

Abstract
The Escherichia coli DNA repair enzyme MutY plays an important role in the prevention of DNA mutations by removing misincorporated adenine residues from 7,8-dihydro-8-oxo-2′-deoxyguanosine:2′-deoxyadenosine (OG:A) mispairs. The N-terminal domain of MutY (Stop 225, Met1–Lys225) has a sequence and structure that is characteristic of a superfamily of base excision repair glycosylases; however, MutY and its homologs contain a unique C-terminal domain. Previous studies have shown that the C-terminal domain confers specificity for OG:A substrates over G:A substrates and exhibits homology to the d(OG)TPase MutT, suggesting a role in OG recognition. In order to provide additional information on the importance of the C-terminal domain in damage recognition, we have investigated the kinetic properties of a form lacking this domain (Stop 225) under multiple- and single-turnover conditions. In addition, the interaction of Stop 225 with a series of non-cleavable substrate and product analogs was evaluated using gel retardation assays and footprinting experiments. Under multiple-turnover conditions Stop 225 exhibits biphasic kinetic behavior with both OG:A and G:A substrates, likely due to rate-limiting DNA product release. However, the rate of turnover of Stop 225 was increased 2-fold with OG:A substrates compared to the wild-type enzyme. In contrast, the intrinsic rate for adenine removal by Stop 225 from both G:A and OG:A substrates is significantly reduced (10- to 25-fold) compared to the wild-type. The affinity of Stop 225 for substrate analogs was dramatically reduced, as was the ability to discriminate between substrate analogs paired with OG over G. Interestingly, similar hydroxyl radical and DMS footprinting patterns are observed for Stop 225 and wild-type MutY bound to DNA duplexes containing OG opposite an abasic site mimic or a non-hydrogen bonding A analog, suggesting that similar regions of the DNA are contacted by both enzyme forms. Importantly, Stop 225 has a reduced ability to prevent DNA mutations in vivo. This implies that the reduced adenine glycosylase activity translates to a reduced capacity of Stop 225 to prevent DNA mutations in vivo.
INTRODUCTION
Reactive oxygen species (ROS) such as superoxide, hydrogen peroxide and hydroxyl radicals are present in the cell as by-products of endogenous reactions or as the result of external sources, such as ionizing radiation (1,2). These ROS can react with DNA to produce a variety of genotoxic lesions that have been implicated in various disease processes (3–5). One of the most common lesions is 7,8-dihydro-8-oxo-2′-deoxyguanosine (OG) (6–8). During DNA replication the presence of OG can result in misincorporation of 2′-deoxyadenosine by DNA polymerase and formation of OG:A base pairs (9,10) which, if not repaired, create permanent G→T transversion mutations in the subsequent replication event. Fortunately, repair systems for OG appear to be present across all phyla (11). In Escherichia coli the ‘GO’ system utilizes the actions of three proteins, MutM, MutY and MutT (12,13). MutM (or Fpg protein) is an OG glycosylase which removes OG from OG:C base pairs, while MutY is an adenine glycosylase which removes misincorporated adenine from OG:A base pairs. MutT hydrolyzes d(OGTP) and therefore prevents its incorporation into DNA (14).
Genetic evidence based on mutation frequencies in E.coli suggests that OG:A mismatches are an important substrate for MutY (15). However, MutY has also been shown to remove adenine in vitro and in vivo from G:A and C:A mismatches (16–18). Using non-cleavable 2′-deoxyadenosine analogs, we and others have shown that MutY has a higher affinity for OG-containing duplexes compared to the corresponding G-containing duplexes (19–23). This difference in affinity is consistent with the OG:A mismatch as the preferred MutY substrate. Furthermore, considerable evidence has shown that MutY has an unusually high affinity for the product of its glycosylase action on an OG:A substrate [an OG:apurinic-apyrimidinic (AP) site]. This has been illustrated by the observation of a defined MutY ‘footprint’ in methidiumpropyl EDTA-Fe(II) [MPE-Fe(II)] hydroxyl radical footprinting experiments on MutY bound to the OG:(AP site) product (19). Moreover, Miller and Michaels reported that in the presence of MutY, MutM is unable to remove OG from the OG:(AP site) product (12), suggesting that MutY remains bound to the product. We have shown previously that the high affinity of MutY for the DNA product affects the relative processing of OG:A and G:A substrates (24). Under conditions where [MutY] < [DNA], the reaction of MutY with both OG:A- and G:A-containing substrates is characterized by biphasic kinetics, displaying an exponential burst followed by a linear steady-state phase of product formation. The observed steady-state rate is considerably smaller for OG:A substrates, likely due to the higher affinity for the OG:(AP site) product. In contrast, the intrinsic rate for adenine removal is considerably larger for OG:A substrates.
MutY is representative of a class of base excision repair (BER) glycosylases that remove a wide variety of inappropriate bases (11). However, MutY is unique in that it catalyzes the removal of an undamaged adenine mispaired with the damaged base OG (12,15). Among the enzymes in the BER superfamily, E.coli MutY exhibits the highest sequence similarity to E.coli endonuclease III (endo III) (25). Endo III displays glycosylase activity for DNA containing damaged pyrimidines such as ring-saturated and ring-fragmented thymine derivatives and cytosine photoproducts (26–28). Based on limited proteolytic digestion with trypsin (29) or thermolysin (30) and sequence homology to endo III (25), MutY may be divided into two domains: an N-terminal domain (Met1–Lys225) that exhibits high sequence homology to endo III (66.3% similar and 23.8% identical over 181 amino acids) and a C-terminal extension domain (Gln226–Val350). Moreover, the crystal structure of the N-terminal portion of MutY reveals an overall structural similarity to endo III (31). Qualitative enzymatic characterization of the truncated enzyme produced by proteolytic digestion (29,30) or overexpression (32) indicates that the general properties of catalytic activity are retained.
Recently, using single-turnover kinetic experiments Noll et al. (33) showed that the N-terminal domain of MutY (cd-MutY, Met1–Gln226) exhibited reduced rates of adenine removal from both OG:A and G:A substrates and reduced preference for an OG:A substrate. Subsequently, using a similar approach, Li et al. (34) reported results that indicated only a decrease in the rate of adenine removal for cd-MutY with an OG:A substrate. Noll et al. (33) also observed that removal of the C-terminal domain results in an increased dissociation of MutY from the DNA product, as determined by gel retardation measurements. These biochemical results suggest that the C-terminal domain plays a role in both substrate and product binding. Moreover, Noll et al. showed that there is sequence homology between the C-terminal domain of MutY and the d(OG)Tpase MutT (33). This has been confirmed by recent NMR structural data on the C-terminal domain (35). Based on these results, it has been suggested that the C-terminal domain may serve as an OG-binding domain and participate in a nucleotide flipping process necessary for efficient adenine removal (33).
