

Abstract
Previous studies on the chemopreventive mechanisms of dietary selenium have focused on its incorporation into antioxidative selenoproteins, such as glutathione peroxidase and thioredoxin reductase. Several studies, however, have revealed that dietary selenium in the form of L-selenomethionine and the 21st amino acid, selenocysteine, also have intrinsic anti-cancer properties. Biochemical mechanisms previously investigated to contribute to their anticancer effects involve β- and γ-lyase reactions. Some pyridoxal 5′-phosphate (PLP)-containing enzymes can catalyze a β-lyase reaction with Se-methyl-L-selenocysteine (MSC) generating pyruvate and ammonia. Other PLP-enzymes can catalyze a γ-lyase reaction with L-selenomethionine (SM) generating α-ketobutyrate and ammonia. In both cases, a purported third product is methylselenol (CH3SeH). Although not directly quantifiable, as a result of its extreme hydrophobicity and high vapor pressure, CH3SeH has been indirectly observed to act through the alteration of protein-sulfhydryl moieties on redox-responsive signal and transcription factors, thereby maintaining a non-proliferative intracellular environment. We have considered the possibility that α-keto acid analogues of MSC (i.e., methylselenopyruvate; MSP) and SM (i.e., α-keto-γ-methylselenobutyrate; KMSB), generated via a transamination and/or L-amino acid oxidase reaction may also be chemoprotective. Indeed, these compounds were shown to increase the level of histone-H3 acetylation in human prostate and colon cancer cells. MSP and KMSB structurally resemble butyrate, an inhibitor of several histone deacetylases. Thus, the seleno α-keto acid metabolites of MSC and SM, along with CH3SeH derived from β- and γ-lyase reactions, may be potential direct-acting metabolites of organoselenium that lead to de-repression of silenced tumor suppressor proteins and/or regulation of genes and signaling molecules.
Keywords: Prostate cancer, Se-Methyl-L-selenocysteine, L-Selenomethionine, Histone deacetylase, Methylselenopyruvate, α-Keto-γ-methylselenobutyrate, Glutamine transaminase K, Glutamine transaminase L, L-Amino acid oxidase
Introduction: chemopreventive properties of naturally occurring organoselenium compounds
Epidemiological data and laboratory investigations strongly support the importance of organoselenium compounds as having a broad spectrum of chemopreventive properties against several human cancer cells (bladder, prostate, breast, ovarian, colon, liver, lung, and leukemic) (Combs 2004; Clark et al. 1996; Rudolf et al. 2008; Sanmartín et al. 2008; Suzana et al. 2009). Over the past several years, important biochemical features of dietary selenium have emerged that relate directly to its anticarcinogenic mechanisms, namely through antioxidant effects, anti-androgen activity, growth inhibitory action by regulating signaling molecules and apoptosis inducers, and by modifying gene expression (Lu and Jiang 2001; Wang et al. 2002; Zeng et al. 2009). These and other anticancer mechanisms ascribed to organoselenium are markedly dependent on its chemical form and metabolic transformations. When consumed in adequate amounts in the diet, organoselenium can manifest its chemopreventive activity either by conversion to selenometabolites or by incorporation into select selenoproteins, such as glutathione peroxidase and thioredoxin reductase. The chemopreventive mechanisms associated with these two redox enzymes and other selenoproteins have been the subject of several reviews (e.g. Ip 1998; Ganther 1999; Papp et al. 2007) and will not be considered further in this manuscript. Instead, our discussion will be limited to the potential anti-cancer properties of metabolites of naturally occurring organoselenium compounds SM and MSC.
Role of methylselenol (CH3SeH) as an anti-cancer agent
In addition to acting as precursors to a diverse number and variety of biologically important selenoproteins, the chemopreventive efficacy and anticancer properties of dietary organoselenium compounds have been postulated to result from in situ formation of CH3SeH. In mammalian cells both thiol and selenol groups are used to maintain reducing intracellular redox states acting as antioxidants and reducing agents in redox signaling with reactive oxygen species. Selenols have a distinct advantage over thiols in having a more facile reducing capacity (Chen and Maret 2001; Lillig and Holmgren 2007) and CH3SeH has been proposed to alter cell-signaling pathways, influence protein kinases and induce cellular apoptosis (Unni et al. 2005; Singh et al. 2008; Zeng et al. 2009). Thus, interaction of selenol with disulfide domains on proteins has been the topic of mechanistic debates to explain how CH3SeH manifests its putative anticancer properties by regulating redox-sensitive effector proteins (Ip et al. 2000). When their disulfide domains become reduced or, conversely, when their thiol domains become oxidized, these modulating proteins act as rheostats or regulatory switches that can transmit controlled responses within a multitude of cell signaling pathways, thus influencing cell proliferation and apoptosis. Two diagrammatic representations of possible interactions of CH3SeH with proteins are depicted below:
or
However, due to its highly volatile and hydrophobic nature, CH3SeH cannot be directly administered to cells, but can be generated endogenously, for example following intracellular reduction of dimethyldiselenide (DMDSe) or methylseleninic acid (MSA) by the redox active tripeptide, glutathione, and conceivably by a variety of protein-sulfhydryl moieties (Spallholz et al. 2001). In fact, MSA has been shown to cause redox changes with proteins and to have antiproliferative effects through induction of inhibitors of cyclin-dependent kinases (Jiang et al. 2002; Gasparian et al. 2002; Venkateswaran et al. 2002). For example, brief exposure of an osteoblast cell line (MC3T3-E1) to MSA decreases the production of inflammatory gene products normally regulated by the redox-sensitive transcription factor, nuclear factor-kappa B (NF-κB). Osteoblasts, when subjected to conditioned media from MDA-MB-231 breast cancer cells, respond by producing inflammatory factors such as interleukin-6, monocyte chemoattractant protein-1, cyclooxygenase-2 and inducible nitric oxide synthase, which, in turn, stimulate osteoclast-induced bone erosion. Chen et al. (2009) showed that this response of osteoblasts was abrogated following treatment with MSA. Presumably, redox-sensitive switching mechanisms are involved following interactions of MSA with sulfhydryl domains on either the ikB-NF-κB protein complex or the upstream iKKα,β,γ inhibitory domains. MSA has also been shown to down regulate expression of hypoxia-inducible factor-1α (a redox-sensitive protein), DNA-binding activity and its downstream targets, VEGF and GLUT1 in hormone refractory prostate cancer cells (Sinha et al. 2009). Thus, mechanisms that govern the interactions of CH3SeH and MSA with sulfur domains on redox-sensitive signal proteins need to be further elucidated. It is unknown whether the chemopreventive efficacies of CH3SeH and MSA are manifested directly by their presence in the cell or through the process by which they become oxidized and reduced, respectively.
Although not directly quantifiable, CH3SeH has been indirectly determined in urine as dimethylselenide and, following methylation, as trimethylselenonium. Unfortunately, these two metabolites of CH3SeH are not useful dietary markers of selenium consumption or following use of micromolar amounts of MSA as they cannot be determined simultaneously under physiological intakes of organoselenium compounds, but only after consumption of toxic to subtoxic doses (Ohta et al. 2009). Thus, the ability to monitor transformation, function and efficacy of organoselenium using metabolites of CH3SeH under physiological conditions is currently unfeasible.
Not only can CH3SeH be generated in vivo from the currently aforementioned synthetic derivatives, DMDSe and MSA, but it can also be generated from MSC and SM. These seleno-amino acids can be converted to CH3SeH by pyridoxal 5′-phosphate (PLP)-dependent γ-lyases (Eq. 1) and β-lyases (Eq. 2) acting on SM and MSC, respectively. For example, Zeng et al. (2009) used L-methionine γ-lyase from Brevibacterium linens BL2 to generate CH3SeH from SM. Exposure of HT1080 tumor cells in situ to this enzyme-generated CH3SeH within the culture medium inhibited cell migration and invasion (Zeng et al. 2009).
| (1) |
| (2) |
Production of CH3SeH from SM and MSC by mammalian liver cystathionine γ-lyase
A γ-lyase that has been shown to catalyze a γ-elimination reaction with SM is mouse liver cystathionine γ-lyase (γ-cystathionase) (Okuno et al. 2005). The net products are α-ketobutyrate [CH3CH2C(O)CO2−], ammonia and CH3SeH (Eq. 1). However, the specific activity of the purified enzyme toward SM is, at best, limited, exhibiting reactivity less than 1% that observed with the natural substrate, cystathionine (Okuno et al. 2005). Cystathionine γ-lyase is very active (when assayed with homoserine or with its natural substrate cystathionine) in rodent liver cytosol and to a lesser extent in kidney cytosol. It is absent, or of very low specific activity, in other organs/tissues.
