

Abstract
Epigenetic reprogramming including demethylation of DNA occurs in mammalian primordial germ cells (PGCs) and in early embryos, and is important for the erasure of imprints and epimutations, and the return to pluripotency1-9. The extent of this reprogramming and its molecular mechanisms are poorly understood. We previously showed that the cytidine deaminases Aid and Apobec1 can deaminate 5-methylcytosine in vitro and in E coli, and in the mouse are expressed in tissues in which demethylation occurs10. Here we profiled DNA methylation throughout the genome by unbiased bisulfite Next Generation Sequencing11-13 (BS-Seq) in wildtype and Aid deficient PGCs at E13.5. Wildtype PGCs revealed dramatic genome-wide erasure of methylation to a level below that of methylation deficient (Np95-/-) ES cells, with female PGCs being less methylated than male ones. By contrast, Aid deficient PGCs were up to three times more methylated than wildtype ones; this substantial difference occurred throughout the genome, with introns, intergenic regions and transposons being relatively more methylated than exons. Relative hypermethylation in Aid deficient PGCs was confirmed by analysis of individual loci in the genome. Our results reveal that erasure of DNA methylation in the germ line is a global process, hence limiting the potential for transgenerational epigenetic inheritance. Aid deficiency interferes with genome-wide erasure of DNA methylation patterns, suggesting that Aid has a critical function in epigenetic reprogramming and potentially in restricting the inheritance of epimutations in mammals.
In plants active demethylation occurs widely (including in imprinted genes) and is carried out by 5meC glycosylases such as Demeter and Demeter like proteins14-16. This class of enzymes does not appear to exist in mammalian genomes, but instead we found that the cytosine deaminases Aid and Apobec1 can deaminate 5meC both in vitro and in E coli10, suggesting deamination of 5meC followed by T:G base excision repair by glycosylases such as Tdg or Mbd4 as an equivalent pathway for demethylation of DNA. Recently it has been shown that co-expression of Aid and Mbd4 in Zebrafish embryos can lead to demethylation of DNA, lending support to the idea that demethylation in animals might proceed by deamination followed by base excision repair17. These interesting results raise the question of whether naturally occurring demethylation in PGCs or early embryos involves deaminases or base excision repair.
We previously found that Aid is expressed in PGCs and in early embryos at a time when demethylation occurs10. Methylation in some single copy genes, differentially methylated regions in imprinted genes, and Line1 repeats has been shown to be substantially erased in PGCs between E11.5 and E13.5, while methylation of IAPs was significantly more resistant to erasure6-9,18. The extent of demethylation of the genome as a whole in PGCs is unknown. In order to understand the dynamics of erasure on a genome-wide scale we carried out unbiased sequencing of bisulfite treated DNA by NGS (BS-Seq), which accurately quantifies whole genome methylation levels11,13. We were able to scale down by 20 fold the construction and analysis of Illumina Solexa libraries to as little as 150 ng of genomic DNA, enabling for the first time a genome-wide view of methylation in fully reprogrammed PGCs at E13.5, isolated by cell sorting of Oct4-Gfp positive germ cells (Fig. 1). Both male and female PGCs were strikingly hypomethylated, with a median methylation level of 16.3% and 7.8% respectively (for comparison, methylation deficient Np95-/- ES cells had a methylation level of 22%). It is interesting to note that female PGCs had a lower methylation level than male PGCs; this is consistent with female ES and EG cells having less methylation than male ones19, suggesting that the proposed effects of two active X chromosomes on decreasing genome methylation might operate in vivo as well as in pluripotent cell lines derived from ICM cells or PGCs. Methylation levels in fetus, ES cells, and sperm were high (73.2-85%), while those in placenta were intermediate (42.3%), consistent with results of previous experiments in which individual gene sequences or transposons were analysed. Notably, genome wide hypomethylation has recently been reported in Arabidopsis endosperm in comparison to the embryo (although the difference, 6%, is considerably smaller than the one we found here in the mammalian system), a tissue that supports embryo development and nutrition in seed plants in analogy to the function of the placenta in mammals15,16.
Figure 1. Genome-wide BS-Seq reveals global hypomethylation in PGCs dependent on Aid.