We have previously exploited non-cleavable substrate analogs (Fig. 1; 19,20,23) as well as pre-steady-state kinetics (24,36,37) to explore factors influencing the recognition and repair of DNA damage by MutY. We have now applied a similar approach to a truncated form of MutY (Met1–Lys225, referred to henceforth as Stop 225). Using both single- and multiple-turnover kinetic experiments we have found that the intrinsic rate of adenine removal from both OG:A and G:A substrates is dramatically reduced with Stop 225. However, multiple turnover experiments revealed that Stop 225 exhibits biphasic kinetic behavior similar to the wild-type enzyme, suggestive of rate-limiting product release after removal of the adenine. The truncated enzyme exhibits a slightly increased turnover rate only with an OG:A substrate, suggesting a reduced affinity for the OG:(AP site) product. Kd measurements revealed that the affinity of MutY for substrate analogs was dramatically reduced by removal of the C-terminal domain. In contrast, the affinity of Stop 225 for a product analog was only slightly reduced relative to the wild-type enzyme. Hydroxyl radical and dimethylsulfate (DMS) footprinting experiments on duplexes containing OG opposite an abasic site mimic or a non-hydrogen bonding A analog indicated no significant differences between the two enzyme forms. The more dramatic effects appear to be in substrate recognition and processing, suggesting that the C-terminal domain plays a significant role in substrate recognition. Moreover, in vivo experiments indicate that Stop 225 has a significantly reduced ability to prevent DNA mutations. This suggests that the reduced binding affinity and processing efficiency for OG:A mismatches observed in the in vitro experiments is reflected in a decrease in the ability to prevent DNA mutations in vivo.
Figure 1.

Substrate, substrate analogs and product analogs for MutY. (A) Structures of an OGsyn:Aanti mispair based on NMR and X-ray crystallographic studies (51,55). (B) Structures of the 2′-deoxyadenosine analogs (F and FA), hydrophobic 2′-deoxyadenosine analog (M) and abasic site mimic (THF) used in this work.
MATERIALS AND METHODS
General methods, bacterial strains, materials and instrumentation
Escherichia coli strains JM101, JM109 and GT100 were used in this work (38). The plasmid containing the mutY gene, pKKYEco, was kindly provided by M. Michaels and J.H. Miller. The JM101 mutY and GT100 mutY::mini-Tn10 mutM E.coli strains have been described previously (15,39,40). All common DNA manipulations were performed using standard protocols (41). All β-cyanoethyl phosphoramidites were purchased from ABI, except the 7,8-dihydro-8-oxo-2′-deoxyguanosine phosphoramidite, which was purchased from Glen Research. DNA oligonucleotides were synthesized by standard phosphoramidite chemistry on an Applied Biosystems model 392 DNA/RNA synthesizer as per the manufacturer’s protocol. The oligonucleotides used for PCR reactions were purified using oligonucleotide purification cartridges (Perkin Elmer). Oligonucleotides for enzyme assays and binding experiments were handled as described previously (19). All buffers and other reagents used were purchased from Fisher, Sigma or US Biochemical. 5′-End-labeling was performed with T4 polynucleotide kinase (New England Biolabs) using [γ-32P]ATP (Amersham Life Sciences). Labeled oligonucleotides were purified using ProbeQuant G-50 microcolumns (Amersham Pharmacia Biotech). UV-visible spectroscopy was performed on a Hewlett Packard 8452A diode array spectrophotometer. Storage phosphor autoradiography was performed using a Molecular Dynamics Storm 840 PhosphorImager. All data fitting was performed using GraFit v.4. PCR reactions were performed in a GeneAmp PCR system 2400 (Perkin Elmer). All electrophoresis was performed using 1× or 0.5× Tris–borate–EDTA (TBE) buffer, pH 8.3, where 1× = 90 mM Tris, 90 mM boric acid, 1 mM EDTA. Chromatography for MutY purification was conducted with a BioLogic chromatography system (Bio-Rad) at 4°C.
Preparation and purification of Stop 225 MutY
Site-directed mutagenesis was performed using a PCR-based method similar to that described previously (40). The stop codon at position 226 and a PstI restriction site were introduced using the appropriate primer (5′-gtg cgc tcc tgc agc gtc tat ttc ggt ttt ttg-3′) and the modified mutY gene was cloned into expression plasmid pKK223-3, producing a new plasmid designated pKKYS225. The E.coli strain JM101 mutY::mini-Tn10 harboring the pKKYS225 plasmid was grown at 37°C in LB medium to an OD600 of 0.9. Expression of Stop 225 was induced by addition of 1 mM IPTG and then the cultures were grown for 3 h at 30°C. The harvested cells were resuspended to a concentration of 10 ml/g of cells in buffer A (50 mM Tris–HCl, pH 8, 2 mM EDTA, 5 mM DTT, 5% glycerol) and disrupted with sonication. The supernatant was removed and streptomycin sulfate was added to 4.75% to precipitate nucleic acids. Subsequently, proteins were precipitated using ammonium sulfate (40%) and the brown pellet was redissolved in 30 ml of de-oxygenated buffer B (20 mM sodium phosphate, pH 7.5, 1 mM EDTA, 1 mM DTT, 5% glycerol). The protein was centrifuged and desalted with a HiPrep 26/10 desalting column (Amersham Pharmacia Biotech) equilibrated with buffer B. The eluted protein was then loaded on a 6 ml UnoS column (Bio-Rad) or High-S econopak (Bio-Rad) equilibrated with buffer B containing 50 mM NaCl. After washing with 30 ml of buffer B, the protein was eluted with a 120 ml linear gradient of NaCl (50–700 mM) in buffer B. The fraction containing MutY was diluted with 5× buffer B and loaded on a 5 ml heparin–Sepharose (Pharmacia) or Hi-Trap heparin (Pharmacia) column equilibrated with buffer B containing 50 mM NaCl. After washing with 30 ml of equilibration buffer, the protein was eluted with a 60 ml linear gradient of NaCl (50–700 mM) in buffer B. The fraction containing MutY was then diluted with an equal volume of glycerol and PMSF was added to 0.1 mM. The protein was then aliquoted and stored in liquid nitrogen. The identical protocol was also used to purify wild-type MutY used in this work, with the exception that the pKKYEco plasmid (containing the wild-type mutY gene) was used.
In vivo activity
The appropriate plasmid (pKK223-3, pKKYEco or pKKYS225) was transformed into E.coli strain GT100 mutY::mini-Tn10 mutM. A minimum of at least eight independent overnight cultures were grown in LB medium containing 100 mg/l ampicillin. To determine the number of viable cells, a 107-fold dilution was plated on LB agar plates containing ampicillin (100 mg/l). A variable volume was also plated on LB agar containing ampicillin (100 mg/l) and rifampicin (100 mg/l) to measure rifampicin revertants (Rifr). After overnight incubation at 37°C, Rifr colonies were counted.
Oligonucleotides
The following DNA duplex sequence was used: d(5′-CGATCATGGAGCCACXAGCTCCCGTTACAG-3′)·d(3′-GCTAGTACCTCGGTGYTCGAGGGCAATGTC-5′), where X is 2′-deoxyguanosine (G) or 7,8-dihydro-8-oxo-2′-deoxyguanosine (OG) and Y is 2′-deoxyadenosine (A), tetrahydrofuran nucleotide (THF), 2′-deoxyformycin A (F), 2′-deoxy-2′-fluoroadenosine (FA), 4-methylindole β-deoxynucleoside (M), purine 2′-deoxynucleoside (P) or 2′-deoxycytidine (C). The F and FA oligonucleotides were prepared form the phosphoramidite monomers as described previously (19,20).
Multiple- and single-turnover kinetics
Kinetic experiments were performed as outlined previously (24). The A-containing strand was 5′-32P-end-labeled with T4 polynucleotide kinase and 2–5% of the end-labeled A-containing strand was added to the unlabeled A-containing strand. The complementary strand was then added in slight excess (10%). Duplex formation was performed by heating to 95°C in annealing buffer (20 mM Tris–HCl, pH 7.6, 10 mM EDTA, 150 mM NaCl) and then slow cooling to room temperature over 3–4 h.
