

Abstract
The t-tubules of mammalian ventricular myocytes are invaginations of the cell membrane that occur at each Z-line. These invaginations branch within the cell to form a complex network that allows rapid propagation of the electrical signal, and hence synchronous rise of intracellular calcium (Ca2+). To investigate how the t-tubule microanatomy and the distribution of membrane Ca2+ flux affect cardiac excitation-contraction coupling we developed a 3-D continuum model of Ca2+ signaling, buffering and diffusion in rat ventricular myocytes. The transverse-axial t-tubule geometry was derived from light microscopy structural data. To solve the nonlinear reaction-diffusion system we extended SMOL software tool (http://mccammon.ucsd.edu/smol/). The analysis suggests that the quantitative understanding of the Ca2+ signaling requires more accurate knowledge of the t-tubule ultra-structure and Ca2+ flux distribution along the sarcolemma. The results reveal the important role for mobile and stationary Ca2+ buffers, including the Ca2+ indicator dye. In agreement with experiment, in the presence of fluorescence dye and inhibited sarcoplasmic reticulum, the lack of detectible differences in the depolarization-evoked Ca2+ transients was found when the Ca2+ flux was heterogeneously distributed along the sarcolemma. In the absence of fluorescence dye, strongly non-uniform Ca2+ signals are predicted. Even at modest elevation of Ca2+, reached during Ca2+ influx, large and steep Ca2+ gradients are found in the narrow sub-sarcolemmal space. The model predicts that the branched t-tubule structure and changes in the normal Ca2+ flux density along the cell membrane support initiation and propagation of Ca2+ waves in rat myocytes.
Author Summary
In cardiac muscle cells, calcium (Ca2+) is best known for its role in contraction activation. A remarkable amount of quantitative data on cardiac cell structure, ion-transporting protein distributions and intracellular Ca2+ dynamics has been accumulated. Various alterations in the protein distributions or cell ultra-structure are now recognized to be the primary mechanisms of cardiac dysfunction in a diverse range of common pathologies including cardiac arrhythmias and hypertrophy. Using a 3-D computational model, incorporating more realistic transverse-axial t-tubule geometry and considering geometric irregularities and inhomogeneities in the distribution of ion-transporting proteins, we analyze several important spatial and temporal features of Ca2+ signaling in rat ventricular myocytes. This study demonstrates that the computational models could serve as powerful tools for prediction and analyses of how the Ca2+ dynamics and cardiac excitation-contraction coupling are regulated under normal conditions or certain pathologies. The use of computational and mathematical approaches will help also to better understand aspects of cell functions that are not currently amenable to experimental investigation.
Introduction
Ventricular cardiac muscle cells have deep invaginations of the extracellular space known as t-tubules [1]–[14]. In rodents, these invaginations branch within the cell to form a complex network that allows rapid propagation of the electrical signal (i.e. the action potential, AP) to the subcellular location (i.e. the sarcoplasmic reticulum, SR) where the intracellular Ca2+ required for the cell contraction is stored [14]. The release of Ca2+ from the SR depends on “trigger Ca2+” entering the cytosol from the extracellular space by activating sarcolemmal Ca2+ channels (L-type Ca2+ channels, LCC) and by Ca2+ entry via Na+/Ca2+ exchanger (NCX), [3], [9], [14], [15]. The trigger Ca2+ activates SR Ca2+ release channels (ryanodine receptors, RyRs) by the process of “Ca2+-induces Ca2+-release” (CICR) which amplifies the modest increase in intracellular Ca2+ concentration ([Ca2+]i) caused by the LCC and NCX influxes to provide sufficient Ca2+ for the proteins regulating muscle force (i.e. troponin C, TN) ), [14]. Thus, by working together, the microanatomy of t-tubules and SR permits spatially homogeneous and synchronized SR Ca2+ release and spatially uniform Ca2+ transients throughout the cell [5], [14], [16]. It has been also observed that the spatially uniform Ca2+ transients might be achieved if the SR Ca2+ release and uptake are abolished [5]. Yet, despite a wealth of information on ventricular cell function and structure, the mechanisms causing the synchrony of activation and the similarity of levels of [Ca2+]i across the myocyte still remain unclear.
In cardiac muscle cells, several computational models have been introduced to investigate the Ca2+ signaling, buffering and diffusion [17]–[19] and Ca2+ wave initialization and propagation [12], [20]–[23]. All these studies, however, are conducted on simplified geometries (such as cylindrical or rectangular shapes) and it has been pointed out that a small geometric change (even in the case the change is uniformly applied) could greatly influence the suggested homogeneous Ca2+ distribution by initiating wave propagation in the computer simulation [20], [22]. Several laboratories, using common pool modeling approaches, have investigated also the effects of LCC and NCX distributions on global [Ca2+]i transients in dyadic, sub-sarcolemmal and cytosol compartments [10], [24], [25]. Recently, to examine how the distribution of Ca2+ flux along the sarcolemma affects Ca2+-entry and Ca2+ diffusion and buffering, we developed a 3-D continuum model in rat ventricular cells [19]. An important limitation of this model is that a cylindrical t-tubule geometry was assumed while several studies have provided evidence that in rodent ventricular myocytes the realistic t-tubule geometry is quite complex (with large local variations in the diameter and transverse-axial anatomies), [9]–[12]. These experimental findings suggest that replacing our idealistic t-tubule model with a realistic geometry is needed. The use of idealistic shapes will change the diffusion distances and realistic Ca2+-transporting protein localizations in plane and depth directions and consequently the predicted [Ca2+]i distributions.
In the present study, we sought to develop a morphologically correct geometric model of the t-tubule and to use this model for computational studies of the intracellular Ca2+ dynamics. We examined the Ca2+ signaling in rat ventricular myocytes that had been treated with ryanodine and thapsigargin to eliminate Ca2+ release and uptake by the SR. By using published electro-physiological data and laser-scanning confocal [Ca2+]i measurements, we were able to analyze several important spatial and temporal features of the Ca2+ signals in these cells. In this context, our goal was at least three-fold. The first aim was to develop a mathematical model that would be in qualitative or quantitative agreement with published experimental measurements on Ca2+ influx, and Ca2+ buffering and diffusion in rat ventricular cells with SR function inhibited [5], [26]. Second, to use the model to investigate the importance of t-tubule ultra-structure and membrane Ca2+ flux distribution for the Ca2+ signals. The third task was to simulate the Ca2+ signals in the absence of fluorescent dye and to study the importance of the mobility of endogenous Ca2+ buffers (ATP and calmodulin) and altered extracellular Na+ ([Na+]e) for the Ca2+ signals. The analysis suggests that the quantitative understanding of the Ca2+ signaling requires more accurate knowledge of the t-tubule microanatomy and Ca2+ flux distribution along the sarcolemma. In agreement with experiment, with 100 µM Fluo-3, the lack of detectible differences in the depolarization-evoked Ca2+ transients was found when the Ca2+ flux was heterogeneously distributed along the sarcolemma. In the absence of Fluo-3, the predicted Ca2+ signals were strongly non-uniform. Even at modest elevation of Ca2+, reached during Ca2+ influx, large and steep Ca2+ gradients may develop in the narrow sub-sarcolemmal space. The model also predicts that branched t-tubule topology and changes in the normal Ca2+ flux density along the cell membrane support Ca2+ waves initiated at the sarcolemma. Preliminary results of this work have been presented to the Biophysical Society in abstract form [27].
Materials and Methods
3-D imaging and geometric modeling of t-tubule microanatomy
Combining light microscopy (LM) and electron microscopy (EM) together with 3-D tomographic reconstruction, Hayashi et al. [6] investigated 3-D topologies of important sub-cellular organelles, including dyadic clefts and t-tubules, in mouse ventricular myocytes. In particular, the use of two-photon microscopy (T-PM) in their studies had provided data showing detailed spatial organization of t-tubules (see Fig. 1A upper panel) that was important for the development of our realistic model for computational studies of intracellular Ca2+ dynamics. The gap between imaging and simulation involves two major steps: (1) extracting features (boundary or skeleton) from imaging data; (2) constructing geometric models (represented by meshes) from the detected features. In addition, image pre-processing is usually necessary for better feature extraction, when the original image is noisy or the contrast between features and background is low. With 3-D T-PM images, Yu and collaborators developed a set of image processing and analysis tools and using the mesh generator called GAMer [28] they were able to generate high-fidelity and quality meshes for 3-D t-tubular systems in mice [29] (see Fig. 1A lower panel).
Figure 1. T-tubular network in mouse ventricular myoyctes, geometric model, and diagram illustrating Ca2+ dynamics.

