtRlg1p may have high affinity to the circular HAC1 intron, so that it remains on the polysomes even after completion of the HAC1i translation. A similar HAC1 intron-ScRlg1p complex was detected when AtRlg1p and ScRlg1p were coexpressed, indicating that the circular HAC1 intron is released from AtRlg1p and reassociated with ScRlg1p, that is, the circular intron dynamically interacts with the Rlg1 proteins and the translational machinery.
Accumulation of the circular intron may not be the only reason why AtRLG1 does not complement UPR defects upon deletion of the chromosomal ScRLG1 gene. First, we found that the circular intron produced by AtRlg1p does not inhibit translation in trans, whereas it can dynamically interact with Rlg1 proteins and translational machinery (see below). Existence of the complex of the HAC1 intron and AtRlg1p is not sufficient to suppress translation of mRNA with the HAC1 5′ UTR. Furthermore, the observation that the GFP reporter mRNA only with the HAC1 5′ UTR is translated less efficiently in the AtRLG1 cells than in the ScRLG1 cells indicates that ScRlg1p has ability to regulate translation of HAC1 mRNA through its action on its 5′ UTR. We noticed that HAC1 deletion in the ScRLG1 cells leads to enhanced translation of the HAC1 5′ UTR-GFP reporter construct (Figure 6B). It is possible that the hac1Δ cells suffer from some perturbation of ER homeostasis to activate other ER-stress–signaling pathway(s) independent of HAC1 (Schröder et al., 2003; Chen et al., 2005). This may indicate a possible signal flow to the 5′ UTR of HAC1 through ScRlg1p. It has been believed that splicing is required and sufficient for release of the HAC1 intron from the 5′ UTR of the HAC1i mRNA to allow Hac1ip translation. However, comparison between ScRlg1p and AtRlg1p now enables us to dissect the splicing and translational restart steps in vivo and reveals that Rlg1p is the factor required for both of the steps.
Involvement of Rlg1p in the translational regulation of Hac1p is further strengthened by the observation that Rlg1p is associated with various HAC1 RNA species that are not bona fide substrates of the ligation reaction and that this association is in part through interaction between Rlg1 proteins and the translational machinery. We first demonstrated interaction between HAC1 RNAs and AtRlg1p by native IP. The specific interactions of ScRlg1p with both HAC1u and HAC1i mRNAs were also observed under the wild-type expression level of Rlg1p by CLIP experiments. These results indicate that 1) Rlg1p recognizes the HAC1u mRNA before recognition by Ire1p, probably during formation of HAC1u mRNP under nonstress conditions, and 2) Rlg1p remains associated with HAC1i mRNA after splicing like in the case of the EJC complex in spliceosomal splicing (Le Hir et al., 2000). Therefore, ScRlg1p is supposed to act not only as a splicing enzyme but also as a marker of completion of splicing and as a translational regulator of the HAC1 mRNA. An attractive model is that ScRlg1p resolves base-pairing between the 5′ UTR and intron of HAC1 and activates its translation after the ligation upon the UPR by itself or by recruiting some RNA remodeling factor(s). AtRlg1p may lack such remodeling or recruiting activities in S. cerevisiae because of the phylogenetic distance between these two organisms. Alternatively, plant cells may not rely on translational regulation of a putative HAC1 counterpart upon the UPR. Indeed, animal cells translate the unspliced XBP1 mRNA, whose product has vital roles on the UPR (Yoshida et al., 2006; Yanagitani et al., 2009). In any cases, substrate recognition of Rlg1p in the UPR is conserved among species, and the plant Ire1p pathway may diverge from that of yeast after this step.
In conclusion, we found that yeast tRNA ligase possesses an unexpected role as a translational regulator in the UPR. Still, we do not know how Rlg1p accomplishes translational restart of HAC1 mRNA after its splicing in detail. When and how is Rlg1p loaded onto HAC1u mRNA? What kinds of structural features of RNA substrates for this unconventional cytoplasmic splicing are recognized by various Rlg1 proteins? Are there any intrinsic signals coming in ScRlg1p to accelerate Hac1p translation and how is this related to the action of an as-yet unidentified trans factor that recognizes the HAC1 5′ UTR? Although our findings provide a clue to understand the precise mechanism of translational regulation coupled with the unconventional splicing during the UPR, at the same time, they raise these new questions for future works.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Kazutoshi Mori and Hiderou Yoshida (Kyoto University) for providing us anti-Hac1p affinity-purified rabbit antibodies and for their critical discussion. We are also most grateful to Dr. Kazuhito Akama (Shimane University) for providing important information about plant splicing enzymes. We appreciate help and suggestions from the present and previous members of our laboratory, especially Drs. Shuh-ichi Nishikawa, Tadashi Makio, Kunio Nakatsukasa, and Eiichi Matsuo for fruitful discussion. This work was supported by Grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by PRESTO from Japan Science and Technology Agency.