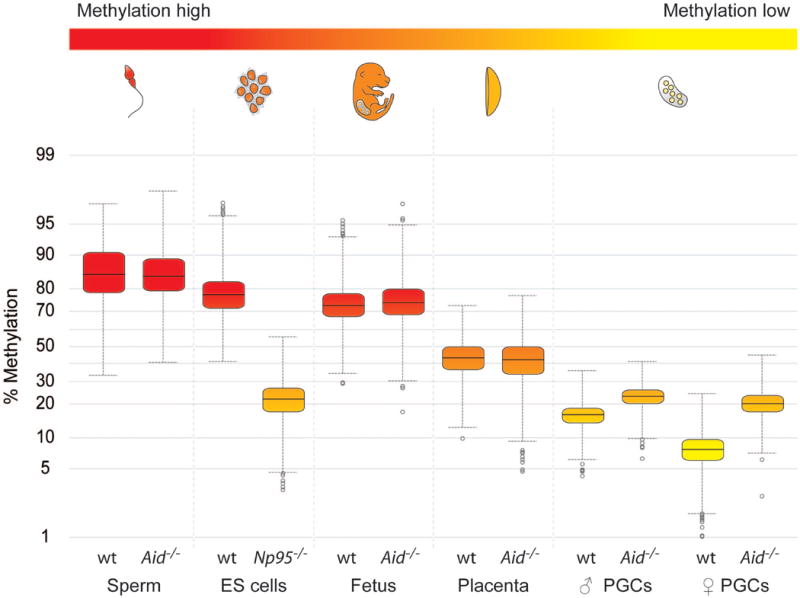
Tissues and cells analysed by BS-Seq are shown in a gradient from red to yellow illustrating methylation levels from high to low. BS-Seq reads were analysed using windows of 250 kilobases across the whole genome. Yellow boxes show the range of the 25-75th quartiles of the data, the line in the middle the median value. Whiskers show either highest and lowest values (if there are no outliers) or upper and lower confidence intervals. Outliers are shown as circles. Placenta, fetal carcass and PGCs were collected at E13.5. Note the global hypomethylation in PGCs, and the substantially higher levels of methylation in Aid-/- PGCs.
We next carried out a more detailed analysis of the patterns of methylation erasure in PGCs in various genomic elements (Figs. 2,3). We assessed levels of methylation of promoters, exons, introns, and intergenic regions (Fig. 2), as well as those of various transposon families (Fig. 3, supplementary Fig. 1; BS-Seq reads of transposons and repeats with identical sequence are eliminated bioinformatically, but BS-Seq reads of transposon family members that map uniquely, due to unique sequence variation, can be assessed). In highly methylated tissues (ES, sperm, fetus), methylation was similarly high in exons (above 70%), and was considerably lower in promoters (35-40%), consistent with a recent whole genome BS-Seq analysis in human ES cells13. Introns and retrotransposons were particularly highly methylated in sperm (approximately 10% higher than in the fetus or ES cells), supporting the notion that de novo methylation in male germ cells, before resumption of mitosis after birth, is important for suppressing transposon mobility in the germ line. In comparison to the highly methylated tissues, the greatest loss of methylation in PGCs (and in placenta) was observed within introns, intergenic regions and repeats, followed by exons, and then promoters. Hence, demethylation in PGCs is indeed global and encompasses genic, intergenic, and transposon sequences. Sequences that retained the highest levels of methylation in the face of reprogramming were LTR-ERV1 and LTR-ERVK elements, approximately 10% of the latter are IAPs. PGCs at the endpoint of reprogramming have therefore attained a unique epigenetic state, with genome-wide demethylation of DNA, and loss of the repressive histone marks H3K9me2 and H2A/H4 R3me2s together with H2AZ, as well as loss of the active histone mark H3K9ac9,20. Thus an epigenetic ground state which is depleted both of key activating and repressive epigenetic marks appears to be uniquely characteristic of reprogrammed PGCs. We carried out a biological replicate of the BS-Seq experiment on E13.5 PGCs isolated by a different method (see Supplementary Methods) which indeed replicated qualitatively all conclusions with the Oct4-Gfp FACS sorted cells, while the base line was shifted to higher levels of methylation, reflecting either a greater contamination with somatic gonadal cells or epigenetic heterogeneity within germ cells that are not Oct4-Gfp positive (Supplementary Fig. 2).