(A, upper panel) Cardiac sarcolemma including t-tubules is visualized using a 2-photon microscopy: external membrane (blue arrows); t-tubules (white arrows); nucleus (Nuc); bar 2 µm. (A, lower panel) Geometric model of the t-tubule sub-system extracted from 2-photon microscopy image. (B) Expanded view of single t-tubule geometry and its surrounding half-sarcomeres. (C) Schematic drawing of Ca2+ entrance and extrusion via the cell membrane and Ca2+ buffering and diffusion inside the myocyte with the SR activity inhibited.
The extreme intricacy of the t-tubule system in mice (with transverse-axial anatomies and large local variations in t-tubule diameter) has been observed in rat ventricular myocytes as well [4], [11], [30]. Because high-fidelity geometric models representing the realistic t-tubule topology in rats are currently not available, in this study we used the geometric model in mice of Yu's et al.
[29]. To investigate the Ca2+ signaling in rat ventricular myocytes, we considered a small compartment containing a single t-tubule and its surrounding half-sarcomeres for two reasons: (a) the entire t-tubular system in a ventricular myocyte forms a roughly periodic pattern corresponding to individual sarcomere; and (b) a small model contains much fewer number of mesh nodes that would render numerical simulation significantly faster and more feasible in ordinary computers. The surrounding half-sarcomeres were modeled as a rectangular-shaped box of 2 µm×2 µm in the plane of external sarcolemma and 5.96 µm in depth (Fig. 1B left panel and Table 1). As Yu's t-tubule model did not include the realistic cell surface, one of the box faces (the top red surface in Fig. 1B) was assumed to be the external cell membrane. The t-tubule inside this compartment was extracted from a sub-volume of the T-PM imaging data corresponding to the region indicated in Fig. 1A lower panel. To make it easier to handle boundary conditions in numerical analysis, we have “closed” the end of each branch, yielding a tree-like t-tubule model (see the yellow mesh in Fig 1B). These added “caps” were treated with the same boundary conditions as for the rest of the t-tubular surface. This simplified treatment clearly could introduce some errors because these “caps” are artificial and no Ca2+-transporting protein should reside there. However, the errors should be negligible as the area of these “caps” is very small, compared to the rest of t-tubular surface. The t-tubule diameter varied from 0.19 µm to 0.469 µm and the t-tubule depth was 5.645 µm. The volume of the model compartment was estimated to be ∼23.31 µm3. The compartment membrane area was ∼9.00 µm2 where the percentage of cell membrane within t-tubule was 64% (∼5.75 µm2) and within the external membrane 36% (∼3.25 µm2), [10], [11], [31], [32]. The accessible volume for Ca2+ was estimated to be ∼35–37% of the total cytosolic volume ( ) ∼12.9–13.6 pL in adult rat ventricular myocytes [33], [34]. The sub-cellular aqueous volume of 35–37% assumes that: (1) myofilaments occupy 47–48% of the cell volume, mitochondria 34–36%, nucleus 0–2%, t-tubule system 0–1.2%, and SR lumen 3.5%; (2) 50% of the myofilament space is accessible for Ca2+ (i.e. contains water); (3) mitochondria and nuclei are not rapidly accessible for Ca2+; (4) the SR lumen is not accessible to Ca2+ in the presence of ryanodine and thapsigargin [18], [33].
) ∼12.9–13.6 pL in adult rat ventricular myocytes [33], [34]. The sub-cellular aqueous volume of 35–37% assumes that: (1) myofilaments occupy 47–48% of the cell volume, mitochondria 34–36%, nucleus 0–2%, t-tubule system 0–1.2%, and SR lumen 3.5%; (2) 50% of the myofilament space is accessible for Ca2+ (i.e. contains water); (3) mitochondria and nuclei are not rapidly accessible for Ca2+; (4) the SR lumen is not accessible to Ca2+ in the presence of ryanodine and thapsigargin [18], [33].
Table 1. Physical constants and cell geometry parameters.
| Symbol | Definition | Value | Ref. |
| F | Faraday constant | 96.5 C mmol−1 | Physical constant |
| T | Temperature | 295 K | Physical constant |
| R | Universal gas constant | 8.314 J mol−1 K−1 | Physical constant |

|
Cell volume | 36.8 pL | [33] |

|
Cell capacitance | 324 pF | [33] |
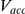
|
Accessible volume for Ca2+ | 12.9–13.6 pL | Estimated |

|
Compartment volume | 23.31 µm3 | Estimated |

|
Compartment surface | 9.00 µm2 | Estimated |
| T-tubule geometry | |||

|
T-tubule radius | 0.19–0.469 µm | [6] |

|
T-tubule depth | 5.645 µm | [6] |
| Rectangular-shaped box geometry | |||

|
Cell surface direction | 2µm | [12] |
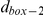
|
Cell surface direction | 2µm | [12] |

|
Depth | 5.96µm | [6] |
Distribution of Ca2+-transporting protein complexes along the cell membrane
Recent immunohistochemical studies have demonstrated that marked variations in the distribution of Ca2+-transporting protein complexes (L-type Ca2+ channel, Na+/Ca2+ exchanger) along the cell membrane probably exist [2], [10], [12], [15], [35]–[43]. The analysis suggests that most of the L-type Ca2+ channels are concentrated in the t-tubules (from 3 to 9 times more in the t-tubule membrane than on the external sarcolemma) [2], [10], [12], [38], [39] and that the concentration of LCC along the t-tubule probably increases toward the center of the cell [36], [40].
Studies on the distribution of the main Ca2+ efflux pathway, the Na+/Ca2+ exchanger, are more controversial. All studies but one [41] have reported NCX to localize both to the external and t-tubule membrane, but most studies suggest that the NCX is 1.7 to 3.5 times more concentrated in the t-tubule membrane [15], [34], [36], [42], [43]. However, Kieval et al. data [43] indicate the NCX is more evenly distributed. In summary, the observed differences in the spatial distribution and molecular architecture of Ca2+ microdomains suggest that significant differences in the excitation-contraction coupling between the cell surface and cell interior may be exist. However how the localization of Ca2+- transporting protein complexes along the sarcolemma regulates the intracellular Ca2+ signaling still remains uncertain.
Reaction-diffusion equations
In the current model, the effects of four exogenous and endogenous Ca2+ buffers (Fluo-3, ATP, calmodulin, troponin C) were considered (Fig. 1C). The endogenous stationary buffer troponin C (TN) was distributed uniformly throughout the cytosol but not on the cell membrane and in the sub-sarcolemmal space (∼40–50 nm in depth). The free Ca2+ and mobile buffers (Fluo-3, ATP, calmodulin) diffuse and react throughout the cytoplasm. The cell membrane and sarcomere box faces are subject to reflective boundary conditions. The nonlinear reaction-diffusion equations describing Ca2+ and buffers dynamics inside the model cell are:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
where: [Bm] represents the concentration of mobile buffer Fluo-3, ATP or calmodulin; [Bs] is the concentration stationary buffer troponin C.
The diffusion coefficients for Ca2+, CaATP, CaCal and CaFluo as well as the total buffer concentrations and buffer rate constants used in the model are shown in Table 2. In the model we also assume: (1) isotropic diffusion for Ca2+ and all mobile buffers [12]; (2) Ca2+ binds to Fluo-3, calmodulin, ATP, and TN without cooperativity; (3) the initial total concentrations of the mobile buffers are spatially uniform; (4) the diffusion coefficients of Fluo-3, ATP or calmodulin with bound Ca2+ are equal to the diffusion coefficients of free Fluo-3, ATP or calmodulin.
Table 2. Calcium and buffer reaction-diffusion parameters.
| Symbol | Definition | Value | Ref. |
| Ca2+ and buffer concentrations | |||

|
Extracellular Ca2+ concentration | 1000 µM | [33] |

|
Resting Ca2+ concentration | 0.1 µM | [33] |
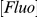
|
Total Fluo-3 concentration | 100 µM | [18] |
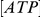
|
Total free ATP concentration | 260 µM | [18] |
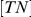
|
Total troponin concentration | 70 µM | [18] |
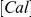
|
Total calmodulin concentration | 24 µM | [18] |
| Diffusion coefficients (at 22°C) | |||

|
Diffusion coefficient for Ca2+ | 0.39 µm2 ms−1 | [18] |

|
Diffusion coefficient for CaFluo | 0.1 µm2 ms−1 | [18] |

|
Diffusion coefficient for CaATP | 0.168 µm2 ms−1 | [18] |
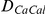
|
Diffusion coefficient for CaCal | 0.025 µm2 ms−1 | [18] |
| Rate coefficients and dissociation constants (at 22°C) | |||

|
Ca2+ on-rate constant for TN | 0.04 µM−1 ms−1 | [19] |

|
Ca2+ off-rate constant for TN | 0.04 ms−1 | [19] |

|
Ca2+ dissociation constant for TN | 1 µM | [19] |
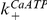
|
Ca2+ on-rate constant for CaATP | 0.225 µM−1 ms−1 | [18] |

|
Ca2+ off-rate constant for CaATP | 45 ms−1 | [18] |

|
Ca2+ dissociation constant for ATP | 200 µM | [18] |

|
Ca2+ on-rate constant for CaFluo | 0.23 µM−1 ms−1 | [18] |
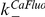
|
Ca2+ off-rate constant for CaFluo | 0.17 ms−1 | [18] |

|
Ca2+ dissociation constant for Fluo | 0.739 µM | [18] |

|
Ca2+ on-rate constant for Cal | 0.125 µM−1 ms−1 | [18] |

|
Ca2+ off-rate constant for Cal | 0.2975 ms−1 | [18] |

|
Ca2+ dissociation constant for Cal | 2.38 µM | [18] |
The total Ca2+ flux (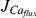 ) throughout the t-tubule and external membrane is:
) throughout the t-tubule and external membrane is:
| (6) |
where: 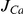 - total LCC Ca2+ influx;
- total LCC Ca2+ influx;  - total NCX Ca2+ flux;
- total NCX Ca2+ flux;  - total membrane Ca2+ leak.
- total membrane Ca2+ leak.
To describe L-type Ca2+ current, Na+/Ca2+ exchanger, Ca2+ leak current densities we used the following expressions:
| (7) |
 |
(8) |
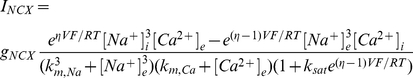 |
(9) |
| (10) |
Flux parameter values were estimated or taken from the literature (see Table 3). In this study, the Ca2+ leak is not actually a particular “leak protein”. The Ca2+ leak was included and adjusted so that at rest Ca2+ influx via Ca2+ leak to match Ca2+ efflux via NCX thus no net movement across the cell membrane to occur.
Table 3. Membrane calcium fluxes parameters.
| Symbol | Definition | Value | Ref. |
| L-type Ca2+ current | |||

|
Constant | 1 | [18] |
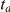
|
Constant | 4 ms | [26] |

|
Constant | 70 ms | [26] |
| Na+/Ca2+ exchange current | |||

|
Extracellular Na+ concentration | 140 mM | [33] |

|
Resting Na+ concentration | 10 mM | [33] |

|
Pump rate of NCX | 38.5 µM ms−1 | [70] |

|
Voltage dependence of NCX control | 0.35 | [70] |

|
Na+ half saturation of NCX | 87.5 mM | [70] |
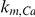
|
Ca2+ half saturation of NCX | 1380 µM | [70] |
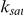
|
Low potential saturation factor of NCX | 0.1 | [70] |
| Membrane Ca2+ leak | |||