For multiple-turnover kinetic experiments, including the active site titration, substrate DNA (20 nM duplex) was equilibrated at 37°C in reaction buffer (20 mM Tris–HCl, pH 7.5, 1 mM EDTA, 15 mM NaCl, 0.1 mg/ml BSA). Enzyme concentrations were adjusted to afford 10–20% product formation for the burst phase of the reaction. Single-turnover experiments were performed in a manner analogous to the multiple turnover experiments, with a Stop 225 protein concentration of 30 nM (as determined by the active site titration).
Equilibrium dissociation constant (Kd) measurements
Kd values were obtained using a gel retardation assay (42) similar to that described previously for MutY with substrate analog duplexes (20). In these experiments only labeled duplex is used, with the estimated upper limit of the duplex concentration based on 100% recovery from the end-labeling procedure. MutY or Stop 225 were diluted just prior to the reaction and added to solutions containing 100 pM DNA duplex (except OG:THF, 5 pM), 20 mM Tris–HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, 0.1 mg/ml BSA. After incubation at 25°C for 30 min the samples were electrophoresed on a 6% non-denaturing polyacrylamide gel (17 × 14 × 0.3 cm) equilibrated with 0.5× TBE buffer at 4°C. The dried gel was exposed to a Molecular Dynamics storage phosphor screen for at least 10 h and the autoradiogram was quantitated using ImageQuant v.4.2a (Molecular Dynamics). Reported Kd values have been corrected for the active enzyme concentration and are the average of at least four separate experiments. It should be noted that for duplexes that exhibited Kd values near that of a non-specific duplex (∼100 nM), more than one band with retarded mobility corresponding to a ‘bound’ complex was observed and all of the retarded bands were considered as ‘bound’ duplex in the apparent Kd determinations.
MPE-Fe(II) and DMS footprinting experiments
A DNA duplex containing either an OG:THF or OG:M base pair was 5′-32P-end-labeled on one of the duplex strands prior to duplex formation. MPE-Fe(II) footprinting experiments were performed as described previously (19) using 10 nM DNA and 0, 100, 200 or 300 nM wild-type or 0, 60, 120 or 180 nM Stop 225 MutY in 10 mM Tris–HCl, 50 mM NaCl, pH 7.4, 0.1 mg/ml BSA, 500 µM calf-thymus DNA. DMS footprinting was performed as described previously (20). Notably, 5 nM OG:THF or OG:M duplex was incubated with 0, 100 or 200 nM wild-type or Stop 225 MutY in buffer containing 50 mM sodium cacodylate, pH 7, 1 mM EDTA. The extent of protection or hyper-reactivity was determined by quantitation of the storage phosphor autoradiogram using ImageQuant software (Molecular Dynamics). The reported quantitation represents the percent cleavage of a given band compared to the total cleaved DNA in each lane. For MPE-Fe(II) footprinting the relative protection shown in histogram form is the sum of this type of analysis of three separate experiments.
RESULTS
Cloning, overexpression and purification of a truncated MutY form
On the basis of the homology of the N-terminal domain of MutY to endo III (25), we initially prepared a truncated form of MutY by introduction of a stop codon at position 217 in the mutY gene which corresponds to the position where endo III ends relative to MutY. However, this form was not over-produced as efficiently as the wild-type enzyme. Therefore, a second truncated form of MutY, analogous to that previously reported by Manuel and Lloyd (32), was prepared by introduction of a stop codon at position 226 in the mutY gene. This truncated form, Stop 225, was overexpressed at levels similar to the wild-type enzyme and was stable to homogeneous purification. The UV-visible absorption spectrum of Stop 225 protein possesses broad absorption features centered at 410 nm similar to those observed with the wild-type enzyme, which are characteristic of thiolate→Fe(III) charge transfer transitions. The ratio A280 nm/A410 nm for Stop 225 of approximately 4–5 is consistent with the expected loss of absorbance at 280 nm of ∼35–40% due to absence of the C-terminal portion of the protein. The Stop 225 enzyme does not exhibit an EPR signal at liquid helium temperatures consistent with the presence of the [4Fe-4S]2+ form of the iron-sulfur cluster as observed in the native enzyme.
Adenine glycosylase activity of Stop 225 with OG:A and G:A substrates
To investigate the activity of Stop 225 in vitro, kinetic studies were performed and compared to the results obtained with wild-type MutY. As shown previously (24), under multiple-turnover conditions ([DNA] > [MutY]) the reaction of wild-type MutY with OG:A and G:A substrates is characterized by biphasic kinetics, displaying an initial exponential burst of product formation followed by a linear steady-state phase. This type of kinetic behavior is characteristic of a slow step occurring subsequent to the chemistry step and is most likely due to slow release of MutY from the DNA product. Based on this behavior, we previously proposed the minimal kinetic mechanism for the wild-type enzyme shown in Scheme 1.

Scheme 1.
The rate constants describing the steps, including chemistry (k2) and product release (k3), can be determined under single- and multiple-turnover conditions, as described in detail previously (24).
The kinetic rate constants were measured using a 30 bp DNA duplex containing either a G:A or OG:A mispair radiolabeled on the strand containing the mispaired adenine. The reaction of the duplex with MutY or Stop 225 produced an apurinic site and subsequent base treatment produces a 14 nt fragment (product) that was separated from the 30 nt strand (substrate) by denaturing PAGE (Fig. 2). The kinetic profiles for conversion of both OG:A and G:A substrates to products by Stop 225 are similar to those observed with the wild-type enzyme (24). Under single-turnover conditions ([DNA] < [MutY]) the binding and chemistry steps of the reaction can be separated from the product release step to allow measurement of k2. Figure 2 shows a comparison of formation of product from an OG:A substrate with Stop 225 and the wild-type enzyme under these conditions. The data obtained from quantitation of the storage phosphor autoradiogram (Fig. 2) were fitted to equation 1 (24), where Ao represents the amplitude of the exponential phase and kobs is the observed rate constant associated with that process.
Figure 2.

A storage phosphor autoradiogram from a single turnover adenine glycosylase assay for MutY. A 30 bp duplex containing a central OG:A mismatch (with A-containing strand 5′-32P-end-labeled) was incubated with wild-type MutY or Stop 225. The reaction was quenched and the abasic site product was cleaved upon addition of NaOH. The substrate (30 nt strand) was resolved from the product (14 nt strand) by denaturing PAGE. The control lane, with no enzyme added, is marked C. A description of the analysis used to determine rate constants is described in the text and the results are listed in Table 1.
[P]t = A0[1 – exp(–kobst)] 1
The observed rate constant under pseudo-first order conditions ([MutY] > [DNA]) is given by equation 2, assuming that enzyme–substrate binding is in rapid equilibrium (k–1 >> k2).
kobs = {[E]0/(Kd + [E]0)}k2 2
Under conditions where the enzyme concentration in the experiment is well above the Kd (k–1/k1), this equation simplifies to kobs ≅ k2. In order to determine whether these kobs values were influenced by a reduced binding affinity of Stop 225 for the substrate, the concentrations of enzyme and DNA were increased 3.5-fold. If DNA binding was a factor, increasing the concentration of both enzyme and DNA would be expected to increase the observed reaction rate. However, the observed rates obtained were similar under these conditions (data not shown), indicating that the kobs = k2 simplification is valid.
The k2 values determined for the OG:A- and G:A-containing substrates with Stop 225 and wild-type MutY are listed in Table 1. These data show that the rate of adenine removal from an OG:A substrate is considerably faster than from a G:A substrate for both truncated and wild-type MutY. Furthermore, these values reveal a dramatic difference between the k2 values for Stop 225 and the wild-type enzyme. For a G:A-containing duplex the rate was 10 times slower with Stop 225 than with wild-type MutY. This difference was even more pronounced for the OG:A-containing duplex. The k2 value for Stop 225 with the OG:A substrate was at least 25 times smaller than the lower limit estimated for the wild-type protein. The k2OG:A/k2G:A ratio for wild-type MutY is more than twice that of Stop 225 (>7 for wild-type and 3 for Stop 225).