Figure 2. Erasure of DNA methylation in different genomic elements in PGCs.
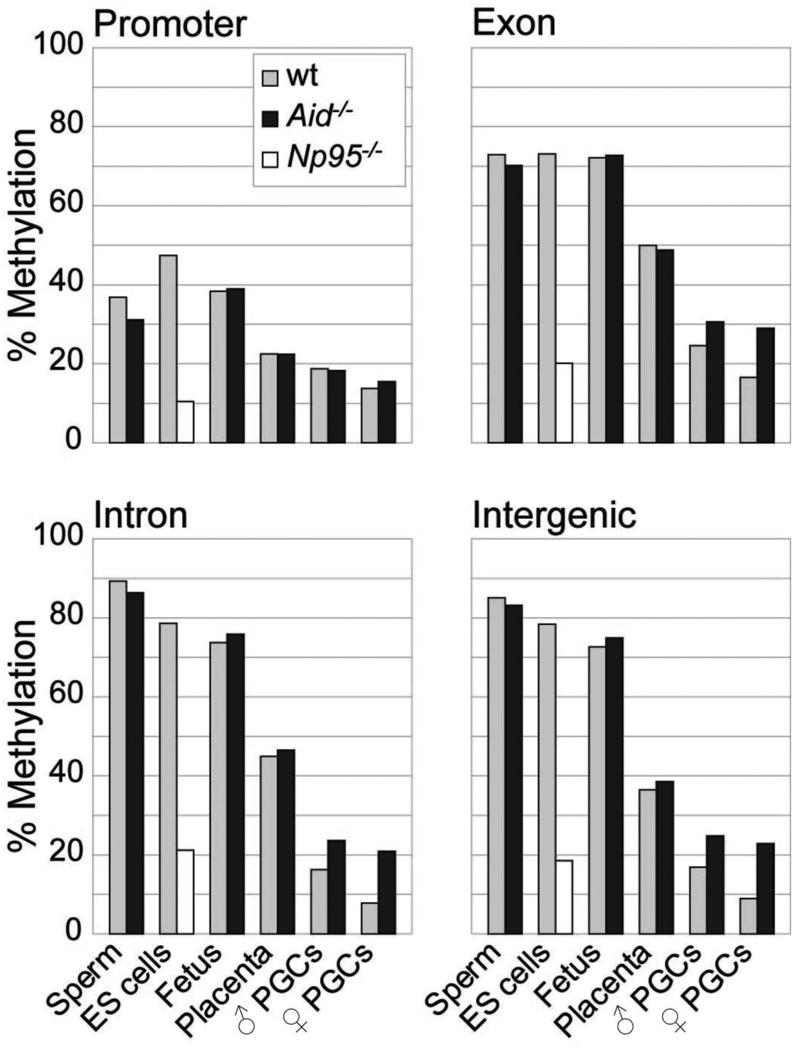
Methylation levels in promoters, exons, introns and intergenic regions in ES cells, Np95-/- ES cells, and various tissues of C57BL/6J and Aid-/- knockout mice are shown based on ratios of methylated to unmethylated BS-Seq reads. Placenta, fetal carcass and PGCs were all collected at E13.5. Note the particularly pronounced effect of Aid deficiency on methylation of introns and intergenic regions.
Figure 3. Erasure of DNA methylation in different classes of transposable elements in PGCs.

Methylation levels of different classes of transposons in ES cells, Np95-/- ES cells, and various tissues of C57BL/6J and Aid-/- knockout mice are shown based on ratios of methylated to unmethylated BS-Seq reads. Placenta, fetal carcass and PGCs were all collected at E13.5. Note that LTR-ERV1 and LTR-ERVK elements retain more methylation in PGCs than any other repeat family.