|
Conductance | 3.4e-6µM mV−1ms−1 | Estimated |
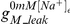
|
Conductance | −6.8e-6µM mV−1ms−1 | Estimated |
In the model, each current density (Ii) was converted to Ca2+ flux (Ji) by using the experimentally suggested surface to volume ratio ( ∼8.8 pF/pL) in adult rat ventricular myocytes [32], [33]:
∼8.8 pF/pL) in adult rat ventricular myocytes [32], [33]:
| (11) |
Then, the total compartment Ca2+ flux (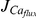 ) was computed by multiplying each total Ji with the model cell volume (
) was computed by multiplying each total Ji with the model cell volume ( ), and distributing
), and distributing 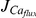 to the external and t-tubule membrane according to the prescribed Ca2+-handling protein concentration ratio.
to the external and t-tubule membrane according to the prescribed Ca2+-handling protein concentration ratio.
The voltage-clamp protocol (holding potential −50mV, electric pulse of 10mV for 70ms) and whole-cell L-type Ca2+ current were derived from Zahradnikova et al. data with the blocked SR activity [26]. In this study, each simulation started with a basal cytosolic Ca2+ of 100 nM, basal cytosolic Na+ of 10 mM and buffers in equilibrium. The extracellular Ca2+ concentration (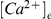 ) was 1 mM and remained constant. Unless specified otherwise in the Figure legends or in the text, the extracellular Na+ concentration (
) was 1 mM and remained constant. Unless specified otherwise in the Figure legends or in the text, the extracellular Na+ concentration ( ) was 140 mM and
) was 140 mM and  3.4−6 µM mV−1 ms−1.
3.4−6 µM mV−1 ms−1.
Numerical algorithms and software
In finite element methods, a complex domain needs to be discretized into a number of small elements (such as triangles or tetrahedra). This process is usually referred to as mesh generation [28], [44]. Although different types of meshes may be generated depending on the numerical solvers to be employed, we restrict ourselves to triangular (surface) and tetrahedral (volumetric) mesh generation as commonly used in biomedical simulation. In the present simulation, the number of finite element nodes and tetrahedral elements are 50,262 and 221,076, respectively.
The nonlinear reaction diffusion system was solved by a finite difference method in time and finite element method in space using our SMOL software tool (Smoluchowski Solver, http://mccammon.ucsd.edu/smol/) with the time step of 4 ms. It takes around 20 minutes to run 400 ms snapshots with a single Intel Xeon X5355 processor. The SMOL program utilizes libraries from the finite element tool kit (FETK), which previously has been used in several molecular level studies [45]–[49]. One bottleneck for dynamic 3-D simulation of nonlinear reaction diffusion system is the computing complexity involved in solving the problem. Here we successfully extended SMOL to solve multiple coupled partial differential equations with nonlinear ordinary equations. Multiple tests demonstrate that our SMOL program is quite robust and flexible for various boundary and initial conditions. The simulation results were visualized using GMV mesh viewers [50]. Post-processing and data analyses were implemented by customized Python, MATLAB 2008b (The MathWorks, Natick, MA) scripts and Xmgrace software [51]. A version control system, subversion, was used to monitor the development of software [52].
Results
Numerical simulation of experimental recordings of Ca2+ influx and Ca2+ concentration changes in rat ventricular myocytes
In agreement with the reported experimental data [2], [10], [12], [36], [38]–[40], the spatial patterns of [Ca2+]i were calculated assuming LCC current density: (1) heterogeneously distributed along the cell surface; (2) six times higher and uniform in the t-tubule membrane; or (3) homogeneously distributed along the sacrolemma. In cases (1–2) the NCX flux density was assumed three times higher in the t-tubule and in case (3) NCX was evenly distributed along the sarcolemma. In this study, Ca2+ leak density was homogeneously distributed along the cell membrane with respect to all distribution choices of LCC and NCX. In case (1), the 3-D distribution of LCC current was computed by combining the cluster density and fluorescent intensity plots, see Fig. 2A. The data were then scaled and fitted by a cubic polynomial:
| (12) |
where: x is the distance from the external cell surface.
Figure 2. L-type Ca2+ current.

(A) The distribution of L-type Ca2+ current is computed (dashed line) by multiplying the experimentally measured cluster density and fluorescent intensity plots (solid line), [36]. (B) The L-type Ca2+ current density is fitted and plotted (solid line) using shape preserving function of the data reported in rats with SR blocked (doted line), [26].
The parameter values of the polynomial (pj, j = 1–4) are shown in Table 4. This polynomial was further scaled by a single factor C (see Table 4) such that the total Ca2+ flux along the t-tubule membrane remained unchanged by redistributing the Ca2+ fluxes. To fit the whole-cell LCC current density to the reported data in rat myocytes with SR release inhibited [26], we used a shape preserving function, (see Eq. 8 and Fig. 2B).
Table 4. Parameter values of the polynomial for L-type Ca2+ current distribution along the t-tubule.
| Symbol | C | p1 | p2 | p3 | p4 |
| L-type Ca2+ current | 0.4515 | −4.1379e-4 | −1.1722e-2 | 1.978e-1 | 1.0033 |
Consistent with the Cheng et al. experiment [5], where the fluorescence signal was recorded along the single scan-lane starting and ending outside the cell and crossing the center of the cell, the model t-tubule was chosen to cross the cell center and the scanned line was located at 200nm away from the t-tubule membrane (see Fig. 1A and Fig. 3). To gain more detailed insights of how the predicted Ca2+ signals are regulated within this geometrically irregular micro-domain we examined [Ca2+]i at two different line-scan positions: 200 nm, angle 120°; 200 nm, angle 60° (see Fig. 3).
Figure 3. Line-scan position.

Diagrams illustrating external membrane, t-tubule mouth, and positions of scanning line (yellow spots) where the changes in local [Ca2+]i have been visualized and examined.
Model results in Figs. 4–5 were computed for conditions approximating those of the experiment by Cheng et al. [5], who examined Ca2+ signals in voltage-clamped rat myocytes in the presence of 100 µM Fluo-3 and pharmacological blockade of the SR (see Fig. 4L). The computed line-scan images and local Ca2+ time-courses are shown in Figs. 4F–4K.
Figure 4. Calcium signals arising from the ionic fluxes via the external and t-tubule membrane in the presence of 100 µM Fluo-3.

(A–B) The voltage-clamp protocol and whole-cell L-type Ca2+ current. (C–E) Predicted global Na+/Ca2+ and Ca2+ leak currents and global Ca2+ transient when no detectible differences in [Ca2+]i are found (see panel F). (F–H) Calcium concentrations visualized as line-scan images in transverse cell direction. (I–K) Local Ca2+ transients taken at three featured spots along the scanning line of interest: 0.17 µm – blue lines; 3.09 µm – green lines; 5.45 µm – red lines. In (F) and (I) the L-type Ca2+ current density followed heterogeneous distribution along the length of t-tubule as shown in Fig. 2A. In (G) and (J) the L-type Ca2+ current density was uniform along the t-tubule and six times higher than in external membrane. In (H) and (K) the L-type Ca2+ current density was homogeneous throughout the cell surface. In (F–G) Na+/Ca2+ flux density was three times higher in the t-tubule and Ca2+ leak homogeneously distributed. In (H) Na+/Ca2+ exchanger and Ca2+ leak were homogeneously distributed via the sarcolemma. (L) Local Ca2+ time-courses with re-plot from experimental data [5]. The re-plots were taken along the scanned line at 0µm (blue), 3.96 µm (green) and 5.65 µm (red) from the near surface location. (M) Estimated SCH values with respect to the three flux distribution choices. In this numerical experiment the line-scan was positioned at 200nm away from the t-tubule membrane at the angle 120°. The scanned line in Cheng et al. experiment was located at 200nm from the surface of the t-tubule.
Figure 5. Calcium concentration distributions at Ca2+ peak in the presence of 100 µM Fluo-3.