Abbreviations used:
- ER
endoplasmic reticulum
- HA
hemagglutinin
- HAC1i
HAC1 induced
- HAC1u
HAC1 uninduced
- mRNP
messenger ribonucleoprotein
- RNP
ribonucleoprotein
- Tm
tunicamycin
- UPR
unfolded protein response.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-08-0693) on September 15, 2010.
REFERENCES
- Aragón T., van Anken E., Pincus D., Serafimova I. M., Korennykh A. V., Rubio C. A., Walter P. Messenger RNA targeting to endoplasmic reticulum stress signaling sites. Nature. 2009;457:736–740. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A., Zhang Y., Hendershot L. M., Harding H. P., Ron D. Dynamic interaction of BiP and the ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Bowring C. E., Llewellyn D. H. Differences in HAC1 mRNA processing and translation between yeast and mammalian cells indicate divergence of the eukaryotic ER stress response. Biochem. Biophys. Res. Commun. 2001;287:789–800. doi: 10.1006/bbrc.2001.5633. [DOI] [PubMed] [Google Scholar]
- Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G., Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Chen Y., Feldman D. E., Deng C., Brown J. A., De Giacomo A. F., Gaw A. F., Shi G., Le Q. T., Brown J. M., Koong A. Identification of mitogen-activated protein kinase signaling pathways that confer resistance to endoplasmic reticulum stress in Saccharomyces cerevisiae. Mol. Cancer Res. 2005;3:669–677. doi: 10.1158/1541-7786.MCR-05-0181. [DOI] [PubMed] [Google Scholar]
- Cox J. S., Shamu C. E., Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–1206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- Cox J. S., Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87:391–404. doi: 10.1016/s0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- Credle J. J., Finer-Moore J. S., Papa F. R., Stroud R. M., Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 2005;102:18773–18784. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver G. M., McCraith S. M., Consaul S. A., Stanford D. R., Phizicky E. M. A 2′-phosphotransferase implicated in tRNA splicing is essential in Saccharomyces cerevisiae. J. Biol. Chem. 1997;272:13203–13210. doi: 10.1074/jbc.272.20.13203. [DOI] [PubMed] [Google Scholar]
- Englert M., Beier H. Plant tRNA ligases are multifunctional enzymes that have diverged in sequence and substrate specificity from RNA ligases of other phylogenetic origins. Nucleic Acids Res. 2005;33:388–399. doi: 10.1093/nar/gki174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englert M., Latz A., Becker D., Gimple O., Beier H, Akama K. Plant pre-tRNA splicing enzymes are targeted to multiple cellular compartments. Biochimie. 2007;89:1351–1365. doi: 10.1016/j.biochi.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Filipowicz W., Shatkin A. J. Origin of splice junction phosphate in tRNAs processed by HeLa cell extract. Cell. 1983;32:547–557. doi: 10.1016/0092-8674(83)90474-9. [DOI] [PubMed] [Google Scholar]
- Gilbert C., Kristjuhan A., Winkler G. S., Svejstrup J. Q. Elongator interactions with nascent mRNA revealed by RNA immunoprecipitation. Mol. Cell. 2004;14:457–464. doi: 10.1016/s1097-2765(04)00239-4. [DOI] [PubMed] [Google Scholar]
- Gonzalez T. N., Sidrauski C., Dörfler S., Walter P. Mechanism of non-spliceosomal mRNA splicing in the unfolded protein response pathway. EMBO J. 1999;18:3119–3132. doi: 10.1093/emboj/18.11.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C., Fink G. R. Methods Enzymology. Vol. 194. San Diego: Academic Press; 1991. Guide to yeast genetics and molecular biology. [PubMed] [Google Scholar]
- Harding H., Zhang Y., Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding H. P., Lackey J. G., Hsu H.-C., Zhang Y., Deng J., Xu R.-M., Damha M. J., Ron D. An intact unfolded protein response in Trpt1 knockout mice reveals phylogenic divergence in pathways for RNA ligation. RNA. 2008;14:225–232. doi: 10.1261/rna.859908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haze K., Yoshida H., Yanagi H., Yura T., Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper A. K., Phizicky E. M. tRNA transfers to the limelight. Genes Dev. 2003;17:162–180. doi: 10.1101/gad.1049103. [DOI] [PubMed] [Google Scholar]
