

We present a genomewide, high-resolution study of nucleolar-associated chromatin using comparative genome hybridization, deep sequencing, and photoactivation microscopy. We show specific regions from most chromosomes associate with nucleoli. These regions are AT-rich, enriched in repressed genes, and overlap with nuclear lamina-associated loci.
Abstract
The nuclear space is mostly occupied by chromosome territories and nuclear bodies. Although this organization of chromosomes affects gene function, relatively little is known about the role of nuclear bodies in the organization of chromosomal regions. The nucleolus is the best-studied subnuclear structure and forms around the rRNA repeat gene clusters on the acrocentric chromosomes. In addition to rDNA, other chromatin sequences also surround the nucleolar surface and may even loop into the nucleolus. These additional nucleolar-associated domains (NADs) have not been well characterized. We present here a whole-genome, high-resolution analysis of chromatin endogenously associated with nucleoli. We have used a combination of three complementary approaches, namely fluorescence comparative genome hybridization, high-throughput deep DNA sequencing and photoactivation combined with time-lapse fluorescence microscopy. The data show that specific sequences from most human chromosomes, in addition to the rDNA repeat units, associate with nucleoli in a reproducible and heritable manner. NADs have in common a high density of AT-rich sequence elements, low gene density and a statistically significant enrichment in transcriptionally repressed genes. Unexpectedly, both the direct DNA sequencing and fluorescence photoactivation data show that certain chromatin loci can specifically associate with either the nucleolus, or the nuclear envelope.
INTRODUCTION
Chromatin occupies a major part of the nuclear space and requires a high level of organization. It is now evident that the higher level organization of the nucleus affects gene function (Cremer et al., 2006; Fraser and Bickmore, 2007; Meaburn et al., 2007; de Wit and van Steensel, 2009; Nunez et al., 2009). Chromosomes are positioned in preferred locations within the nucleus, so-called chromosome territories (CTs; Cremer and Cremer, 2001), which seem to correlate with gene density, where gene-poor chromosomes tend to localize more at the nuclear periphery and gene-rich chromosomes more in the nuclear interior (Croft et al., 1999), and/or with chromosome size (Sun and Yokota, 1999). Despite their preferred position in CTs, chromosomes do intermingle (Branco and Pombo, 2006), and chromosome regions can loop out to interact with other loci both in cis and in trans (Chuang and Belmont, 2007; Misteli, 2007; de Laat et al., 2008; Gondor and Ohlsson, 2009), or to bring them in close proximity to nuclear bodies.
Indeed, specific genomic regions can associate with particular nuclear bodies or compartments, e.g., U1 and U2 snRNA genes, the U3 snoRNA gene, and histone genes associate with Cajal bodies (Gall et al., 1981; Frey and Matera, 1995; Smith et al., 1995; Gao et al., 1997; Frey and Matera, 2001), transcriptionally active genomic regions associate with promyelocytic leukemia (PML) bodies (Wang et al., 2004), heat-shock granules form around regions of chromosomes 9,12, and 15 (Denegri et al., 2002), Oct1 PTF transcription (OPT) domains form around loci on chromosomes 6 and 7 (Pombo et al., 1998), paraspeckles are often located next to the NEAT1 locus on chromosome 11 (Bond and Fox, 2009; Clemson et al., 2009), and the perinucleolar compartment is reported to be directly associated with a DNA locus that still has to be identified (Norton et al., 2009). However, the first and most extensively studied example is the tandemly repeated ribosomal DNA (rDNA) genes called nucleolar organizer regions (NORs), which initiate formation of the nucleolus. In humans, these NORs are present on the acrocentric (i.e., with short p-arm) chromosomes 13, 14, 15, 21, and 22 (reviewed in McStay and Grummt, 2008).
The nucleolus is highly dynamic and in mammals it disassembles during M-phase and reassembles during telophase (Leung et al., 2004; Angelier et al., 2005; McStay and Grummt, 2008). In addition to the main function of the nucleolus in ribosome subunit biogenesis, recently additional functions have been described, such as a role in cell cycle progression and the stress response (Pederson, 1998; Rubbi and Milner, 2003; Raska et al., 2006; Boisvert et al., 2007; Sirri et al., 2008; Pederson and Tsai, 2009).
Nucleoli are surrounded by a shell of chromatin, which mainly consists of late-replicating highly condensed heterochromatic DNA (Ferreira et al., 1997; Sadoni et al., 1999). This chromatin can affect the organization of both rDNA repeat units and the nucleolus itself (Espada et al., 2007; Peng and Karpen, 2007). However, the nature of these nucleolar-associated heterochromatic sequences is mostly unknown, and it has not been addressed whether the same or random sequences associate with nucleoli from one cell cycle to the next.
Previous studies identified specific chromosomal regions, including centromeres, that are positioned near the nucleolus in different cell types and species (Manuelidis, 1984; Manuelidis and Borden, 1988; Haaf and Schmid, 1989, 1991; Billia and Deboni, 1991; Ochs and Press, 1992; Leger et al., 1994; Carvalho et al., 2001). In addition, the entire human Y chromosome and the human chromosomal 1q42-44 region encoding for the 5S DNA are reported to associate with nucleoli, as well as some telomeres in root tips of Pisum sativum and Vicia faba (Comings, 1980; Rawlins and Shaw, 1990). Moreover, the inactive X chromosome (Xi) can associate with nucleoli in S phase to maintain its repressive chromatin state (Zhang et al., 2007). Finally, tRNA genes are known to cluster at nucleoli in Saccharomyces cerevisiae (Thompson et al., 2003).
We present here a genome-wide, high-resolution analysis of both unique and repetitive sequences that have a high probability to associate endogenously and copurify with nucleoli (nucleolar-associated domains [NADs]) in human cells, using a combination of fluorescence comparative genome hybridization (CGH), Illumina deep DNA sequencing (http://www.illumina.com/) and fluorescence photoactivation.
MATERIALS AND METHODS
Cell Culture
Early passage HT1080 cells (ATCC, Manassas, VA; p5) were cultured in Dulbecco's modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin-streptomycin in a humidified incubator at 37°C with 5% CO2. A HeLa cell line stably expressing photoactivatable green fluorescent protein (PA-GFP)-H2B was established and grown in DMEM supplemented with 10% FBS, 100 U/ml penicillin-streptomycin, and 200 μg/ml G418, as previously described (Trinkle-Mulcahy et al., 2003).
Immunocytochemistry and Antibodies
For immunolabeling, cells were grown on glass coverslips and fixed for 10 min in 3.7% paraformaldehyde. After a 10-min permeabilization with 1% Triton X-100 in phosphate-buffered saline (PBS), cells were blocked with 1% goat serum + 0.1% Tween-20 for 15 min and then incubated with primary antibodies for 1 h, washed, and incubated with secondary antibodies for 1 h. If required, cells were stained with DAPI (0.3 μg/ml; Sigma, St. Louis, MO). After a final set of washes, cells were mounted in Vectashield media (Vector Laboratories, Burlingame, CA).
The primary antibody rabbit anti-nucleolin (Abcam, Cambridge, MA) and secondary goat anti-rabbit-Cy5 (Jackson ImmunoResearch, West Grove, PA) were diluted 1:300 and 1:100, respectively.
Fluorescence In Situ Hybridization
Fluorescence in situ hybridization (FISH) on Interphase Cells.
Cells were cultured on microscope slides, permeabilized in 0.5% Triton X-100 in CSK buffer (10 mM PIPES, pH 6.8, 10 mM NaCl, 300 mM sucrose, 3 mM MgCl2, and 2 mM EDTA) for 10 min at room temperature (RT), and fixed in methanol:acetic acid (3:1) for 30 min at RT. Slides were incubated in 70% ethanol overnight at 4°C, dehydrated, air-dried, and then prewarmed for 5 min at 37°C. FISH on metaphase spreads: human male lymphocyte metaphase spreads were obtained from Abbott Molecular (Maidenhead, United Kingdom). Slides were air-dried and prewarmed for 5 min at 37°C.
FISH Probes.
BAC clones RP11-42M14 (chromosome 17q22) and RP11-266I24 (chromosome 17q21) were obtained from BACPAC (Oakland, CA). One microgram of BAC clone DNA and total HT1080 genomic and HT1080 nucleolar DNA were labeled using a nick translation kit (GE Healthcare Life Sciences, Piscataway, NJ), according to the manufacturer's instructions. Fluorescent dCTPcy3, dCTPcy5 (GE Healthcare Life Sciences) and dUTP-green (all at 1 mM; Abbott Molecular) were used for probe labeling. Each labeled probe, 150 ng, was precipitated in ethanol and 0.3 M sodium acetate together with 10 μg COT1 carrier DNA (Invitrogen) and dissolved in 10 μl hybridization buffer (Hybrisol, Abbott Molecular). Directly labeled fluorescent telomeric probes were obtained from Cytocell (Cambridge, United Kingdom). Probes were prewarmed for 5 min at 37°C.