(A–C) 3-D views of the Ca2+ concentration distribution at Ca2+ peak of 76 ms. In (D) the spatial profiles at Ca2+ peak along the scanning line of interest are compared. L-type Ca2+ flux density heterogeneously distributed along the t-tubule membrane (A and black plots in D). L-type Ca2+ flux density six times higher and uniform via t-tubule membrane (B and red plots in D). L-type Ca2+ flux density uniformly distributed via the sarcolemma (C and green plots in D). In (A–B) Na+/Ca2+ flux density was three times higher in the t-tubule and Ca2+ leak homogeneously distributed. In (C) Na+/Ca2+ exchanger and Ca2+ leak were homogeneously distributed via the sarcolemma.
These results demonstrate that with LCC heterogeneous or LCC six times higher in the t-tubule: (1) predicted Ca2+ concentration profiles were non-uniform when t<100 ms but the variations in [Ca2+]i seem to be within the range of experimental noise in Fig. 4L; (2) [Ca2+]i was more evenly distributed when t>100 ms, ( Figs. 4F–G, Figs. 4I–J, Video S1). To delineate further the suggested spatial differences in [Ca2+]i (see Figs. 4F–H), we introduced a quantity called ‘spatial Ca2+ heterogeneity’ (SCH). The SCH is defined to be the difference of the maximal and minimal [Ca2+]i values, normalized by the maximal value at given reference point along the line-scan (0.17 µm, 3.09 µm, 5.45 µm) in given moment tj, (see Figs. 4I–K). High SCH suggests non-uniform [Ca2+]i distribution and SCH of zero indicates spatially uniform [Ca2+]i distribution. The histogram in Fig. 4M shows that assuming LCC heterogeneous versus LCC 6 times higher in the t-tubule decreased SCH(tIca-peak) by 1.6 folds, SCH(t70 ms) by 2.29 folds, SCH([Ca]i-peak) by 2.34 folds, SCH(t100ms) by 2.87 folds, and SCH(t200ms) by 8.45 folds. These findings demonstrate that the predicted [Ca2+]i distribution with LCC heterogeneous more closely resembles the Chang et al. experimental findings [5], (compare Figs. 4I and 4L). Finally, the model predicts strongly non-uniform Ca2+ transients when the LCC, NCX and Ca2+ leak fluxes were uniformly distributed throughout the cell surface (Figs. 4H, 4K, 4M). In addition, Video S1 (see right panel) demonstrates that here the Ca2+ signal spreads from the external membrane to the cell center as a continuum wave but after LCC channel closing (t∼72 ms) this wave faltered.
Predicted global [Ca2+]i transient, Na+/Ca2+exchanger, and Ca2+ leak currents with LCC pathways heterogeneously distributed (as in Fig. 4F) are shown in Figs. 4E and 4C–D. Figure 4C demonstrates that: (1) the depolarization of cell membrane reversed the rest exchanger's direction (i.e. in the interval 0ms–70ms NCX operated in Ca2+ entry mode) while when repolarization occurred the flow of Ca2+ through NCX was reversed again (i.e. in the interval 70ms–400 ms NCX operated in Ca2+ exit mode); (2) upon returning to resting voltage of −50mV the exchanger's rate rapidly increased ( ) while
) while  rate remained unchanged (note
rate remained unchanged (note  is not voltage-dependent) thus causing fast extrusion of Ca2+ out of the cell and subsequent sudden drop in the local and global [Ca2+]i. Figures 4F–K illustrate also that the global and all local Ca2+ transients reached the peak after ∼76 ms and that [Ca2+]i levels were higher near the t-tubule mouth because the density of t-tubule branches was higher in this region and close topological proximity of the external membrane additionally increased the relative amount of the entering Ca2+. Due to the higher [Ca2+]i gradient under the outer cell edge (t∼70ms) Ca2+ diffused toward the cell center and when
is not voltage-dependent) thus causing fast extrusion of Ca2+ out of the cell and subsequent sudden drop in the local and global [Ca2+]i. Figures 4F–K illustrate also that the global and all local Ca2+ transients reached the peak after ∼76 ms and that [Ca2+]i levels were higher near the t-tubule mouth because the density of t-tubule branches was higher in this region and close topological proximity of the external membrane additionally increased the relative amount of the entering Ca2+. Due to the higher [Ca2+]i gradient under the outer cell edge (t∼70ms) Ca2+ diffused toward the cell center and when  ratio along the cell membrane became approximately equal to
ratio along the cell membrane became approximately equal to  ratio a new equilibrium level of [Ca2+]i (∼0.16 µM) was reached. Intracellular Ca2+ equilibrated faster when Ca2+ flux was more concentrated in the t-tubule membrane because [Ca2+]i gradient near the t-tubule mouth was lower there than [Ca2+]i gradient with Ca2+transporting complexes distributed homogeneously. Additional reasons for the observed rapid equilibrium of [Ca2+]i may be that [Na+]i was kept constant (in contrast to existing evidence for the presence of local sub-sarcolemmal [Na+]i gradients on the action potential time-scale [33], [53]) or that the realistic distribution of NCX flux may be differ as assumed in the model. Finally, the results demonstrate that the computed average [Ca2+]i peak of 160–185 nM (see Figs. 4E, 4I–4J), is comparable with the measured one of about 163 nM when the SR release and uptake were inhibited [5].
ratio a new equilibrium level of [Ca2+]i (∼0.16 µM) was reached. Intracellular Ca2+ equilibrated faster when Ca2+ flux was more concentrated in the t-tubule membrane because [Ca2+]i gradient near the t-tubule mouth was lower there than [Ca2+]i gradient with Ca2+transporting complexes distributed homogeneously. Additional reasons for the observed rapid equilibrium of [Ca2+]i may be that [Na+]i was kept constant (in contrast to existing evidence for the presence of local sub-sarcolemmal [Na+]i gradients on the action potential time-scale [33], [53]) or that the realistic distribution of NCX flux may be differ as assumed in the model. Finally, the results demonstrate that the computed average [Ca2+]i peak of 160–185 nM (see Figs. 4E, 4I–4J), is comparable with the measured one of about 163 nM when the SR release and uptake were inhibited [5].
This model is also able to predict how the Ca2+ transients are regulated at different line-scan positions within this geometrically irregular micro-domain. Note, due to the technical limitations the Cheng et al. experiment is not able to suggest where exactly the scanned line is positioned with regard to the specific t-tubule, but the Cheng et al. measurements [5] strongly suggest the similarity of [Ca2+]i at the peripheral and deeper cytoplasm when the SR activity is abolished. For this reason, we examined the Ca2+ profiles (LCC heterogeneous along the t-tubule, Fig. 4F) positioning the line-scan at 200 nm and angle 60° (see Fig. 3) or positioning the line-scan at 50, 100, 200, 300 or 400 nm at different angles. No visible differences in the visualized spatial Ca2+ profiles were found (data not shown).
The 3-D Ca2+ concentration distributions and spatial Ca2+ profiles at Ca2+ peak (76 ms) are shown in Figs. 5A–D. These model results demonstrate that the Ca2+ concentration near the external membrane decreased while [Ca2+]i in the cell interior increased when Ca2+ transporters were uniformly distributed and after that heterogeneously redistributed. The jumps in Fig. 5D show the predicted local Ca2+ flux ( ) at [Ca2+]i peak in the regions where the scanning line of interest is crossing the t-tubule membrane.
) at [Ca2+]i peak in the regions where the scanning line of interest is crossing the t-tubule membrane.
Additional interesting model findings are that: (1) large and steep [Ca2+]i gradients were predicted inside the sub-sarcolemmal 3-D space (see Video S1); (2) the global Ca2+ time-course and time to [Ca2+]i peak did not depend on whether Ca2+ fluxes are distributed homogeneously or heterogeneously along the sarcolemma (data not shown); (3) redistributing NCX flux uniformly via the sarcolemma was not able to alter significantly the predicted Ca2+ signals in Fig. 4F (data not shown).
In this study the value of 390 µm2 s−1 for diffusion coefficient of free Ca2+ and published buffer diffusion coefficients and parameters were used to compare the calculated Ca2+ signals with the Cheng's et al. fluorescence Ca2+ signals recorded in rats [5]. It has been suggest, however, that the effective diffusion of free Ca2+ in the cytosol ( ) will be slowed down because the exogenous and endogenous Ca2+ buffers and free Ca2+ concentrations are able to affect Ca2+ diffusion strikingly [18], [19], [54]–[62]. The measurements of Allbritton et al. [54] report a value of 5–21 µm2 s−1 for
) will be slowed down because the exogenous and endogenous Ca2+ buffers and free Ca2+ concentrations are able to affect Ca2+ diffusion strikingly [18], [19], [54]–[62]. The measurements of Allbritton et al. [54] report a value of 5–21 µm2 s−1 for  at low free
at low free 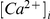 when a value of ∼223 µm2 s−1 is assumed for
when a value of ∼223 µm2 s−1 is assumed for  . During Allbritton's et al. in vitro experiments Ca2+ sequestration by the SR, mitochondria and ATP was inhibited and only mobile calmodulin and stationary troponin C were present in the cytosolic extract. It is possible to estimate
. During Allbritton's et al. in vitro experiments Ca2+ sequestration by the SR, mitochondria and ATP was inhibited and only mobile calmodulin and stationary troponin C were present in the cytosolic extract. It is possible to estimate  (0 µM Fluo-3, 0 µM ATP), using a simplified equation (see Eq. 13), because in this study the predicted maximal Ca2+ elevations were sufficiently small (
(0 µM Fluo-3, 0 µM ATP), using a simplified equation (see Eq. 13), because in this study the predicted maximal Ca2+ elevations were sufficiently small (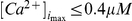 ) and the diffusion coefficients for Ca2+-bound and free mobile buffer forms were assumed equal [18], [19], [58], [59]. Our calculations predict a value of ∼8 µm2 s−1 for
) and the diffusion coefficients for Ca2+-bound and free mobile buffer forms were assumed equal [18], [19], [58], [59]. Our calculations predict a value of ∼8 µm2 s−1 for  when
when  was 390 µm2 s−1 and
was 390 µm2 s−1 and 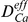 ∼6 µm2 s−1 when
∼6 µm2 s−1 when  was 223 µm2 s−1. Therefore, the estimated value for
was 223 µm2 s−1. Therefore, the estimated value for  (∼8 µm2 s−1) is in reasonable agreement with the experimental observation.
(∼8 µm2 s−1) is in reasonable agreement with the experimental observation.
 |
(13) |
We could not find experimental data suggesting 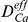 (260 µM ATP, 70 µM TN, 24 µM Cal) or
(260 µM ATP, 70 µM TN, 24 µM Cal) or 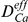 (100 µM Fluo-3, 260 µM ATP, 70 µM TN, 24 µM Cal) in the solution. Therefore, we used published concentrations of Ca2+ binding proteins and published diffusion and dissociation constants (
(100 µM Fluo-3, 260 µM ATP, 70 µM TN, 24 µM Cal) in the solution. Therefore, we used published concentrations of Ca2+ binding proteins and published diffusion and dissociation constants ( ) to estimate the effective diffusion constants of free Ca2+ in the presence of ATP or Fluo-3 and ATP in the cytosol. Our calculations indicate that adding 260 µM ATP in the solution accelerated
) to estimate the effective diffusion constants of free Ca2+ in the presence of ATP or Fluo-3 and ATP in the cytosol. Our calculations indicate that adding 260 µM ATP in the solution accelerated  (∼10.4 µm2 s−1) and that
(∼10.4 µm2 s−1) and that  increased additionally when 100 µM Fluo-3 was added (
increased additionally when 100 µM Fluo-3 was added (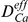 ∼66 µm2 s−1). Furthermore, our studies suggest that in the presence of 100 µM Fluo-3 and LCC heterogeneous Ca2+ binding and diffusion of ATP and calmodulin could not affected significantly the predicted [Ca2+]i distributions (data not shown).
∼66 µm2 s−1). Furthermore, our studies suggest that in the presence of 100 µM Fluo-3 and LCC heterogeneous Ca2+ binding and diffusion of ATP and calmodulin could not affected significantly the predicted [Ca2+]i distributions (data not shown).
During simulations of SR Ca2+ release into the diadic cleft, a major effect of the stationary phospholipids Ca2+ binding sites has been suggested [17], [18]. To examine the impact of the phospholipids on the much smaller Ca2+ signals (arising from Ca2+ influx via Ca2+ current only), we included this buffer in our model. The results demonstrated that the phospholipids had only a limited effect on the calculated Ca2+ signals in the sub-sarcolemmal region (0 µM Fluo-3, 260 µM ATP, 24 µM calmodulin) and that this effect was even smaller when 100 µM Fluo-3 was included (data not shown).
Model estimations of Ca2+ distribution under varying conditions
Ca2+ concentration changes in the absence of fluorescence dye and Ca2+ pathways heterogeneously distributed via the sarcolemma
This model is also able to predict the spatial [Ca2+]i signals that would occur in the absence of Fluo-3 (note experiments without fluorescent dye cannot be performed because of technical reasons). Since it has been suggested that the dye could not affect Ca2+ entry via L-type channels [63], [64], the same global LCC flux (as in Figs. 4–5) was used during this numerical experiment. Figures 6E–I show predicted Ca2+ signals arising from the ionic influx via L-type Ca2+ channels during voltage-clamp stimulation with LCC and NCX pathways heterogeneously distributed (as in Fig. 4F). The calculated global NCX and Ca2+ leak currents are shown in Figs. 6C–D, respectively.
Figure 6. Model predictions in the absence of Fluo-3 and Ca2+ pathways heterogeneously distributed via the cell membrane.