Table 1. Kinetic parameters for wild-type and Stop 225 MutY with OG:A- and G:A-containing duplexes at 37°Ca.
| BPc | Wild-type MutYb | Truncated MutY (Stop 225) | ||||
| STO | MTO | STO | MTO | |||
| |
k2 (min–1) |
kobs (min–1)d |
k3 (min–1) |
k2 (min–1) |
kobs (min–1)d |
k3 (min–1) |
| OG:A | >10 | NRe | 0.004 ± 0.002 | 0.39 ± 0.07 | 0.5 ± 0.2 | 0.010 ± 0.003 |
| G:A | 1.6 ± 0.4 | 1.8 ± 0.6 | 0.03 ± 0.01 | 0.15 ± 0.01 | 0.23 ± 0.07 | 0.03 ± 0.01 |
aAll values are reported as averages of at least four separate experiments, errors are reported as one standard deviation from the average.
bThese values are as reported in Porello et al. (24).
cBP indicates central base pair in the 30 bp duplex substrate.
dkobs ≅ k2. See text for details.
eNR, not reported. Rate constants from the burst phase are not reported due to large errors.
Adenine excision from OG:A and G:A substrates by Stop 225: determination of k3
We have previously shown that appropriate fitting of the data obtained under conditions of multiple turnover ([MutY] < [DNA]) with both OG:A and G:A substrates allows for determination of both k2 and k3 (24). Thus, similar experiments with the Stop 225 enzyme were performed in a manner analogous to those with the wild-type enzyme in order to determine the turnover rate k3. This will provide additional information as to the effects of removal of the C-terminal domain on steps subsequent to base removal. The observed formation of product (nM) as a function of time (Fig. 3) reveals biphasic behavior for Stop 225 similar to that observed for wild-type MutY (24). By fitting the data to equation 3 (24), several kinetic parameters were determined.
Figure 3.

Kinetic properties of Stop 225 with OG:A- and G:A-containing duplexes under multiple turnover conditions ([DNA] > [Stop 225]). Experiments were performed using 20 nM duplex DNA substrate containing either a G:A (closed circles) or OG:A (open circles) mismatch and Stop 225 (10 nM by Bradford determination). Based on the amplitude of the burst with the OG:A duplex, the active site concentration of Stop 225 was 2.1 nM. The k3 values for these particular experiments were 0.03 and 0.007 min–1 for the G:A and OG:A duplexes, respectively.
[P]t = A0{1 – exp(–kobst)} + ksst 3
A0 represents the amplitude of the burst phase. The rate constant kobs represents the rate of the exponential phase, while kss corresponds to the slope of the linear phase. In terms of the microscopic rate constants in Scheme 1, Ao, kobs and kss are defined by equations 4–7. Equation 7 defines the effective rate constant k′ for the process MutY + (DNA)S → MutY·(DNA)P.
A0 = {k′/(k′ + k3)}2[MutY]0 4
kobs = k′ + k3 5
kss = k′k3/(k′ + k3)[MutY]0 6
where
k′ = 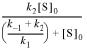 7
7
If k′ >> kss, as with the wild-type enzyme with both OG:A and G:A substrates and Stop 225 with OG:A substrates, these equations reduce to A0 = [MutY]0, kobs = k′ and kss = k3[MutY]0, respectively. In the case of G:A substrates with Stop 225, k′ > kss and, therefore, the expression for k3 is as shown in equation 8.
k3 = (ksskobs)/(A0kobs + kss) 8
In addition, assuming that k–1 >> k2 and k1 >> k2, equation 7 will also further reduce such that k′ ≅ k2. In previous work with the wild-type enzyme (24) this approximation was found to provide values of the k2 rate constants that were comparable to those determined under single-turnover conditions.
In Figure 3 the activity of Stop 225 with the G:A substrate is compared to that with an OG:A substrate under multiple-turnover conditions. The observed rates for the exponential burst phase for both the G:A and OG:A substrates were determined from several experiments and provided k2 rate constants that were similar to those obtained from the single-turnover experiments (Table 1). A comparison of all of the k3 values (Table 1) indicates that removal of the C-terminal domain increases the k3 value two-fold for the OG:A substrate. In contrast, both enzyme forms exhibit similar k3 values with G:A substrates. With both enzyme forms the k3 values are smaller with the OG:A substrate relative to the G:A substrate. These data suggest that, as with the wild-type enzyme, product release is the rate-limiting step for the reaction of Stop 225 with both OG:A and G:A substrates; however, the rate of product release from OG:A substrates is slightly increased by removal of the C-terminal domain.
Due to the very slow turnover rate (k3) of wild-type MutY with an OG:A-containing duplex, the amplitude of the burst phase A0 obtained from the multiple-turnover studies approximates the active enzyme concentration (A0 ≅ [MutY]0) (24). This approximation holds for Stop 225 since the value of k3 is only slightly increased. The amplitude of the burst for Stop 225 shown in Figure 3 with an OG:A-containing substrate is 2.1 nM, which provides a percent active site concentration of 21%, based on the total protein concentration calculated by the Bradford assay. The average active site concentration determined from several preparations of Stop 225 was 23%, which is slightly lower than the active site concentrations routinely obtained with different preparations of wild-type MutY. The active site concentrations were used to ensure that the concentration of active MutY was greater than [DNA] in single-turnover experiments and to correct dissociation constant (Kd) values to allow for comparison with data previously reported with the wild-type enzyme.
Purine glycosylase activity of wild-type and Stop 225 MutY
The X-ray crystal structure of cd-MutY complexed to adenine reveals an extensive array of hydrogen bond contacts between residues in the N-terminal domain of MutY and the adenine base (31). In particular, the exocyclic N6 amino group of the adenine participates in specific hydrogen bonds with both Glu37 and Gln182. This exocyclic N6 amino group is also involved in base pairing to OGsyn in an OGsyn:Aanti mismatch (Fig. 1). For these reasons it seemed likely that removal of the N6 amino group by replacement of A with P in DNA substrates may perturb the recognition and repair properties of MutY. Furthermore, since Stop 225 was found to have a reduced preference for OG:A relative to G:A substrates, we desired to determine if differences in the processing of a P-containing substrate relative to those containing adenine would be observed for Stop 225 in comparison to the wild-type enzyme. Duplexes containing central OG:P and G:P base pairs were analyzed for purine glycosylase activity by wild-type and Stop 225 MutY and the resulting k2 and k3 rate constants were determined (Table 2).
Table 2. Kinetic parameters for wild-type and Stop 225 MutY on OG:P- and G:P-containing duplexes at 37°Ca.
| Substrate | Wild-type MutY | Truncated MutY (Stop 225) | ||
| |
k2 (min–1) |
k3 (min–1) |
k2 (min–1) |
k3 (min–1) |
| OG:P | 4.7 ± 0.5 | 0.003 ± 0.001 | 0.12 ± 0.01 | 0.02 ± 0.01 |
| G:P | 0.6 ± 0.1 | 0.04 ± 0.01 | 0.06 ± 0.02 | 0.04 ± 0.02 |
aAll values are reported as averages of at least four separate experiments; errors are reported as one standard deviation from the average.