- Inada T., Winstall E., Tarun S. Z., Jr, Yates J. R., III, Schieltz D., Sachs A. B. One-step affinity purification of the yeast ribosome and its associated proteins and mRNAs. RNA. 2002;8:948–958. doi: 10.1017/s1355838202026018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara T., Yanagi H., Yura T., Mori K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol. Biol. Cell. 1997;8:1845–1862. doi: 10.1091/mbc.8.10.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y., Ishiwata-Kimata Y., Yamada S., Kohno K. Yeast unfolded protein response pathway regulates expression of genes for anti-oxidative stress and for cell surface proteins. Genes Cells. 2006;11:59–69. doi: 10.1111/j.1365-2443.2005.00921.x. [DOI] [PubMed] [Google Scholar]
- Kuhn K. M., DeRisi J. L., Brown P. O., Sarnow P. Global and specific translational regulation in the genomic response of Saccharomyces cerevisiae to a rapid transfer from a fermentable to a nonfermentable carbon source. Mol. Cell. Biol. 2001;21:916–927. doi: 10.1128/MCB.21.3.916-927.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.P.K., Dey M., Neculai D., Cao C., Dever T. E., Sicheri F. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell. 2008;132:89–100. doi: 10.1016/j.cell.2007.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H., Moore M. J., Maquat L. E. Pre-mRNA splicing alters mRNP composition: evidence for stable association of proteins at exon-exon junctions. Genes Dev. 2000;14:1098–1108. [PMC free article] [PubMed] [Google Scholar]
- Mori K., Ma W., Gething M.-J., Sambrook J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell. 1993;74:743–756. doi: 10.1016/0092-8674(93)90521-q. [DOI] [PubMed] [Google Scholar]
- Mori K., Kawahara T., Yoshida H., Yanagi H., Yura T. Signaling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells. 1996;1:803–817. doi: 10.1046/j.1365-2443.1996.d01-274.x. [DOI] [PubMed] [Google Scholar]
- Murray E. L., Schoenberg D. R. Assays for determining poly(A) tail length and the polarity of mRNA decay in mammalian cells. Methods Enzymol. 2008;448:483–504. doi: 10.1016/S0076-6879(08)02624-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh S.-J., Kwon C. S., Chung W.-I. Characterization of two homologs of Ire1p, a kinase/endoribonuclease in yeast, in Arabidopsis thaliana. Biochim. Biophys. Acta. 2002;1575:130–134. doi: 10.1016/s0167-4781(02)00237-3. [DOI] [PubMed] [Google Scholar]
- Oikawa D., Kimata Y., Kohno K. Self-association and BiP dissociation are not sufficient for activation of the ER stress sensor Ire1. J. Cell Sci. 2007;120:1681–1688. doi: 10.1242/jcs.002808. [DOI] [PubMed] [Google Scholar]
- Papa F. R., Zhang C., Shokat K., Walter P. Bypassing a kinase activity with an ATP-competitive drug. Science. 2003;302:1533–1537. doi: 10.1126/science.1090031. [DOI] [PubMed] [Google Scholar]
- Perkins K. K., Furneaux H., Hurwitz J. Isolation and characterization of an RNA ligase from HeLa cells. Proc. Natl. Acad. Sci. USA. 1985;82:684–688. doi: 10.1073/pnas.82.3.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky E. M., Schwartz R. C., Abelson J. Saccharomyces cerevisiae tRNA ligase. Purification of the protein and isolation of the structural gene. J. Biol. Chem. 1986;261:2978–2986. [PubMed] [Google Scholar]
- Phizicky E. M., Consaul S. A., Nehrke K. W., Abelson J. Yeast tRNA ligase mutants are nonviable and accumulate tRNA splicing intermediates. J. Biol. Chem. 1992;267:4577–4582. [PubMed] [Google Scholar]
- Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rüegsegger U., Leber J. H., Walter P. Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell. 2001;107:103–114. doi: 10.1016/s0092-8674(01)00505-0. [DOI] [PubMed] [Google Scholar]
- Salgia S. R., Singh K, S., Gurha P., Gupta R. Two reactions of Haloferax volcanii RNA splicing enzymes: joining of exons and circularization of introns. RNA. 2003;9:319–330. doi: 10.1261/rna.2118203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saloheimo M., Valkonen M., Penttilä M. Activation mechanisms of the HAC1-mediated unfolded protein response in filamentous fungi. Mol. Microbiol. 2003;47:1149–1161. doi: 10.1046/j.1365-2958.2003.03363.x. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Russell D. W., editors. Molecular Cloning, A Laboratory Manual. 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Scheuner D., Song B., McEwen E., Liu C., Laybutt R., Gillespie P., Saunders T., Bonner-Weir S., Kaufman R. J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Schröder M., Clark R., Kaufman R. J. IRE1- and HAC1-independent transcriptional regulation in the unfolded protein response of yeast. Mol. Microbiol. 2003;49:591–606. doi: 10.1046/j.1365-2958.2003.03585.x. [DOI] [PubMed] [Google Scholar]
- Schröder M. Endoplasmic reticulum stress responses. Cell Mol. Life Sci. 2007;65:862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B., Sawaya R., Ho C. K., Shuman S. Portability and fidelity of RNA-repair systems. Proc. Natl. Acad. Sci. USA. 2004;101:2788–2793. doi: 10.1073/pnas.0305859101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamu C., E., Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996;15:3028–3039. [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C., Cox J. S., Walter P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell. 1996;87:405–413. doi: 10.1016/s0092-8674(00)81361-6. [DOI] [PubMed] [Google Scholar]
- Tanner N. K., Hanna M. M., Abelson J. Binding interactions between yeast tRNA ligase and a precursor transfer ribonucleic acid containing two photoreactive uridine analogues. Biochemistry. 1988;27:8852–8861. doi: 10.1021/bi00424a025. [DOI] [PubMed] [Google Scholar]
- Travers K. J., Patil C. K., Wodicka L., Lockhart D. J., Weissman J. S., Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Wang L. K., Schwer B., Englert M., Beier H., Shuman S. Structure-function analysis of the kinase-CPD domain of yeast tRNA ligase (Trl1) and requirements for complementation of tRNA splicing by a plant Trl1 homolog. Nucleic Acids Res. 2006;34:517–527. doi: 10.1093/nar/gkj441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzer S., Martinez J. The human RNA kinase hClp1 is active on 3′ transfer RNA exons and short interfering RNAs. Nature. 2007;447:222–226. doi: 10.1038/nature05777. [DOI] [PubMed] [Google Scholar]
- Yanagitani K., Imagawa Y., Iwawaki T., Hosoda A., Saito M., Kimata Y., Kohno K. Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol. Cell. 2009;34:191–200. doi: 10.1016/j.molcel.2009.02.033. [DOI] [PubMed] [Google Scholar]
- Ye J., Rawson R. B., Komuro R., Chen X., Dave U. P., Prywes R., Brown M. S., Goldstein J. L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Okada T., Haze K., Yanagi H., Yura T., Negishi M., Mori K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000;20:6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Oku M., Suzuki M., Mori K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell Biol. 2006;172:565–575. doi: 10.1083/jcb.200508145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihisa T., Yunoki-Esaki K., Ohshima C., Tanaka N., Endo T. Possibility of cytoplasmic pre-tRNA splicing: the yeast tRNA splicing endonuclease mainly localizes on the mitochondria. Mol. Biol. Cell. 2003;14:3266–3279. doi: 10.1091/mbc.E02-11-0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihisa T., Ohshima C., Yunoki-Esaki K., Endo T. Cytoplasmic splicing of tRNA in Saccharomyces cerevisiae. Genes Cells. 2007;12:285–297. doi: 10.1111/j.1365-2443.2007.01056.x. [DOI] [PubMed] [Google Scholar]
- Zhou J., Liu C. Y., Back S. H., Clark R. L., Peisach D., Xu Z., Kaufman R. J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA. 2006;103:14343–14348. doi: 10.1073/pnas.0606480103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zillmann M., Gorovsky M. A., Phizicky E. M. Conserved mechanism of tRNA splicing in eukaryotes. Mol. Cell. Biol. 1991;11:5410–5416. doi: 10.1128/mcb.11.11.5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.