(A–B) The voltage-clamp protocol and whole-cell L-type Ca2+ current used in this set of simulations. (C–D) The predicted global Na+/Ca2+ and Ca2+ leak currents. (E–F) The global Ca2+ transient and Ca2+ concentrations visualized as line-scan image in the transverse cell direction. (G) Local Ca2+ transients taken at three featured spots along the scanning line (0.17 µm – blue lines, 3.09 µm – green lines, 5.45 µm – red lines). In (H–I) the spatial profile of Ca2+ along the scanning line and 3-D [Ca2+]i distribution at Ca2+ peak of 76 ms are shown. In this numerical experiment the scanned line was positioned at 200nm away from the t-tubule membrane at the angle 120°.
The model predicts here that the spread and buffer capacity of 100 µM Fluo-3 were able to mask completely the pronounced spatial non-uniformities in [Ca2+]i distribution that will occur during the Ca2+ influx when the SR Ca2+ metabolism is inhibited [5]. Figure 6 also illustrates, that detectible differences in [Ca2+]i are found when t<150 ms and that [Ca2+]i was more evenly distributed when t>150 ms, (see also Video S2, left panel). The removing of the mobile Ca2+ dye from the solution increased SCH(tIca-peak) by 1.635 folds, SCH(t70 ms) by 2.63 folds, SCH([Ca]i-peak) by 2.72 folds, SCH(t100ms) by 4.46 folds, and SCH(t200ms) by 8.65 folds (compare Fig. 6G with Fig. 4I where Fluo-3 was 100 µM). Additional findings are that: (1) upon depolarization NCX operated again in Ca2+ entry mode; (2) the reversal of current of NCX upon returning to resting voltage of −50mV increased additionally the current rate (compare Fig. 6C with Fig. 4C); (3) the global and local [Ca2+]i peaks increased while the times to peak remained unchanged, i.e. ∼76ms (compare Figs. 6E and 6G with Figs. 4E and 4I); (4) the predicted 3-D [Ca2+]i distribution at Ca2+ peak was strongly non-uniform (compare Fig. 6I and Fig. 5A); (5) upon repolarization initially [Ca2+]i rapidly dropped, the following [Ca2+]i decay was slow and the equilibrium was not reached even after 400 ms (Fig. 6E and Fig. 6G). Interesting model prediction is that larger, steeper and heterogeneous [Ca2+]i gradients with regard to those predicted in the presence of 100 µM Fluo-3 could be expected in the narrow sub-sarcolemmal space (compare left panel in Video S1 with left panel in Video S2). Interestingly, no visible differences in the local Ca2+ time-courses were found again (as with 100 µM Fluo-3) when the line-scan was positioned 200 nm away from the t-tubule surface at angle 60° or at 50, 100, 200, 300 or 400 nm at different angles (data not shown).
Endogenous buffer mobility
The conjecture made in the present model, that the endogenous calmodulin and ATP are mobile Ca2+ buffers, allowed us to examine how the mobility of these buffers would affect the Ca2+ dynamics and membrane flux time-courses within this irregular micro-domain. Figure 7 shows a simulation in which ATP (as CaATP) and calmodulin (as CaCal) were immobilized and Fluo-3 indicator removed from the solution. Under these conditions Ca2+ diffuses more slowly toward the center of the cell during the analyzed interval, resulting in a higher Ca2+ concentrations near the outer cell edge (compare line-scan images in Fig. 7F and Fig. 6F, see
Video S2). Figure 7 also demonstrates that the peak of local Ca2+ transient near the external membrane increased while [Ca2+]i peaks in the featured spots, 3.09 µm and 5.45 µm, decreased (see dashed lines in Fig. 7G). Here analysis suggests that assuming CaATP and CaCal stationary versus CaATP and CaCal mobile increased SCH(tIca-peak) by 1.17 folds, SCH(t70 ms) by 1.47 folds, SCH(t[Ca]i-peak) by 1.49 folds, SCH(t100ms) by 1.92 folds, and SCH(t200 ms) by 3.54 folds (see Fig. 7G). The results also indicate that the predicted local changes in [Ca2+]i gradients affected global NCX, Ca2+-leak and [Ca2+]i time-courses: (1) upon repolarization global 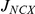 slightly decreased while
slightly decreased while  slightly increased; (2) [Ca2+]i peak decreased but the time to peak remained unchanged (∼76 ms), (see dashed lines in Figs. 7C–E). Finally, these simulations demonstrated that under these conditions (i.e. immobilizing all endogenous Ca2+ buffers) Ca2+ could escape the tight control of membrane voltage in some sub-cellular regions giving rise of cytosolic Ca2+ wave initiated at the sarcolemma (see Video S2
right panel - near the outer cell edge Ca2+ wave was initiated (t∼60 ms) but very soon after ∼12ms this wave faltered).
slightly increased; (2) [Ca2+]i peak decreased but the time to peak remained unchanged (∼76 ms), (see dashed lines in Figs. 7C–E). Finally, these simulations demonstrated that under these conditions (i.e. immobilizing all endogenous Ca2+ buffers) Ca2+ could escape the tight control of membrane voltage in some sub-cellular regions giving rise of cytosolic Ca2+ wave initiated at the sarcolemma (see Video S2
right panel - near the outer cell edge Ca2+ wave was initiated (t∼60 ms) but very soon after ∼12ms this wave faltered).
Figure 7. Effects of ATP and camodulin mobility on subcellular [Ca2+]i signals.

(A–B) The voltage-clamp protocol and whole-cell L-type Ca2+ current. (C–E) Quantitative comparison of the effects of buffer mobility on the global Na+/Ca2+ and Ca2+ leak currents and global Ca2+ transient (solid lines - CaATP and CaCal mobile, dashed lines - CaATP and CaCal stationary). (F) Line-scan image with CaATP and CaCal immobilized. (G) Quantitative comparison of the effects of buffer mobility on the local Ca2+ transients. During this numerical experiment Fluo-3 was zero, Ca2+ fluxes heterogeneously distributed via the sarcolemma, line-scan positioned at 200nm away from the t-tubule membrane at the angle 120°. Along the scanning line the featured spots were chosen to be the same as in Fig. 6G.
Contribution of Na+-Ca2+ exchange to sub-cellular excitation-contraction coupling
In this study, we also examined the effects of NCX inhibition on the voltage-clamp induced Ca2+ signals in the absence of fluorescent indicator. The inhibition of NCX forward mode was achieved by removing extracellular sodium (i.e. 0 mM [Na+]e). To adjust Ca2+ flux via Ca2+ leak to match Ca2+ influx via NCX, so that at rest no net movement across the cell membrane to occur, we estimated Ca2+ leak constant ( ) assuming [Na+]e zero (see Table 3). Under these conditions NCX operated only in Ca2+ entry mode while membrane leak pumped Ca2+ out of the cell (see Figs. 8C–D
dashed lines). Figures 8F–G (see dashed lines) show the predicted local [Ca2+]i transients and the corresponding line-scan image. The line scan-image demonstrates that [Ca2+]i distribution was again non-uniform but rather different compared to that predicted with 140 mM [Na+]e (compare Fig. 8F with Fig. 6F). The results in Fig. 8G demonstrate that: (1) local [Ca2+]i peaks in the featured spots (0.17 µm, 3.09 µm and 5.45 µm) increased and that this increase was more pronounced near t-tubule mouth; (2) upon repolarization [Ca2+]i suddenly dropped because
) assuming [Na+]e zero (see Table 3). Under these conditions NCX operated only in Ca2+ entry mode while membrane leak pumped Ca2+ out of the cell (see Figs. 8C–D
dashed lines). Figures 8F–G (see dashed lines) show the predicted local [Ca2+]i transients and the corresponding line-scan image. The line scan-image demonstrates that [Ca2+]i distribution was again non-uniform but rather different compared to that predicted with 140 mM [Na+]e (compare Fig. 8F with Fig. 6F). The results in Fig. 8G demonstrate that: (1) local [Ca2+]i peaks in the featured spots (0.17 µm, 3.09 µm and 5.45 µm) increased and that this increase was more pronounced near t-tubule mouth; (2) upon repolarization [Ca2+]i suddenly dropped because  rate decreased while
rate decreased while  rate remained unchanged; (3) the decay of Ca2+ transient near the outer cell edge was slow; (4) [Ca2+]i in the featured spots, 3.09 µm and 5.45 µm, begun slowly to increase when t>70 ms because
rate remained unchanged; (3) the decay of Ca2+ transient near the outer cell edge was slow; (4) [Ca2+]i in the featured spots, 3.09 µm and 5.45 µm, begun slowly to increase when t>70 ms because  rate remained unchanged but
rate remained unchanged but  rate slightly decreased. With [Na+]e zero versus 140 mM [Na+]e SCHs increased: SCH(tIca-peak) by 1.29 folds; SCH(t70 ms) by 1.4 folds; SCH(t[Ca]i-peak) by 1.43 folds; SCH(t100ms) by 1.65 folds; and SCH(t200ms) by 3.04 folds (see Fig. 8G). Furthermore, the NCX inhibition had visible effect on the global [Ca2+]i transient, i.e. global Ca2+ peak increased and during membrane repolarization initially [Ca2+]i suddenly decreased and after that rapidly equilibrated. In agreement with experimental data reported previously [65], the model also predicts increase in global [Ca2+]i peak and no changes in the time to peak when [Na+]e is completely substituted with 140 mM Li+, (see dashed line in Fig. 8E). New finding is that, with zero extracellular Na+, the Ca2+ signal spreads from the external membrane to the cell center as continuum Ca2+ wave initiated after 56 ms but very soon this wave faltered (see Video S3, right panel).
rate slightly decreased. With [Na+]e zero versus 140 mM [Na+]e SCHs increased: SCH(tIca-peak) by 1.29 folds; SCH(t70 ms) by 1.4 folds; SCH(t[Ca]i-peak) by 1.43 folds; SCH(t100ms) by 1.65 folds; and SCH(t200ms) by 3.04 folds (see Fig. 8G). Furthermore, the NCX inhibition had visible effect on the global [Ca2+]i transient, i.e. global Ca2+ peak increased and during membrane repolarization initially [Ca2+]i suddenly decreased and after that rapidly equilibrated. In agreement with experimental data reported previously [65], the model also predicts increase in global [Ca2+]i peak and no changes in the time to peak when [Na+]e is completely substituted with 140 mM Li+, (see dashed line in Fig. 8E). New finding is that, with zero extracellular Na+, the Ca2+ signal spreads from the external membrane to the cell center as continuum Ca2+ wave initiated after 56 ms but very soon this wave faltered (see Video S3, right panel).
Figure 8. Effects of reduced extracellular [Na+] on subcellular [Ca2+]i signals.