We next examined the effects of Aid deficiency on erasure of methylation in PGCs. We introgressed the Oct4-Gfp transgene into Aid-/- knockout mice21 (both on a C57Bl/6J inbred genetic background). PGCs from Aid-/- knockout mice showed substantially higher levels of methylation than wildtype ones; this difference was more pronounced in the female than in the male (Figs. 1-3), in which the mean methylation level was more than 2.5 fold higher in the Aid-/- knockout than the wildtype. Aid deficient PGCs were particularly more methylated than wildtype ones in introns and transposons (more than a 3 fold difference in LINE-L2 and in SINE-B4 elements, supplementary Fig. 1), followed by exons, while no differences were found in promoters. We were unable to analyse PGCs by BS-Seq prior to genome-wide erasure (E10.5, 11.5) because the numbers of cells that can be obtained are not currently sufficient for this technique. Previous analyses of individual gene loci and of transposons found that the methylation profile of PGCs prior to erasure is similar to that of embryonic somatic cells6-9,18, while another study suggested the overall levels to be slightly lower20. Hence, while Aid-/- PGCs are hypermethylated in comparison to wildtype ones, a significant level of demethylation also occurs in Aid-/- PGCs the extent of which depends on the precise genomic levels prior to erasure. Notably, an effect of Aid deficiency on genome-wide methylation levels was only found in primordial germ cells, and not in the fetus, the placenta, or in sperm (Figs. 1-3). These observations were again qualitatively replicated in an independent set of BS-Seq experiments using an alternative method of isolation of Aid-/- PGCs (Supplementary Fig. 2). Indeed, the ratios of the percentages of methylation between wildtype and Aid-/- PGCs were very similar when comparing the datasets obtained with the two different methods of isolation (Figs. 2,3, Supplementary Fig. 2).
The depth of sequencing in our experiments does not currently allow the comparison of methylation levels of individual gene loci between the various tissues analysed. However, given the global differences observed between wildtype and Aid-/- PGCs we compared methylation levels in randomly selected genomic loci (R1 and R2 are located in intergenic regions, R3 in the first intron of Foxo1, and R4 in the seventh exon of Xirp2), promoters or differentially methylated regions (DMRs) of genes including imprinted ones that are known to become demethylated during PGC development (Dazl, H19, Lit1), and retrotransposons by Sequenom bisulfite assay (Fig. 4, Supplementary Fig. 3). Where methylation differences were observed it was always the Aid-/- PGCs that were more highly methylated, confirming in an independent assay that Aid-/- PGCs remained relatively hypermethylated. This assay also confirmed that IAPs retained relatively high levels of methylation in PGCs (note that IAPs are a subfamily of LTR-ERVK elements and that the absolute values of methylation cannot therefore be compared between the Sequenom and BS-Seq assays). We also analysed methylation by Sequenom bisulfite assay in E12.5 PGCs, and found again relative hypermethylation of several loci in Aid-/- in comparison to wildtype PGCs (data not shown). More detailed analyses at single CpG resolution of regions that were found hypermethylated in Aid-/- PGCs (Fig. 4) revealed that predominantly there was a rather homogeneous increase in methylation across the whole of the regions analysed (Supplementary Fig. 3).
Figure 4. Analysis of DNA methylation of individual genomic loci in E13.5 PGCs by Sequenom MassArray.
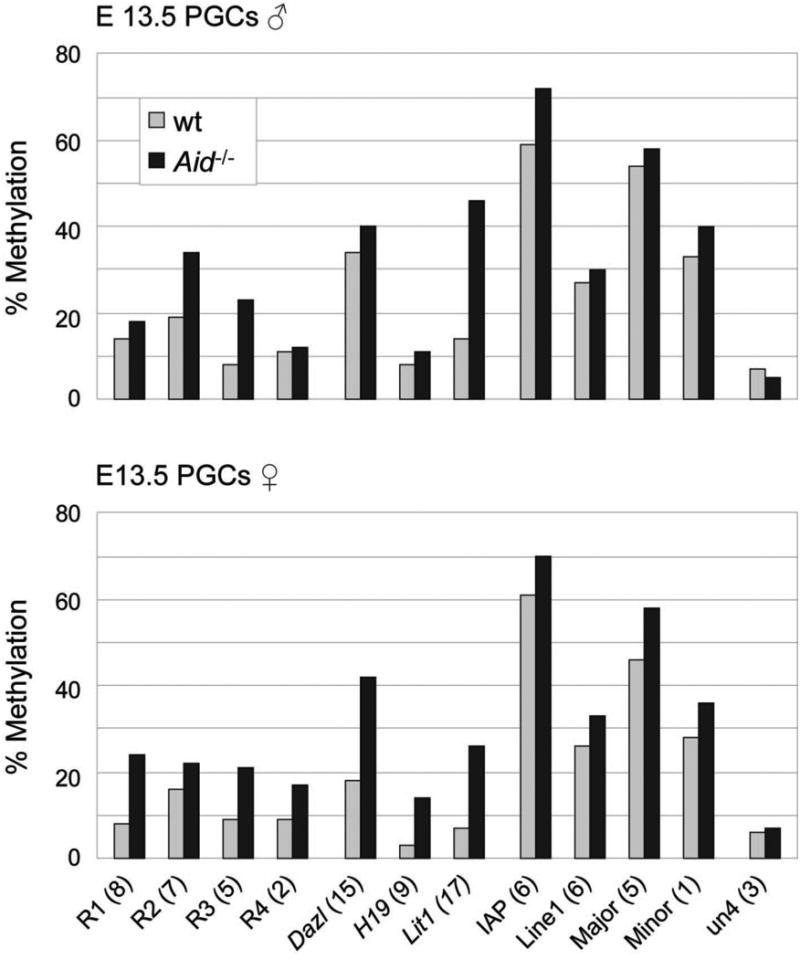
Methylation levels of individual genomic loci (R1-4, randomly selected sequences, located in intergenic regions, the first intron of Foxo1 and the seventh exon of Xirp2, respectively; the Dazl amplicon is located in the promoter region and the H19 and Lit1 amplicons are located in the differentially methylated regions), and of transposons and satellite repeats in wildtype and Aid-/- E13.5 PGCs are shown based on Sequenom Massarray analysis. The number of CpGs analysed for each region is stated in brackets. Un4, unmethylated control located in the Hoxc cluster. Note substantially increased levels of methylation in many genomic loci in Aid-/- PGCs.
Our results show that the vast majority of genomic DNA methylation is erased during normal development of PGCs. This limits the scope in mammals for inheritance of epigenetic marks (based on methylation) across generations, which if it occurred would be of potential evolutionary significance as well as affecting disease risk in humans22. Indeed in the mouse the only two well documented examples of transgenerational inheritance involve alleles of genes with an insertion of an IAP element whose DNA methylation alters the expression of the neighbouring gene22, consistent with our observation that LTR retrotransposons including IAPs are the genomic elements that are most resistant to erasure. This is in marked contrast to seed plants in which reprogramming of DNA methylation does not appear to occur in germ cells themselves23, and in which stable inheritance of epialleles across generations is more common24. Our observation that erasure of DNA methylation is less efficient in Aid-/- PGCs (especially in the female germ line) suggests that the extent to which epigenetic information is heritable in mammals might be under regulation by genetic factors. We observed in crosses between Aid deficient and wildtype parents significant effects on growth of offspring at birth as well as on litter size. Notably, Aid deficient mothers did not regulate the size of their offspring in an inverse relationship to litter size, as wildtype mothers do (Supplementary Fig. 4). Second, litter sizes from both Aid deficient mothers and fathers in crosses with wildtype animals were significantly larger than in wildtype or homozygous Aid-/- crosses (Supplementary Fig. 4). Detailed epigenetic analyses need to be carried out on mature Aid deficient germ cells (particularly oocytes) and offspring in order to examine if heritable epimutations are indeed the basis for these significant reproductive phenotypes in Aid-/- knockout mice. It cannot be excluded that kinetics of methylation reprogramming are shifted in Aid deficient animals or that further reprogramming steps occur after E13.5 in Aid-/- knockout PGCs which could further modify the differences observed here. This needs substantially increased depth of sequencing on much reduced numbers of cells which awaits further technical improvements and cost savings in BS-Seq technology.