It has long been known that, with an appropriate substrate, cystathionine γ-lyase can catalyze β-elimination reactions in addition to γ-lyase reactions (Cavallini et al. 1960). For example, the enzyme can catalyze β-elimination reactions involving cysteine and cystine (Cavallini et al. 1960). We have now found that MSC is an excellent β-lyase substrate of highly purified rat liver cystathionine γ-lyase (Krasnikov, Cooper, Pinto, unpublished data). In fact, the enzyme catalyzed a β-elimination reaction with 10 mM MSC at approximately the same rate as it catalyzes a γ-elimination from 10 mM cystathionine (Krasnikov, Cooper, Pinto, unpublished data). Interestingly, the enzyme is incapable of catalyzing a β-lyase reaction with S-methyl-L-cysteine (Cooper and Pinto 2005). The ability of cystathionine γ-lyase to cleave the C–Se bond of MSC but not the C–S bond of S-methyl-L-cysteine is due to the greater leaving group propensity of the –SeCH3 moiety relative to that of the –SCH3 group (see the discussion below).
CH3SeH production from MSC by glutamine transaminase K
Glutamine transaminase K is widely expressed in rat tissues. In the rat, the highest specific activity is in kidney, but appreciable activity is also present in liver and, to a lesser extent, in other organs (Cooper 1988; Cooper and Meister 1981). The rat kidney enzyme has broad specificity (as an aminotransferase) toward glutamine, methionine (and other sulfur-containing amino acids), aromatic amino acids, and the α-keto acid analogs of most amino acid substrates (Cooper 1998; Cooper and Meister 1981; Cooper and Anders 1990; Commandeur et al. 2000).
It is of considerable interest that rat kidney GTK also exhibits appreciable cysteine S-conjugate β-lyase activity with cysteine S-conjugates that contain a good leaving group (nucleofuge) in the β position (Cooper 1998, 2004; Stevens et al. 1986; Commandeur et al. 2000). The net reaction is shown in Eq. 3, where XS- becomes the sulfhydryl leaving group.
| (3) |
This ability of rat kidney GTK to catalyze β-elimination reactions was exploited by Commandeur et al. (2000). These authors showed that highly purified rat kidney GTK catalyzes competing transaminating and β-lyase reactions with a large number of L-selenocysteine Se-conjugates. The β-lyase reaction with these conjugates is shown in Eq. 4.
| (4) |
MSC may be regarded as a special case of a selenocysteine Se-conjugate in which X = CH3– and the eliminated Se-containing fragment is CH3SeH. MSC was among the large number of selenocysteine Se-conjugates shown to be effective transaminase/β-lyase substrates of highly purified rat kidney GTK by Commandeur et al. (2000). We have found that MSC is likewise an effective transamination/β-lyase substrate of highly purified recombinant human GTK (Cooper et al. 2008).
Intriguingly, Commandeur et al. (2000) showed that the rate of transamination and β-elimination reactions with L-selenocysteine Se-conjugates catalyzed by highly purified rat kidney GTK in most cases was at least an order of magnitude greater than that exhibited with the corresponding L-cysteine S-conjugates. The remarkable increase in reactivity of the selenocysteine Se-conjugate versus the corresponding cysteine S-conjugate may be due to subtle differences between the chemistry of sulfur and selenium (Commandeur et al. 2000; Rooseboom et al. 2002). For a β-lyase reaction to proceed with a cysteine (or selenocysteine) conjugate, breakage of the C–S (or C–Se) bond must occur. It is well known that the C–Se bond is considerably weaker than the C–S bond (Wessjohann et al. 2007), thereby facilitating elimination of CH3SeH from the selenocysteine Se-conjugate relative to elimination of CH3SH from the corresponding cysteine S-conjugate. Moreover, both the transamination reaction and the β-elimination reaction require the breakage of the α C–H bond of the conjugate. This bond appears to be weaker (i.e., more easily cleaved) in the selenocysteine Se-conjugates relative to that in the cysteine S-conjugates (Commandeur et al. 2000), thereby facilitating both transamination and β-elimination reactions. It should also be pointed out that the pKa value of the –SeH group of selenocysteine (pKa = 5.2) is considerably lower than that of the –SH group of cysteine (pKa = 8.3), and that selenocysteine is a stronger nucleophile that enhances its reactivity relative to cysteine (Huber and Criddle 1967; Wessjohann et al. 2007).
The transamination reaction catalyzed by GTK is discussed in more detail below. It is important to note, however, that if an α-keto acid co-substrate is omitted from the reaction mixture a half transamination reaction will convert the PLP form of the co-enzyme to the pyridoxamine 5′-phosphate (PMP) form. The PLP form, but not the PMP form, can support a β-lyase reaction. Therefore, in order for GTK to continue to catalyze an effective β-lyase reaction with MSC an α-keto acid co-substrate (or adequate PLP) must be present to ensure conversion of the PMP form of the enzyme to the PLP form (Cooper 1998; Stevens et al. 1986). In the case of GTK, a particularly effective α-keto acid substrate is α-keto-γ-methiolbutyrate, the α-keto acid analogue of methionine (Cooper 1998; Cooper and Meister 1981).
Human GTK catalyzes effective transamination with MSC but not with SM
We have investigated the ability of purified recombinant human GTK to catalyze transamination reactions with MSC. In our original report, for ease of assay, we used phenylpyruvate as the α-keto acid co-substrate (Cooper et al. 2008). However, we have now shown that the enzyme readily catalyzes transamination between MSC and α-keto-γ-methiolbutyrate to yield β-methylselenopyruvate (MSP) and L-methionine (Eq. 5).
| (5) |
Since L-methionine and S-methyl-L-cysteine are substrates of purified rat kidney GTK (Cooper and Meister 1981), we reasoned that L-methionine should also be a good substrate of highly purified recombinant human GTK. This was shown to be the case (Cooper et al. 2008). Since L-methionine and MSC are good substrates of human GTK, we anticipated that SM should also be a good aminotransferase substrate. Much to our surprise, SM was an exceedingly poor substrate of human GTK (<0.1% the activity exhibited with L-methionine) (Cooper et al. 2008). We will revisit this point later when we discuss the metabolism of MSC and SM in human prostate cells and the chemopreventive properties of SM relative to that of MSC. The ability of human GTK to catalyze effective transamination with L-methionine, S-methyl-L-cysteine and MSC, but not with SM underscores yet again the subtle differences between the chalcogenide chemistry of S and Se. The covalent radius of Se (1.17 Å) is about 13% larger than that of S (1.04 Å) (Moeller 1963), which may be just enough to prevent the binding of SM, but not that of methionine or even MSC, which is a single methylene group shorter than SM, to the active site of human GTK.
Transamination of SM by a bovine liver glutamine transaminase
SM has been reported to be a good substrate of bovine liver glutamine transaminase (Blarzino et al. 1994). At first glance, this finding appears to contradict our finding that GTK (a glutamine transaminase) does not effectively catalyze transamination with SM. The apparent contradiction may be due to species differences (rat enzyme versus bovine enzyme). More likely, however, the difference in catalytic specificity is due to the fact that there are at least two distinct glutamine transaminases. Although rat tissue homogenates were long known to catalyze transamination of glutamine (Meister et al. 1952), it was not until some time later that rat tissues were shown to contain at least two glutamine transaminases (Cooper and Meister 1981). As noted above, in the rat, GTK is most active in kidney, but is widely distributed in other tissues. On the other hand, rat liver possesses a distinct glutamine transaminase, named glutamine transaminase L (GTL). Unlike GTK activity which is widespread, this enzyme appears to be largely confined to the liver (Cooper 1988: Cooper and Meister 1981). Unlike GTK, which has been widely studied as an aminotransferase and cysteine S-conjugate β-lyase, very few studies have been conducted with GTL. Our laboratory has recently demonstrated that GTL can catalyze transamination of SM with α-keto-γ-methiolbutyrate to give α-keto-γ-methylselenobutyrate (KMSB) and L-methionine (Eq. 6) (unpublished data). Unlike rat and human GTK where the aminotransferase activity toward MSC is much greater than that toward SM, rat liver GTL exhibits greater aminotransferase activity toward SM than toward MSC.
| (6) |
Are MSC and SM substrates of mammalian L-amino acid oxidase (LAAO)?
In addition to generation through transamination reactions, MSP and KMSB may theoretically also arise from MSC and SM by LAAO reactions (Eqs. 7, 8), respectively.
| (7) |
| (8) |
Mammalian tissues contain at least three LAAOs that potentially may convert one or both selenoamino acids to their corresponding α-keto acids. The first to be discovered is of highest activity in kidney and liver. The enzyme activity is catalyzed as a side reaction of L-hydroxy acid oxidase B (Duley and Holmes 1976). The enzyme has a pH maximum (≥9.0) and is active with L-methionine and other large neutral amino acids. A second LAAO, purified from mouse milk (Sun et al. 2002), reacts with a broad range of amino acids in the order of Phe > Met, Tyr > Cys, Leu > His. Due to its dose and time-dependent reactivity with these amino acids to produce H2O2, the secreted enzyme was shown to be crucial for host defense against bacterial infection in the mammary gland (Nagaoka et al. 2009). Most recently, Mason et al. (2004) demonstrated that a mouse B lymphocyte protein, IL4I1, has unique LAAO properties. Interestingly, the same protein, isolated from a mediastinal lymphoma B-cell line, was shown to be secreted (Boulland et al., 2007) and to exhibit LAAO activity which was optimal at physiological pH and primarily directed toward phenylalanine. Immunohistochemical analysis of secondary lymphoid organs showed LAAO staining within germinal center macrophages and inflammatory myeloid cells with the highest staining in mature dendritic cells. Carbonnelle-Puscian et al. (2009) found that expression of IL4I1 in tumors was very frequent and that it was detectable in tumor-associated macrophages and in neoplastic cells from follicular lymphoma, classic and nodular lymphocyte predominant Hodgkin's lymphoma and small lymphocytic lymphoma. These findings raise the possibility that organoseleno amino acids have the potential of being converted to their corresponding α-keto acids at or within lymphoid cells. The possibility of IL4I1 functioning as a pro-activator of selenoamino acids to their corresponding keto acid forms is currently under investigation in our laboratory. Thus, at present, it remains uncertain as to the extent to which the leukocyte LAAOs can catalyze in vivo oxidative deamination of MSC and SM to MSP and KMSB, respectively.
In situ formation of MSP and KMSB with Crotalus adamanteus venom LAAO
Purified LAAO from the venom of the diamondback rattlesnake (C. adamanteus) has a remarkably broad specificity toward amino acids (Lichtenberg and Wellner 1968). The enzyme is, however, most active with large hydrophobic L-amino acids. The most active substrate is L-methionine (Lichtenberg and Wellner 1968). Based on this specificity, we predicted that MSC and SM would be excellent substrates, and this was found to be the case (Lee et al. 2009). The snake venom LAAO can be used as a convenient reagent to prepare both MSP and KMSB from MSC and SM, respectively. This enzyme is used to produce batch quantities of both MSP and KMSB for studies involving mechanisms of the histone deacetylase inhibitory effects of seleno α-keto acids (Lee et al. 2009; Nian et al. 2009b).
MSP and KMSB as histone deacetylase (HDAC) inhibitors
Research in the area of histone acetylation and deacetylation has evolved into a fascinating theme that encompasses regulation of numerous molecular targets that control cancer proliferation (Marks and Xu 2009). The literature is replete with reviews and studies that demonstrate the role of histone proteins in modulating chromatin plasticity, facilitating protein:DNA interactions and thus manifesting transcriptional control (Giacinti et al. 2008; Keppler and Archer 2008). Concurrent with the interest in histone acetylation and de-acetylation, research has focused on identifying compounds that are able to accelerate histone acetylation or inhibit histone de-acetylation (Vigushin and Coombes 2004; Bolden et al. 2006; Dokmanovic et al. 2007; Frew et al. 2009).
In the latter case, the emergence of a number of tools that facilitate the screening of both molecular targets and therapeutic candidates has led to identifying a variety of compounds that are associated with inhibition of histone de-acetylation, including short-chain fatty acids (e.g., butyrate), hydroxamic acids (e.g., SAHA and Trichostatin), epoxyketones (e.g., trapoxin), benzamides (Lane and Chabner 2009; Shankar and Srivastava 2008; Richon et al. 2000), and a variety of other chemical families including organosulfur (Nian et al. 2008, 2009a; Myzak et al. 2004) and recently organoselenium compounds (Lee et al. 2009; Nian et al. 2009b). The structural multiplicity of HDAC inhibitors reflects both the diversity of substrates for HDACs and the heterogeneity of tumor cell phenotypes.
Since MSP and KMSB strongly resemble a major class of HDAC inhibitors, namely short-chain fatty acids and in particular, butyric acid, we examined whether seleno α-keto acids possess selectivity as HDAC inhibitors. Eleven of the eighteen recognized HDACs harbor a coordinating zinc atom within their active site (Buggy et al. 2000; Vanommeslaeghe et al. 2005). Thus, an intriguing feature of an inhibition by the seleno α-keto acids would be the proximity and juxtaposition of the highly electronegative selenium moiety with the active-site zinc atom which may facilitate disruption of the charge relay system within the HDAC pocket (Nian et al. 2009a, b; Lee et al. 2009).
HDAC inhibitors show promise clinically for the treatment of several human cancers, and several mechanisms have been proposed (Ungerstedt et al. 2005; Insinga et al. 2005; Xu et al. 2007). The challenge to identifying the role of organoselenium compounds in HDAC inhibition is to attribute specific HDAC complexes to cellular function and to identify which organoselenium metabolite is able to block the activity of these functions. Meeting the challenge of chemoprevention through nutritional management is likely to produce dietary regimens involving naturally occurring organoselenium and organosulfur compounds (Myzak et al. 2004; Nian et al. 2009a, b; Lee et al. 2009) coupled with combinations of other nutrients such as vitamins A and D (Puccetti et al. 2002). These dietary constituents share co-repressor activity and selectivity not currently developed with existing cytostatic drugs. Controlled combinations of these nutrients would have the ability to target the expression of selected genes and sequester factors involved in the differentiation signaling pathways usually reserved for biological agents.
The possible metabolic routes for SM and MSC are shown in Fig. 1 and illustrate that the chemoprotective properties of seleno amino acids reside in the cell's ability to convert them via lyase reactions to CH3SeH and through transamination or L-amino acid oxidation to their corresponding seleno α-keto acids metabolites, KMSB and MSP, both of which are now known to function as effective HDAC inhibitors. Current studies with KMSB and MSP in both colon (Nian et al. 2009b) and human prostate cancer cells (Lee et al. 2009) by our group reveal robust, concentration-dependent, HDAC inhibition by seleno α-keto acids and, coupled with molecular modeling studies, support a mechanism that involves reversible competitive inhibition. In addition, both cell types have been examined for changes in growth, apoptosis and expression of p21. In view of the recently aborted Selenium and Vitamin E Cancer Prevention Trial (SELECT) study (Lippman et al. 2009), a human prostate cancer prevention trial (see below for more details), this review will focus on prostate cancer cells rather than on colon cancer cells to illustrate the need to understand the specificity and selectivity of the biochemical transformations of organoselenium compounds.
Fig. 1.
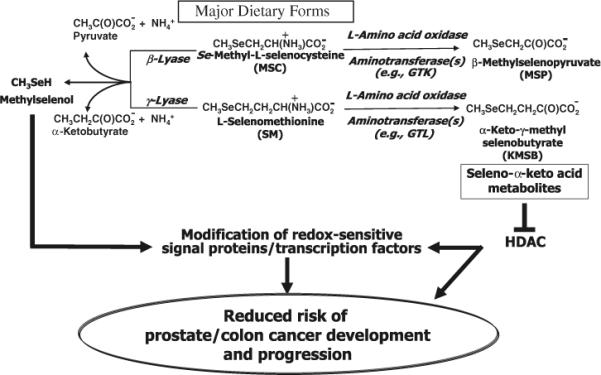
Proposed metabolic pathways for naturally occurring organoselenium compounds. Dietary seleno amino acids can undergo either β-/γ-elimination reactions with formation of CH3SeH or transamination/oxidative deamination reactions with formation of seleno α-keto acids. CH3SeH from the former reaction is a putative anticancer metabolite of organoselenium compounds which can react with redox sensitive signal proteins. The α-keto acid metabolites from the latter reaction exhibit HDAC inhibitory properties in human prostate and colon cancer cells which is symbolized by ⊥
Human prostate cells are targets for HDAC inhibition
Treatments for prostate cancer include hormone ablation and endocrine therapy that lead to temporary palliation of the disease, but relapse usually occurs 1–2 years after treatment (Scher et al. 1996). Although the androgen receptor has been the primary target governing therapeutic strategies for prostate cancer, androgen-independent mechanisms are involved in the progression and transformation of prostate cancer and may underlie the cause of the lingering complications in traditional androgen deprivation therapy (Shah et al. 2004). Thus, there remains a continuing need to identify novel targets and to develop convenient dietary agents that may eliminate or slow progression and transformation of both androgen receptor-positive and -negative prostate cancer cells.