The processing of OG:P and G:P substrates by wild-type and Stop 225 MutY exhibited similar kinetic profiles. In particular, under conditions of [MutY] < [DNA] biphasic behavior was observed with both enzymes with the purine opposite OG and G. The k3 values for OG:P- and G:P-containing substrates were quite similar to those measured with the corresponding OG:A and G:A substrates, indicating that modification of the base removed by MutY does not affect enzymatic turnover. This is consistent with the notion that the k3 rate constants are dominated by dissociation from the DNA product. However, measurements of the intrinsic rate of base removal (k2) in the single-turnover experiments revealed a diminished ability of both wild-type and Stop 225 to remove purine relative to adenine opposite OG and G. For both enzymes the decrease was ∼2- to 3-fold for substitution of purine for adenine opposite both OG and G (Tables 1 and 2). The fact that purine can be removed by MutY indicates that contacts with the N6 amino group are not absolutely required for removal of A. Importantly, the relative changes in k3 and k2 values on substitution of purine for adenine were similar for Stop 225 and the wild-type enzyme, suggesting that the C-terminal domain does not participate directly in removal of the adenine.
In vivo activity of Stop 225
MutY prevents G:C→T:A transversion mutations in vivo and therefore it is possible to evaluate the in vivo activity of wild-type MutY and modified MutY enzymes (Table 3). These experiments were performed in an E.coli strain lacking both mutY and mutM (GT100 mutY::mini-Tn10 mutM) (15). The mutation rate was judged by determining the number of colonies able to grow on rifampicin (Rifr). When this E.coli strain is transformed with the pKKYEco plasmid to supply wild-type MutY, a significantly reduced number of colonies are observed (2 ± 1) compared to the corresponding transformation using a plasmid lacking mutY (980 ± 160). However, in the corresponding experiment with Stop 225, significantly more colonies were observed (350 ± 140) compared to the wild-type enzyme. These data indicate that the mutation frequency is decreased at least 500-fold with the wild-type enzyme, but only ∼3-fold with the truncated enzyme. Similar experiments reported recently by Li et al. (34) using an E.coli strain lacking only mutY revealed a decrease in the mutation frequency of 70-fold with the wild-type enzyme and 2-fold with the truncated enzyme. Thus, both studies show that removal of the C-terminal domain significantly reduced the efficiency of suppression of DNA mutations.
Table 3. Mutation frequency of E.coli GT100 mutY::mini-Tn10 mutM.
| |
pKK223–3 |
pKKYEco |
PKKYS225 |
| No. of Rifr colonies per 108 cells | 980 ± 160 | 2 ± 1 | 350 ± 140 |
| Decrease in mutation frequency (n-fold) relative to pKK223-3 | 500 | 3 |
Dissociation constant (Kd) measurements
In order to determine the effect of removal of the C-terminal domain on the recognition properties of MutY, equilibrium dissociation constants (Kd) were determined for Stop 225 with a series of 30 bp duplexes (Table 4). We have previously shown that DNA duplexes containing the 2′-deoxyadenosine analogs FA and F opposite OG and G are effective substrate mimics for wild-type MutY (Fig. 1; 19,20). In addition, wild-type and Stop 225 bind tightly to duplexes containing M (Fig. 1) opposite OG (23). The FA, F and M nucleotides are resistant to the glycosylase action of MutY and therefore remove complications associated with enzymatic turnover during measurements of DNA binding. DNA duplexes containing the abasic site mimic THF opposite G or OG have been previously shown to bind tightly to wild-type MutY as well as serve as general inhibitors of BER glycosylases (43). The Kd values with various DNA duplexes with Stop 225 were measured using a gel retardation assay under conditions similar to those used previously (19,20) and these values were compared to those for the wild-type and Stop 225 enzymes previously reported (Table 4).
Table 4. Equilibrium dissociation constants (Kd) for wild-type and Stop 225 MutY with substrate and product analogsa.
| Central base pair |
Wild-type MutY Kd
(nM) |
Stop 225 MutY Kd
(nM) |
| OG:FA | 0.12 ± 0.02b | 90 ±10b |
| G:FA | 5.8 ± 0.6b | 80 ± 10b |
| OG:F | 0.28 ± 0.02c | 80 ± 30 |
| G:F | 15 ± 3c | 140 ± 50 |
| G:M | 40 ± 6d | 20 ± 10d |
| OG:M | 0.17 ± 0.05d | 1.4 ± 0.3d |
| OG:THF | <0.03 | 0.15 ± 0.08 |
| G:THF | 3 ± 1 | 2 ± 1 |
| G:C | 150 ± 60c | 130 ± 30d |
aAll values are reported as averages of at least four separate experiments; errors are reported as one standard deviation from the average. Measurements were performed at 25°C.
bThese values were previously reported in Chepanoske et al. (20).
cThese values were previously reported in Porello et al. (19).
dThese values were previously reported in Chepanoske et al. (23).
The Kd values for Stop 225 with OG:FA and OG:F duplexes (Kd = 90 ± 10 and 80 ± 30 nM, respectively) were found to be remarkably similar to the Kd value measured for wild-type MutY with a non-specific duplex (Kd = 150 ± 60 nM). A comparison of these values with the corresponding measurements with wild-type MutY (Kd = 0.12 ± 0.02 and 0.28 ± 0.02 nM for the FA and F analogs, respectively) indicates a significantly reduced affinity of Stop 225 for these substrate analogs. Indeed, Stop 225 binds to the FA- and F-containing duplexes with ∼750- and 300-fold reduced affinity, respectively, relative to the wild-type enzyme. In the case of the wild-type enzyme there is significant discrimination between OG- and G-containing substrates; however, with Stop 225 the Kd value for the G:FA duplex (Kd = 80 ± 10 nM) is essentially identical to that for the OG:FA duplex (Kd = 90 ± 10 nM) and ∼14-fold higher than that observed with the wild-type enzyme. Similarly, the Kd values for Stop 225 with both the OG:F- and G:F-containing duplexes are within experimental error. The severely reduced affinity of Stop 225 for the substrate analog duplexes containing FA and F does not appear to be related to a decrease in non-specific DNA binding since the Kd value for Stop 225 with a non-mismatch-containing duplex (Kd = 130 ± 30 nM) is similar to that observed with the wild-type enzyme (Kd = 150 ± 60 nM) (19).
We have previously shown that Stop 225 binds much more tightly to duplexes containing the M analog than the FA analog (23). The M analog is incapable of hydrogen bonding to the base opposite and, therefore, we hypothesized that the improved recognition of this analog by Stop 225 may be due to the more facile extrusion of this analog from the DNA helix. Consistent with this idea, Stop 225 was found to exhibit relatively high affinity for the product mimic OG:THF duplex (Kd = 0.15 ± 0.08 nM). Moreover, Kd values for Stop 225 and the wild-type enzyme with the G:THF duplex were essentially identical (Table 4).
MPE-Fe(II) footprinting
We have previously shown that duplexes containing a central OG:F or OG:FA base pair are excellent templates for MPE-Fe(II) hydroxyl radical footprinting with wild-type MutY (19,20). The small difference in Kd values of Stop 225 for F- and FA-containing duplexes relative to a non-specific duplex suggested that footprinting experiments with the substrate analogs would be difficult. Indeed, we were unable to identify conditions for Stop 225 under which we could observe protection from hydroxyl radical cleavage in the region surrounding the OG:F/FA base pair without also observing protection of the entire DNA duplex. However, the affinity of Stop 225 for the OG:M and OG:THF duplexes was significantly higher than for the non-specific duplex and therefore hydroxyl radical footprinting experiments were performed with these duplexes. A representative storage phosphor autoradiogram of hydroxyl radical footprinting using MPE-Fe(II) footprinting with Stop 225 and wild-type MutY bound to an OG:THF duplex is shown in Figure 4. Quantitation of autoradiograms from at least three separate experiments for both the OG:THF- and OG:M-containing duplexes is shown in Figure 5.