Hybridization and Washing.
Probes were added to prewarmed slides, after which the slides were sealed with a coverslip and rubber cement and denatured for 2 min at 75°C. Slides were hybridized in a wet chamber overnight at 37°C. After removing the coverslips, the slides were washed in 0.4× SSC for 2 min at 72°C, followed by 2× SSC/0.05% Tween-20 for 1 min at RT. Subsequent immunostaining was performed as described.
Time-Lapse Imaging and Microscopy
Imaging was done on a DeltaVision Core widefield microscope (Applied Precision, Issaquah, WA) mounted on an Olympus IX70 inverted stand (Tokyo, Japan). An Olympus 60×, 1.4 NA plan apo oil immersion lens was used (Olympus). Images were collected on a Coolsnap HQ camera (Roper Scientific, Tucson, AZ) using a 2 × 2 bin. Image acquisition and analysis was done using SoftWorx software (Applied Precision), and image restoration was done with a proprietary iteratively constrained deconvolution algorithm (Applied Precision).
HeLa cells stably expressing PA-GFP-H2B were transiently transfected with mCherry-B23 to mark the nucleoli. Cells were incubated with Hoechst 33342 (25 ng/ml, Sigma) for 20 min at 37°C, washed with PBS, and subsequently imaged in CO2-independent DMEM (Invitrogen). Early prophase cells expressing mCherry-B23 were selected based on their condensed DNA pattern.
Photoactivation was done using a Quantifiable Laser Module (Applied Precision), which focuses a diffraction limited laser spot into the center of the field of view. A region was photoactivated by placing the region of interest (ROI) in the cell into the center of the field of view, in this case either a region overlapping a nucleolus or separate from nucleoli. PA-GFP was then activated using a 20-mW 406-nm continuous wave blue diode laser at 10% power (Point Source iFlex 2000; Point Source, Southampton, United Kingdom). z-stacks at 12 μm were subsequently imaged at 1.2-μm spacing every 20 min for 20–23 h. Excitation light was delivered using a 300-W Xenon lamp with a 360/40-nm (for Hoechst, but 360/40 nm light is only used to select early prophase cells and not for time-lapse imaging), 555/28-nm (for mCherry), and 490/20-nm (for activated GFP) bandpass excitation filter, and a Sedat QUAD dichroic mirror and an 0% neutral density filter (Chroma Technology, Brattleboro, VT).
Cell Fractionation and DNA Isolation
Nucleoli were isolated as previously described (Andersen et al., 2005), but using a 0.35 M sucrose buffer supplemented with 2.5 mM MgCl2. The final nucleolar pellet was resuspended in 0.35 M sucrose with 0.5 mM MgCl2. DNA was isolated from both purified nucleoli and total HT1080 cells (n = 12 and one 14-cm confluent culture dishes, respectively), using the DNeasy blood and tissue kit (Qiagen, Chatsworth, CA) according to the manufacturer's instructions.
The enrichment for rDNA in the nucleolar fraction was confirmed on Southern blot. Nucleolar and total genomic DNA, 100 ng, were digested with HindIII and subsequently loaded onto a 0.8% TAE agarose gel. After blotting, the membrane was hybridized with a 11.9-kbp EcoRI rDNA probe (kind gift from Dr. B. McStay, Galway, Ireland), as described (Lemmers et al., 2007). Isolated DNA was sheared using a Biorupter (Diagenode, Liège, Belgium) for 15 min (30 s on/off) to obtain fragments of approximately 200 bp and subsequently were used for FISH procedures and deep sequencing.
Image Analysis
Image analysis was done using ImageJ (http://rsb.info.nih.gov/ij/) software to quantify the amount of the green PA-GFP-H2B signal colocalized to the red B23-staining signal in the daughter cells. For this purpose, the regions of the high PA-GFP-H2B signal were identified by image threshold segmentation on green intensity. The intensity of red signal was then measured for these regions. The values for the intensities of the red and green signals were normalized by the mean red and green signal intensities, respectively, for the whole image. Then the ratio of red to green signal was calculated. A Mann-Whitney test was used to compare the distributions of intensity ratios in daughter cells of cells where the PA-GFP-H2B was activated in a region overlapping with high red signal and daughter cells where PA-GFP-H2B was activated separate from regions of high red signal.
ImageJ was also used to analyze the FISH data. An in-house developed plug-in automatically detects the cell nuclei in the image from the 2D DAPI channel and automatically segments the image into subimages for each nucleus. The subimages were thresholded to detect nucleoli and probe locations. The program subsequently detects the periphery of the nucleoli and the FISH signals and measures the shortest distance between the boundaries of each FISH spot to the nearest nucleolar edge.
Illumina Deep Sequencing
The sample preparation was performed using the Illumina DNA sample preparation kit, according to the manufacturer's instruction (New England Biolabs). The samples were blunt-ended with T4 DNA polymerase, Klenow polymerase, and T4 polynucleotide kinase. A dA (deoxyadenosine) residue was added to the 3′ end of each strand with Klenow (exo-) polymerase. Subsequently, adapters designed for library construction were ligated to the PCR fragments, ligation products were gel-purified to select for ∼180–300-bp fragments, and PCR amplification was performed to enrich for the ligated fragments. The amplified library was quantified by a lab-on-a-chip bioanalyzer (Agilent Technologies, Amstelveen, The Netherlands), and cluster generation was performed after applying a 6-μl volume of a 4 pM solution of each sample to the individual lanes of the Illumina 1G flowcell. After hybridization of the sequencing primer to the single-stranded products, 36 cycles of base incorporation were carried out on the Illumina Genome Analyzer according to the manufacturer's instructions. Image analysis and base-calling were performed using the Illumina Pipeline 1.3.2, where sequence tags were obtained after purity filtering.
Paired end sequences were generated by The Gene Pool (Edinburgh, United Kingdom) according to their standard procedures (http://genepool.bio.ed.ac.uk/illumina/index.html).
Analysis of Unique DNA Sequence Data
Short-read data were obtained from nucleolar-associated and genomic DNA in two replicates with both lanes of each replicate (one nucleolar, one genomic) being sequenced on the same slide. Results from replicate 2 are consistent with those from replicate 1; in particular, prominent peaks in the nucleolar-genomic ratio are found at the same loci. A third, independent biological replicate, which will be reported in detail separately, was obtained during the course of submission. This additional replicate shows very consistent results to replicates 1 and 2 (see Results). Summaries for these four datasets are shown in Table 1. The figures in this study focus on data from replicate 1 and are consistent with the results from all datasets.
Table 1.
Statistical properties of the short read sequence data
| Replicate no. | Set origina | Total no. of reads | No. of clipped and filtered readsb | Mean length of a read (bp) | Genome coverage (%) | No. of hits matching human genome | No. of unique hits | Unique hits in filtered sample (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | Gen | 13,825,368 | 4,708,244 | 25.9 | 4.26 | 4,625,587 | 3,219,863 | 69.6 |
| 1 | Nuc | 12,075,449 | 5,286,354 | 28.0 | 5.18 | 5,006,543 | 3,526,616 | 70.4 |
| 2 | Gen | 16,268,206 | 1,699,835 | 22.2 | 1.32 | 1,667,205 | 942,805 | 56.6 |
| 2 | Nuc | 16,595,964 | 1,560,377 | 22.5 | 1.23 | 1,487,844 | 648,863 | 43.5 |
| 3 | Gen | 6,496,895 | 6,496,895 | 50 | 11.6 | 5,427,359 | 5,194,347 | 95.7 |
| 3 | Nuc | 7,539,495 | 7,539,495 | 50 | 13.5 | 5,515,177 | 5,063,940 | 91.8 |
a Gen, genomic; Nuc, nucleolar.
b No clipping was done for the third replicate.
Short read sequences were quality clipped to remove erroneous base calls by truncating each sequence when the average quality score in a three-base pair moving window dropped below a threshold level, in this case 20. All clipped reads shorter than 16 base pairs (bp) and poly-A sequences were rejected.
Quality clipped sequences were matched against the human genome (assembly build GRCh37) using Bowtie alignment program version 0.12.3 (http://bowtie-bio.sourceforge.net/index.shtml; Langmead et al., 2009), allowing for up to two mismatches. All nonunique hits were kept separate at this stage. Unique sequence hits were counted in 250-kbp bins and normalized to the mean count rate integrated over all chromosomes. This corrects for the different overall quantity of unique matches observed in the genomic and nucleolar experiments.
Sequence Property Calculations.
The same bins were used for calculating the AT fraction, the gene density, and gene expression levels. Gene density was determined by querying the Ensembl database for genes annotated to that bin (http://www.ensembl.org/). Gene expression per bin was determined by obtaining the only three datasets for HT-1080 cells in the ArrayExpress public expression data repository (http://www.ebi.ac.uk/arrayexpress/; Parkinson et al., 2009; Accession no. E-MTAB-37, Experiment ID no. 1691329576). The expression data contain results from 54,675 probes, of which we have identified 27,587 individual genes, where their unique Ensembl identifiers were available. In some cases several probes were linked to the same gene, in which case the strongest signal was selected, consistent with analysis of peak data. The processed data set comprises three individual replicates per gene (three expression signal values), for which the mean signal has been calculated. Chromosomal positions of the genes were obtained via Ensembl, and the signals were binned to form chromosomal histograms.
Normalization of Nucleolar to Genomic Ratios.
The ratio of the nucleolar sequence read counts to those for genomic (nucleolar-genomic) was determined for each bin. The nucleolar-genomic ratios were then normalized to establish an appropriate background level. For this, chromosomal regions with no obvious nucleolar signal were selected as background and the nucleolar-genomic ratios were divided by the mean ratio in these regions. The background regions were found by selecting the bottom 10% of bins with lowest nucleolar-genomic ratio in each chromosome separately. This way, the nucleolar-genomic peaks in all chromosomes are presented with respect to the constant background level of 1, as marked with dashed lines in Figures 2B, 7, and 8. It should be noted that the results of the statistical analysis carried out below do not depend on the background level selection.
Figure 2.

DNA deep sequencing analysis of nucleolar-associated chromatin.(A) Schematic showing of comparative sequencing strategy used for Illumina analysis of nucleolar-associated (red) and total genomic (red plus green) DNA. (B) Graphs show the nucleolar-to-genomic ratios for all chromosomes on the same horizontal scale (bin size 250 kbp), representing the position along each chromosome with position 0 starting on the p-arms of the chromosomes (Mbp). The ratios are calculated the same way as in Figure 7, III. Horizontal lines depict the scaled background of 1. The vertical scale is linear and ranges from 0 to 12 in each panel. The flat horizontal sections of zeroes correspond to nonsequenced regions of the human genome.
Figure 7.

Deep sequencing data analysis of nucleolar-associated chromatin for acrocentric chromosome 15 and nonacrocentric chromosome 17. Left and right figures show histograms (bin size 250 kbp) along chromosomes 15 (left) and 17 (right) (Mbp), respectively. Panels show from top to bottom: (I) genomic and (II) nucleolar count rate, both normalized to chromosomal mean; (III) normalized ratio of nucleolar to genomic rates (see Materials and Methods). Panels show the following: (IV) the fraction of A and T nucleotides in each bin; (V) known gene count; (VI) gene expression amplitude; and (VII) correlation between nucleolar-genomic ratios and AT fraction (red), gene count (blue) and gene expression amplitude (green) calculated as a Pearson's r in a running 20-Mbp window. The red dashed line in III shows the (background) value of 1 for guidance. The red circles in I–III indicate peaks that correspond to an expression amplitude above an arbitrary threshold of 2000 (VI, dashed line). Sites of LADs are indicated by blue boxes in III, where light and dark boxes correspond to LAD scores below or above 0.95, respectively.
Figure 8.

Investigation of the relationship between nucleolar-genomic ratios and gene expression levels for each somatic chromosome. Nucleolar-genomic ratios plotted as a function of gene expression level for each somatic chromosome (expression data were not available for the X and Y chromosomes). Each point corresponds to the same 250-kbp bins as used in Figures 2B and 7 and Figure S2. The dashed line indicates the background level, normalized to a value of 1.0 (as described in Materials and Methods).
Significance Analysis.
The statistical significance of the relationship between nucleolar association and expression signal was explored with a Monte Carlo approach. The score
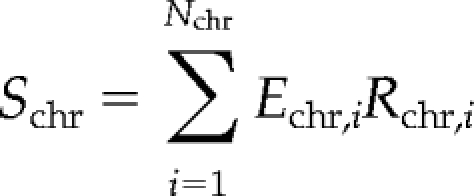 |
was defined for each chromosome (chr), where i is the bin number, Nchr is the number of bins in chromosome chr, Echr,i and Rchr,i represent expression signal and nucleolar-genomic ratio, respectively, in bin i of chromosome chr. This simple quantity has a desired selective property: the better alignment of expression peaks with nucleolar-genomic troughs, the smaller Schr. Hence, if the observed alignment is significant, the actual measured Schr should be small in comparison with a set of randomly drawn alignments.
Random alignments (n = 100,000) were drawn for each chromosome. Each individual alignment was a random permutation of signal strengths with respect to chromosomal positions (strengths were simply shuffled between gene loci). Scores were calculated for each random shuffle, Sp, and their probability distribution was determined. This was compared with the observed score, Schr, which is the statistical significance expressed as the probability of finding a chance alignment with a smaller score, P(Sp < Schr). In some cases (chromosomes 1–3, 12, and 19) these probabilities could not be found directly from drawn distributions, because Schr < Sp for all randomly drawn alignments. In these cases mean and SD of the drawn scores were determined, assumed to be representative of normally distributed data, and the Z-score and probability derived from a Gaussian distribution of the same mean and SD. The results are shown in Table 3.
Table 3.
Significance of gene expression with genomic/nucleolar ratios
| Chromosome | P(Sp < Schr) | Z | PGauss |
|---|---|---|---|
| 1 | — | 4.1 | 2.2 × 10−5 |
| 2 | — | 4.0 | 3.8 × 10−5 |
| 3 | — | 4.7 | 1.2 × 10−6 |
| 4 | 1.5 × 10−4 | 2.8 | 0.0024 |
| 5 | 4.7 × 10−4 | 2.8 | 0.0023 |
| 6 | 2.7 × 10−4 | 2.7 | 0.0038 |
| 7 | 0.008 | 2.2 | 0.014 |
| 8 | 1 × 10−5 | 3.4 | 2.9 × 10−4 |
| 9 | 6.5 × 10−4 | 2.6 | 0.0043 |
| 10 | 4 × 10−5 | 3.1 | 9.1 × 10−4 |
| 11 | 1 × 10−5 | 3.4 | 2.1 × 10−4 |
| 12 | — | 4.2 | 1.5 × 10−5 |
| 13 | 0.053 | 1.5 | 0.067 |
| 14 | 5 × 10−5 | 3.0 | 0.0012 |
| 15 | 0.38 | 0.4 | 0.35 |
| 16 | 0.013 | 1.9 | 0.027 |
| 17 | 2.4 × 10−4 | 2.6 | 0.0047 |
| 18 | 9.4 × 10−4 | 2.5 | 0.059 |
| 19 | — | 3.6 | 1.4 × 10−4 |
| 20 | 0.0012 | 2.7 | 0.0031 |
| 21 | 1.2 × 10−4 | 3.1 | 8.2 × 10−4 |
| 22 | 3.3 × 10−4 | 2.6 | 0.0044 |
Significance was calculated by randomly shuffling expression values between gene loci for each chromosome. The second column gives the probability of chance alignment between gene expression peaks and nucleolar/genomic troughs, derived by comparing the actual alignment with random shuffles. When the actual alignment was outside the random distribution, the probability was estimated from a Gaussian distribution with the same mean and SD as in the random distribution. The third and fourth columns give the Gaussian Z-score and the corresponding probability. —, not.
Analysis of Repetitive Sequences
Repetitive genome hits, constituting ∼30% of all reads, were not included in the analysis of nucleolar association. We analyzed the content of these reads separately.
First, we created a simulated data set in silico by randomly drawing reads from the reference genome assembly. These reads had the same length distribution (i.e., number of reads for a given length) as the genomic set. Then, we mapped simulated reads to the human genome in the same way as for the experimental data. Interestingly, this gave 72.7% of unique hits (for all lengths 16–36 bp), a number very similar to the results from our experimental data (see Table 1), which argues against any sampling bias inherent in our isolation and sequencing procedure.
We used the genomic, nucleolar, and simulated data sets to study the nature of the reads with unique and multiple hits. We limited these calculations to the longest reads (36 bp) and mapped them to the reference genome using the Bowtie alignment tool reporting all possible hits (up to two mismatches allowed). We then searched the Ensembl database for annotations of repeat sequences of both unique and multiple hit locations from all three datasets.
RESULTS
Comparative Genome Hybridization Shows Non-rDNA Regions to be Nucleolar-associated
Fluorescence CGH was used to address whether nucleolar-associated chromatin corresponds to regions from acrocentric chromosomes only or whether it also includes loci from other chromosomes. FISH probes were prepared using either total nuclear DNA (genomic), or DNA isolated from purified nucleoli derived from the human fibrosarcoma cell line HT1080 (shown schematically in Figure 1A, with DAPI-stained images of purified nucleoli in Supplementary Figure S1A). DNA blot analysis confirmed that the nucleolar FISH probe was enriched for rDNA sequences relative to the genomic probe (Figure S1B).
Figure 1.

Comparative genome hybridization for nucleolar versus total genomic DNA. (A) Schematic showing of the preparation of nucleolar-associated (red) and total genomic (red plus green) DNA used for generating FISH probes for comparative genome hybridization analysis of metaphase chromosome spreads and interphase nuclei. (B) FISH analyses of interphase (top panels) and metaphase (bottom panels) cells. Probes labeled either total genomic DNA (panels I and VI), nucleolar-associated DNA (panels II and VII) or rDNA (blue staining, shown as a merge with DAPI in IX). Total DNA was counterstained with DAPI (V and X), nucleoli labeled using an anti-nucleolin antibody (IV). Merged images of genomic and nucleolar probes are shown in III and VIII. In interphase panels, red arrows show nucleoli. In metaphase panels, yellow arrows show rDNA repeat unit clusters, the purple arrow shows the Y chromosome, white arrowheads show centromeres, blue arrowheads show interstitial loci on nonacrocentric chromosomes, and green arrowheads show telomeres. Scale bars, 10 μm.
The genomic and nucleolar FISH probes were each used to label both interphase nuclei and metaphase chromosome spreads, which then were analyzed by fluorescence microscopy (Figure 1B). In interphase cells, the genomic probe labeled extensively throughout the nucleus (Figure 1B, I), whereas the nucleolar probe predominantly labeled nucleoli (Figure 1B, II, red arrows), similar to the pattern seen with an anti-nucleolin antibody (Figure 1B, IV, red arrows). The genomic FISH probe labeled all chromosomes in metaphase spreads (Figure 1B, VI). The nucleolar FISH probe labeled most chromosomes, but only at a restricted set of loci (Figure 1B, VII). This included, as expected, the rDNA repeat clusters at all the acrocentric chromosomes, which was confirmed by independently labeling metaphase spreads with an rDNA-specific FISH probe (Figure 1B, IX, yellow arrows). In addition, the entire Y chromosome was labeled (Figure 1B, VI–VIII, purple arrow). The nucleolar FISH probe labeled most centromeres, a subset of telomeres and a number of internal loci from nonacrocentric chromosomes (Figure 1B, VII and VIII; cf. loci shown by white, green, and blue arrowheads, respectively).
In summary, most if not all human chromosomes contain regions that associate with nucleoli, including loci from nonacrocentric chromosomes distinct from centromeres and telomeres. Although CGH allows both comparisons between total genomic and nucleolar-derived DNA and mapping of associated regions to specific chromosomes, it lacks the resolution to identify individual loci. Note that the more intensively stained chromosome regions in the CGH images mainly represent repetitive regions, as these amplified sequences will be readily detected with this technique.
Deep Sequencing Reveals That Most Chromosomes Have Unique Nucleolar-associated Regions
A deep-sequencing strategy (Figure 2A) was used to obtain a genome-wide high-resolution map of individual NADs. This approach uniquely and directly identifies positions of endogenous chromosome loci, without the need to carry out modifications or enzymatic reactions.
DNA was deep-sequenced from the same genomic and nucleolar preparations used to derive the FISH probes (see Figure 1). Total genomic DNA was used to control for possible nonspecific associations, caused either by the sample preparation method or by the sequencing methodology. Initially, the entire deep-sequencing analysis was performed in duplicate, using two independent preparations of genomic and nucleolar DNA from HT1080 cells (biological replicates; Table 1). The separate biological replicate data sets independently identified the same NADs, providing a strong control for the specificity and reproducibility of the results presented.
More than 3 million unique hits, each 16–36 bp long, were mapped onto the respective human chromosome sequences, and count rates for both the nucleolar and genomic reads in 250-kbp bins were calculated and converted to normalized ratios. A graph of these ratios reveals distinct peaks corresponding to regions of potential nucleolar association along each chromosome (Mbp, Figure 2B). We note that the data correspond to statistical average values based on the analysis of chromosomes isolated from ∼107 cells. We also would like to point out that in a recent study of nucleolar-associated DNA reported by Nemeth et al. (2010), they selected a defined set of NADs by employing an arbitrary threshold in nucleolar count rates. We note that the peaks in our ratio plots (e.g., Figure 7) reveal a continuous distribution of peaks with varying degrees of nucleolar association, instead of well-defined nucleolus-associated regions on the nonassociated background. Any particular selection of well-defined NADs from this smooth distribution is thus necessarily arbitrary.
A third independent biological replicate (see Materials and Methods and Discussion) was used for comparison. It contains ∼6.5 and 7.5 million paired-end, 50-bp reads for genomic and nucleolar sets, respectively. We processed these reads in a similar manner to the main data, except that no quality clipping was done. Instead, Bowtie was set to use quality data directly to accept or reject mismatches. This resulted in ∼96 and 92% of unique genomic and nucleolar hits, respectively. Hits were counted in the same 250-kbp bins, and this identified normalized nucleolar-to-genomic ratios, as in the main data. The third replicate gives almost identical results to the two initial replicates used here (Figure 3).
Figure 3.

Biological triplicates show robustness in deep sequencing technique. Nucleolar-to-genomic ratio of three replicates, listed in Table 1, presented for chromosome 17. Other chromosomes also show a similar level of agreement between the three replicates. Because of background correction, absolute values of these ratios vary from sample to sample. However, the shape, relative height, and location of the peaks in all three panels are very similar. This confirms robustness of nucleolar association presented in this paper.
In summary, this genome-wide deep-sequencing approach provides high-resolution information about which unique chromatin loci are associated with nucleoli in a population of cells grown in culture. The results are consistent with the CGH in showing that most human chromosomes contain regions that preferentially associate with nucleoli. Furthermore, we show that three separate biological replicates analyzed using two different sequence technologies provide near identical results.
Analysis of “Multiple-Hit” Reads Shows Mostly rRNA Genes and Satellite Sequences to be Nucleolar-associated
About 30% of the reads, including sequences within repetitive DNA elements, gave multiple hits to the genome on different chromosomes and hence could not be included in a statistical evaluation of nucleolar associations at the level of specific chromosome loci. However, we analyzed these multiple-hit reads to understand their origin and to check for any systematic bias in these data by comparing the experimental data sets with a computationally randomly generated simulated data set (for details see Materials and Methods). To do so, we first showed that the unique hit reads, both in the nucleolar and genomic data sets, have a similar distribution of matching classes as the random simulated set. This proves that neither the sample preparation method, nor the sequencing methodology, introduced significant bias in the distribution of reads. The observed distribution thus simply reflects the frequency of repeat sequences in the human genome. About a third of the unique hit reads is associated with LINE/L1 sequences (Figure 4A), and the majority of the multiple hits matched repeat classes such as LINE/L1, SINE/Alu, low-complexity regions and tandem repeats (98.3% of all multiple hits; Figure 4B).
Figure 4.

Distribution of unique and multiple hits to repeat sequence classes.(A) The classes are as defined in the Ensembl database. Dust and trf are low-complexity regions and tandem repeats, respectively. Each matching locus in the reference genome reported by Bowtie was searched against Ensembl for overlapping repeat sequences. Counts for entire classes of overlapping sequences were accumulated, and a fraction of total counts was found for each class. This has been done independently for genomic, nucleolar, and simulated data sets. Only the top 12 classes sorted by the number of genomic hits are shown. They contain 96.7% of all genomic hits. (B) As in A, but for multiple hits reported by Bowtie. Note that the y-axis is in logarithmic scale. The top 12 classes sorted by the number of genomic hits are shown. They contain 99.87% of all genomic hits.
There are, however, a few differences. Satellite sequences are more abundant in genomic (factor ∼10) and nucleolar (factor ∼25) experimental data sets with respect to the simulated data. This is due to the fact that centromeres, consisting largely of satellite DNA, are not sequenced in the reference genome and hence are underrepresented in the simulated data.
We also compared the frequency of matches to the repeat sequences from genomic and nucleolar sets. Figure 5 shows the most pronounced classes, with a nucleolar-genomic hit ratio greater than 2. These include satellite repeats and rRNA genes. This distribution was expected, because NORs are clustered around the short arms of the acrocentric chromosomes, which contain tandem repeats of rRNA genes and satellite DNA. The clear enrichment of rRNA genes confirms the separation of nucleolar from total genomic DNA in these experiments.
Figure 5.

Comparison of nucleolar-genomic ratio count rates for different repeat sequences with either multiple or unique hits. Each bar shows the nucleolar-genomic ratio of unique or multiple hit fraction to repeat sequences (see Figure 4A for details). Only repeat classes with nucleolar-genomic ratio greater than 2 are shown. Most notably, rRNA genes are significantly enriched in the nucleolar set.
Single-cell FISH Confirms the Localization of Specific DNA Regions near Nucleoli
The DNA sequencing data identified specific regions within human chromosomes that associate with nucleoli in cell populations, and interestingly, some chromosomes show preferential nucleolar association of the telomere from one chromosome arm. For example, the telomere from the p arm of chromosome 4 is more strongly associated with nucleoli than the 4q telomere (Figure 2B and Figure S2, III). This conclusion was confirmed independently at the single cell level by FISH analysis, using a specific 4p and 4q telomere probe (Figure 6, I, yellow arrow), with nucleoli labeled using an anti-nucleolin antibody (Figure 6, I, white arrowheads). In contrast, FISH probes for the chromosome 2 telomeres, which are not associated with nucleoli in deep sequencing, also showed no preferential nucleolar association as judged by FISH (Figure 6, II, white arrows). Statistical analysis on the FISH data using the Fisher's exact test shows a highly significant association of the 4p telomere over the 4q telomere (p = 0.017). No significant association was obtained between the 2q and 2p telomeres (p = 0.25). All single-cell FISH analyses were done with more than 160 FISH spots (Table 2) and were statistically analyzed using a Fisher's exact test. The nucleolar-association of an internal region from chromosome 17 (q22), which showed association with nucleoli in deep sequencing, was also confirmed at the single-cell level by DNA FISH. A BAC clone was used to make a chromosome 17q22 FISH probe, and its specificity was confirmed by metaphase spread analysis (Figure 6, III, white arrows). The same probe showed preferential association with nucleoli identified by anti-nucleolin antibody staining (Figure 6, IV, associated 17q22 locus indicated by yellow arrow and nucleoli by white arrowheads). In contrast, a control probe from a nearby region of chromosome 17 (q21), which did not show nucleolar association by deep sequencing, also did not show a significant association with nucleoli in the FISH analysis (Figure 6, IV, white arrows). Comparison of FISH data confirmed that the differences in nucleolar association of the Chr17q22 versus Chr17q21 were statistically highly significant using the Fisher's exact test (p = 3.10−6; Table 2). We point out that in most cases it seems to be only one of the two alleles that show an enriched nucleolar association, resulting in a maximum hypothetical association of 50%. In this context, it is clear that the detected association frequency of at least 26% is statistically highly significant, as supported by the calculated p value.
Figure 6.

FISH analysis of chromosomal regions that were identified by Illumina deep sequencing to be associated with nucleoli. FISH analysis was carried out on interphase HT1080 cells (I, II, and IV) or on a human lymphocyte metaphase chromosome spread (panel III). Probes detected either 4p telomere (green) and 4q telomere (red; I), 2p telomere (green) and 2q telomere (red; (II) or chr17q22 (green) and chr17q21 (red; III and IV). Nucleoli were labeled with an anti-nucleolin antibody and shown in blue (white arrowheads, I, II, and IV). Total DNA was stained with DAPI in the metaphase spread (gray, III). Nucleolar-associated loci are indicated with yellow arrows and nonassociated loci with white arrows. Scale bars, 10 μm.
Table 2.
Statistical analysis of FISH spots
| Spots | 17q21 | 17q22 | p |
|---|---|---|---|
| Assoc | 33 (15) | 74 (34) | 3 × 10−6 |
| Nonassoc | 192 (85) | 145 (66) | |
| Total | 225 (100) | 219 (100) | |
| Spots | 4q tel | 4p tel | p |
| Assoc | 21 (12) | 41 (23) | 0.017 |
| Nonassoc | 148 (88) | 140 (77) | |
| Total | 169 (100) | 181 (100) | |
| Spots | 2q tel | 2p tel | p |
| Assoc | 24 (15) | 26 (14) | 0.25 |
| Nonassoc | 136 (85) | 164 (86) | |
| Total | 160 (100) | 190 (100) |
Values are the number of spots, with the percentages of FISH spots that do associate (assoc) and do not associate (nonassoc) with nucleoli in parentheses (see Figure 6); corresponding p values are shown.
In summary, the peaks in the plots of nucleolar-genomic count rate ratios shown by statistical evaluation of the DNA sequence data correspond to chromosome regions that preferentially associate with nucleoli in situ.
Bioinformatic Analysis Identifies a Strong Correlation of Nucleolar-associated Regions with AT-Rich Sequence Elements, Low Gene Density, and Transcriptionally Repressed Genes
All chromosome regions showing preferential nucleolar association in the unique hit data sets were compared with search for common features; however, no simple sequence motif was identified between these unique regions. Interestingly, a more general comparison revealed a clear enrichment in AT-rich sequence elements within the nucleolar-associated loci, as illustrated for both the acrocentric chromosome 15 and the nonacrocentric chromosome 17 (Figure 7) and for all chromosomes in Figure S2, (panel IV in each figure). Analysis of gene density along each chromosome also showed a strong negative correlation with the regions empirically identified as nucleolar-associated (Figure 7 and Figure S2, panel V in both).
Furthermore, genes identified within the NADs showed a striking and statistically significant positive correlation with transcriptionally repressed genes previously identified in HT1080 cells by microarray analysis (for details see Materials and Methods; Figure 7 and Figure S2, VI, and Table 3). Thus, highly expressed genes are mostly found in the regions of low nucleolar-genomic ratio, as marked with red circles in Figure 7 and Figure S2 (I–III). The inverse is also seen: regions of strong nucleolar association, represented by peaks in nucleolar-genomic ratio, contain very few, if any, highly expressed genes. This is true for all chromosomes, including chromosomes 1–14, where nucleolar-genomic peaks are not as distinct, and is directly illustrated by plotting nucleolar-genomic ratios versus expression levels for all chromosomes (Figure 8). Correlation coefficients between nucleolar-genomic ratios and either AT fraction (red), gene density (blue) or gene expression level (green), are shown by correlation coefficients in Figure 7, VII (shown for all chromosomes in Figure S2, VII).
Unexpectedly, analysis of deep sequencing data detected a partial, but clear, overlap between the NADs and loci previously reported to associate with the nuclear envelope (lamina associated domains (LADs; Guelen et al., 2008; Figure 7 and Figure S2, III, underlined blue boxes). Both the Guelen et al., study and our data involved high-throughput analysis of cell populations, and the overlap in loci suggests the possibility that specific regions could alternate between associating with the nucleolus and the nuclear periphery either in different cells, or at different times.
In summary, our sequence analysis reveals a clear and statistically significant correlation between regions of nucleolar-associated chromatin with AT-rich sequence elements, low gene density, and transcriptionally repressed genes. In addition, we report an overlap in nucleolar-associated loci (NADs) with regions previously reported to associate with the nuclear periphery (LADs).
Photoactivation Shows Nucleolar Chromatin Returns Either to Nucleoli or to the Nuclear Periphery, in Daughter Cells
To analyze whether chromosome loci associated with nucleoli in one cell cycle either return to nucleoli, move to the nuclear periphery, or become randomly distributed throughout the nucleoplasm in daughter nuclei, we used a photoactivation and time-lapse fluorescence microscopy assay. A HeLa cell line was established that stably expresses PA-GFP fused to the carboxy terminus of histone H2B. It is known that most histones largely remain associated with a single locus throughout interphase, as shown by a low rate of exchange between histones and chromatin using FRAP experiments (Gerlich and Ellenberg, 2003; Cvackova et al., 2009). In agreement with these previous data, HeLa cells that failed to enter mitosis in our photoactivation experiments also showed minimal movement of chromatin and little exchange of histones (data not shown). Using photoactivation of PA-GFP-H2B, as opposed to photobleaching of GFP-H2B, facilitated this analysis of chromatin during mitosis because enough PA-GFP-H2B signal remains as a detectable spot after imaging for more than 20 h to clearly identify the marked regions in daughter cells.
To identify nucleoli in live cells, the nucleolar marker protein B23 was fused to mCherry and transiently expressed in the HeLa PA-GFP-H2B stable cell line. A ROI either overlapping the nucleolus (Figure 9, I, blue arrow) or in the nucleoplasm separate from the nucleolus (Figure 9, III, blue arrow) was photoactivated using laser excitation at 406 nm and imaged using time-lapse microscopy. The chromatin either just above, or below, the nucleolus was also activated to some extent. To control for these additional activated regions, daughter cells were also analyzed where the activated region was separate from the nucleolus (Figure 9, IV).
Figure 9.

Photoactivation of nucleolar-associated chromatin to compare its localization in parental and daughter cells following mitosis. Photoactivation of PA-GFP-H2B stably expressed in HeLa cells. ROIs were either nucleolar-associated (I and II) or separate from nucleoli in the nucleoplasm (III and IV). These ROIs were photoactivated at 406 nm (blue arrows), and representative midplane deconvolved images are shown from cells either before (I and III) or after (II and IV) mitosis. Both postmitotic images show daughter cells 4.5 h after the onset of telophase. PA-GFP-H2B is shown in green, and nucleoli are labeled with mCherry-B23 (red). White arrows mark photoactivated chromatin that relocalizes at nucleoli in daughter cells; yellow arrowheads point at chromatin that relocalizes at the nuclear periphery in daughter cells. Scale bars, 10 μm.
Images were recorded in both red and green channels every 20 min for 20–23 h to follow cells throughout mitosis. Figure 9, II and IV, show representative postmitotic images of both daughter cells 4.5 h after the onset of telophase.
The frequency of photoactivated chromatin located either at, or close to, nucleoli was higher in daughter nuclei from a cell where the photoactivated ROI before mitosis overlapped nucleoli, as opposed to when it was in the nucleoplasm separate from nucleoli (Figure 9, II and IV, white arrows). Quantitation of the respective red and green signals and statistical analysis comparing the distribution of photoactivated chromatin using a Mann-Whitney test (see Materials and Methods and Figure S3), showed that the differences in daughter nuclei for cells with a ROI either overlapping, or separate from nucleoli, are statistically highly significant (p = 0.0045). Our results are in agreement with a recent publication in which the photoconvertible H4-Dendra2 was used in HePG2 cells to study nucleolar-associated chromatin (Cvackova et al., 2009).
We note that because of the size of the photoactivated area, not only nucleolar-associated chromatin, but also adjacent chromatin regions that are positioned close to the nucleolus in that particular cell, are inevitably photoactivated as well. Hence, only a subset of the overall photoactivated regions can be expected to return to nucleoli.
In addition to regions returning to nucleoli after cell division, photoactivated chromatin was also observed at the nuclear periphery in daughter cells when the ROI had overlapped the nucleolus in the parental cell (Figure 9, II, yellow arrowheads). In contrast, little or no association of photoactivated loci with either nucleoli, or the nuclear periphery, was observed in cells when the ROI in the parental cell was separate from the nucleolus (Figure 9, IV).
In summary, at least a subset of chromosomal loci associated with nucleoli in one cell cycle return to associate with nucleoli in daughter cells after disassembly and reassembly of the nucleolus. In addition, the photoactivation data combined with the overlap observed between the LADs and the nucleolar-associated loci in our deep sequencing analyses provide evidence that some DNA regions can associate with either nucleoli, or the nuclear periphery, after cell division.
DISCUSSION
The three different approaches we have used to analyze in detail DNA regions that associate with nucleoli are highly complementary. CGH analysis showed non-rDNA regions to be associated with nucleoli on human metaphase spreads (Figure 1B). On interphase cells, the nucleolar DNA probe shows a predominant hybridization in the nucleolus, confirming a clear enrichment in nucleolar purified DNA.
Because this technique is limited to showing mainly repetitive regions, with little or no detailed information about individual loci, we went on to use deep DNA sequencing to provide a comprehensive and quantitative characterization of chromatin regions associated with nucleoli in populations of cultured cells. This genome-wide analysis proved to be specific and reproducible because three independent biological replicates identified the same NADs. We identified a clear overlap between some NADs and previously reported LADs (Guelen et al., 2008). Fluorescence photoactivation of nucleolar-associated chromatin confirmed that after cell division some chromosomal loci associated with nucleoli in the mother cell either return to nucleoli, or associate with the nuclear periphery, in the daughter cells.
We emphasize that repetitive DNA sequences had to be removed from the final evaluation of the deep sequencing analysis and mapping of chromosome associations, although some of the repetitive sequences are strongly associated with the nucleolus. This is because these repetitive sequences gave multiple hits to the genome on different chromosomes, and therefore could not be included in a statistical evaluation of nucleolar associations at the level of specific chromosome loci. Hence the data from the CGH, the photoactivation and DNA sequencing experiments are consistent and provide a detailed picture of the composition of nucleolar-associated chromatin.
The repositioning of some of the nucleolar-associated chromatin to the nuclear periphery, as revealed by fluorescence photoactivation, supports the observed overlap between NADs and LADs in the deep sequencing data sets and argues against a contamination in our sample preparation. This is consistent with previous reports that describe the localization of centromeric regions of chromosomes 1 and 9 as well as the entire inactive X chromosome (Xi) to either nucleoli or to the nuclear periphery (Manuelidis and Borden, 1988; Zhang et al., 2007). Additionally, both the nucleolar-associated and nuclear periphery–associated chromatin domains exhibit common features, being highly condensed and known to replicate preferentially at late stages of S phase.
The preferential association of specific chromatin regions with nucleoli, including individual telomeres, centromeres, and internal chromosome loci, may contribute to the subnuclear organization of chromosomes (Figure 10). In 1885, Rabl published the nonrandom but polarized position of chromosomes within the nucleus, known as the Rabl orientation. In this model, centromeres are located at the opposite side of the nucleus to telomeres (reviewed in Comings, 1980). Even though this exact orientation is not commonly found in human cells, our data support the hypothesis of using anchoring points within the nucleus to polarize chromosomes, as was previously suggested (Comings, 1980; Parada et al., 2004).
Figure 10.

Nucleolar-associated chromatin regions. A schematic showing of the association of a few representative chromosome loci with nucleoli, such as rDNA regions, telomeres, centromeres, and interstitial regions. Chromosomes shown both as solid and dashes lines, represent dual localization patterns. It illustrates a potential role for the nucleolus in the global organization of chromosomes within the nucleus.
The statistically significant correlation we observe between transcriptionally repressed genes and DNA regions displaying nucleolar association is consistent with the highly condensed nature of the chromatin that surrounds nucleoli and copurifies with them (Boisvert et al., 2007). In general, heterochromatic regions are strongly associated with gene silencing (reviewed in Cockell and Gasser, 1999; Dundr and Misteli, 2001 and references therein).
We hypothesize additional roles for the nucleolus in human cells both in nuclear architecture and in sequestering chromosomal loci to remove them from the transcriptionally active environment throughout the rest of the nucleoplasm. This is consistent with recent evidence showing that tethering ectopically expressed genes to the nuclear periphery can affect gene regulation (Finlan et al., 2008; Kumaran and Spector 2008). The statistically significant correlation between NADs and transcriptional repression supports the hypothesis that specific loci may either remain at, or be recruited to, the nucleolus, in order to be silenced. Future work will determine whether nucleolar association of chromatin can contribute to gene regulation and whether the nucleolar-associated loci vary between cell types and/or in response to major cell changes, such as oncogenic transformation.
The overlap between loci that associate with nucleoli and those previously detected at the nuclear periphery could thus provide a higher order gene regulatory mechanism. It would be interesting to see whether genes located in chromosome regions that are neither associated with nucleoli, nor with the nucleolar periphery are subject to a more dynamic regulation of gene expression.
During the course of submission of this study, an article entitled “Initial Genomics of the Human Nucleolus” appeared that also analyzed nucleolar-associated chromatin, though in HeLa and IMR90 cells rather than the HT1080 cells studied here (Nemeth et al., 2010). Many of the conclusions in this article and in our study are in agreement. For example, both identified a nucleolar enrichment of chromatin regions containing olfactory receptor genes, zinc finger genes, immunoglobulin gene families, and 5S RNA genes. However, the data also show several important differences. First, we do not observe any clear enrichment of tRNA genes associated with the nucleolus as reported by Nemeth et al. Second, we demonstrate a highly significant correlation between NADs and both loci containing AT-rich sequences and loci shown to have low levels of gene expression. Third and possibly most important, we observe in all three independent biological replicates of the nucleolar sequence analysis, a clear overlap between NADs and a subset of the regions previously reported to be associated with the nuclear lamina (Guelen et al., 2008; see also Figure 7 and Figure S2B). In contrast, Nemeth et al. (2010) report that NADs differ markedly from LADs in their analysis of HeLa and IMR90 cells. Our study of HT1080 cells not only revealed the overlap between NADs and LADs in three separate biological replicates using two distinct sequencing technologies, but confirmed this also using live cell time-lapse fluorescence microscopy. Thus we observed by photoactivation of nucleolar-associated chromatin specific relocation either back to nucleoli, or to the nuclear lamina, in daughter cells after mitosis (see Figure 9). This apparent discrepancy could reflect a difference in the loci associated with nucleoli in different human cell lines. It will be interesting in the future to perform further studies on other cell lines to investigate in more detail differences in chromosome loci associated with nucleoli as well as the potential functional relationship between this association and gene expression. Alternatively, the much higher density of sequence coverage (>20 times higher) obtained in our present study, combined with the analysis of parallel genomic control samples, may have revealed here an overlap between NADs and LADs that was not obvious in the datasets reported by Nemeth et al.
Supplementary Material
ACKNOWLEDGMENTS
We thank our colleagues for helpful advice and for comments on the manuscript and Dr. Sam Swift and staff in the CLS Microscopy Facility for help and advice. We thank Sheilagh Boyle and Wendy Bickmore for help with the FISH analysis and Brian McStay for advice on comparative genome hybridization and for providing rDNA probes. This work was supported by a Wellcome Trust Programme Grant (073980/Z/03/Z) to A.I.L., by a Wellcome Trust Strategic Award to the Centre for Gene Regulation and Expression (WTWT083524MA), and by a Rubicon grant from the Netherlands Organisation for Scientific Research (NWO) to S.v.K. Part of the Illumina deep sequencing was supported by a small research TENOVUS grant to S.v.K. A.I.L. is a Wellcome Trust Principal Research Fellow.
Abbreviations used:
- CGH
comparative genome hybridization
- CT
chromosome territory
- FISH
fluorescence in situ hybridization
- LAD
lamina-associated domain
- NAD
nucleolar-associated domain
- PA-GFP
photoactivatable green fluorescent protein
- ROI
region of interest.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-06-0508) on September 8, 2010.
REFERENCES
- Andersen J. S., Lam Y. W., Leung A. K., Ong S. E., Lyon C. E., Lamond A. I., Mann M. Nucleolar proteome dynamics. Nature. 2005;433:77–83. doi: 10.1038/nature03207. [DOI] [PubMed] [Google Scholar]
- Angelier N., Tramier M., Louvet E., Coppey-Moisan M., Savino T. M., De Mey J. R., Hernandez-Verdun D. Tracking the interactions of rRNA processing proteins during nucleolar assembly in living cells. Mol. Biol. Cell. 2005;16:2862–2871. doi: 10.1091/mbc.E05-01-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billia F., Deboni U. Localization of Centromeric satellite and telomeric DNA sequences in dorsal root ganglion neurons, in vitro. J. Cell Sci. 1991;100:219–226. doi: 10.1242/jcs.100.1.219. [DOI] [PubMed] [Google Scholar]
- Boisvert F. M., van Koningsbruggen S., Navascues J., Lamond A. I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007;8:574–585. doi: 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- Bond C. S., Fox A. H. Paraspeckles: nuclear bodies built on long noncoding RNA. J. Cell Biol. 2009;186:637–644. doi: 10.1083/jcb.200906113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco M. R., Pombo A. Intermingling of chromosome territories in interphase suggests role in translocations and transcription–dependent associations. Plos Biol. 2006;4:780–788. doi: 10.1371/journal.pbio.0040138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho C., Pereira H. M., Ferreira J., Pina C., Mendonca D., Rosa A. C., Carmo-Fonseca M. Chromosomal G-dark bands determine the spatial organization of centromeric heterochromatin in the nucleus. Mol. Biol. Cell. 2001;12:3563–3572. doi: 10.1091/mbc.12.11.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang C. H., Belmont A. S. Moving chromatin within the interphase nucleus–controlled transitions? Semin. Cell Dev. Biol. 2007;18:698–706. doi: 10.1016/j.semcdb.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson C. M., Hutchinson J. N., Sara S. A., Ensminger A. W., Fox A. H., Chess A., Lawrence J. B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell. 2009;33:717–726. doi: 10.1016/j.molcel.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockell M., Gasser S. M. Nuclear compartments and gene regulation. Curr. Opin. Genet. Dev. 1999;9:199–205. doi: 10.1016/S0959-437X(99)80030-6. [DOI] [PubMed] [Google Scholar]
- Comings D. E. Arrangement of chromatin in the nucleus. Hum. Genet. 1980;53:131–143. doi: 10.1007/BF00273484. [DOI] [PubMed] [Google Scholar]
- Cremer T., Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001;2:292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- Cremer T., Cremer M., Dietzel S., Muller S., Solovei I., Fakan S. Chromosome territories—a functional nuclear landscape. Curr. Opin. Cell Biol. 2006;18:307–316. doi: 10.1016/j.ceb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Croft J. A., Bridger J. M., Boyle S., Perry P., Teague P., Bickmore W. A. Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 1999;145:1119–1131. doi: 10.1083/jcb.145.6.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvackova Z., Masata M., Stanek D., Fidlerova H., Raska I. Chromatin position in human HepG2 cells: Although being non-random, significantly changed in daughter cells. J. Struct. Biol. 2009;165:107–117. doi: 10.1016/j.jsb.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat W., Klous P., Kooren J., Noordermeer D., Palstra R. J., Simonis M., Splinter E., Grosveld F. Three-dimensional organization of gene expression in erythroid cells. Red Cell Dev. 2008;82:117–139. doi: 10.1016/S0070-2153(07)00005-1. [DOI] [PubMed] [Google Scholar]
- de Wit E., van Steensel B. Chromatin domains in higher eukaryotes: insights from genome-wide mapping studies. Chromosoma. 2009;118:25–36. doi: 10.1007/s00412-008-0186-0. [DOI] [PubMed] [Google Scholar]
- Denegri M., Moralli D., Rocchi M., Biggiogera M., Raimondi E., Cobianchi F., De Carli L., Riva S., Biamonti G. Human chromosomes 9, 12, and 15 contain the nucleation sites of stress-induced nuclear bodies. Mol. Biol. Cell. 2002;13:2069–2079. doi: 10.1091/mbc.01-12-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr M., Misteli T. Functional architecture in the cell nucleus. Biochem. J. 2001;356:297–310. doi: 10.1042/0264-6021:3560297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espada J., Ballestar E., Santoro R., Fraga M. F., Villar-Garea A., Nemeth A., Lopez-Serra L., Ropero S., Aranda A., Orozco H., Moreno V., Juarranz A., Stockert J. C., Langst G., Grummt I., Bickmore W., Esteller M. Epigenetic disruption of ribosomal RNA genes and nucleolar architecture in DNA methyltransferase 1 (Dnmt1) deficient cells. Nucleic Acids Res. 2007;35:2191–2198. doi: 10.1093/nar/gkm118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira J., Paolella G., Ramos C., Lamond A.I. Spatial organization of large-scale chromatin domains in the nucleus: A magnified view of single chromosome territories. J. Cell Biol. 1997;139:1597–1610. doi: 10.1083/jcb.139.7.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlan L. E., Sproul D., Thomson I., Boyle S., Kerr E., Perry P., Ylstra B., Chubb J. R., Bickmore W. A. Recruitment to the nuclear periphery can alter expression of genes in human cells. Plos Genet. 2008;4:e1000039. doi: 10.1371/journal.pgen.1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser P., Bickmore W. Nuclear organization of the genome and the potential for gene regulation. Nature. 2007;447:413–417. doi: 10.1038/nature05916. [DOI] [PubMed] [Google Scholar]
- Frey M. R., Matera A. G. Coiled bodies contain U7 small nuclear RNA and associate with specific DNA sequences in interphase human cells. Proc. Nat. Aced. Sci USA. 1995;92:5915–5919. doi: 10.1073/pnas.92.13.5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey M. R., Matera A. G. RNA-mediated interaction of Cajal bodies and U2 snRNA genes. J. Cell Biol. 2001;154:499–509. doi: 10.1083/jcb.200105084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall J. G., Stephenson E. C., Erba H. P., Diaz M. O., Barasacchipilone G. Histone genes are located at the sphere loci of newt Lampbrush chromosomes. Chromosoma. 1981;84:159–171. doi: 10.1007/BF00399128. [DOI] [PubMed] [Google Scholar]
- Gao L. I., Frey M. R., Matera A. G. Human genes encoding U3 snRNA associate with coiled bodies in interphase cells and are clustered on chromosome 17p11.2 in a complex inverted repeat structure. Nucleic Acids Res. 1997;25:4740–4747. doi: 10.1093/nar/25.23.4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlich D., Ellenberg J. Dynamics of chromosome positioning during the cell cycle. Curr. Opin. Cell Biol. 2003;15:664–671. doi: 10.1016/j.ceb.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Gondor A., Ohlsson R. Chromosome crosstalk in three dimensions. Nature. 2009;461:212–217. doi: 10.1038/nature08453. [DOI] [PubMed] [Google Scholar]
- Guelen L., Pagie L., Brasset E., Meuleman W., Faza M. B., Talhout W., Eussen B. H., de Klein A., Wessels L., de Laat W., van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–U983. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Haaf T., Schmid M. Centromeric association and non-random distribution of centromeres in human tumor cells. Hum. Genet. 1989;81:137–143. doi: 10.1007/BF00293889. [DOI] [PubMed] [Google Scholar]
- Haaf T., Schmid M. Chromosome topology in mammalian interphase nuclei. Exp. Cell Res. 1991;192:325–332. doi: 10.1016/0014-4827(91)90048-y. [DOI] [PubMed] [Google Scholar]
- Kumaran R. I., Spector D. L. A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J. Cell Biol. 2008;180:51–65. doi: 10.1083/jcb.200706060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger I., Guillaud M., Krief B., Brugal G. Interactive computer-assisted analysis of chromosome-1 colocalization with nucleoli. Cytometry. 1994;16:313–323. doi: 10.1002/cyto.990160405. [DOI] [PubMed] [Google Scholar]
- Lemmers R.J.L.F., Wohlgemuth M., van der Gaag K. J., van der Vliet P. J., van Teijlingen C.M.M., de Knijff P., Padberg G. W., Frants R. R., van der Maarel S. M. Specific sequence variations within the 4q35 region are associated with Facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2007;81:884–894. doi: 10.1086/521986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung A. K., Gerlich D., Miller G., Lyon C., Lam Y. W., Lleres D., Daigle N., Zomerdijk J., Ellenberg J., Lamond A. I. Quantitative kinetic analysis of nucleolar breakdown and reassembly during mitosis in live human cells. J. Cell Biol. 2004;166:787–800. doi: 10.1083/jcb.200405013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L. Different central nervous system cell types display distinct and nonrandom Arrangements of satellite DNA sequences. Proc. Natl. Acad. Sci. USA. 1984;81:3123–3127. doi: 10.1073/pnas.81.10.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L., Borden J. Reproducible compartmentalization of individual chromosome domains in human CNS cells revealed by in situ hybridization and 3-dimensional reconstruction. Chromosoma. 1988;96:397–410. doi: 10.1007/BF00303033. [DOI] [PubMed] [Google Scholar]
- Mcstay B., Grummt I. The epigenetics of rRNA genes: from molecular to chromosome biology. Ann. Rev. Cell Dev. Biol. 2008;24:131–157. doi: 10.1146/annurev.cellbio.24.110707.175259. [DOI] [PubMed] [Google Scholar]
- Meaburn K.J., Misteli T., Soutoglou E. Spatial genome organization in the formation of chromosomal translocations. Semin. Cancer Biol. 2007;17:80–90. doi: 10.1016/j.semcancer.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- Nemeth A., Conesa A., Santoyo–Lopez J., Medina I., Montaner D., Peterfia B., Solovei I., Cremer T., Dopazo J., Langst G. Initial genomics of the human nucleolus. Plos Genet. 2010;6:e1000889. doi: 10.1371/journal.pgen.1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton J. T., Wang C., Gjidoda A., Henry R. W., Huang S. The perinucleolar compartment is directly associated with DNA. J. Biol. Chem. 2009;284:4090–4101. doi: 10.1074/jbc.M807255200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez E., Fu X. D., Rosenfeld M. G. Nuclear organization in the 3D space of the nucleus—cause or consequence? Curr. Opin. Genet. Dev. 2009;19:424–436. doi: 10.1016/j.gde.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs R. L., Press R. I. Centromere autoantigens are associated with the nucleolus. Exp. Cell Res. 1992;200:339–350. doi: 10.1016/0014-4827(92)90181-7. [DOI] [PubMed] [Google Scholar]
- Parada L. A., Sotiriou S., Misteli T. Spatial genome organization. Exp. Cell Res. 2004;296:64–70. doi: 10.1016/j.yexcr.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Parkinson H., Kapushesky M., Kolesnikov N., Rustici G., Shojatalab M., Abeygunawardena N., Berube H., Dylag M., Emam I., Farne A., Holloway E., Lukk M., Malone J., Mani R., Pilicheva E., Rayner T. F., Rezwan F., Sharma A., Williams E., Bradley X. Z., Adamusiak T., Brandizi M., Burdett T., Coulson R., Krestyaninova M., Kurnosov P., Maguire E., Neogi S. G., Rocca-Serra P., Sansone S. A., Sklyar N., Zhao M., Sarkans U., Brazma A. ArrayExpress update from an archive of functional genomics experiments to the atlas of gene expression. Nuc. Acids Res. 2009;37:D868–D872. doi: 10.1093/nar/gkn889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson T. The plurifunctional nucleolus. Nuc. Acids Res. 1998;26:3871–3876. doi: 10.1093/nar/26.17.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson T., Tsai R.Y.L. In search of nonribosomal nucleolar protein function and regulation. J. Cell Biol. 2009;184:771–776. doi: 10.1083/jcb.200812014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J. C., Karpen G. H. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat. Cell Biol. 2007;9:25–U24. doi: 10.1038/ncb1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo A., Cuello P., Schul W., Yoon J. B., Roeder R. G., Cook P. R., Murphy S. Regional and temporal specialization in the nucleus: a transcriptionally-active nuclear domain rich in PTF, Oct1 and PIKA antigens associates with specific chromosomes early in the cell cycle. EMBO J. 1998;17:1768–1778. doi: 10.1093/emboj/17.6.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raska I., Shaw P. J., Cmarko D. Structure and function of the nucleolus in the spotlight. Curr. Opin. Cell Biol. 2006;18:325–334. doi: 10.1016/j.ceb.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Rawlins D. J., Shaw P. J. Localization of ribosomal and telomeric DNA sequences in intact plant nuclei by in situ hybridization and 3-dimensional optical microscopy. J. Microsc. 1990;157:83–89. doi: 10.1111/j.1365-2818.1990.tb02949.x. [DOI] [PubMed] [Google Scholar]
- Rubbi C. P., Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003;22:6068–6077. doi: 10.1093/emboj/cdg579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoni N., Langer S., Fauth C., Bernardi G., Cremer T., Turner B. M., Zink D. Nuclear organization of mammalian genomes: polar chromosome territories build up functionally distinct higher order compartments. J. Cell Biol. 1999;146:1211–1226. doi: 10.1083/jcb.146.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirri V., Urcuqui-Inchima S., Roussel P., Hernandez-Verdun D. Nucleolus: the fascinating nuclear body. Histochem. Cell Biol. 2008;129:13–31. doi: 10.1007/s00418-007-0359-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K. P., Carter K. C., Johnson C. V., Lawrence J. B. U2 and U1 snRNA gene loci associate with coiled bodies. J. Cell. Biochem. 1995;59:473–485. doi: 10.1002/jcb.240590408. [DOI] [PubMed] [Google Scholar]
- Sun H. B., Yokota H. Correlated positioning of homologous chromosomes in daughter fibroblast cells. Chrom. Res. 1999;7:603–610. doi: 10.1023/a:1009279918034. [DOI] [PubMed] [Google Scholar]
- Thompson M., Haeusler R. A., Good P. D., Engelke D. R. Nucleolar clustering of dispersed tRNA genes. Science. 2003;302:1399–1401. doi: 10.1126/science.1089814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L., Andrews P. D., Wickramasinghe S., Sleeman J., Prescott A., Lam Y. W., Lyon C., Swedlow J. R., Lamond A. I. Time-lapse imaging reveals dynamic relocalization of PP1 gamma throughout the mammalian cell cycle. Mol. Biol. Cell. 2003;14:107–117. doi: 10.1091/mbc.E02-07-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Shiels C., Sasieni P., Wu P. J., Islam S. A., Freemont P. S., Sheer D. Promyelocytic leukemia nuclear bodies associate with transcriptionally active genomic regions. J. Cell Biol. 2004;164:515–526. doi: 10.1083/jcb.200305142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. F., Huynh K. D., Lee J. T. Perinucleolar targeting of the inactive X during S phase: Evidence for a role in the maintenance of silencing. Cell. 2007;129:693–706. doi: 10.1016/j.cell.2007.03.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.