HDACs are under active investigation as novel enzymatic targets that lend themselves to development of both drug and dietary strategies against androgen-dependent and -resistant prostate cancers. HDAC inhibitors not only abrogate transcription of androgen receptor proteins, but also function as transcription inhibitors of target genes of the androgen receptor thus making them effective against androgen-responsive and -refractory cells (Welsbie et al. 2009). Using a combination of DNA microarray, in situ hybridization, and immunohistochemical techniques, Wang et al. (2009) demonstrated that HDAC levels are dysregulated in prostate cancer and that HDAC 1–5 show increased expression. Clark and Cooper (2009) and Huang and Waknitz (2009) demonstrated that high expression of HDAC 1 in prostate cancers correlates with expression of gene fusion between erythroblastosis virus E26 transforming sequence (ETS) and androgen-regulated, prostate specific transmembrane protease serine 2 (TMPRSS2). Thus, for approximately 50% of the prostate cancer patients who harbor ETS gene fusions, targeting HDAC 1 would be a definite therapeutic possibility. HDAC3, but not HDAC 1 or 2, interacts with FOXO1, a transcription factor that functions downstream of the tumor suppressor, phosphatase and tensin homolog (PTEN) and controls expression of genes involved in apoptosis and cell cycle progression. HDAC 3 inhibitors can attenuate FOXO1-mediated inhibition of the androgen receptor (Liu et al. 2008; Yang et al. 2005). A bitter melon protein that contains Type I ribosome-inactivating proteins inhibits HDAC 1 activity, promotes histone-3 and -4 protein acetylation, and promotes PTEN expression in prostatic intraepithelial neoplasia (PIN) and prostate cancer cell lines resulting in increased apoptosis (Xiong et al. 2009). The chemopreventive phytochemical, phenethyl isothiocyanate (PEITC), a constituent of cruciferous vegetables, induces growth arrest and apoptosis in human prostate cancer cells, and in rodent xenograft models of prostate cancer, inhibits testosterone-mediated prostatic growth by regulating the androgen receptor and cell cycle progression. PEITC enhances histone acetylation and induces selective modification of histone methylation for chromatin remodeling. The effect of chromatin unfolding thus permits transcription activation of the tumor suppressor gene, p21 (Wang et al. 2008). Thus, of the variety of aberrant epigenomic covalent alterations that occurs in prostate cancer and contributes to gene silencing and carcinogenesis, deficient histone acetylation involving excessive HDAC activity is a prominent feature. This aberrant increase in histone deacetylation appears to underlie disruption of the epigenetic control of gene expression in normal prostate cells and plays a major part in the multistep process of carcinogenesis.
Our recent discovery that KMSB and MSP have potent HDAC inhibitory properties expands our understanding of the chemopreventive efficacy of organoselenium compounds (Lee et al. 2009). In addition, insights are emerging that different tissues may show distinctive responses to, or may have selectivity to metabolize their precursor amino acids namely, SM and MSC. For example in liver, SM would be rapidly metabolized by GTL to KMSB but only marginally metabolized to CH3SeH by cystathionine-γ-lyase. In kidney, SM would be marginally metabolized to KMSB by GTK and marginally converted to CH3SeH by the low level of cystathionine-γ-lyase. In the case of exposure to MSC, liver would marginally convert MSC to MSP by GTL and GTK but rapidly convert it to CH3SeH by the action of cystathionine-γ-lyase. In kidney, MSC would be marginally converted by GTK and cystathionine-γ-lyase to MSP and CH3SeH, respectively. Table 1 provides a brief outline of the enzymes including their relative activities that catalyze reactions with SM and MSC. By contrast to the substrate selectivity of the glutamine transaminases and cystathionine-γ-lyase in liver and kidney, which favor one or the other of SM or MSC, tissues such as lymphocytes that contain an LAAO may have the potential to convert both SM and MSC to their corresponding α-keto acids. Thus, LAAOs in certain tissues, milk and activated lymphocytes show promise in oxidizing both MSC and SM to their corresponding keto acids which then may show selectivity and/or synergy toward different HDAC enzymes with sensitivity to MSP or KMSB or both. Lastly, the ability of cells to metabolize either SM or MSC or both may reflect the difference between toxicity and chemopreventive efficacy of the dietary source. For example, selenium-enriched garlic contains predominately MSC, which can be metabolized via transamination in most tissues that contain GTK to MSP. In addition, MSC can be converted to CH3SeH in a competing reaction catalyzed by GTK and by cystathionine-γ-lyase in the liver. Whereas selenium-enriched yeast which contains predominately SM can be metabolized to KMSB by liver which possesses high levels of GTL and only marginally to CH3SeH by hepatic cystathionine-γ-lyase.
Table 1.
Enzymes, their location, relative specific activity and relative amounts of products derived from L-selenomethionine (SM) and Se-methyl-L-selenocysteine (MSC)
| Enzyme | Location/relative specific activity with natural substrate | Products derived from SM relative to natural substrates (+++++) | Products derived from MSC relative to natural substrates (+++++) |
|---|---|---|---|
| Cystathionine γ-lyase | Liver (+++++); kidney (+++); other tissues (+ or ND) | α-Ketobutyrate and CH3SeH (+) | Pyruvate and CH3SeH (++++) |
| Glutamine transaminase K (GTK) | Kidney (+++++); liver (+++); other tissues (++) | KMSB (±)a | Pyruvate and CH3SeH (+++)b MSP (+++)a |
| Glutamine transaminase L (GTL)c | Liver (+++++) | KMSB (++++) | MSP (++) |
| L-Amino acid oxidase | Snake venom (+++++) Crotalus adamanteus | KMSB (+++++) | MSP (+++++) |
+++++, greatest amount; +, least amount
ND not detectable, KMSB α-Keto-γ-methylselenobutyrate, MSP β-Methylselenopyruvate
<0.1% the activity exhibited with methionine
GTK catalyzes competing β-elimination and transamination reactions. The products are pyruvate plus CH3SeH and MSP, respectively
GTL is also theoretically capable of catalyzing a β-elimination reaction with MSC. However, this possibility has not yet been investigated
Prostate cancer cells metabolize MSC but not SM
We have shown that human prostate cancer cells (LNCaP, LNCaP C4-2, PC-3 and DU 145) possess GTK activity as assessed by the standard phenylalanine—α-keto-γ-methiolbutyrate transaminase assay (Lee et al. 2009). However, we were unable to detect cystathionine γ-lyase activity or L-amino acid oxidase activity. These findings suggest that human prostate cancer cells in culture are able to metabolize MSC but not SM. This was found to be the case. With the aid of an HPLC system equipped with a Coul-Array detector, we showed that human prostate cells in culture metabolize MSC to MSP. The rate of disappearance of MSC was enhanced in the presence of α-keto-γ-methiolbutyrate. The disappearance of MSC in the presence of this α-keto acid was associated with appearance of L-methionine as expected from our specificity studies with purified recombinant human GTK (Cooper et al. 2008). Also, as expected from these specificity studies, the cells were unable to catalyze transamination of SM with α-keto-γ-methiolbutyrate even over a 24-h period.
When MSC and SM are separately added to cell culture medium, MSC-treated cells express increased levels of acetylated histone 3 after 5 h incubation. The finding that LNCaP cells demonstrated greater accumulation of acetylated histone 3 in the presence of MSC at 50 μM concentration after 5 h incubation than observed in other prostate cancer cell lines may reflect the higher specific activity of GTK for MSC in LNCaP compared to LNCaP C4-2 and PC3 cells (Lee et al. 2009). However, cells incubated for up to 48 h with SM failed to increase acetylated histone levels in any of the cell lines tested, illustrating further that SM is not transaminated in human prostate cancer cells. By contrast, direct treatment of cells with the α-keto acid forms of MSC and SM, namely MSP and KMSB, results in elevation of acetylated histone 3 in all cell lines within 5 h at 50 μM concentration, supporting our hypothesis that the increased level of acetylated histone 3 following MSC treatment is due to its metabolite, MSP. Thus, the seleno amino acids, MSC and SM do not affect total histone 3 levels but the corresponding seleno α-keto acids, however, increase the ratio of acetylated to total histone 3.
Although CH3SeH is regarded as an important chemopreventive metabolite of both SM and MSC, our data indicate that the corresponding α-keto acid metabolites (especially that derived from MSC) through their properties of a having HDAC inhibitory effects may be just as important, or perhaps even more so (see below). Our findings implicate the central role of GTK as an in situ generator and contributor to the pool of MSP and a potential source of CH3SeH. The beneficial effect of MSC toward the prostate may thus be correlated with the presence of GTK in this organ, which has the potential to endogenously generate two chemoprotective species, namely CH3SeH and the α-keto acid analogue of MSC (i.e., MSP). We have shown that the α-keto acid analogue of SM (i.e., KMSB) also has the potential to be chemoprotective via inhibition of HDACs. However, since the prostate cells cannot generate KMSB from SM, for KMSB to be a useful chemoprotectant it would have to be generated exogenously (perhaps in the liver, kidney or lymphocytes) and transported to the prostate cells. Work is ongoing in our laboratory to investigate this possibility.
From a comparison of our studies (Cooper and Pinto 2005; Pinto et al. 2007; Sinha et al. 2008; Cooper et al. 2008; Lee et al. 2009) with those of Suzuki et al. (2008) and Tsuji et al. (2009), we suggest that (a) the α-keto analogues MSP and KMSB of MSC and SM, respectively, have marked utility as selective HDAC inhibitors, are measurable within cancer cell models and are at least as effective as CH3SeH as anticancer and chemopreventive agents, and (b) MSC, because of its ability to be transaminated to an α-keto acid in a greater variety of tissues than is SM may be a better candidate as an anti-cancer agent in the whole body than is SM for which transamination may be limited to the liver. Biologically, this is in accord with the widespread occurrence of GTK, the ability of GTK to catalyze transamination of MSC, the inability of GTK to catalyze transamination of SM, and the lack of effective cystathionine γ-lyase and GTL activities in extrahepatic/extrarenal tissue. These findings corroborate those of Li et al. (2008) who demonstrated a dose-dependent growth inhibition of prostate cancer xenograft models using MSC and MSA in a well-tolerated dose range of 3 mg/kg body weight. Similar to our findings in cell culture systems, SM at the same dosage in the xenograft models was not growth inhibitory. Interestingly, these authors found differential targeting responses between MSC and MSA treatments in Du-145 and PC-3 xenografts. Accordingly, despite their presumed common CH3SeH metabolite, MSC and MSA exhibit differences in apoptotic and angiogenic indices in Du-145 xenografts while MSC had no effect on these indices in PC-3 xenografts. These apparently divergent responses may be reflective of differences in GTK activities expressed in Du-145 and PC-3 cells and their ability to convert portions of the endogenous levels of MSC to MSP thus contributing to chemopreventive activity through HDAC inhibition.
Overall, our findings together with those of Li et al. (2008) have important implications when considering the relative chemopreventive activity and mechanisms of action elicited by MSC, SM as well as MSA and also the efficacy of other dietary sources of organoselenium as chemopreventive agents.
Enzymological profiles are necessary when assessing epidemiological and clinical findings with seleno amino acids
In earlier studies, designed to determine the effect of selenium on the incidence of recurrent nonmelanoma skin cancer in high-risk individuals, the results of a secondary endpoint for the study showed that consumption of selenium-enriched yeast caused a 63% reduction in overall incidence of prostate cancer (Clark et al. 1996; Duffield-Lillico et al. 2003). This sparked the initiation of multiple trials worldwide to elucidate the role of selenium (in various forms and doses) in the prevention of prostate cancer. Two large-scale double-blind, placebo-controlled clinical intervention trials that involved selenium supplementation and the incidence of prostate cancer in healthy men have been conducted: SELECT in the United States (Klein et al. 2001, 2003; Lippman et al. 2005) and, in Europe, the Danish Prevention of Cancer by Intervention by Selenium (PRECISE) (Ravn-Haren et al. 2007).
SELECT examined the use of SM (200 μg/day) alone or in combination with vitamin E (400 IU/day) to monitor development of prostate cancer in healthy men above the age of 50 years with no history of cancer (Lippman et al. 2009). With regard to the PRECISE study, after 5 years of intervention using selenium-enriched yeast, in which the majority of selenium was in the form of SM, no effect of Se supplementation was observed on glutathione metabolizing and selenium-dependent proteins in plasma, RBC and thrombocytes. Neither were changes observed in gene expression for NAD(P)H:quinone oxidoreductase and aryl hydrocarbon receptor repressor factors that contribute to chemoprotection against cancer and chemical toxicity by natural or synthetic compounds (Ravn-Haren et al. 2007).
Early data analysis from the SELECT study showed a non-significant rise in prostate cancer incidence in men taking only vitamin E. SM alone had no effect on the incidence of prostate cancer, but did cause a non-significant rise in the incidence of diabetes (Lippman et al. 2009). Although high serum selenium concentrations are associated with higher prevalence of diabetes coupled with higher fasting plasma glucose and glycosylated hemoglobin levels (Laclaustra et al. 2009), the mechanisms by which SM possibly increased diabetes in the SELECT population requires further exploration. The SELECT study was halted in October 2008, but subjects will be continuously followed over the remaining 2 years of the study. These results do not prove that there is a risk from selenium supplements, but rather point to the lack of understanding of the organ specificity, selectivity, and sensitivity to different dietary forms of organoselenium as well as the enzymology controlling their metabolism. The insignificant increase in prostate cancer in the vitamin E arm and the insignificant increase in diabetes in the selenium arm of the study may be “due to chance” as stressed by the co-chairperson of the SELECT scientific steering committee [National Cancer Institute Cancer Bulletin, November 4, 2008] and the corresponding author on the respective publication (Lippman et al. 2009). Also, as discussed by El-Bayoumy (2009) the negative results of the SELECT study do not necessarily discredit the use of organoselenium in trials of cancer prevention. Questions may arise regarding differences in metabolic in situ conversion of SM or MSC within pancreas, liver, or prostate. To what extent are these amino acids converted to their corresponding α-keto acids, KMSB or MSP, which function as HDAC inhibitors or to CH3SeH which potentially modifies redox- responsive signal and transcription factors?
As cited previously, differences in tissue distribution of GTK and GTL as well as differences in their specificities toward MSC and SM may account for the fact that MSC appears to be a better chemopreventive agent than is SM. Prostate cancer cells lack the ability to convert SM to its α-keto acid derivative at doses that can be achieved physiologically (Li et al. 2008) and this seems to correlate with the lack of inhibitory effect that SM has on growth and proliferation in transformed cells. Unfortunately, this information was not available prior to initiating the SELECT study and these findings might have influenced selection of the dietary form of organoselenium. Our findings emphasize the importance of understanding the enzymological profile of seleno amino acids in order to correlate their efficacy of intake with successful clinical outcomes or with individuals who would benefit from their consumption, information that was indirectly derived after the SELECT study (Lippman et al. 2009; Hatfield and Gladyshev 2009).
Conclusion
Research over the past several decades provides convincing evidence that supports the hypothesis that diets rich in organoselenium compounds may be protective against the risk of different types of cancers. Evidence in support of the protective effect of selenium-enriched vegetables such as garlic and broccoli as well as those of selenized yeast against cancer risk has been derived from population-based case–control studies as well as from animal and cell culture models. These studies have prompted intense research aimed at (1) elucidating mechanisms by which organoselenium-enriched foods and food components may prevent cancer, (2) identifying the forms of organoselenium components responsible for the anticancer effects, and (3) determining their efficacy for prevention of cancer in animal and cell culture models. The bioactive components responsible for cancer chemopreventive effects of selenium-enriched vegetables have, for the most part, been identified as MSC and SM. Selenium-enriched wheat, maize, rice, and yeast contain the bulk of organoselenium as SM whereas selenized garlic, onions, broccoli florets, broccoli sprouts and wild leeks contain MSC and an MSC precursor, γ-glutamyl-Se-methylselenocysteine (Whanger 2004). Since the chalcogenides, selenium and sulfur, demonstrate similar chemistries, it is likely that the number of naturally occurring organoselenium compounds may approach that of their organosulfur homologues. The efficacy of these organoselenium compounds in chemoprevention requires further study.
Unlike cancer chemotherapeutic drugs, which have the effect of causing cell retribution, organoselenium compounds can selectively target cancer cells at several pathways that involve antiproliferative and pro-apoptotic processes (Sinha and El-Bayoumy 2004). Whether a unified molecular theory exists that selectivity explains the anticancer effects of organoselenium compounds is currently unknown and must await further research. In the meantime, the overall chemopreventive effects of organoselenium compounds appear to be multifaceted.
Thus, dietary organoselenium compounds exhibit several chemopreventive functions. In addition to being incorporated into selenoproteins which exhibit their own intrinsic qualities as modifiers of redox active signal proteins (thioredoxin reductase) and in an anti-peroxidative capacity (glutathione peroxidase), they can be metabolized to direct acting metabolites that exhibit chemopreventive properties by targeting multiple signal transduction pathways (CH3SeH) and, as recently shown, through HDAC inhibition (MSP and KMSB). The observational and experimental evidence in support of organoselenium compounds as chemopreventive agents in a variety of cancers has been encouraging and will be invaluable to support future clinical trials necessary to determine the efficacy of selenium-enriched vegetables for prevention of human cancers.