Figure 4.
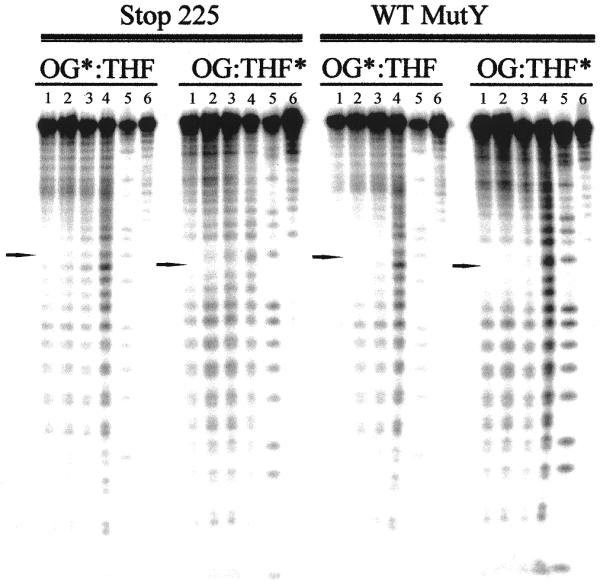
Storage phosphor autoradiogram of hydroxyl radical footprinting using MPE-Fe(II) of Stop 225 and wild-type MutY with an OG:THF-containing DNA duplex. Lanes 1–3, DNA, MPE-Fe(II) and enzyme at 100, 200 and 300 (wild-type) or 60, 120 and 180 nM (Stop 225), respectively; lane 4, DNA and MPE-Fe(II); lane 5, Maxam–Gilbert G+A reaction; lane 6, DNA only. The 32P-end-labeled strand is indicated with an asterisk (*). The location of the OG or THF is indicated by an arrow. It should be noted that the active concentration of Stop 225 used in this experiment was less than that of wild-type MutY and therefore a lesser degree of protection was observed.
Figure 5.

Histograms of hydroxyl radical and DMS footprinting experiments of Stop 225 and wild-type MutY with OG:M- and OG:THF-containing DNA duplexes. The duplex sequences containing either OG:M (A and B) and OG:THF (C and D) are shown with a histogram indicating the relative protection from hydroxyl radical cleavage for wild-type MutY (A and C) and Stop 225 MutY (B and D). The G residue shown to be hyper-reactive to DMS footprinting is shown in red and underlined. The data were obtained by quantitation of the storage phosphor autoradiograms from at least three separate experiments. The heights of the bars indicate the relative protection from hydroxyl radical cleavage. Both wild-type and Stop 225 MutY produce larger extents of protection of the OG:THF duplex than the OG:M duplex, therefore, the heights of the bars for the OG:THF duplex have been decreased by a factor of two. The observed protection patterns are strikingly similar for Stop 225 compared to the wild-type.
The results of the MPE-Fe(II) footprinting experiments with the OG:M duplex revealed a similar pattern of protection from hydroxyl radical cleavage surrounding the OG:M base pair with both wild-type and Stop 225 MutY (Fig. 5) as observed previously with wild-type MutY bound to OG:F (19) and OG:FA duplexes (20). With both enzymes high degrees of protection were observed for two nucleotides on the 5′-side and five nucleotides on the 3′-side of the M analog. The protection offered by wild-type and Stop 225 MutY on the OG-containing strand was also similar: three nucleotides on the 5′-side and four nucleotides on the 3′-side of the OG were protected. An interesting difference in the footprinting experiments between the two enzyme forms was the greater degree of protection at the OG residue provided by the wild-type enzyme. Indeed, wild-type MutY provides 4-fold greater protection of the OG nucleotide than Stop 225. This difference may be due to a lower affinity of Stop 225 for the mismatch and lends additional support for an OG recognition role for the C-terminal domain of MutY.
MPE-Fe(II) footprinting experiments on the OG:THF duplex provided an opportunity to compare the footprinting patterns of wild-type and Stop 225 MutY on a product-like duplex (Figs 4 and 5). As with the OG:M duplex, the observed patterns of protection of the OG:THF duplex from hydroxyl radical cleavage by both enzyme forms were similar. In both cases the THF nucleotide was strongly protected from cleavage. Appreciable degrees of protection were observed for three nucleotides on the 5′-side of the THF nucleotide with both enzyme forms; however, on the 3′-side of the THF three nucleotides were significantly protected with Stop 225 while four nucleotides were similarly protected with the wild-type enzyme. Overall, similar trends were observed on the OG-containing strand. The OG nucleotide was the most strongly protected position, while a lesser degree of protection was observed for three nucleotides on the 5′-side of the OG with both enzymes. On the 3′-side of the OG five nucleotides were protected with Stop 225 and six nucleotides were protected with the wild-type enzyme. The slight differences between the two enzyme forms may be attributed to the lower affinity of Stop 225 for the OG:THF duplex.
The differences in footprinting between wild-type and Stop 225 MutY are less obvious than the differences in the patterns observed for substrate analog (OG:M) versus product analog (OG:THF) duplexes (Fig. 5). Indeed, the shape and symmetry of the two footprinting patterns are distinct. With both enzyme forms more nucleotides are protected on the M strand compared to the THF strand and the degrees of protection of each nucleotide differ significantly between the two strands in the two duplexes. For example, the THF nucleotide is significantly more protected than the M nucleotide relative to protection of the surrounding nucleotides. Slight differences also exist in the footprinting patterns on the OG-containing strand of the OG:M- and OG:THF-containing duplexes. For example, both enzymes provide more protection of nucleotides on both sides of the OG in the OG:THF duplex, while more significant protection is observed on the 5′-side of the OG in the OG:M duplex. The observed footprinting pattern with the OG:THF duplex was also distinctly different from that previously observed with an OG:F- or OG:FA-containing duplex (19,20). These results indicate that the interaction of MutY is different with the product analog duplex compared to the substrate analog duplexes.
DMS footprinting
We have previously used DMS footprinting to provide additional information pertaining to the interaction of wild-type MutY with substrate analog duplexes (19,20). Results with wild-type MutY bound to an OG:FA-containing duplex (20) revealed two guanines adjacent to the FA that were hyper-reactive to the DMS reagent compared to the duplex alone, while there was a guanine residue on the OG-containing strand that was protected from reaction compared to the free duplex. The presence of hypersensitive guanine residues was consistent with a model wherein binding of MutY to its target mismatch results in distortion of the DNA duplex.
In order to further characterize the interaction of Stop 225 with the OG:M- and OG:THF-containing duplexes relative to the wild-type enzyme, DMS footprinting experiments were performed in a similar fashion as with the OG:F and OG:FA duplexes. Surprisingly, wild-type and Stop 225 MutY did not enhance the DMS reactivity of G bases proximal to the M of the OG:M duplex as had been observed with the OG:F and OG:FA duplexes. However, significant hyper-reactivity of the G residue located on the 3′-side of the THF in the OG:THF-containing duplex was observed (Fig. 5). The hyper-reactive guanine is ∼2-fold more reactive in the presence of both wild-type and Stop 225 MutY than in the control lane. This result also indicates that there are no significant differences in the conformational properties of the product DNA when bound by either full-length or truncated MutY.