(A–B) The voltage-clamp protocol and whole-cell L-type Ca2+ current. (C) Quantitative comparison of the effects of changes in [Na+]e on the global Na+/Ca2+ flux (solid lines - [Na+]e 140 mM, dashed lines – zero [Na+]e). (D) Predicted Ca2+ leak with zero [Na+]e. (E) Quantitative comparison of the effects of changes in [Na+]e on the global Ca2+ transient. (F) Calcium concentrations visualized as line-scan images in transverse cell direction with zero [Na+]e. (G) Quantitative comparison of the effects of changes in [Na+]e on local Ca2+ transients. In this numerical experiment Fluo-3 was zero, Ca2+ fluxes heterogeneously distributed via the sarcolemma, the line-scan positioned at 200nm away from the t-tubule membrane at the angle 120°. Along the scanning line the featured spots were chosen to be the same as in Fig. 6G.
Discussion
The model with realistic transverse-axial t-tubule microanatomy provides new insights on how the Ca2+ signaling is regulated in rats
The current study attacks a difficult problem on how to incorporate the structural-based biological information, critical for the subcellular and cellular function, into sophisticated computational investigations. Pursuing this goal we developed a 3-D continuum model of Ca2+ signaling in rat ventricular cells that incorporates the realistic transverse-axial t-tubule topology and considers geometric irregularities and inhomogeneities in the distribution of ion-transporting proteins. The t-tubule micro-architecture was extracted from the Hayashi et al. two-photon imaging data in mice [6]. Because currently high-fidelity geometric models representing the realistic t-tubule micro-architecture in rats are not available, in this study we used the Yu's et al. geometric model in mice [28], [29]. On the basis of experimental data in rats the aqueous sub-cellular volume, accessible to Ca2+, was estimated to be ∼35–37% [33]. Since the Ca2+ signaling in cells is largely governed by Ca2+ diffusion and binding to mobile and stationary Ca2+ buffers [17]–[21], [23], [54]–[62], [66], [67], the effect of four Ca2+ buffers (Fluo-3, ATP, calmodulin, TN) was considered.
The model was validated against published experimental data on Ca2+ influx, membrane protein distributions and Ca2+ diffusion in rat cells treated with ryanodine and thapsigargin to inhibit the SR Ca2+ metabolism [2], [5], [10], [12], [15], [26], [35]–[43]. We found that with 100 µM Fluo-3 the results more closely resemble the Cheng's et al. experimental data [5] when the LCC density increases ∼1.7 fold along the t-tubule length and the NCX density is assumed three times higher in the t-tubule. An interesting finding is that with LCC six times and NCX three times higher and uniform in the t-tubule, the predicted fluctuations in the [Ca2+]i profiles were within the range of experimental noise [5]. Strongly non-uniform spatial Ca2+ gradients and propagation of Ca2+ wave are predicted, not observed in Cheng et al. experiment, when the LCC and NCX were uniformly distributed along the sarcolemma.
The model studies with 100 µM Fluo-3 indicate also that the [Ca2+]i gradients depend on the diffusion distances in the axial and cell surface directions. Thus, when the LCC were distributed uniformly the local Ca2+ peak in radial depth (5.96 µm) decreased from ∼1.5 fold while in the other cell directions (1 µm×1 µm) no significant changes were found. Redistributing the amount of Ca2+ pumped via the cell membrane (i.e. increasing LCC current density along the t-tubule) while keeping total Ca2+ flux unchanged, lowered Ca2+ gradients near the surface membrane and increased Ca2+ levels in the cell interior (see Video S1). The results also showed that with 100 µM Fluo-3 and Ca2+ flux heterogeneously distributed along the sarcolemma, the computed average [Ca2+]i peak (160–185 nM) is comparable to the measured of about 163 nM [5] and that the NCX redistribution alone yields to qualitatively similar [Ca2+]i profiles.
It should be noted, that in our previous work we used the simplified t-tubule geometry (assuming cylindrical shape) to simulate the Ca2+ dynamics in rats [19]. This idealistic t-tubule model also predicts the lack of systematic differences in the fluorescence Ca2+ signal when the Ca2+ transporters were distributed heterogeneously along the sarcolemma. Thus, the following question arises: How these new computational studies based on more realistic t-tubule structural model will further advance our current knowledge on the cell excitability and Ca2+ cycling in rats? First, in agreement with experiment [2]–[12] current study confirms that due to the branched t-tubule microstructure high and steep sub-sarcolemmal [Ca2+]i gradients could occur throughout the whole cell volume [2]–[6], [33], (see Video S1). Note, Lu et al. idealistic t-tubule model predicts high and steep sub-sarcolemmal [Ca2+]i gradients only in the transverse cell direction [19]. Second, our realistic t-tubule model predicts non-uniformities in [Ca2+]i distribution along the depth of the t-tubule when t<100 ms (see Fig. 4F and Video S1 left panel) while this was not the case when the t-tubule geometry is assumed cylindrical (Fig. 4g in Lu et al., [19]). Third, interesting finding is that no visible differences in the local Ca2+ profiles are predicted when the line-scan was positioned at different. Note, due to the technical limitations the Cheng et al. experiment is not able to suggest where and how exactly the scanned line is positioned with regard to the specific t-tubule [5].
A surprising and important finding of this study is that the spread and buffer capacity of 100 µM Fluo-3 were able to mask completely the pronounced spatial [Ca2+]i non-uniformities that would occur during the Ca2+ influx in the absence of dye (see Video S2 left panel - SR Ca2+ metabolism inhibited, LCC and NCX transporters heterogeneously distributed). Here the simulations demonstrated that with zero Fluo-3 the local and global Ca2+ peaks increased while the time of Ca2+ rise remained unchanged. The predicted sub-sarcolemmal [Ca2+]i gradients were heterogeneous along the cell membrane and larger and steeper compared to those with 100 µM Fluo-3. The NCX and Ca2+ leak time-courses were also affected due to increased local free [Ca2+]i levels. It is interesting that under these conditions no differences in the local Ca2+ time-courses were found (as with 100 µM Fluo-3) when the line-scan was positioned at different angles and distances. To test further the model we also examined how the mobility of endogenous Ca2+ buffers (ATP and calmodulin) and altered extracellular Na+ ([Na+]e) would affect the Ca2+ signals in the absence of fluorescence dye when the Ca2+-transportes are heterogeneously distributed. The results showed that when ATP and calmodulin were immobilized Ca2+ diffuses slowly toward the center of the cell, resulting in higher Ca2+ concentrations near the outer cell edge. When the NCX forward mode was inhibited (assuming [Na+]e = 0 mM) the local [Ca2+]i peaks increased and this increase was more pronounced near the outer cell edge. New findings are that under these conditions near the outer cell edge Ca2+ wave was initiated while this was not the case when ATP and calmodulin were mobile and [Na+]e 140 mM (see Video S2 and Video S3).
Taken together, these studies provide compelling evidence that (1) the exogenous Fluo-3 acts as a significant buffer and carrier for Ca2+, and that (2) the use of 100 µM Fluo-3 during the experiment can sensitively alter the realistic Ca2+ distribution. A new the question, however, arises: Based on the above model findings what could be the underlying mechanism(s) for the predicted heterogeneous Ca2+ concentrations gradients in the absence of Fluo-3? A reasonable answer is that the Ca2+ movement and distribution inside the cell rely strongly not only on the specific cell micro-architecture and Ca2+ transporters distribution but also on the presence of endogenous mobile and stationary Ca2+ buffers (ATP, calmodulin, troponin C - known to affect strikingly the Ca2+ dynamics) [12], [17]–[21], [23], [27], [67]. In support of this hypothesis, our simulations studies revealed that in the absence of Fluo-3: (1) the stationary Ca2+ buffer troponin C imposed stronger diffusion barrier for Ca2+ that resulted in larger and steeper sub-sarcolemmal Ca2+ gradients; (2) in the cell interior, owing on their sheer buffering capacity, Ca2+ buffers (troponin C, ATP, calmodulin) tended to slow down additionally the propagation of Ca2+ so that ATP and calmodulin spreading alone was not able to contribute the spatially uniform Ca2+ profiles to be achieved; (3) immobilizing the endogenous Ca2+ buffers slowed down the Ca2+ movement from the cell periphery to the center leading to build-up of large sub-sarcolemmal Ca2+ gradients and subsequent initiation of Ca2+ wave. It is important to mention that the Lu et al. idealistic t-tubule model predicts completely different 3-D [Ca2+]i distributions with zero Fluo-3, SR Ca2+ metabolism inhibited and Ca2+ transporters heterogeneously distributed [19].
Limitations of the model and future directions
Important limitations of the current modeling approach are: (1) the relatively small size of the model compartment that contains only a single realistic t-tubule shape and spans by just a half-sarcomere inside the ventricular myocyte; and (2) the assumption that the model compartment is a repeating unit inside the cell. The structural studies, however, provide evidence that in rodent ventricular myocytes the realistic t-tubule network is quite complex, (see Fig. 1A), [6]. The above limitations can be overcome in the future by extending the current geometric model toward more realistic models containing several t-tubules, whole-cell t-tubule network or other sub-cellular organelles (including mitochondria, SR, nuclei). This would allow building an improved geometric models representing more correctly the cell segment of interest and help to gain further insights of how the Ca2+-signaling in rat ventricular myocytes is regulated in the absence or presence of SR Ca2+ release and uptake [20]–[23], [66], [67]. However, it is equally important to mention here, that although the limitations (1–2) this model in a first approximation may yield insights across the whole-cell scale of biological organization. It allows simulating the global Ca2+ signal (computed from the line-scan image in Fig. 4F) that roughly would reproduce the whole-cell Ca2+ transient in the Cheng et al. experiment due to observed spatial similarities in [Ca2+]i (see Fig. 1B–1C in [5]). This assumption enables also investigating how the whole-cell Ca2+ signal is regulated by the realistic t-tubule microanatomy, by 3-D distributions of ion-transporting proteins, by mobile dye or endogenous mobile and stationary Ca2+ buffers. It should be noted, that the common pool modeling approaches could not be used to investigate these effects [10], [24], [25].
Important limitation of this study is also that we assume that the ion flux pathways are continuously distributed throughout the t-tubule membrane. Immunohistochemical studies, however, suggest that L-type Ca2+ channels appear to be concentrated as discrete clusters in the dyadic clefts (narrow spaces between LCC and RyR) distributed regularly along the t-tubule membrane at relatively small distances of ∼0.68 µm, [12], [38], [68]. It is interesting to mention that in contrast to Soeller and collaborators data in rats [12], Hayashi et al. data in mice [6] suggest that the dyadic clefts are distributed irregularly along the t-tubule branches. In addition, NCX appears to be absent from the longitudinal tubules [42]. Thus, the above data clearly imply that localized concentration of LCC or NCX flux pathways could result in larger sub-sarcolemmal Ca2+ gradients and local membrane currents that will affect differently the spatial Ca2+ profiles as predicted with the current model. Further extending of our current t-tubule model toward more realistic geometric models containing dyadic cleft topology and L-type Ca 2+ channel clustering could help to better understand how the Ca2+ signaling is regulated in the heart.
Finally, in the present model the effects of two endogenous Ca2+ mobile buffers (ATP, calmodulin) and one stationary Ca2+ buffer (troponin C) were considered only. The ventricular cells, however, contain other stationary Ca2+ buffers (including phospholipids, myosin, calsequestrin) or small and high mobile Ca2+ binding molecules (ADP, AMP) that were not included in the model (or may be other stationary and mobile buffers that have not been identified yet), [18], [24], [69].
Conclusions
Simulations presented in this study demonstrate that the more accurate knowledge of transverse-axial t-tubule microanatomy and protein distributions along the sarcolemma is important to better understand the mechanisms regulating the excitation-contraction coupling in rat ventricular myocytes. The results demonstrate that Ca2+ movement from the cell membrane to the cell interior relies also strongly on the presence of mobile and stationary Ca2+ buffers, including the Ca2+ indicator dye. The key findings are: (1) the model predicts lack of detectible differences in the fluorescence Ca2+ signals at the peripheral and deep myoplams when the membrane Ca2+ flux is heterogeneously distributed along the sarcolemma; (2) 100 µM mobile Fluo-3 is able to mask the pronounced spatial non-uniformities in the [Ca2+]i distribution that would occur when the SR Ca2+ metabolism is inhibited; (3) during the Ca2+ influx alone, large and steep Ca2+ gradients are predicted in the narrow sub-sarcolemmal space (∼40–50 nm in depth); (4) in rodents the branched t-tubule topology, the punctuate spatial distribution of Ca2+ flux along the sarcolemma and the endogenous Ca2+ buffers actually function to inhibit Ca2+ waves. Improved functional and structural computational models are needed to guide the experiments and to test further our understanding of how the t-tubule microanatomy and protein distributions regulate the normal cell function or cell cycle under certain pathologies. To our best knowledge, this study is the first attempt to use the finite element methods to investigate the intracellular Ca2+ responses in physiologically realistic transverse-axial t-tubule geometry.
Supporting Information
100 µM Fluo-3: Calcium flux heterogeneously distributed (left panel); Calcium flux higher and uniform in the t-tubule (middle panel); Calcium flux uniformly distributed (right panel).
(5.45 MB AVI)
Zero Fluo-3 and calcium flux heterogeneously distributed: ATP and calmodulin mobile (left panel); ATP and calmodulin immobile (right panel).
(4.05 MB AVI)
Zero Fluo-3 and calcium flux heterogeneously distributed: Extracellular sodium 140 mM (left panel); Zero extracellular sodium (right panel.)
(4.07 MB AVI)
Acknowledgments
The authors thank Shaoying Lu (Urbana-Champaign, University of Illinois) for the valuable advices and discussions and Yuhan Fu for technical assistance. We also thank the reviewers of the manuscript for useful suggestions. The SMOL and FETK source codes are available to download from http://mccammon.ucsd.edu/smol and http://www.fetk.org, respectively.
Footnotes
The authors have declared that no competing interests exist.
This work was supported by the National Biomedical Computational Resource (NIH grant 5P41 RR08605-16, http://www.nih.gov/). Dr. McCammon acknowledges additional support from NIH (R01 GM31749, http://www.nih.gov/), NSF (MCB-0506593, http://www.nsf.gov/), HHMI (http://www.hhmi.org/), and CTBP (NSF, PHY-0822283, http://www.nsf.gov/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Asghari P, Schulson M, Scriven DR, Martens G, Moore ED. Axial tubules of rat ventricular myocytes form multiple junctions with the sarcoplasmic reticulum. Biophys J. 2009;96:4651–4660. doi: 10.1016/j.bpj.2009.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brette F, Orchard C. T-tubule function in mammalian cardiac myocytes. Circ Res. 2003;92:1182–1192. doi: 10.1161/01.RES.0000074908.17214.FD. [DOI] [PubMed] [Google Scholar]
- 3.Brette F, Sallé L, Orchard CH. Quantification of calcium entry at the T-tubules and surface membrane in rat ventricular myocytes. Biophys J. 2006;90:381–389. doi: 10.1529/biophysj.105.069013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brette F, Orchard C. Resurgence of Cardiac T-Tubule Research. Physiology. 2007;22:167–173. doi: 10.1152/physiol.00005.2007. [DOI] [PubMed] [Google Scholar]
- 5.Cheng H, Cannell MB, Lederer WJ. Propagation of excitation-contraction coupling into ventricular myocytes. Pflugers Arch. 1994;428:415–417. doi: 10.1007/BF00724526. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi T, Martone ME, Yu Z, Thor A, Doi M, et al. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Science. 2009;122:1005–1013. doi: 10.1242/jcs.028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martone ME, Edelman VM, Thor A, Young SJ, Lamont SP, et al. Three dimensional analysis of transverse tubules in normal and failing heart: A combined confocal and high voltage electron microscope study. Microscopy and Microanalysis. 1997;3:231–232. [Google Scholar]
- 8.Orchard C, Brette F. t-Tubules and sarcoplasmic reticulum function in cardiac ventricular myocytes. Cardiovasc Res. 2008;77:237–244. doi: 10.1093/cvr/cvm002. [DOI] [PubMed] [Google Scholar]
- 9.Orchard CH, Pásek M, Brette F. The role of mammalian cardiac t-tubules in excitation-contraction coupling: experimental and computational approaches. Exp Physiol. 2009;94:509–519. doi: 10.1113/expphysiol.2008.043984. [DOI] [PubMed] [Google Scholar]
- 10.Pásek M, Brette F, Nelson A, Pearce C, Qaiser A, et al. Quantification of t-tubule area and protein distribution in rat cardiac ventricular myocytes. Prog Biophys Mol Biol. 2008;96:244–257. doi: 10.1016/j.pbiomolbio.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 11.Soeller C, Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image – processing techniques. Circ Res. 1999;84:266–275. doi: 10.1161/01.res.84.3.266. [DOI] [PubMed] [Google Scholar]
- 12.Soeller C, Jayasinghe ID, Li P, Holden AV, Cannell MB. Three-dimensional high-resolution imaging of cardiac proteins to construct models of intracellular Ca2+ signalling in rat ventricular myocytes. Exp Physiol. 2009;94:496–508. doi: 10.1113/expphysiol.2008.043976. [DOI] [PubMed] [Google Scholar]
- 13.Song LS, Guatimosim S, Gómez-Viquez L, Sobie EA, Ziman A, et al. Calcium biology of the transverse tubules in heart. Ann N Y Acad Sci. 2005;1047:99–111. doi: 10.1196/annals.1341.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 15.Despa S, Brette F, Orchard CH, Bers DM. Na/Ca exchange and Na/K-ATPase function are equally concentrated in transverse tubules of rat ventricular myocytes. Biophys J. 2003;85:3388–3396. doi: 10.1016/S0006-3495(03)74758-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berridge MJ, Bootman MD, Roderick HL. Calcium signaling: dynamics, homeostasis and remodeling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 17.Soeller C, Cannell MB. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophys J. 1997;73:97–111. doi: 10.1016/S0006-3495(97)78051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michailova A, DelPrincipe F, Egger M, Niggli E. Spatiotemporal features of Ca2+ signaling, buffering and diffusion in atrial myocytes with inhibited sarcoplasmic reticulum. Biophys J. 2002;83:3134–3151. doi: 10.1016/S0006-3495(02)75317-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu S, Michailova A, Saucerman JJ, Cheng Y, Yu Z, et al. Multi-Scale Modeling in Rodent Ventricular Myocytes: Contributions of structural and functional heterogeneities to excitation-contraction coupling. IEEE Eng Med Biol. 2009;28:46–57. doi: 10.1109/MEMB.2009.931787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Izu L, Wier WG, Balke C. Evolution of cardiac calcium waves from stochastic calcium sparks. Biophys J. 2001;80:103–120. doi: 10.1016/S0006-3495(01)75998-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Izu LT, Means SA, Shadid NJ, Chen-Izu Ye, Balke CW. Interplay of Ryanodine Receptor Distribution and Calcium Dynamics. Biophys J. 2006;91:95–112. doi: 10.1529/biophysj.105.077214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koh X, Srinivasan B, Ching H, Levchemko A. A 3-D Monte Carlo analysis of role of dyadic space geometry in spark generation. Biophys J. 2006;90:1999–2014. doi: 10.1529/biophysj.105.065466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keizer J, Smith GD, Ponce-Dawson S, Pearson JE. Saltatory Propagation of Ca2+ Waves by Ca2+ Sparks. Biophys J. 1998;75:595–600. doi: 10.1016/S0006-3495(98)77550-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shannon TR, Wang F, Puglisi J, Weber C, Bers DB. A Mathematical Treatment of Integrated Ca Dynamics within the Ventricular Myocyte. Biophys J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sher AA, Noble PJ, Hinch R, Gavaghan DJ, Noble D. The role of the Na+/Ca2+ exchangers in Ca2+ dynamics in ventricular myocytes. Prog Biophys Mol Biol. 2008;96:377–398. doi: 10.1016/j.pbiomolbio.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 26.Zahradnikova A, Kubalova Z, Pavelková J, Györke S, Zahradník I. Activation of calcium release assessed by calcium release-induced inactivation of calcium current in rat cardiac myocytes. Am J Physiol Cell Physiol. 2004;286:C330–C341. doi: 10.1152/ajpcell.00272.2003. [DOI] [PubMed] [Google Scholar]
- 27.Cheng Y, Yu Z, Hoshijima M, Holst M, McCulloch A, et al. Effects of membrane calcium flux localizations and realistic t-tubule geometry on cardiac excitation-contraction coupling. Biophys J 98, Supplement. 2010;1:551a. [Google Scholar]
- 28.Yu Z, Holst M, Cheng Y, McCammon JA. Feature-Preserving Adaptive Mesh Generation for Molecular Shape Modeling and Simulation. J Mol Graph Model. 2008;26:1370–1380. doi: 10.1016/j.jmgm.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 29.Yu Z, Holst MJ, Hayashi T, Bajaj CL, Ellisman MH, et al. Three-dimensional geometric modeling of membrane-bound organelles in ventricular myocytes: Bridging the gap between microscopic imaging and mathematical simulation. J Structural Biology. 2008;164:304–313. doi: 10.1016/j.jsb.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leeson TS. The transverse tubular (T) system of rat cardiac muscle fibers as demonstrated by tannic acid mordanting. Can J Zool. 1978;56:1906–1916. doi: 10.1139/z78-259. [DOI] [PubMed] [Google Scholar]
- 31.Pásek M, Simurda J, Christé G, Orchard CH. Modelling the cardiac transverse-axial tubular system. Prog Biophys Mol Biol. 2008;96:226–243. doi: 10.1016/j.pbiomolbio.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 32.Satoh H, Delbridge LM, Blatter LA, Bers DM. Surface:volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitance measurements: species-dependence and developmental effects. Biophys J. 1996;70:1494–1504. doi: 10.1016/S0006-3495(96)79711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bers DM. Excitation-contraction coupling and cardiac contractile force. Dortrecht, Boston, London: Kluwer Academic Press; 2001. [Google Scholar]
- 34.Page E. Quantitative ultrastructural analysis in cardiac membrane physiology. Am J Physiol. 1978;235:C146–C158. doi: 10.1152/ajpcell.1978.235.5.C147. [DOI] [PubMed] [Google Scholar]
- 35.Scriven DR, Dan P, Moore EW. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scriven DR, Klimek A, Lee KL, Moore EW. The molecular architecture of calcium microdomains in rat cardiomyocytes. Ann NY Acad Sci. 2002;976:488–499. doi: 10.1111/j.1749-6632.2002.tb04783.x. [DOI] [PubMed] [Google Scholar]
- 37.Jayasinghe ID, Cannell MB, Soeller C. Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys J. 2009;97:2664–2673. doi: 10.1016/j.bpj.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brette F, Leroy J, Le Guennec JY, Sallé L. Ca2+ currents in cardiac myocytes: Old story, new insights. Prog Biophys Mol Biol. 2006;91:1–82. doi: 10.1016/j.pbiomolbio.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 39.Leach RN, Desai JC, Orchard CH. Effect of cytoskeleton disruptors on L-type Ca channel distribution in rat ventricular myocytes. Cell Calcium. 2005;38:515–526. doi: 10.1016/j.ceca.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 40.Scriven DR, Klimek A, Asghari P, Bellve K, Moore EW. Caveolin-3 is adjacent to a group of extradyadic ryanodine receptors. Biophys J. 2005;89:1893–1901. doi: 10.1529/biophysj.105.064212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Z, Pascarel C, Steele DS, Komukai K, Brette F, et al. Na+-Ca2+ exchange activity is localized in the T-tubules of rat ventricular myocytes. Circ Res. 2002;91:315–322. doi: 10.1161/01.res.0000030180.06028.23. [DOI] [PubMed] [Google Scholar]
- 42.Thomas MJ, Sjaastad I, Andersen K, Helm PJ, Wasserstrom JA, et al. Localization and function of the Na+/Ca2+-exchanger in normal and detubulated rat cardiomyocytes. J Mol Cell Cardiol. 2003;35:1325–1337. doi: 10.1016/j.yjmcc.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 43.Kieval RS, Bloch JR, Lindenmayer JE, Ambesi A, Lederer WJ. Immunofluorescence localization of the Na-Ca exchanger in heart cells. Am J Physiol. 1992;263:C545–C550. doi: 10.1152/ajpcell.1992.263.2.C545. [DOI] [PubMed] [Google Scholar]
- 44.Yu Z, Holst M, McCammon JA. High-Fidelity Geometric Modelling for Biomedical Applications. Finite Elem Anal Des. 2008;44:715–723. [Google Scholar]
- 45.Bank R, Holst M. A New Paradigm for Parallel Adaptive Meshing Algorithms. SIAM Review. 2003;45:291–323. [Google Scholar]
- 46.Holst M. Adaptive numerical treatment of elliptic systems on manifolds. Adv Comput Math. 2001;15:139–191. [Google Scholar]
- 47.Cheng Y, Suen JK, Zhang D, Bond SD, Zhang Y, et al. Finite Element Analysis of the Time-Dependent Smoluchowski Equation for Acetylcholinesterase Reaction Rate Calculations. Biophys J. 2007;92:3397–3406. doi: 10.1529/biophysj.106.102533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng Y, Suen JK, Radić Z, Bond SD, Holst M, et al. Continuum Simulations of Acetylcholine Diffusion with Reaction-determined Boundaries in Neuromuscular Junction Models. Biophys Chem. 2007;127:129–139. doi: 10.1016/j.bpc.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng Y, Chang C, Yu Z, Zhang Y, Sun M, et al. Diffusional Channeling in the Sulfate Activating Complex: Combined Continuum Modeling and Coarse-grained Brownian Dynamics Studies. Biophys J. 2008;95:4659–4667. doi: 10.1529/biophysj.108.140038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ortega F. The General Mesh Viewer. 1996. Available: http://www.generalmeshviewer.com/. Accessed 1 September 2010.
- 51.Vaught A. Graphing with Gnuplot and Xmgr. 1996. Linux J. Available: http://www.linuxjournal.com/issue/28. Accessed 1 September 2010.
- 52.Collins-Sussman B, Fitzpatrick BW, Pilato CM. Version Control with Subversion. 2002. Available: http://subversion.tigris.org/. Accessed 1 September 2010.
- 53.Bers DM, Barry WH, Despa S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res. 2003;57:897–912. doi: 10.1016/s0008-6363(02)00656-9. [DOI] [PubMed] [Google Scholar]
- 54.Allbritton NL, Meyer T, Stryer L. Range of messenger action of calcium ion and inositol 1,4,5-triphosphate. Science. 1992;258:1812–1815. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]
- 55.Kushmerick MJ, Podolsky RJ. Ionic mobility in muscle cells. Science. 1969;166:1297–1298. doi: 10.1126/science.166.3910.1297. [DOI] [PubMed] [Google Scholar]
- 56.Safford RE, Bassingthwaighte EA, Bassingthwaighte JB. Diffusion of water in cat ventricular myocardium. J Gen Physiol. 1978;72:513–538. doi: 10.1085/jgp.72.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Z, Neher E. Mobile and immobile calcium buffers in bovine adrenal chromaffin cells. J Physiol. 1993;469:245–273. doi: 10.1113/jphysiol.1993.sp019813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wagner J, Keizer J. Effects of rapid buffers on Ca2+ diffusion and Ca2+ oscillations. Biophys J. 1994;67:447–456. doi: 10.1016/S0006-3495(94)80500-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gabso M, Neher E, Spira ME. Low mobility of the Ca2+ buffers in axons of cultured Aplysia neurons. Neuron. 1997;18:473–481. doi: 10.1016/s0896-6273(00)81247-7. [DOI] [PubMed] [Google Scholar]
- 60.Baylor SM, Hollingworth S. Model of sarcomeric Ca2+ movements, including ATP Ca2+ binding and diffusion, during activation of frog skeletal muscle. J Gen Physiol. 1998;112:297–316. doi: 10.1085/jgp.112.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang Y, Schlumpberger T, Kim T, Lueker M, Zucker RS. Effects of mobile buffers on facilitation: Experimental and computer studies. Biophys J. 2000;78:2735–2751. doi: 10.1016/s0006-3495(00)76819-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramay HR, Jafri MS, Lederer WJ, Sobie EA. Predicting local SR Ca2+ dynamics during Ca2+ wave propagation in ventricular myocytes. Biophys J. 2010;98:2515–2523. doi: 10.1016/j.bpj.2010.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soeller C, Crossman D, Gilbert R, Cannell MB. Analysis of ryanodine receptor clusters in rat and human cardiac myocytes. Proc Natl Acad Sci USA. 2007;104:14958–14963. doi: 10.1073/pnas.0703016104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grantham CJ, Cannell MB. Ca2+ influx during the cardiac action potential in guinea pig ventricular myocytes. Circ Res. 1996;79:194–200. doi: 10.1161/01.res.79.2.194. [DOI] [PubMed] [Google Scholar]
- 65.Sheehan KA, Blatter LA. Regulation of junctional and non-junctional sarcoplasmic reticulum calcium release in excitation-contraction coupling in cat atrial myocytes. J Physiol. 2003;546:119–135. doi: 10.1113/jphysiol.2002.026963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Niggli E. Ca(2+) Sparks in Cardiac Muscle: Is There Life Without Them? News Physiol Sci. 1999;14:129–134. doi: 10.1152/physiologyonline.1999.14.4.129. [DOI] [PubMed] [Google Scholar]
- 67.Jafri MS, Keizer J. On the roles of Ca2+ diffusion, Ca2+ buffers, and the endoplasmic reticulum in IP3-induced Ca2+ waves. Biophys J. 1995;69:2139–2153. doi: 10.1016/S0006-3495(95)80088-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pásek M, Simurda J, Orchard CH, Christé G. A model of the guinea-pig ventricular cardiac myocyte incorporating a transverse-axial tubular system. Prog Biophys Mol Biol. 2008;96:258–280. doi: 10.1016/j.pbiomolbio.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 69.Michailova A, Lorentz W, McCulloch AD. Modeling transmural heterogeneity of KATP current in rabbit ventricular myocytes. Am J Physiol. 2007;293:C542–C557. doi: 10.1152/ajpcell.00148.2006. [DOI] [PubMed] [Google Scholar]
- 70.Hinch R, Greenstein JL, Tanskanen AJ, Xu L, Winslow RL. A simplified local control model of calcium induced calcium release in cardiac ventricular myocytes. Biophys J. 2004;87:3723–3736. doi: 10.1529/biophysj.104.049973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
100 µM Fluo-3: Calcium flux heterogeneously distributed (left panel); Calcium flux higher and uniform in the t-tubule (middle panel); Calcium flux uniformly distributed (right panel).
(5.45 MB AVI)
Zero Fluo-3 and calcium flux heterogeneously distributed: ATP and calmodulin mobile (left panel); ATP and calmodulin immobile (right panel).
(4.05 MB AVI)
Zero Fluo-3 and calcium flux heterogeneously distributed: Extracellular sodium 140 mM (left panel); Zero extracellular sodium (right panel.)
(4.07 MB AVI)