This review provides a summary of the current knowledge on: (1) the key enzymes and tissue specific sites that metabolize both SM and MSC to proximate bio-active derivatives, CH3SeH, MSP and KMSB; (2) the function of these metabolites in cancer prevention; and (3) in human prostate cancer cells, the complexity of the cell signaling pathways and the multiplicity of chemopreventive possibilities through HDAC inhibition inherent in the seleno α-keto acid metabolites, MSP and KMSB.
Acknowledgments
This work was supported in part by NIH grants CA111842 (to JTP & RS) and ES8421 (to AJLC).
Abbreviations
- DMDSe
Dimethyldiselenide
- KMSB
α-Keto-γ-methylselenobutyrate
- GTK
Glutamine transaminase K
- GTL
Glutamine transaminase L
- HDAC
Histone deacetylase
- LAAO
L-Amino acid oxidase
- MSC
Se-Methyl-L-selenocysteine
- MSA
Methylseleninic acid
- MSP
β-Methylselenopyruvate
- NaB
Sodium butyrate
- PBS
Phosphate buffered saline
- PLP
Pyridoxal 5′-phosphate
- PMP
Pyridoxamine 5′-phosphate
- SM
L-Selenomethionine
Footnotes
This article is published as part of the Special Issue on sulfur- and seleno-containing amino acids.
References
- Blarzino C, Coccia R, Pensa B, Cini C, De Marco C. Selenomethionine as substrate for glutamine transaminase. Biochem Mol Biol Int. 1994;32:79–86. [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Boulland ML, Marquet J, Molinier-Frenkel V, Möller P, Guiter C, Lasoudris F, Copie-Bergman C, Baia M, Gaulard P, Leroy K, Castellano F. Human IL4I1 is a secreted L-phenylalanine oxidase expressed by mature dendritic cells that inhibits T-lymphocyte proliferation. Blood. 2007;110:220–227. doi: 10.1182/blood-2006-07-036210. [DOI] [PubMed] [Google Scholar]
- Buggy JJ, Sideris ML, Mak P, Lorimer DD, McIntosh B, Clark JM. Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem J. 2000;350:199–205. [PMC free article] [PubMed] [Google Scholar]
- Carbonnelle-Puscian A, Copie-Bergman C, Baia M, Martin-Garcia N, Allory Y, Haioun C, Crémades A, Abd-Alsamad I, Farcet JP, Gaulard P, Castellano F, Molinier-Frenkel V. The novel immunosuppressive enzyme IL4I1 is expressed by neoplastic cells of several B-cell lymphomas and by tumor-associated macrophages. Leukemia. 2009;23:952–960. doi: 10.1038/leu.2008.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallini D, De Marco C, Moldovi B, Mori BG. The cleavage of cystine by cystathionase and the transsulfuration of hypotaurine. Enzymologia. 1960;22:161–173. [PubMed] [Google Scholar]
- Chen Y, Maret W. Catalytic selenols couple the redox cycles of metallothionein and glutathione. Eur J Biochem. 2001;268:3346–3353. doi: 10.1046/j.1432-1327.2001.02250.x. [DOI] [PubMed] [Google Scholar]
- Chen YC, Sosnoski DM, Gandhi UH, Novinger LJ, Prabhu KS, Mastro AM. Selenium modifies the osteoblast inflammatory stress response to bone metastatic breast cancer. Carcinogenesis. 2009;30:1941–1948. doi: 10.1093/carcin/bgp227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JP, Cooper CS. ETS gene fusions in prostate cancer. Nat Rev Urol. 2009;6:429–439. doi: 10.1038/nrurol.2009.127. [DOI] [PubMed] [Google Scholar]
- Clark LC, Combs GF, Jr, Turnbull BW, Slate EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG, Krongrad A, Lesher JL, Jr, Park HK, Sanders BB, Jr, Smith CL, Taylor JR. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276:1957–1963. [PubMed] [Google Scholar]
- Combs GF., Jr Status of selenium in prostate cancer prevention. Br J Cancer. 2004;91:195–199. doi: 10.1038/sj.bjc.6601974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commandeur JNM, Andreadou I, Rooseboom M, Out M, de Leur LJ, Groot E, Vermeulen NPE. Bioactivation of selenocysteine Se-conjugates by a highly purified rat renal cysteine conjugate β-lyase/glutamine transaminase K. J Pharmacol Exp Ther. 2000;294:753–761. [PubMed] [Google Scholar]
- Cooper AJL. Glutamine aminotransferases and ω-amidases. In: Kvamme E, editor. Glutamine and glutamate in mammals. CRC Press; Boca Raton: 1988. pp. 33–52. [Google Scholar]
- Cooper AJL. Mechanisms of cysteine S-conjugate β-lyases. Adv Enzymol Relat Areas Mol Biol. 1998;72:199–238. doi: 10.1002/9780470123188.ch6. [DOI] [PubMed] [Google Scholar]
- Cooper AJL. The role of glutamine transaminase K (GTK) in sulfur and α-keto acid metabolism in the brain, and in the possible bioactivation of neurotoxicants. Neurochem Int. 2004;44:557–577. doi: 10.1016/j.neuint.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Cooper AJL, Anders MW. Glutamine transaminase K and cysteine conjugate β-lyase. Ann N Y Acad Sci. 1990;585:118–127. doi: 10.1111/j.1749-6632.1990.tb28048.x. [DOI] [PubMed] [Google Scholar]
- Cooper AJL, Meister A. Comparative studies of glutamine transaminases from rat tissues. Comp Biochem Physiol. 1981;69B:137–145. [Google Scholar]
- Cooper AJL, Pinto JT. Aminotransferase, L-amino acid oxidase and β-lyase reactions involving L-cysteine S-conjugates found in allium extracts. Relevance to biological activity? Biochem Pharmacol. 2005;69:209–220. doi: 10.1016/j.bcp.2004.08.034. [DOI] [PubMed] [Google Scholar]
- Cooper AJL, Pinto JT, Krasnikov BF, Niatsetskaya ZV, Han Q, Li J, Vauzour D, Spencer JPE. Substrate specificity of human glutamine transaminase K as an aminotransferase and as a cysteine S-conjugate β-lyase. Arch Biochem Biophys. 2008;474:72–81. doi: 10.1016/j.abb.2008.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- Duffield-Lillico AJ, Dalkin BL, Reid ME, Turnbull BW, Slate EH, Jacobs ET, Marshall JR, Clark LC, Nutritional Prevention of Cancer Study Group Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: an analysis of the complete treatment period of the Nutritional Prevention of Cancer Trial. BJU Int. 2003;91:608–612. doi: 10.1046/j.1464-410x.2003.04167.x. [DOI] [PubMed] [Google Scholar]
- Duley JA, Holmes RS. L-α-Hydroxyacid oxidase isozymes. Purification and molecular properties. Eur J Biochem. 1976;63:163–173. doi: 10.1111/j.1432-1033.1976.tb10219.x. [DOI] [PubMed] [Google Scholar]
- El-Bayoumy K. The negative results of the SELECT study do not necessarily discredit the selenium-cancer prevention hypothesis. Nutr Cancer. 2009;61:285–286. doi: 10.1080/01635580902892829. [DOI] [PubMed] [Google Scholar]
- Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett. 2009;280:125–133. doi: 10.1016/j.canlet.2009.02.042. [DOI] [PubMed] [Google Scholar]
- Ganther HE. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: complexities with thioredoxin reductase. Carcinogenesis. 1999;20:1657–1666. doi: 10.1093/carcin/20.9.1657. [DOI] [PubMed] [Google Scholar]
- Gasparian AV, Yao YJ, Lü J, Yemelyanov AY, Lyakh LA, Slaga TJ, Budunova IV. Selenium compounds inhibit IκB kinase (IKK) and nuclear factor-κB (NF-κB) in prostate cancer cells. Mol Cancer Ther. 2002;1:1079–1087. [PubMed] [Google Scholar]
- Giacinti L, Vici P, Lopez M. Epigenome: a new target in cancer therapy. Clin Ter. 2008;159:347–360. [PubMed] [Google Scholar]
- Hatfield DL, Gladyshev VN. The Outcome of Selenium and Vitamin E Cancer Prevention Trial (SELECT) reveals the need for better understanding of selenium biology. Mol Interv. 2009;9:18–21. doi: 10.1124/mi.9.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Waknitz M. ETS gene fusions and prostate cancer. Am J Transl Res. 2009;1:341–351. [PMC free article] [PubMed] [Google Scholar]
- Huber RE, Criddle RS. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch Biochem Biophys. 1967;122:164–173. doi: 10.1016/0003-9861(67)90136-1. [DOI] [PubMed] [Google Scholar]
- Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, Altucci L, Nervi C, Minucci S, Pelicci PG. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11:71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- Ip C. Lessons from basic research in selenium and cancer prevention. J Nutr. 1998;128:1845–1854. doi: 10.1093/jn/128.11.1845. [DOI] [PubMed] [Google Scholar]
- Ip C, Thompson HJ, Shu Z, Ganther HE. In vitro and in vivo studies of methylseleninic acid: evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000;60:2882–2886. [PubMed] [Google Scholar]
- Jiang C, Wang Z, Ganther H, Lü J. Distinct effects of methylseleninic acid versus selenite on apoptosis, cell cycle, and protein kinase pathways in DU145 human prostate cancer cells. Mol Cancer Ther. 2002;1:1059–1066. [PubMed] [Google Scholar]
- Keppler BR, Archer TK. Chromatin-modifying enzymes as therapeutic targets—Part 2. Expert Opin Ther Targets. 2008;12:1457–1467. doi: 10.1517/14728222.12.11.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein EA, Thompson IM, Lippman SM, Goodman PJ, Albanes D, Taylor PR, Coltman C. SELECT: the next prostate cancer prevention trial Selenum and Vitamin E Cancer Prevention Trial. J Urol. 2001;166:1311–1315. doi: 10.1016/s0022-5347(05)65759-x. [DOI] [PubMed] [Google Scholar]
- Klein EA, Thompson IM, Lippman SM, Goodman PJ, Albanes D, Taylor PR, Coltman C. SELECT: the selenium and vitamin E cancer prevention trial. Urol Oncol. 2003;21:59–65. doi: 10.1016/s1078-1439(02)00301-0. [DOI] [PubMed] [Google Scholar]
- Laclaustra M, Navas-Acien A, Stranges S, Ordovas JM, Guallar E. Serum selenium concentrations, diabetes in U.S. adults: National Health, Nutrition Examination Survey (NHANES) 2003–2004. Environ Health Perspect. 2009;117:1409–1413. doi: 10.1289/ehp.0900704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- Lee JI, Nian H, Cooper AJL, Sinha R, Dai J, Bisson WH, Dashwood RH, Pinto JT. α-Keto acid metabolites of naturally occurring organoselenium compounds as inhibitors of histone deacetylase in human prostate cancer cells. Cancer Prev Res. 2009;2:683–693. doi: 10.1158/1940-6207.CAPR-09-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GX, Lee HJ, Wang Z, Hu H, Liao JD, Watts JC, Combs GF, Jr, Lü J. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis. 2008;29:1005–1012. doi: 10.1093/carcin/bgn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenberg LA, Wellner D. A sensitive fluorometric assay for amino acid oxidase. Anal Biochem. 1968;26:313–319. doi: 10.1016/0003-2697(68)90343-6. [DOI] [PubMed] [Google Scholar]
- Lillig CH, Holmgren A. Thioredoxin and related molecules—from biology to health and disease. Antioxid Redox Signal. 2007;9:25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- Lippman SM, Goodman PJ, Klein EA, Parnes HL, Thompson IM, Jr, Kristal AR, Santella RM, Probstfield JL, Moinpour CM, Albanes D, Taylor PR, Minasian LM, Hoque A, Thomas SM, Crowley JJ, Gaziano JM, Stanford JL, Cook ED, Fleshner NE, Lieber MM, Walther PJ, Khuri FR, Karp DD, Schwartz GG, Ford LG, Coltman CA., Jr Designing the Selenium and Vitamin E Cancer Prevention Trial (SELECT) J Natl Cancer Inst. 2005;97:94–102. doi: 10.1093/jnci/dji009. [DOI] [PubMed] [Google Scholar]
- Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, Parnes HL, Minasian LM, Gaziano JM, Hartline JA, Parsons JK, Bearden JD, 3rd, Crawford ED, Goodman GE, Claudio J, Winquist E, Cook ED, Karp DD, Walther P, Lieber MM, Kristal AR, Darke AK, Arnold KB, Ganz PA, Santella RM, Albanes D, Taylor PR, Probstfield JL, Jagpal TJ, Crowley JJ, Meyskens FL, Jr, Baker LH, Coltman CA., Jr Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Li S, Gan L, Kao TP, Huang H. A transcription-independent function of FOXO1 in inhibition of androgen-independent activation of the androgen receptor in prostate cancer cells. Cancer Res. 2008;68:10290–10299. doi: 10.1158/0008-5472.CAN-08-2038. [DOI] [PubMed] [Google Scholar]
- Lu J, Jiang C. Antiangiogenic activity of selenium in cancer chemoprevention: metabolite-specific effects. Nutr Cancer. 2001;40:64–73. doi: 10.1207/S15327914NC401_12. [DOI] [PubMed] [Google Scholar]
- Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009;107:600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JM, Naidu MD, Barcia M, Porti D, Chavan SS, Chu CC. IL-4-induced gene-1 is a leukocyte L-amino acid oxidase with an unusual acidic pH preference and lysosomal localization. J Immunol. 2004;173:4561–4567. doi: 10.4049/jimmunol.173.7.4561. [DOI] [PubMed] [Google Scholar]
- Meister A, Sober HA, Tice SV, Fraser PE. Transamination and associated deamidation of asparagine and glutamine. J Biol Chem. 1952;197:319–330. [PubMed] [Google Scholar]
- Moeller T. Inorganic chemistry: an advanced textbook. Wiley; New York: 1963. p. 135. [Google Scholar]
- Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- Nagaoka K, Aoki F, Hayashi M, Muroi Y, Sakurai T, Itoh K, Ikawa M, Okabe M, Imakawa K, Sakai S. L-Amino acid oxidase plays a crucial role in host defense in the mammary glands. FASEB J. 2009;23:2514–2520. doi: 10.1096/fj.08-126466. [DOI] [PubMed] [Google Scholar]
- Nian H, Delage B, Pinto JT, Dashwood RH. Allyl mercaptan, a garlic-derived organosulfur compound, inhibits histone deacetylase and enhances Sp3 binding on the P21WAF1 promoter. Carcinogenesis. 2008;29:1816–1824. doi: 10.1093/carcin/bgn165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nian H, Delage B, Ho E, Dashwood RH. Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: studies with sulforaphane and garlic organosulfur compounds. Environ Mol Mutagen. 2009a;50:213–221. doi: 10.1002/em.20454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nian H, Bisson WH, Dashwood W-M, Pinto JT, Dashwood RH. α-Keto acid metabolites of organoselenium compounds inhibit histone deacetylase activity in human colon cancer cells. Carcinogenesis. 2009b;30:1416–1423. doi: 10.1093/carcin/bgp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Kobayashi Y, Konishi S, Hirano S. Speciation analysis of selenium metabolites in urine and breath by HPLC- and GC-inductively coupled plasma-MS after administration of selenomethionine and methylselenocysteine to rats. Chem Res Toxicol. 2009;22:1795–1801. doi: 10.1021/tx900202m. [DOI] [PubMed] [Google Scholar]
- Okuno T, Motobayashi S, Ueno H, Nakamuro K. Identification of mouse selenomethionine α, γ-elimination enzyme: cystathionine γ-lyase catalyzes its reaction to generate methylselenol. Biol Trace Elem Res. 2005;108:245–257. doi: 10.1385/BTER:108:1-3:245. [DOI] [PubMed] [Google Scholar]
- Papp LV, Lu J, Holmgren A, Khanna KK. From selenium to selenoproteins: synthesis, identity, and their role in human health. Antioxid Redox Signal. 2007;9:775–806. doi: 10.1089/ars.2007.1528. [DOI] [PubMed] [Google Scholar]
- Pinto JT, Sinha R, Papp K, Facompre ND, Desai D, El-Bayoumy K. Differential effects of naturally occurring and synthetic organoselenium compounds on biomarkers in androgen responsive and androgen independent human prostate carcinoma cells. Int J Cancer. 2007;120:1410–1417. doi: 10.1002/ijc.22500. [DOI] [PubMed] [Google Scholar]
- Puccetti E, Obradovic D, Beissert T, Bianchini A, Washburn B, Chiaradonna F, Boehrer S, Hoelzer D, Ottmann OG, Pelicci PG, Nervi C, Ruthardt M. AML-associated translocation products block vitamin D3-induced differentiation by sequestering the vitamin D3 receptor. Cancer Res. 2002;62:7050–7058. [PubMed] [Google Scholar]
- Ravn-Haren G, Krath BN, Overvad K, Cold S, Moesgaard S, Larsen EH, Dragsted LO. Effect of long-term selenium yeast intervention on activity and gene expression of antioxidant and xenobiotic metabolizing enzymes in healthy elderly volunteers from the Danish Prevention of Cancer by Intervention by Selenium (PRECISE) pilot study. Br J Nutr. 2007;99:1190–1198. doi: 10.1017/S0007114507882948. [DOI] [PubMed] [Google Scholar]
- Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooseboom M, Vermeulen NPE, Durgut F, Commandeur JNM. Comparative study on the bioactivation mechanisms and cytotoxicity of Te-phenyl-L-tellurocysteine, Se-phenyl-L-selenocysteine, and S-phenyl-L-cysteine. Chem Res Toxicol. 2002;15:1610–1618. doi: 10.1021/tx020034f. [DOI] [PubMed] [Google Scholar]
- Rudolf E, Králová V, Cervinka M. Selenium and colon cancer—from chemoprevention to new treatment modality. Anticancer Agents Med Chem. 2008;8:598–602. [PubMed] [Google Scholar]
- Sanmartín C, Plano D, Palop JA. Selenium compounds and apoptotic modulation: a new perspective in cancer therapy. Mini Rev Med Chem. 2008;8:1020–1031. doi: 10.2174/138955708785740625. [DOI] [PubMed] [Google Scholar]
- Scher HI, Mazumdar M, Kelly WK. Clinical trials in relapsed prostate cancer: Defining the target. J Natl Canc Inst. 1996;88:1623–1634. doi: 10.1093/jnci/88.22.1623. [DOI] [PubMed] [Google Scholar]
- Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA, Kim R, Rubin MA, Pienta KJ. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- Shankar S, Srivastava RK. Histone deacetylase inhibitors: mechanisms and clinical significance in cancer: HDAC inhibitor-induced apoptosis. Adv Exp Med Biol. 2008;615:261–298. doi: 10.1007/978-1-4020-6554-5_13. [DOI] [PubMed] [Google Scholar]
- Singh U, Null K, Sinha R. In vitro growth inhibition of mouse mammary epithelial tumor cells by methylseleninic acid: involvement of protein kinases. Mol Nutr Food Res. 2008;52:1281–1288. doi: 10.1002/mnfr.200700356. [DOI] [PubMed] [Google Scholar]
- Sinha R, El-Bayoumy K. Apoptosis is a critical cellular event in cancer chemoprevention and chemotherapy by selenium compounds. Curr Cancer Drug Targets. 2004;4:13–28. doi: 10.2174/1568009043481614. [DOI] [PubMed] [Google Scholar]
- Sinha R, Pinto JT, Facompre N, Kilheffer J, Baatz JE, El-Bayoumy K. Effects of naturally occurring and synthetic organoselenium compounds on protein profiling in androgen responsive and androgen independent human prostate cancer cells. Nutr Cancer. 2008;60:267–275. doi: 10.1080/01635580701630479. [DOI] [PubMed] [Google Scholar]
- Sinha R, Sinha I, Null K, King T, Wolter W, Suckow M. Methylseleninic acid inhibits HIF-1α in hormone refractory prostate cancer. 100th Annual Meeting of the American Association for Cancer Research; Denver, CO. 18–22 April 2009; Philadelphia: AACR; 2009. Abstract. Abstract # 5583. [Google Scholar]
- Spallholz JE, Shriver BJ, Reid TW. Dimethyldiselenide and methylseleninic acid generate superoxide in an in vitro chemiluminescence assay in the presence of glutathione: implications for the anticarcinogenic activity of L-selenomethionine and L-Se-methylselenocysteine. Nutr Cancer. 2001;40:34–41. doi: 10.1207/S15327914NC401_8. [DOI] [PubMed] [Google Scholar]
- Stevens JL, Robbins JD, Byrd RA. A purified cysteine conjugate β-lyase from rat kidney cytosol Requirement for an α-keto acid or an amino acid oxidase for activity and identity with soluble glutamine transaminase K. J Biol Chem. 1986;261:15529–15537. [PubMed] [Google Scholar]
- Sun Y, Nonobe E, Kobayashi Y, Kuraishi T, Aoki F, Yamamoto K, Sakai S. Characterization and expression of L-amino acid oxidase of mouse milk. J Biol Chem. 2002;277:19080–19086. doi: 10.1074/jbc.M200936200. [DOI] [PubMed] [Google Scholar]
- Suzana S, Cham BG, Ahmad Rohi G, Mohd Rizal R, Fairulnizal MN, Normah H, Fatimah A. Relationship between selenium and breast cancer: a case-control study in the Klang Valley. Singapore Med J. 2009;50:265–269. [PubMed] [Google Scholar]
- Suzuki KT, Tsuji Y, Ohta Y, Suzuki N. Preferential organ distribution of methylselenol source Se-methylselenocysteine relative to methylseleninic acid. Toxicol Appl Pharmacol. 2008;227:76–83. doi: 10.1016/j.taap.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Tsuji Y, Suzuki N, T Suzuki K, Ogra Y. Selenium metabolism in rats with long-term ingestion of Se-methylselenocysteine using enriched stable isotopes. J Toxicol Sci. 2009;34:191–200. doi: 10.2131/jts.34.191. [DOI] [PubMed] [Google Scholar]
- Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, Marks PA. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102:673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unni E, Koul D, Yung W-K, Sinha R. Se-methylselenocysteine inhibits phosphatidylinositol 3-kinase activity of mouse mammary epithelial tumor cells in vitro. Breast Cancer Res. 2005;7:R699–R707. doi: 10.1186/bcr1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K, Loverix S, Geerlings P, Tourwé D. DFT-based ranking of zinc-binding groups in histone deacetylase inhibitors. Bioorg Med Chem. 2005;13:6070–6082. doi: 10.1016/j.bmc.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Venkateswaran V, Klotz LH, Fleshner NE. Selenium modulation of cell proliferation and cell cycle biomarkers in human prostate carcinoma cell lines. Cancer Res. 2002;62:2540–2545. [PubMed] [Google Scholar]
- Vigushin DM, Coombes RC. Targeted histone deacetylase inhibition for cancer therapy. Curr Cancer Drug Targets. 2004;4:205–218. doi: 10.2174/1568009043481560. [DOI] [PubMed] [Google Scholar]
- Wang Z, Jiang C, Lu J. Induction of caspase-mediated apoptosis and cell-cycle G1 arrest by selenium metabolite methylselenol. Mol Carcinog. 2002;34:113–120. doi: 10.1002/mc.10056. [DOI] [PubMed] [Google Scholar]
- Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, Feng J, Liu D, Chiao JW. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int J Oncol. 2008;33:375–380. [PubMed] [Google Scholar]
- Wang L, Zou X, Berger AD, Twiss C, Peng Y, Li Y, Chiu J, Guo H, Satagopan J, Wilton A, Gerald W, Basch R, Wang Z, Osman I, Lee P. Increased expression of histone deacetylases (HDACs) and inhibition of prostate cancer growth and invasion by HDAC inhibitor SAHA. Am J Transl Res. 2009;1:62–71. [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, Sawyers CL. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009;69:958–966. doi: 10.1158/0008-5472.CAN-08-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessjohann LA, Schneider A, Abbas M, Brandt W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol Chem. 2007;388:997–1006. doi: 10.1515/BC.2007.138. [DOI] [PubMed] [Google Scholar]
- Whanger PD. Selenium and its relationship to cancer: an update. Br J Nutr. 2004;91:11–28. doi: 10.1079/bjn20031015. [DOI] [PubMed] [Google Scholar]
- Xiong SD, Yu K, Liu XH, Yin LH, Kirschenbaum A, Yao S, Narla G, DiFeo A, Wu JB, Yuan Y, Ho SM, Lam YW, Levine AC. Ribosome-inactivating proteins isolated from dietary bitter melon induce apoptosis and inhibit histone deacetylase-1 selectively in premalignant and malignant prostate cancer cells. Int J Cancer. 2009;125:774–782. doi: 10.1002/ijc.24325. Erratum in: Int J Cancer (2009) 125:1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- Yang Y, Hou H, Haller EM, Nicosia SV, Bai W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005;24:1021–1032. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Wu M, Botnen JH. Methylselenol, a selenium metabolite, induces cell cycle arrest in G1 phase and apoptosis via the extracellular-regulated kinase 1/2 pathway and other cancer signaling genes. J Nutr. 2009;139:1613–1618. doi: 10.3945/jn.109.110320. [DOI] [PubMed] [Google Scholar]