DISCUSSION
A detailed investigation of the functional properties of a truncated form of MutY has been performed in order to determine the effects of removal of the C-terminal domain on MutY substrate processing. Our results indicate that the C-terminal domain plays an important role in recognition and repair of damaged and mismatched DNA by MutY. Specifically, we found that truncation dramatically reduced the intrinsic rate of adenine removal from both OG:A and G:A substrates. Indeed, the truncated form exhibits a 10-fold slower rate with a G:A-containing substrate and at least a 25-fold slower rate with an OG:A-containing substrate. In contrast, the Stop 225 enzyme exhibits a 2-fold increased ability to turnover an OG:A substrate, as judged by the increased k3 value. The slower rate of the glycosylase reaction and increased turn over ability of Stop 225 with OG:A substrates may be due to a loss of OG-specific DNA binding affinity for both the substrate and the product. Importantly, there is also a reduced ability of the Stop 225 enzyme to prevent DNA mutations in vivo. This suggests that the reduction in the rate of adenine removal in vitro translates into a reduced ability of Stop 225 to prevent DNA mutations in vivo. Thus, though the truncated enzyme retains the ability to recognize and remove adenine from OG:A and G:A mispairs, this activity is dramatically affected by absence of the C-terminal domain.
The magnitude of the reduction in the k2 rate constants for Stop 225 relative to the wild-type enzyme for both substrates observed herein are completely consistent with previous measurements by Noll et al. (33). The inconsistencies between our results and those reported by Li et al. (34) are likely due to errors associated with determining rate constants of reactions that have not proceeded to completion. Under single-turnover conditions, reactions would be expected to approach completion and failure to do so is indicative of incomplete DNA duplex formation or low active enzyme concentrations. We have previously discussed in detail how such analyses can be misleading with the wild-type enzyme (24).
The observed trends in Kd values of Stop 225 with substrate and product analogs are consistent with a decrease in binding affinity of the truncated form for both the substrate and product. However, Stop 225 and wild-type MutY have similar affinities for a non-specific DNA duplex. The most dramatic decrease in binding affinity for Stop 225 relative to the wild-type enzyme is observed with the substrate analogs F and FA. Indeed, the observed Kd values with these two substrate analogs are close to that for a duplex lacking a mismatch. However, Stop 225 does exhibit relatively high affinity for the hydrophobic substrate analog M, which is incapable of hydrogen bonding to OG. This suggests that the absence of hydrogen bonding between OG and the A analog M may eliminate the need for the C-terminal domain. In a similar vein, the absence of the C-terminal domain does not dramatically affect product binding, as evidenced by the relatively small decrease in affinity of Stop 225 for the OG:THF duplex compared to the wild-type enzyme. Furthermore, Stop 225 and wild-type MutY bind with essentially the same affinity to the G:THF duplex.
The steady-state turnover rates (k3) for Stop 225 with OG:A and G:A substrates indicate a two-fold increase for Stop 225 with OG:A substrates relative to the wild-type enzyme. We have previously interpreted the slow steady-state turnover to be a consequence of slow product release (24). This suggests that removal of the C-terminal domain slightly decreases affinity for the OG:(AP site) product. In addition, k3 values for the purine substrates mirror those obtained with adenine, indicating that the nature of the base removed does not affect turnover (k3). Recently, Noll et al. (33) reported direct measurements of the dissociation rate of wild-type and cd-MutY from the product (kdiss) using a gel retardation method. Based on our proposed kinetic scheme for MutY (Scheme 1), we would expect that the k3 values from multiple turnover kinetics experiments would be the same as kdiss values. Using k3 for the wild-type enzyme with OG:A, the calculated half-life of 2.9 h is remarkably similar to the half-life of the MutY–OG:(AP site) DNA complex of 2.6 h determined by Zharkov and Grollman (44) using sodium borohydride trapping experiments. In addition, the kdiss value for dissociation of the wild-type enzyme from the OG:(AP site) product (33; S.L.Porello and S.S.David, unpublished results) and the k3 value are quite similar, consistent with DNA product release as the rate limiting step. However, the k3 values for Stop 225 with OG:A and G:A substrates reported herein, as well as the k3 value for the wild-type enzyme with a G:A duplex reported previously, are considerably smaller than the kdiss values determined with the corresponding AP site products by gel retardation methods (33). These differences may be a result of the different conditions and methods used. In particular, others have noted that there are potential errors associated with the gel retardation method for measuring dissociation rates (45). Interestingly, the relative trends in k3 are consistent with the relative changes in Kd of the product mimic THF-containing duplexes. For example, similar Kd values for Stop 225 and wild-type MutY with the G:THF duplex were observed, consistent with the similar k3 values. It is also possible that differences between k3 and kdiss may be due to differences in rate-limiting steps and, therefore, experiments to address this issue in more detail are currently underway.
The kinetic data and the Kd measurements show that the C-terminal domain of MutY is involved in specific recognition of a DNA mismatch. The dramatic decrease in k2 for Stop 225 with both G:A and OG:A substrates and the reduced affinity of Stop 225 for the substrate analogs suggest that the C-terminal domain may be involved in orienting or positioning the N-terminal portion of MutY for efficient recognition of the mismatch and catalysis. Moreover, there may be a conformational change that is required subsequent to the initial DNA binding step which is important for orienting MutY/Stop 225 onto its substrate. Recall that our analysis is based on a simplistic mechanistic scenario (Scheme 1) where k2 represents the overall process of conversion MutY*(DNA)s→MutY*(DNA)p. This process may contain more than a single step and therefore the k2 value may be composed of more than one rate constant (24). Indeed, there may be a conformational step occurring after initial binding of MutY to the substrate which is required for efficient hydrolysis of the N-glycosyl bond of 2′-deoxyadenosine.
A similar kinetic scheme was proposed for uracil-DNA glycosylase based on fluoresence experiments with substrates and substrate analogs (46). These experiments provided evidence for an enzyme-assisted mechanism for uracil flipping in which local destabilization of the DNA facilitates extrusion of the uracil. A requirement for a conformational change may also explain the relatively weak binding of Stop 225 with the substrate analogs F and FA. Tight binding of MutY to these substrate analogs may require a conformational change and this may be reflected in the relative Kd values. In contrast, Stop 225 exhibits a higher affinity for the non-hydrogen bonding analog M than F or FA. Since the M analog is incapable of hydrogen bonding, the improved affinity of Stop 225 for OG:M duplexes may be due to the ease of a conformational change in the DNA, such as nucleotide flipping, provided with this analog.
Nucleotide flipping appears to be a common feature of enzymes involved in BER (47,48). A nucleotide flipping mechanism has been proposed for MutY based on crystallographic characterization of the N-terminal domain of MutY in the presence of adenine (31). In addition, Noll et al. (33) proposed that the C-terminal domain is directly involved in OG-specific contacts based on the sequence similarity of this domain to the d(OG)TP hydrolase MutT. This idea is further supported by NMR studies of the C-terminal domain (35) that show striking similarities to the solution structure of MutT. Thus, on the basis of these results it has been suggested that MutY may use a ‘double-flip’ mechanism and contain both OG- and A-specific binding pockets (35). This idea is also consistent with previous work from our laboratory using substrate analogs to evaluate substrate recognition that showed that the affinity of the wild-type enzyme is sensitive to modification of both the OG and A of the mismatch (19). This is also illustrated by the rate constant measurements of the glycosylase activity of MutY; replacement of A with P and OG with G results in a reduced rate for the glycosylase reaction.
Previously, Noll et al. (33) observed that removal of the C-terminal domain produced a decrease in substrate specificity for OG:A relative to G:A. Additional evidence for the role of the C-terminal domain in OG recognition is provided herein. Specifically, this is reflected in the ratio k2(OG:A):k2(G:A), which is at least 2-fold greater for wild-type MutY compared to Stop 225. In addition, the Kd measurements showed that wild-type MutY discriminates more strongly between OG and G duplexes than Stop 225. For example, wild-type MutY binds ∼50-fold tighter to an OG:FA duplex than to the corresponding G:FA duplex, while Stop 225 binds these duplexes with similar affinities. The slightly reduced magnitude of protection offered by Stop 225 from hydroxyl radical cleavage in the MPE-Fe(II) footprinting experiments is also consistent with a reduced affinity of Stop 225 for OG-containing duplexes.
X-ray structural analyses of both E.coli endo III (49) and the N-terminal domain of MutY (31) revealed a similar overall structure for both proteins. Notably, the conserved regions include the helix–hairpin–helix and the [4Fe-4S]2+ cluster loop motifs, which have been implicated in DNA binding (26–28,36,49). Interestingly, endo III, which does not contain an analogous C-terminal domain, catalyzes removal of a distinctly different type of DNA damage. Substrates for endo III such as thymine glycol may be more easily promoted into an extra-helical conformation. Indeed, NMR studies of a thymine glycol-containing DNA duplex indicate that the presence of thymine glycol induces significant and highly localized structural alterations in the duplex, such that the thymine glycol is predominantly extra-helical (50). In contrast, OG:A base pairs have been shown to exhibit stabilities rivaling their Watson–Crick counterparts (51). Promoting an extra-helical conformation of the target A nucleotide from an OG:A base pair may require considerable protein–DNA interactions, thus requiring the presence of the C-terminal domain. In addition, the larger magnitude decrease in k2 for Stop 225 relative to the wild-type enzyme with OG:A substrates over G:A substrates may be related to the higher stability of DNA duplexes containing an OG:A mismatch (51). A role of the C-terminal domain in disrupting the base pair is also consistent with the higher relative affinity of the wild-type enzyme compared to Stop 225 for the duplexes containing the substrate analogs F and FA over the non-hydrogen bonding substrate analog M and product analog THF.
It is also possible that the C-terminal domain may facilitate recognition of OG indirectly or that interactions with OG may occur transiently as a prelude to nucleotide flipping. Determination of the theoretical pI indicates that the N-terminal domain is much more positively charged under physiological conditions (pI = 9.4) than the C-terminal domain (pI = 5.2) (52,53). This suggests that the C-terminal domain may not be directly involved in interacting with DNA, but rather may play a more subtle and indirect role in damage recognition by MutY. Indeed, the C-terminal domain could promote protein conformational changes that are needed to facilitate nucleotide flipping. This idea is also supported by the similarity in the footprinting patterns between wild-type and Stop 225 MutY observed with the OG:M and OG:THF duplexes. We have recently found that Lys142 of MutY can be cross-linked to OG of the OG:(AP site) product by oxidation of OG with Na2IrCl6 (54). This suggests that this residue, located in the N-terminal domain of MutY, is in close proximity to OG, however, this result does not preclude additional contacts of the OG with residues within the C-terminal domain. Studies to directly address this issue are presently in progress.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Mark Michaels for generously providing the E.coli strains used in this work. We also thank Dr Silvia Porello for her help and advice on performing kinetic experiments with MutY. This work was supported by the National Institutes of Health (CA 67985), the Huntsman Cancer Institute, the University of Utah and the Department of Chemistry of the University of Utah. S.S.D. is an A.P. Sloan Research Fellow (1998–2000). DNA sequencing was performed by the DNA sequencing facility at the Huntsman Cancer Institute, University of Utah, which is supported in part by NCI grant 5P30CA43014.
References
- 1.Cadet J., Berger,M., Douki,T. and Ravanat,J.L. (1997) Oxidative damage to DNA: formation, measurement and biological significance. Rev. Physiol. Biochem. Pharmacol., 131, 1–87. [DOI] [PubMed] [Google Scholar]
- 2.Henle E.S. and Linn,S. (1997) Formation, prevention and repair of DNA damage by iron/hydrogen peroxide. J. Biol. Chem., 272, 19095–19098. [DOI] [PubMed] [Google Scholar]
- 3.Ames B.N. and Gold,L.S. (1991) Endogenous mutagens and the causes of aging and cancer. Mutat. Res., 250, 121. [DOI] [PubMed] [Google Scholar]
- 4.Gowen L.C., Avrutsky,A.V., Latour,A.M., Koller,B.H. and Leadon,S.A. (1998) BRCA1 required for transcription-coupled repair of oxidative DNA damage. Science, 281, 1009. [DOI] [PubMed] [Google Scholar]
- 5.Lu R., Nash,H.M. and Verdine,G.L. (1997) A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr. Biol., 7, 397–407. [DOI] [PubMed] [Google Scholar]
- 6.Shingenaga M.K., Park,J.W., Cundy,K.C., Gimeno,C.J. and Ames,B.N. (1990) 8-Hydroxy-2′-deoxyguanosine as a biological marker of in vivo oxidative damage. Methods Enzymol., 186, 521–530. [DOI] [PubMed] [Google Scholar]
- 7.Cadet J. (1999) Hydroxy radicals and DNA base damage. Mutat. Res., 424, 9–21. [DOI] [PubMed] [Google Scholar]
- 8.Beckman K.B. and Ames,K.B. (1997) Oxidative decay of DNA. J. Biol. Chem., 272, 19633–19636. [DOI] [PubMed] [Google Scholar]
- 9.Wood M.L., Esteve,A., Morningstar,M.L., Kuziemko,G.M. and Essigmann,J.M. (1992) Genetic effects of oxidative DNA damage—comparative mutagenesis of 7,8-dihydro-8-oxoguanine and 7,8-dihydro-8-oxoadenine in E.coli. Nucleic Acids Res., 20, 6023–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Insertion of specific bases during DNA synthesis past the oxidation damaged base 8-oxodG. Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 11.David S.S. and Williams,S.D. (1998) Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev., 98, 1221–1261. [DOI] [PubMed] [Google Scholar]
- 12.Michaels M.L., Tchou,J., Grollman,A.P. and Miller,J.H. (1992) A repair system for 8-oxo-7,8-dihydrodeoxyguanosine. Biochemistry, 31, 10964–10968. [DOI] [PubMed] [Google Scholar]
- 13.Grollman A.P. and Moriya,M. (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends. Genet., 9, 246–249. [DOI] [PubMed] [Google Scholar]
- 14.Maki H. and Sekiguchi,M. (1992) MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature, 355, 273–275. [DOI] [PubMed] [Google Scholar]
- 15.Michaels M.L., Cruz,C., Grollman,A.P. and Miller,J.H. (1992) Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl Acad. Sci. USA, 89, 7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Au K.G., Clark,S., Miller,J.H. and Modrich,P. (1989) Escherichia coli mutY gene encodes an adenine glycosylase active on G-A mispairs. Proc. Natl Acad. Sci. USA, 86, 8877–8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Radicella J.P., Clark,E.A. and Fox,M.S. (1988) Some mismatch repair activities in Escherichia coli. Proc. Natl Acad. Sci. USA, 85, 9674–9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu A.-L., Tsai-Wu,J.-J. and Cillo,J. (1995) DNA determinants and substrate specificities of Escherichia coli MutY. J. Biol. Chem., 270, 23582–23588. [DOI] [PubMed] [Google Scholar]
- 19.Porello S.L., Williams,S.D., Kuhn,H., Michaels,M.L. and David,S.S. (1996) Recognition of substrate analogs by the DNA mismatch repair enzyme MutY. J. Am. Chem. