

Abstract
We report a series of inhibitors of secreted phospholipases A2 (sPLA2s) based on substituted indoles, 6,7-benzoindoles, and indolizines derived from LY315920, a well-known indole-based sPLA2 inhibitor. Using the human group X sPLA2 crystal structure, we prepared a highly potent and selective indole-based inhibitor of this enzyme. Also, we report human and mouse group IIA and IIE specific inhibitors and a substituted 6,7-benzoindole that inhibits nearly all human and mouse sPLA2s in the low nanomolar range.
Introduction
Secreted phospholipases A2 (sPLA2s)a are a family of disulfide-rich, Ca2+-dependent enzymes that hydrolyze the sn-2 position of glycero-phospholipids to release a fatty acid and a lysophospholipid.1 The mouse genome encodes 10 sPLA2s (groups IB, IIA, IIC, IID, IIE, IIF, III, V, X, XIIA), whereas the human genome encodes all of these except the group IIC enzyme, which occurs as a pseudogene.2,3 More than a decade ago there was interest in human group IIA sPLA2 (hGIIA) as an anti-inflammatory drug target because it was found at high concentrations in synovial fluid from arthritis patients,4 although a clinical trial with an inhibitor against hGIIA failed to show efficacy in the treatment of rheumatoid arthritis.5 Interest in inhibitors of sPLA2s has remained because of the possible involvement of these enzymes in inflammation. For example, studies with mGX- and mGV-deficient mice show that these sPLA2s contribute to airway inflammation in a mouse model of allergic asthma.6,7 Studies with macrophages from mGV-deficient mice show a partial reduction in eicosanoid production in response to agonists.8
Substituted indoles and indolizines first reported by workers at Lilly and Shionogi are the most potent sPLA2 inhibitors and the ones with drug potential in terms of pharmacokinetic profiles. Compounds in this group include the indolizine Indoxam and the substituted indoles Me-Indoxam and 1 (LY315920; Figure 1).9–12 The development of these compounds is an early example of structure-guided improvement of binding potency starting from a lead compound obtained through high-throughput screening13 and making use of the X-ray structure of hGIIA.14
Figure 1.

Substituted indole and indolizine sPLA2 inhibitors.
With the availability of the full set of mouse and human recombinant sPLA2s, it has been recently possible to explore the specificity of these compounds against all mammalian family members.15–17 For example, Me-Indoxam inhibits hGIIA, mGIIA, mGIIC, hGIIE, mGIIE, hGV, and mGV sPLA2s with low nanomolar potency, is less potent on hGIB, mGIB, hGX, and mGX, and inhibits hGIID, mGIID, hGXIIA, and mGXIIA only at micromolar concentrations.15 Compound 1 potently inhibits hGIIA, mGIIA, hGIIE, mGIIE, hGX, and mGX enzymes and is less potent on the other mammalian sPLA2s.17 In the current study we have taken a structure-guided approach using the X-ray structure of hGX16,18 to obtain inhibitors in the class shown in Figure 1 that are highly specific for hGX. Along the way we also obtained a highly specific inhibitor that binds only to hGIIA, mGIIA, hGIIE, and mGIIE as well as a broadly potent inhibitor that shows strong inhibition against human and mouse GIB, GIIA, GIID, GIIE, GIIF, GV and GX sPLA2s. These compounds may be useful in the study of the role of various mammalian sPLA2s in cellular and whole animal responses.
Chemistry
Reported compounds were prepared using slightly modified routes.9–12,17,19 The substituted indole and 6,7-benzoindole inhibitors were prepared using analogous routes starting from 2-carbomethoxy-4-methoxy-indole 4a and 2-carbomethoxy-4-methoxy-6,7-benzoindole 4b, respectively. However, because 4b could not be purchased commercially, it was prepared from commercially available 3-methoxy-2-naphthalenemethanol 2a (Scheme 1). 3-Methoxy-2-naphthalenemethanol (2a) was oxidized with PCC to form the aldehyde 2b. The aldehyde was treated with methyl azidoacetate and sodium methoxide to form the azidocinamate 3. Ring closure of 3 was achieved via the Hemetsberger reaction to give 2-carbomethoxy-4-methoxy-6,7-benzoindole 4b.
Scheme 1a.

aReagents and conditions: (a) PCC, NaAcetate in CH2Cl2; (b) methyl azidoacetate, NaOMe in THF; (c) xylene or toluene, reflux.
Indole-based inhibitors 11c, 11d, 12a, and 12b were prepared by N-1 benzylation of commercially available 4a using sodium hydride as the base to yield 5a (Scheme 2). The methyl ester was saponified to form the 2-carboxylic acid indole 6a. The 2-acetyl indole 7a was formed by treatment of 6a with methyllithium. Reduction of the ketone was carried out with NaBH4 to yield 8a. Deoxygenation of 8a was achieved using a mixture of NaBH4 and trifluoroacetic acid to give 9a. The 2-isobutyl indole intermediate 9b was prepared in a similar fashion as 9a except isobutyllithium was used in place of methyllithium to form 7b with subsequent transformations to give 9b. Compounds 10a–d were prepared by first deprotecting the 4-methoxy substituent on 9a and 9b using BBr3 followed by addition of the appropriate alkyl bromoactetate or 2-bromo-N-(arylsulfonyl)acetamide with sodium hydride as the base. Addition of the oxalamide group to the indole was carried out by treating 10a–d with oxalyl chloride followed by addition of ammonia gas to give compounds 11a–d. Deprotection of the indole esters 11a and 11b was carried out with NaOH to give 12a or with trifluoroacetic acid to yield 12b.
Scheme 2a.

aReagents and conditions: (a) NaH, BnBr in DMF; (b) 30% KOH/MeOH/THF (2:1:1), reflux; (c) MeLi or isopropyllithium in THF; (d) NaBH4 in EtOH/THF; (e) NaBH4, TFA in CH2Cl2; (f) (1) BBr3 in CH2Cl2, (2) NaH R2COCHR3Br in DMF; (g) (1) oxalyl chloride in CH2Cl2, (2) NH3(g); (h) 1.5 M NaOH in MeOH/THF or 20% TFA in CH2Cl2.
Preparation of the 6,7-benzoindole inhibitors 11g, 11h, 12e, and 12f was done using identical routes described for the substituted indole inhibitors (Scheme 2). Compounds 14a and 14b, N-methyl amides 15a and 15b, and all 11d derivatives were prepared using analogous steps to those outlined in Scheme 2. All Indoxam derivatives (15c and 16a–c) were prepared using similar techniques to those already described.12
Results and Discussion
Molecular Modeling
We recently reported that compound 1 was 30 times more potent than the 2-methyl indole against hGX.17 We explored this gain in selectivity by docking indole compounds with larger 2-alkyl groups into the hGIIA and hGX sPLA2 active sites of existing X-ray crystal structures13,16 using the FLO/QXP docking program.20 An overlay of the hGIIA and hGX enzyme structures (rms Cα = 0.98 Å) revealed a region of extra space in the hGX active site not present in hGIIA. This difference in hydrophobic space results mostly from a change in one amino acid residue. hGIIA has an isoleucine whereas hGX has a valine in the active site region which is contacted by the 2-position substituent on the indole ring (Figure 2). Larger 2-alkyl substituents would clash with this portion of the hGIIA active site but not in the case of hGX. Our designs were supported by data from workers at Shionogi showing that 2-isobutyl indole and indole-like inhibitors selectively inhibited the hGX enzyme.21 However, this report only included IC50 values for these compounds against hGIB, hGIIA, hGV, and hGX. As a group X specific inhibitor would be extremely useful, we wanted to test 2-isobutyl indole derivatives against all human and mouse sPLA2 enzymes.
Figure 2.

An overlay of hGIIA (green) and hGX (magenta) with 12b docked into the active site. The isoleucine of hGIIA, but not the valine of hGX, provides extra hydrophobic bulk near the 2-isobutyl group on the indole ring and presumably excludes the 2-isobutyl indole from the active site.
Also, in attempts to increase hydrophobicity of these compounds in order to make them more cell permeable, docking studies revealed that larger substituents such as arylsulfonamides or alkylsulfonamides could replace the carboxylic acid OH group on the indole scaffold. In our previous studies, addition of a methyl group to the 6-position on the indole scaffold did not affect inhibition potency against the various sPLA2s tested.17 Larger groups including a benzene ring fused to the 6,7-position of the indole scaffold were also docked into the active site without affecting key binding interactions.
In Vitro Inhibiton
Using a fluorometric sPLA2 assay,16 the substituted indoles, 6,7-benzoindoles, and indolizines were tested against the full panel of human and mouse sPLA2 enzymes, with the exception of mGIIC (because humans contain a group IIC pseudogene) and mGXIIA, which has 94% sequence identity to hGXIIA.15 All reported compounds in this study except 13a–i, 14b, and 15a–c were tested against hGIII and hGXIIA sPLA2 enzymes, and gave <50% inhibition for both enzymes at 1.6 µM concentrations. The active sites of GIII and GXIIA sPLA2 are predicted to be significantly different than those of the other mammalian sPLA2s, and this probably explains why the indole/indolizine set of inhibitors lack potency on GIII and GXIIA enzymes. IC50 values generated against hGIID were obtained using the [3H]oleic acid-labeled E. coli membrane assay, which was preferred for this enzyme because of the higher sensitivity achieved over the fluorometric assay. Data in Table 1 show that 11d and 12b are highly selective for hGX over all other human and mouse sPLA2s. Thus, the large isobutyl group is well tolerated only by hGX, which is consistent with modeling studies. Interestingly, these compounds lack potency against mGX despite the fact that hGX and mGX share 72% sequence identity. Structural alignment reveals that mGX does not contain a valine in the active site region that contacts the indole 2-position like hGX, but rather a leucine. This extra hydrophobic bulk sterically excludes the 2-isobutyl indoles from the mGX active site in similar fashion as with GIIA. Other sPLA2s such as GIB, GIIE, and GV also have an isoleucine in this region like the GIIA enzyme. However, GIID and GIIF have a valine in this region like human GX, which supports the fact that the 2-isobutyl compounds 11h and 12f display somewhat increased potency against GIID and GIIF enzymes.
Table 1.
IC50 Values of Substituted Indole Inhibitors against Human and Mouse sPLA2sa
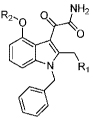 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50(nM) | ||||||||||||||||
| comp | R1 | R2 | hGIB | mGIB | hGIIA | mGIIA | hGIIDc | mGIID | hGIIE | mGIIE | hGIIF | mGIIF | hGV | mGV | hGX | mGX |
| 1b | 750±150 | 140±75 | 125±20 | 70±20 | 60±10 | 430±90 | 50±20 | 75±20 | 130±30 | >1600 | 500±50 | 750±100 | 75±10 | 75±10 | ||
| 11c | 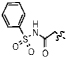 |
>1600 | 950±80 | 250±20 | 60±10 | 170±30 | 890±45 | 7±1 | 12±2 | 1100±60 | >1600 | 100±20 | 60±7 | 15±3 | 30±7 | |
| 11d |  |
>1600 | >1600 | >1600 | >1600 | 700±230 | >1600 | >1600 | >1600 | >1600 | >1600 | >1600 | >1600 | 21±7 | >1600 | |
| 12a |  |
80±20 | 100±30 | 170±40 | 60±10 | 120±40 | >1600 | 9±4 | 140±50 | 90±30 | 470±190 | 44±9 | 57±20 | 22±2 | 20±5 | |
| 12b | >1600 | 580±10 | >1600 | >1600 | 500±90 | >1600 | 1300±18 | >1600 | 550±100 | >1600 | >1600 | >1600 | 50±16 | >1600 | ||
IC50 values are reported as the mean of duplicate or triplicate analysis with standard deviations. Each compound was screened at 1660 nM and reported as >1600 nM if the inhibition was <50%.
IC50 values for GIB, GIIA, GIIE, GV, and GX from ref 17. Retest of this compound against hGV and mGV gave 110 ± 30 and 160 ± 20 nM, respectively.
IC50 values obtained using E. coli membrane assay. Each compound was screened at 1330 nM and reported as >1300 nM if the inhibition was <50%.
A small subset of 11d derivatives were synthesized and tested against hGX sPLA2 (Table 2). As initial docking studies predicted that the phenylsulfonamide group would extend out of the active site, it was surprising to see a 38-fold difference in inhibition for compounds 13b–d when the phenyl ring was substituted with a chlorine at the para-, meta-, and ortho-positions (Table 2). Compounds 13d, and 13f, with substitutions at the ortho-position with a chloro- or methyl- group, resulted in higher inhibition potency over 11d (Table 2). It is possible that the extra methyl or chlorine groups pack into a small pocket of the active site, which would increase the binding affinity. However, replacing the phenylsulfonamide on 11d with a methylsulfonamide (13h) also increases potency against hGX (Table 2). Without a crystal structure, it is difficult to conclude how this pheynlsulfonamide is contacting the enzyme active site.
Table 2.
IC50 Values of 11d Derivatives against hGX sPLA2a
 | ||
|---|---|---|
| hGX | ||
| Comp | R | IC50 (nM) |
| 13a | 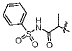 |
80±10 |
| 13b | 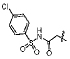 |
540±60 |
| 13c | 140±30 | |
| 13d | 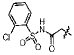 |
14±3 |
| 13e | 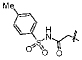 |
320±30 |
| 13f | 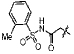 |
11±1 |
| 13g | 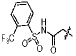 |
70±20 |
| 13h | 7±2 | |
| 13i | 30±10 | |
IC50 values are reported as the mean of duplicate or triplicate analysis with standard deviations.
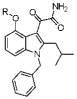
The 6,7-benzoindole inhibitors display general potency against all tested human and mouse sPLA2 enzymes (Table 3). Because the extra hydrophobic bulk is predicted not to make direct contact with the enzyme, the increased potency is likely due to increased partitioning of the inhibitor into the phospholipid substrate vesicles, which increases the ratio of XI/KI* (XI is the mole fraction of inhibitor in the interface and KI* is the interfacial dissociation constant).22,23 Of particular note is compound 12e that inhibited human and mouse groups IB, IIA, IID, IIE, IIF, V, and X sPLA2s with an IC50 of less than 350 nM (Table 3). We also sought structurally similar compounds that would be devoid of sPLA2 binding activity because such compounds are useful as controls in cellular studies. The X-ray structure of an Indoxam analogue bound to hGIIA and Me-Indoxam bound to hGX show that the carboxyl group of the substituent at the 4-position of the indole directly coordinates to the active site Ca2+.16,24 We thus synthesized 14a and 14b with only a methoxy group at the 4-position to remove the interaction made between the inhibitor and Ca2+. Surprisingly, while 14b (Figure 3) gave an IC50 of 1000 nM against hGX (data not included in table), 14a had an IC50 of 14 and 34 nM against human and mouse GIIA, respectively (Table 3). Compound 14a was also potent against hGIIE and mGIIE, consistent with trends observed for other potent group IIA indole-based inhibitors. Poor inhibiting control compounds were successfully designed by introduction of an N-methyl group on the oxalamide of the indole scaffold to give compounds 15a–15c (Figure 3). Analysis of the co-crystal structure containing Me-Indoxam in the hGX active site reveals that the introduced N-methyl group disrupts a key hydrogen bond with either a histidine or aspartate residue, while also introducing extra hydrophobic bulk into the active site.16 All N-methyl oxalamide control compounds had IC50 values that were >30-fold higher than their parent compound (Figure 3).
Table 3.
IC50 Values of Substituted Benzo-Fused Indole Inhibitors against Human and Mouse sPLA2sa
 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nm) | ||||||||||||||||
| comp | R1 | R2 | hGIB | mGIB | hGIIA | mGIIA | hGIIDb | mGIID | hGIIE | mGIIE | hGIIF | mGIIF | hGV | mGV | hGX | mGX |
| 11g | 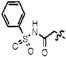 |
1300±290 | 1000±40 | 100±20 | 90±20 | 35±10 | 1000±590 | 16±1 | 48±9 | 550±120 | >1600 | 140±6 | 70±10 | 90±20 | 35±5 | |
| 11h | 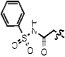 |
>1600 | 920±180 | >1600 | >1600 | 270±150 | 530±30 | 840±290 | >1600 | 290±50 | 450±120 | >1600 | >1600 | 30±4 | >1600 | |
| 12e | 84±3 | 160±40 | 40±2 | 30±1 | 7±3 | 320±5 | 7±2 | 18±2 | 50±3 | 170±33 | 35±7 | 20±1 | 20±3 | 6±1 | ||
| 12f | 1400±40 | 290±20 | >1600 | >1600 | 80±20 | 640±190 | 260±10 | 1500±170 | 90±10 | 130±20 | >1600 | >1600 | 10±2 | >1600 | ||
| 14a | 810±80 | 1600±30 | 14±2 | 34±1 | 240±4 | >1600 | 20±6 | 150±4 | >1600 | >1600 | >1600 | >1600 | 1500±270 | >1600 | ||
IC50 values are reported as the mean of duplicate or triplicate analysis with standard deviations. Each compound was screened at 1660 nM and reported as >1600 nM if the inhibition was <50%.