Together with work just published in which it was shown that Aid is required in ES cells in order to demethylate pluripotency genes during reprogramming of a somatic genome by cell fusion25, our study is the first to show that Aid has a role in epigenetic reprogramming in mammals. Off-target effects of Aid have recently been described in the immune system26; in this respect it is perhaps not surprising that Aid has also evolved roles outside of the immune system. Aid might exert its substantial effect on genome-wide demethylation of DNA in PGCs through its established function as a cytosine27 or 5meC (refs. 10,28) deaminase. This can be tested genetically by examining the base excision repair pathways that are expected to be downstream of Aid, including Tdg and Mbd4. Of particular interest is a recent report in Zebrafish suggesting that Aid and Mbd4 are involved cooperatively in demethylation of DNA17; this link might also involve Gadd45 which has been implicated previously in demethylation of DNA. Notably, the effect of Aid deficiency on methylation in PGCs was considerably more pronounced than that of deficiency of the 5-methylcytosine glycosylase Demeter in the Arabidopsis endosperm15,16. Alternatively, Aid might be required more indirectly as an essential component in the pathway that regulates erasure of methylation in PGCs. Other deaminases such as Apobec1-3 might also have roles in demethylation of DNA, especially since our results show that Aid deficiency does interfere with, but does not abrogate, erasure of methylation in PGCs. It is interesting to note that in plants Demeter is specifically required for erasure of methylation in imprinted genes, while Demeter like genes are responsible for more general reprogramming of methylation patterns14. Other pathways to deamination of 5meC such as one involving the de novo methyltransferases Dnmt3a and b are less likely to operate in PGCs since Dnmt3a is not expressed and Dnmt3b is excluded from the nucleus at the time of erasure of methylation6. Finally, our results do not exclude the existence of other pathways to demethylation of DNA in mammals including oxidation of 5meC by the recently described TET family of 5meC hydroxylases29,30, or removal of 5meC by glycosylases.
Methods Summary
Mouse tissues, including male and female PGCs at E13.5, were isolated from C57BL/6J, C57BL/6J mice transgenic for Oct4-Gfp, or Aid-/- knockout mice bred into a C57BL/6J background for 7 generations prior to this study. The Oct4-Gfp transgene was subsequently bred into the Aid-/- knockout. PGCs were isolated on a FACS-Aria cell sorter by sorting for green fluorescence; the isolated cell populations were > 98% pure. DNA was isolated, bisulfite converted, and prepared for Illumina Solexa libraries. Each Illumina Solexa library was sequenced in a single end read, except for Oct4-Gfp isolated PGC libraries which were sequenced in two single end reads each; subsequently, a published highly customized software package was used to carry out Gaussian basecalling and sequence alignment for bisulfite converted reads against the mouse genome11. On average, around 1.5 million aligned 27 bp reads (5.4 million 50 bp reads for the PGC libraries) were obtained for each library. For methylation analysis, bases 6 to 22 in the 27 bp reads (bases 15 to 41 in the 50 bp reads) were used, and CpGs were base called as methylated or unmethylated, respectively. Genome-wide averages of DNA methylation of individual samples, or averages of methylation in promoters, exons, introns, remainder of the genome, and different classes of transposons, were bioinformatically determined. For Sequenom MassArray, bisulfite converted DNA was amplified and subjected to quantitative analysis of methylation by masspectrometry.
Methods
Mice and isolation of tissue and DNA samples
Mice deficient for Aid have been described previously21 and were kindly provided by Dr T. Honjo. These were backcrossed for 7 generations into the C57BL/6J strain during the course of this study. C57BL/6J mice or C57BL/6J mice carrying an Oct4-Gfp transgene were used as controls throughout. The Oct4-Gfp transgene was bred into the Aid-/- knockout following backcrossing into the C57BL/6J background. Epididymal sperm was collected from fertile adult males. PGCs were isolated on a FACS-Aria cell sorter by sorting for green fluorescence; the isolated cell populations were > 98%. Placentas and carcasses were taken from fetuses used for PGC collection. Genomic DNA from wild-type (E14) and Np95-/- mouse ES cells was kindly provided by Amander Clarke.
Shotgun bisulfite sequencing and computational analysis of data
Genomic DNA extracted from various mouse tissues with the Qiagen blood and tissue kit were treated with sodium bisulfite and then used to generate Illumina/Solexa sequencing libraries as described previously11, except that fewer cycles of PCR amplification were used (15 cycles instead of 18 cycles) in order to optimize the base composition of the libraries. For PGC samples, due to the limited sources of tissue, the input DNA amount for libraries had to be reduced to as low as 150 ng. Therefore, the enzymatic reaction steps used for library construction (including reagents and adapters for PCR) were scaled down to accommodate the reduced input amount. On the other hand, more DNA template (in volume) was used in each PCR reaction and more duplicate PCR reactions were performed in parallel in order to obtain equivalent amounts of product as for the other libraries. The libraries were sequenced on an Ultra-high-throughput Illumina/Solexa 1G Genome Analyzer following manufacturer's instructions. Initial sequencing data analysis was performed using version 0.3 of the Illumina/Solexa Analysis Pipeline; subsequently, a previously published highly customized software package was used to carry out Gaussian basecalling and sequence alignment for bisulfite converted reads against the mouse genome11. Around 5.4 million aligned 50 bp reads were obtained for PGC libraries isolated by Oct4-Gfp FACS sorting, while sequencing of all other libraries yielded on average 1.5 million aligned 27 bp reads. For methylation status analysis, bases 15 to 41 in the 50bp reads and bases 6 to 22 in the 27 bp reads were used, equalling a coverage of around 5.8% and 1% respectively. Methylated cytosines were identified as cytosines (or guanines as appropriate) in sequencing reads aligned to genomic cytosines, while unmethylated cytosines were identified as thymines (or adenines as appropriate) in sequencing reads aligned to genomic cytosines. Bisulfite conversion efficiency was always above 95% as judged by conversion of cytosines in CHG and CHH contexts (data not shown). The mapped bisulfite sequences were split into three groups. Sequences not spanning a CpG were discarded, and separate lists were made for sequences showing complete methylation or complete demethylation. In the very small number of cases where the same sequence showed both methylation and demethylation it was added to both lists. Where there were multiple datasets for the same sample the methylated and unmethylated lists were merged. Analysis of the data was performed using SeqMonk (www.bioinformatics.bbsrc.ac.uk/projects/seqmonk). The methylated and unmethylated lists were merged together with the methylation status being encoded in the strand of the read (methylated=forward, unmethylated=reverse). A tile of 250 kilobase regions was overlaid on the genome and the methylation status of each tile was calculated. Tiles containing less than 10 reads were discarded, as were tiles where there were 5 or more reads with exactly the same mapped position. The methylation status was calculated as the log2 ratio of the methylated:unmethylated counts. The distribution of values showed a normal distribution and a comparison between tissues was made using a boxwhisker plot which showed the median, upper and lower quartiles and extremities (median +/- 2 × interquartile range). Any values outside this range were plotted individually as outliers. To calculate the methylation levels in specific genomic regions (promoters, genes, introns, exons, transposon families) SeqMonk was used to generate probe regions using the Ensembl features from the annotated NCBIM37 genome as a template. Total counts of overlapping reads in all of these regions across the genome were made and a single methylated:unmethylated ratio was produced. The positions of all repeats in the NCBIM37 mouse genome were extracted from Ensembl and classified into families based on their annotation. A count was made of reads which overlapped with all of these repeat regions and these counts were combined across all members of each family. A single measure per family was then made of the log2 ratio of methylated:unmethylated reads. All repeat families shown are represented by more than 1000 CpG containing reads in each dataset.
Methylation analysis by Sequenom MassArray
DNA from FACS-sorted PGCs was extracted using the AllPrep DNA/RNA Micro Kit (Qiagen). The DNA was then treated with bisulfite reagent using the two-step modification procedure outlined in the Imprint DNA Modification kit (Sigma). Primer pairs were designed using the MethPrimer program (http://www.urogene.org/methprimer/index1.html). A complete list of primers used for analysis is available on request (primers for IAPs were based on the consensus sequence of IAPLTR1a repeats which represent approximately 1.5 % of the ERV-K family). Amplification of the bisulfite converted DNA and preparation of PCR products for quantitative analysis of methylation as detected by the MassArray system was according to the protocol provided by the manufacturer.
Supplementary Material
Acknowledgments
We would like to thank Hugh Morgan for his contributions to some of the early analysis of Aid-/- mice, Anne Segonds-Pichon for help with statistical evaluation and Jonathan Hetzel for assisting in preparing the Illumina Solexa libraries and their sequencing. We also thank Svend Petersen-Mahrt, Cristina Rada, and Fatima Santos for advice and discussions. C.P. was a Boehringer-Ingelheim predoctoral Fellow. S.F. is a Howard Hughes Medical Institute Fellow of the Life Science Research Foundation. S.E.J. is an investigator of the Howard Hughes Medical Institute. This work was supported by BBSRC, MRC, EU NoE The Epigenome, and TSB (to W.R.) and by HHMI, NSF Plant Genome Research Programme, and NIH (to S.E.J.).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author contributions C.P. and W.D. isolated tissue samples and PGCs, assessed the purity of the samples, and prepared DNA. C.P. undertook genetic crosses and determined weights of mouse pups. S.F. constructed bisulfite libraries and did Illumina Solexa sequencing, S.J.C., S.A., and M.P. carried out mapping, base-calling, and computational analyses. C.P., W.D., S.F., S.J.C., S.A., M.P., S.E.J and W.R. analysed data. C.P., W.D., S.F., S.E.J and W.R. designed experiments; S.E.J and W.R. designed and directed the study. C.P. and W.R. wrote the manuscript.
Author information All sequencing files have been deposited on GEO under accession codes XXX. Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 2.Sasaki H, Matsui Y. Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nature Rev Genet. 2008;9:129–140. doi: 10.1038/nrg2295. [DOI] [PubMed] [Google Scholar]
- 3.Oswald J, et al. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000;10:475–478. doi: 10.1016/s0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- 4.Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature. 2000;403:501–501. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 5.Dean W, et al. Conservation of methylation reprogramming in mammalian development: aberrant reprogramming in cloned embryos. Proc Natl Acad Soc USA. 2001;98:13734–13734. doi: 10.1073/pnas.241522698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hajkova P, et al. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- 7.Lee J, et al. Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development. 2002;129:1807–1817. doi: 10.1242/dev.129.8.1807. [DOI] [PubMed] [Google Scholar]
- 8.Yamazaki Y, et al. Reprogramming of primordial germ cells begins before migration into the genital ridge, making these cells inadequate donors for reproductive cloning. Proc Natl Acad Sci USA. 2003;100:12207–12212. doi: 10.1073/pnas.2035119100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hajkova P, et al. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452:877–881. doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 11.Cokus SJ, et al. Shotgun bisulfite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meissner A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gehring M, Reik W, Henikoff S. DNA demethylation by DNA repair. Trends Genet. 2009;25:82–90. doi: 10.1016/j.tig.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Gehring M, Bubb KL, Henikoff S. Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science. 2009;324:1447–1451. doi: 10.1126/science.1171609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsieh TF, et al. Genome-wide demethylation of Arabidopsis endosperm. Science. 2009;324:1451–1454. doi: 10.1126/science.1172417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rai K, et al. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lane N, et al. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis. 2003;35:88–93. doi: 10.1002/gene.10168. [DOI] [PubMed] [Google Scholar]
- 19.Zvetkova I, et al. Global hypomethylation of the genome in XX embryonic stem cells. Nature Genet. 2005;37:1274–1279. doi: 10.1038/ng1663. [DOI] [PubMed] [Google Scholar]
- 20.Seki T, et al. Cellular dynamics associated with the genome-wide epigenetic reprogramming in migrating primordial germ cells in mice. Development. 2007;134:2627–2638. doi: 10.1242/dev.005611. [DOI] [PubMed] [Google Scholar]
- 21.Muramatsu M, et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potent RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 22.Whitelaw NC, Whitelaw E. Transgenerational epigenetic inheritance in health and disease. Curr Opin Genet Dev. 2008;18:273–279. doi: 10.1016/j.gde.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 23.Slotkin RK, et al. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell. 2009;136:461–472. doi: 10.1016/j.cell.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teixeira FK, et al. A role for RNAi in the selective correction of DNA methylation defects. Science. 2009;323:1600–1604. doi: 10.1126/science.1165313. [DOI] [PubMed] [Google Scholar]
- 25.Bhutani N, et al. Reprogramming towards pluripotency requires Aid-dependent DNA demethylation. Nature. 2010 doi: 10.1038/nature08752. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robbiani DF, et al. Aid produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell. 2009;36:631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neuberger MS, Harris RS, Di Noia J, Petersen-Mahrt SK. Immunity through DNA deamination. Trends Biochem Sci. 2003;28:305–312. doi: 10.1016/S0968-0004(03)00111-7. [DOI] [PubMed] [Google Scholar]
- 28.Larijani M, et al. Methylation protects cytidines from Aid-mediated deamination. Mol Immunol. 2005;42:599–604. doi: 10.1016/j.molimm.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 29.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.