IC50 values obtained using E. coli membrane assay. Each compound was screened at 1330 nM and reported as >1300 nM if the inhibition was <50%.
Figure 3.

Control compounds designed by removing the functionality from the 4-postion (14b) or by methylating the oxalamide (15a–c). Control compounds are >30-fold less potent than their parent compound (listed below the compound in parenthesis) when tested against hGX (14b and 15a), human and mouse GIIA, GV, and GX (15b), or human GIIA (15c).
The 2-isobutyl Indoxam derivative 16a was synthesized and found to poorly inhibit sPLA2 enzymatic activity (Table 4). Since Indoxam does not inhibit hGX in the low nanomolar range (Table 4), it is not surprising that 16a fails to inhibit hGX. This result suggests that poor inhibition of hGX activity by Indoxam or it derivatives has more to do with the indolizine heterocyle and not the substituents present on the ring. Interestingly, the 8-oxopropanone derivative 16b and the 8-methoxy derivative 16c were selectively potent against hGIIA and hGIIE which was similar to the gain in selectivity displayed by 14a. We also prepared 15c (Figure 3), which did not significantly inhibit hGIIA at concentrations below 1600 nM.
Table 4.
IC50 Values of Substituted Indolizine Inhibitors against Human and Mouse sPLA2sa
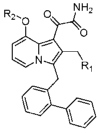 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nm) | ||||||||||||||||
| comp | R1 | R2 | hGIB | mGIB | hGIIA | mGIIA | hGIIDb | mGIID | hGIIE | mGIIE | hGIIF | mGIIF | hGV | mGV | hGX | mGX |
| Indoxam | 700±30 | 1000±60 | 60±10 | 150±40 | >1300 | >1600 | 10±2 | 35±15 | >1600 | >1600 | 100±5 | 170±10 | >1600 | 900±300 | ||
| 16a | >1600 | >1600 | >1600 | >1600 | >1300 | >1600 | >1600 | >1600 | 970±50 | 1100±200 | >1600 | >1600 | >1600 | >1600 | ||
| 16b | >1600 | 1200±110 | 30±10 | >1600 | >1300 | >1600 | 90±15 | 410±20 | >1600 | >1600 | >1600 | >1600 | >1600 | >1600 | ||
| 16c | >1600 | 320±20 | 35±2 | >1600 | >1300 | >1600 | 50±10 | 230±110 | >1600 | >1600 | >1600 | >1600 | >1600 | >1600 | ||
IC50 values are reported as the mean of duplicate or triplicate analysis with standard deviations. Each compound was screened at 1660 nM and reported as >1600 nM if the inhibition was <50%.
IC50 values obtained using E. coli membrane assay. Each compound was screened at 1330 nM and reported as >1300 nM if the inhibition was <50%.
Conclusion
A series of indole- and indolizine-based compounds were synthesized and tested against the full set of human and mouse sPLA2 enzymes. Compound 11d was found to be selectively potent against hGX over all other human and mouse sPLA2 enzymes. Derivatives of 11d, such as 13h, were also found to bind with higher affinity to the hGX enzyme active site and may help in further studies of hGX sPLA2 function. An inhibitor selective for mouse and human GIIA and GIIE sPLA2 (14a) as well as selective human GIIA and GIIE inhibitors (16b and 16c) were also identified from this group of compounds. Compound 12e is potent against human and mouse groups IB, IIA, IID, IIE, IIF, V, and X and is the most generally potent sPLA2 inhibitor reported to date. It is also the first reported potent inhibitor of groups IID and IIF sPLA2s. The inhibitors we describe may be useful in probing the roles of sPLA2s in inflammatory diseases such as asthma and arthritis.
Experimental Section
Enzyme Inhibition Assays
For compounds with IC50s <1600 nM in the fluorometric assay or <1300 nM in the E. coli membrane assay, inhibitor concentrations were varied with five different concentrations used to determine IC50 values. All IC50 values were obtained by nonlinear regression curve-fitting of percent inhibition versus log [inhibitor] using the Kaleidagraph software.
Fluorometric Assay
Microtiter plate assay of sPLA2s using pyrene-labeled phosphatidylglycerol as the substrate was performed as described previously16 with the exception that seven wells were used per assay instead of eight.
E. coli Membrane Assay
IC50 values calculated for hGIID were done using a modified procedure from that reported previously.25 See Supporting Information for details.
Synthesis
All reagents were purchased from Sigma-Aldrich and used directly unless otherwise stated. Reactions were performed under an atmosphere of dry nitrogen in oven-dried glassware. Reactions were monitored for completeness by thin layer chromatography (TLC) using Merck 60F254 silica plates, and column chromatography was done with 60 Å silica gel purchased from Silicycle. 1H NMR spectra were recorded on dilute solutions in CDCl3, CD3OD, or DMSO-d6. NMR spectra were obtained on a Bruker AC-300 (300 MHz) and electrospray ionization mass spectra were acquired on a Bruker Esquire LC00066 for all compounds. Preparative reverse phase HPLC was performed on an automated Varian Prep Star system using a YMC S5 ODS column (20 × 100 mm, Waters Inc.).
Representative Procedure for Synthesis of Substituted 6,7-Benzoindole Inhibitors (Compound 12e): Preparation of 1-Benzyl-2-carbomethoxy-4-methoxy-6,7-benzoindole (5b)
Compound 4b (synthesis described in Supporting Information; 800 mg, 3.14 mmol) was dissolved in 10 mL dry DMF and stirred at 0 °C and sodium hydride (140 mg, 5.5 mmol) was added. After stirring for five minutes at 0 °C, benzylbromide (820 uL, 6.90 mmol) was added and the reaction was stirred for 30 min at room temperature. The reaction mixture was poured onto 20 mL of H2O and 20 mL of EtOAc in a separatory funnel. The layers were separated and the organic layer was washed with 3 × 10 mL H2O and the combined aqueous layer was back-extracted with 1 × 20 mL EtOAc. The combined organic layer was dried over MgSO4 and filtered, and the solvent was removed by rotary evaporation. The crude white solid was purified by column chromatography on silica gel (20% EtOAc/80% hexanes) to give a white solid (820 mg, 75% yield). 1H NMR (300 MHz, CDCl3) δ 3.85 (s, 3H), 4.06 (s, 3H), 6.34 (bs, 2H), 6.77 (s, 1H), 7.09 (d, J = 7.2 Hz, 2H), 7.16–7.31 (m, 4H), 7.37 (t, J = 7.2 Hz, 1H), 7.68 (s, 1H), 7.78 (d, J = 8.1 Hz, 1H), 8.06 (d, J = 8.4 Hz, 1H).
Preparation of 1-Benzyl-2-carboxylic acid-4-methoxy-6,7-benzoindole (6b)
Compound 5b (485 mg, 1.41 mmol) was suspended in 15 mL of 30% KOH/MeOH/THF (2:1:1) and refluxed for 2.0 h (all the solid dissolved during reflux). After refluxing, the reaction was cooled on ice and the pH was made acidic using 2 N HCl, causing the product to precipitate. The white solid was collected by vacuum filtration and washed with 1 × 10 mL of cold water and 2 × 10 mL of cold hexanes to give a white solid (400 mg, 86% yield). 1H NMR (300 MHz, DMSO-d6) δ 4.02 (s, 3H), 6.41 (bs, 2H), 6.98 (m, 3H), 7.20–7.26 (m, 2H), 7.32 (t, J = 7.5 Hz, 2H), 7.39 (t, J = 8.1 Hz, 1H), 7.49 (s, 1H), 7.86 (d, J = 7.5 Hz, 1H), 8.12 (d, J = 8.4 Hz, 1H).
Preparation of 1-Benzyl-2-acetyl-4-methoxy-6,7-benzoindole (7c)
Compound 6b (920 mg, 1.12 mmol) was dissolved in 40 mL of dry THF to which 6.6 mL of 1.25 M MeLi in diethyl ether was added dropwise and stirred at room temperature for 2.5 h. Saturated NH4Cl (8 mL) was added followed by the addition of 2 N HCl until the mixture had an acidic pH. The reaction mixture was then poured onto 30 mL of EtOAc and 30 mL of H2O in a separatory funnel. The layers were separated and the water phase was washed with 2 × 20 mL of EtOAc. The organic layers were combined, dried over MgSO4 and filtered, and the solvent was removed by rotary evaporation. The crude white solid was triturated in 15 mL of 1:1 EtOAc/hexanes and separated from the solvent by vacuum filtration. The white solid collected via vacuum filtration was washed with 2 × 10 mL of 1:1 EtOAc/hexanes giving 6b. Additional product could be purified from the combined filtrate and washings by removing the solvent and repeating the trituration step described above followed by chromatography on silica gel (20% EtOAc/80% hexanes) of the filtrate and washings combined together from the second trituration step. The purified product was combined to afford a white solid (366 mg, 40% yield). 1H NMR (300 MHz, CDCl3) δ 2.63 (s, 3H), 4.08 (s, 3H), 6.35 (bs, 2H), 6.77 (s, 1H), 7.07 (d, J = 6.9 Hz, 2H), 7.20–7.31 (m, 4H), 7.39 (t, J = 8.1 Hz, 1H), 7.67 (s, 1H), 7.78 (d, J = 8.1 Hz, 1H), 8.09 (d, J = 8.7 Hz, 1H).
Preparation of 1-(1-Benzyl-4-methoxy-1H-6,7-benzoindol-2-yl)-ethanol (8c)
Compound 7c (366 mg, 1.11 mmol) was dissolved in 30 mL of 75% EtOH/25% THF, and NaBH4 (100 mg, 3.33 mmol) was added to the mixture and stirred at room temperature for 16 h. The reaction mixture was then poured onto 30 mL EtOAc and 30 mL H2O in a separatory funnel. The layers were separated and the water phase was washed with 2 × 20 mL of EtOAc. The organic layers were combined and washed with 2 × 20 mL of H2O and 1 × 20 mL of satd NaCl. The organic layer was dried over MgSO4 and filtered, and the solvent was removed by rotary evaporation to give 8c as a white solid that was used without further purification (355 mg, 96% yield). 1H NMR (300 MHz, CDCl3) δ 1.72 (d, J = 6.3 Hz 3H), 4.08 (s, 3H), 4.99 (m, 1H), 5.96 (d, J = 20.7 Hz, 1H), 6.09 (d, J = 20.7 Hz, 1H), 6.81 (s, 1H), 6.85 (s, 1H), 7.05 (d, J = 6.9 Hz, 2H), 7.15 (t, J = 6.9 Hz, 1H), 7.20–7.31 (m, 4H), 7.79 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 8.7 Hz, 1H).
Preparation of 1-Benzyl-2-ethyl-4-methoxy-1H-6,7-benzoindole (9c)
Compound 8c (420 mg, 1.26 mmol) was dissolved in 20 mL of dry CH2Cl2 and added dropwise to a mixture of 14 mL of 99% trifluoroacetic acid (TFA) and NaBH4 (243 mg, 6.3 mmol) at 20 °C (prepare TFA/NaBH4 mixture in an ice bath by careful dropwise addition of TFA to NaBH4 and let stir until NaBH4 fully dissolves before raising temperature). The reaction mixture was stirred at room temperature for 30 min and then poured onto 30 mL of satd NaHCO3 and 30 mL of CH2Cl2 in a separatory funnel. Once bubbling ceased, the layers were separated and the water phase was washed with 2 × 20 mL of CH2Cl2. The organic layers were combined, dried over MgSO4 and filtered, and the solvent was removed by rotary evaporation. The crude material was purified by column chromatography on silica gel (20% EtOAc/80% hexanes) to afford a white solid (335 mg, 84% yield). 1H NMR (300 MHz, CDCl3) δ 1.36 (t, J = 7.5 Hz 3H), 2.75 (q, J = 7.5 Hz 2H), 4.07 (s, 3H), 5.77 (s, 2H), 6.56 (s, 1H), 6.80 (s, 1H), 7.05 (d, J = 6.9 Hz, 2H), 7.13 (t, J = 6.9 Hz, 1H), 7.20–7.31 (m, 4H), 7.79 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 8.7 Hz, 1H).
Preparation of Methyl 2-(1-Benzyl-2-ethyl-1H-6,7-benzoindol-4-yloxy)acetate (10e)
Compound 9c (80 mg, 0.25 mmol) was dissolved in 8 mL of dry CH2Cl2 and stirred at 0 °C. BBr3 (1.0 M in CH2Cl2; 635 µL, 0.635 mmol) was added in portions over 5 min to the reaction mixture at 0 °C, and the reaction mixture was stirred for 3 h at room temperature or until product formation was complete as indicated by TLC. H2O (8 mL) was added to the reaction mixture to quench excess BBr3, and the reaction mixture was poured onto 20 mL of CH2Cl2 and 20 mL of H2O in a separatory funnel. The layers were separated and the water phase was washed with 2 × 20 mL of CH2Cl2. The organic layers were combined, dried over MgSO4 and filtered, and the solvent was removed by rotary evaporation to give the 4-hydroxy-6,7-benzoindole intermediate, which is unstable and decomposes giving a purple color upon exposure to air. After drying in vacuo for 30 min, this compound was then immediately dissolved in 4 mL of DMF and stirred in an ice bath. Sodium hydride (10.3 mg, 0.41 mmol) was added to the reaction mixture and stirred 5 min at 0 °C, with subsequent addition of methyl bromoacetate (40 µL, 0.456 mmol). The reaction was stirred at room temperature for 30 min. Additional portions of sodium hydride were added at 0 °C until the reaction was shown to be complete by TLC. The reaction mixture was then poured onto 20 mL of H2O and 20 mL of EtOAc in a separatory funnel. The layers were separated and the organic layer was washed with 4 × 10 mL of H2O, and the combined aqueous layer was back-extracted with 1 × 20 mL of EtOAc. The combined organic layers were dried over MgSO4 and filtered, and the solvent was removed by rotary evaporation. The crude material was purified by column chromatography on silica gel (20% EtOAc/80% hexanes) to afford a white solid (23 mg, 24% yield over two steps). 1H NMR (300 MHz, CDCl3) δ 1.37 (t, J = 7.5 Hz 3H), 2.75 (q, J = 7.5 Hz 2H), 3.85 (s, 3H), 4.90 (s, 2H), 5.76 (s, 2H), 6.69 (s, 1H), 6.76 (s, 1H), 7.05 (d, J = 6.9 Hz, 2H), 7.15 (t, J = 6.9 Hz, 1H), 7.20–7.31 (m, 4H), 7.75 (d, J = 8.1 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H).
Preparation of Methyl-2-(3-(2-amino-2-oxoacetyl)-1-benzyl-2-ethyl-1H-6,7-benzoindol-4-yloxy)acetate (11e)
Compound 10e (22.4 mg, 0.06 mmol) was dissolved in 7 mL of dry CH2Cl2 and added dropwise to 14 mL of dry CH2Cl2 containing oxalyl chloride (26 µL, 0.30 mmol) at room temperature. The reaction mixture was stirred overnight at room temperature. Ammonia gas was then bubbled into the reaction mixture for five minutes. The reaction mixture was then poured into a separatory funnel containing 20 mL of 2 N HCl. The layers were separated and the aqueous layer was extracted with 2 × 10 mL of CH2Cl2. The organic layers were combined, dried over MgSO4 and filtered, and the solvent was removed by rotary evaporation. The crude mixture was purified by column chromatography over silica gel (70% EtOAc/30% hexanes) to give a white yellow solid (10.9 mg, 41% yield). 1H NMR (300 MHz, CDCl3) δ 1.23 (t, J = 7.5 Hz 3H), 2.94 (q, J = 7.5 Hz 2H), 3.81 (s, 3H), 4.88 (s, 2H), 5.42 (bs, 1H), 5.81 (s, 2H), 6.72 (bs, 1H), 6.81 (s, 1H), 7.10 (d, J = 6.9 Hz, 2H), 7.17 (t, J = 6.9 Hz, 1H), 7.20–7.31 (m, 4H), 7.74 (d, J = 7.8 Hz, 1H), 7.92 (d, J = 8.4 Hz, 1H). MS (ESI, pos. ion) m/z: 467 (M + Na+).
Preparation of 2-(3-(2-Amino-2-oxoacetyl)-1-benzyl-2-ethyl-1H-6,7-benzoindol-4-yloxy)acetic Acid (12e)
Compound 11e (10.9 mg, 0.024 mmol) was dissolved in 5 mL of MeOH/THF (5:1) with 0.5 mL of 1.5 M NaOH added to the reaction mixture and stirred for 2.5 h at room temperature. The reaction mixture was then poured onto 20 mL of 2 N HCl and 20 mL of CH2Cl2 in a separatory funnel. The layers were separated and the aqueous layer was extracted with 2 × 10 mL of CH2Cl2. The combined organic layer was dried over MgSO4 and filtered, and the solvent was removed by rotary evaporation to yield 12e quantitatively. A portion of 12e was purified by HPLC using the following program (eluting solvents each contained 0.08% TFA): 0–5 min 30% MeOH/70% H2O, 5–30 min 30% MeOH/70% H2O–70% MeOH/30% H2O, 30–32 min 70% MeOH/30% H2O–100% MeOH, 32–35 min 100% MeOH. The product eluted at 24.5 min and the solvent was removed by Speed-Vac to afford a white/yellow solid (4.9 mg). 1H NMR (300 MHz, MeOD) δ 1.26 (t, J = 7.2 Hz 3H), 3.04 (q, J = 7.5 Hz 2H), 4.93 (s, 2H), 5.99 (s, 2H), 6.96 (s, 1H), 7.15–7.23 (m, 3H), 7.27–7.39 (m, 4H), 7.83 (d, J = 6.9 Hz, 1H), 8.07 (d, J = 8.4 Hz, 1H). MS (ESI, pos. ion) m/z: 431 (M+).
Supplementary Material
Acknowledgment
The authors thank Justin Siegel and Drs. Brian Smart and Zhanglin Ni for helpful discussions about the inhibitors used in this work and Dr. Christophe Verlinde for modeling discussions. This work was supported by an NIH Molecular Biophysics Training Grant (R.C.O.), the Mary Gates Foundation (N.C.), and a Merit Award from the National Institutes of Health (M.H.G.; R37HL036235).
Footnotes
Abbreviations: hGIIA, human group IIA secreted phospholipase A2 (likewise for other group names); mGIIA, mouse group IIA secreted phospholipase A2 (likewise for other group names); sPLA2, secreted phospholipase A2.
Supporting Information Available: Details of synthetic methods, including NMR and MS data, for all other described compounds, HPLC traces showing purity of key target compounds, molecular modeling details, and E. coli membrane enzyme assay procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lambeau G, Gelb MH. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 2008 doi: 10.1146/annurev.biochem.76.062405.154007. in press. [DOI] [PubMed] [Google Scholar]
- 2.Valentin E, Lambeau G. Increasing molecular diversity of secreted phospholipases A2 and their receptors and binding proteins. Biochim. Biophys. Acta. 2000;1488:59–70. doi: 10.1016/s1388-1981(00)00110-4. [DOI] [PubMed] [Google Scholar]
- 3.Six DA, Dennis EA. The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim. Biophys. Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 4.Pruzanski W. Phospholipase A2: quo vadis. J. Rheumatol. 2005;32:400–402. [PubMed] [Google Scholar]
- 5.Bradley JD, Dmintrienko AA, Kivitz AJ, Gluck OS, Weaver AL, Wiesenhutter C, Myers SL, Sides GD. Randomized, double-blinded, placebo-controlled clinical trial of LY333013, a selective inhibitor of group II secretory phospholipases A2, in the treatment of rheumatoid arthritis. J. Rheumatol. 2005;32:417–423. [PubMed] [Google Scholar]
- 6.Henderson WR, Jr, Chi EY, Bollinger JG, Tien YT, Ye X, Castelli L, Rubtsov YP, Singer AG, Chiang GK, Nevalainen T, Rudensky AY, Gelb MH. Importance of group X-secreted phospholipase A2 in allergen-induced airway inflammation and remodeling in a mouse asthma model. J. Exp. Med. 2007;204:865–877. doi: 10.1084/jem.20070029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munoz NM, Meliton AY, Arm JP, Bonventre JV, Cho W, Leff AR. Deletion of secretory group V phospholipase A2 attentuates cell migration and airway hyperresponsiveness in immunosensitized mice. J. Immunol. 2007;179:4800–4807. doi: 10.4049/jimmunol.179.7.4800. [DOI] [PubMed] [Google Scholar]
- 8.Satake Y, Diaz BL, Balestrieri B, Lam BK, Kanaoka Y, Grusby MJ, Arm JP. Role of group V phospholipase A2 in zymosan-induced eicosanoid generation and vascular permeability revealed by targeted gene disruption. J. Biol. Chem. 2004;279:16488–16494. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dillard RD, Bach NJ, Draheim SE, Berry DR, Carlson DG, Chirgadze NY, Clawson DK, Hartley LW, Johnson LM, Jones ND, McKinney ER, Mihelich ED, Olkowski JL, Schevitz RW, Smith AC, Snyder DW, Sommers CD, Wery JP. Indole inhibitors of human nonpancreatic secretory phospholipase A2. 1. Indole-3-acetamides. J. Med. Chem. 1996;39:5119–5136. doi: 10.1021/jm960485v. [DOI] [PubMed] [Google Scholar]
- 10.Dillard RD, Bach NJ, Draheim SE, Berry DR, Carlson DG, Chirgadze NY, Clawson DK, Hartley LW, Johnson LM, Jones ND, McKinney ER, Mihelich ED, Olkowski JL, Schevitz RW, Smith AC, Snyder DW, Sommers CD, Wery JP. Indole inhibitors of human nonpancreatic secretory phospholipase A2. 2. Indole-3-acetamides with additional functionality. J. Med. Chem. 1996;39:5137–5158. doi: 10.1021/jm960486n. [DOI] [PubMed] [Google Scholar]
- 11.Draheim SE, Bach NJ, Dillard RD, Berry DR, Carlson DG, Chirgadze NY, Clawson DK, Hartley LW, Johnson LM, Jones ND, McKinney ER, Mihelich ED, Olkowski JL, Schevitz RW, Smith AC, Snyder DW, Sommers CD, Wery JP. Indole inhibitors of human nonpancreatic secretory phospholipase A2. 3. Indole-3-glyoxamides. J. Med. Chem. 1996;39:5159–5175. doi: 10.1021/jm960487f. [DOI] [PubMed] [Google Scholar]
- 12.Hagishita S, Yamada M, Shirahase K, Okada T, Murakami Y, Ito Y, Matsuura T, Wada M, Kato T, et al. Potent inhibitors of secretory phospholipase A2: Synthesis and inhibitory activities of indolizine and indene derivatives. J. Med. Chem. 1996;39:3636–3658. doi: 10.1021/jm960395q. [DOI] [PubMed] [Google Scholar]
- 13.Schevitz RW, Bach NJ, Carlson DG, Chigadze SE, Hartley LW, Jones ND, Mehilich ED, et al. Structure-based design of the first potent and selective inhibitor of human non-pancreatic secretory phospholipase A2. Nat. Struct. Biol. 1995;2:458–465. doi: 10.1038/nsb0695-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott DL, White SP, Browning JL, Rosa JJ, Gelb MH, Sigler PB. Structures of free and inhibited human secretory phospholipase A2 from inflammatory exudate. Science. 1991;254:1007–1010. doi: 10.1126/science.1948070. [DOI] [PubMed] [Google Scholar]
- 15.Singer AG, Ghomashchi F, Le Calvez C, Bollinger J, Bezzine S, Rouault M, Sadilek M, Nguyen E, Lazdunski M, Lambeau G, Gelb MH. Interfacial kinetic and binding properties of complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J. Biol. Chem. 2002;277:48535–48549. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- 16.Smart BP, Pan YH, Weeks AK, Bollinger JG, Bahnson BJ, Gelb MH. Inhibition of the complete set of mammalian secreted phospholipases A2 by indole analogs: A structure-guided study. Bioorg. Med. Chem. 2004;12:1737–1749. doi: 10.1016/j.bmc.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 17.Smart BP, Oslund RC, Walsh LA, Gelb MH. The first potent inhibitor of mammalian group X secreted phospholipase A2: Elucidation of sites for enhanced binding. J. Med. Chem. 2006;49:2858–2860. doi: 10.1021/jm060136t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan YH, Yu BZ, Singer AG, Ghomashchi F, Lambeau G, Gelb MH, Jain MK, Bahnson BJ. Crystal structure of human group X secreted phospholipase A2. Electrostatically neutral interfacial surface targets zwitterionic membranes. J. Biol. Chem. 2002;277:29086–29093. doi: 10.1074/jbc.M202531200. [DOI] [PubMed] [Google Scholar]
- 19.Sawyer JS, Beight DW, Smith EC, Snyder DW, Chastain MK, Tielking RL, Hartley LW, Carlson DG. Carbocyclic[g]indole inhibitors of human nonpancreatic s-PLA2. J. Med. Chem. 2005;48:893–896. doi: 10.1021/jm0401309. [DOI] [PubMed] [Google Scholar]
- 20.McMartin C, Bohacek RS. QXP: Powerful, rapid computer algorithms for structure-based drug design. J. Comput.-Aided Mol. Des. 1997;11:333–344. doi: 10.1023/a:1007907728892. [DOI] [PubMed] [Google Scholar]
- 21.Ogawa T, Seno K, Hanasaki K, Ikeda M, Ono T. Compounds exhibiting X-type sPLA2 inhibiting effect. 7026318B2. U.S. Patent. 2006
- 22.Jain MK, Yuan W, Gelb MH. Competitive inhibition of phospholipase A2 in vesicles. Biochemistry. 1989;28:4135–4139. doi: 10.1021/bi00436a002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain MK, Yu B-Z, Gelb MH, Berg OG. Assay of phospholipase A2 and their inhibitors by kinetic analysis in the scooting mode. Med. Inflamm. 1992;1:85–100. doi: 10.1155/S0962935192000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitadokoro K, Hagishita S, Sato T, Ohtani M, Miki K. Crystal structure of human secretory phospholipase A2-IIA complex with the potent indolizine inhibitor 120–1032. J. Biochem. 1998;123:619–623. doi: 10.1093/oxfordjournals.jbchem.a021982. [DOI] [PubMed] [Google Scholar]
- 25.Ancian P, Lambeau G, Lazdunski M. Multifunctional activity of the extracellular domain of the M-type (180 kDa) membrane receptor for secretory phospholipases A2. Biochemistry. 1995;34:13146–13151. doi: 10.1021/bi00040a028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.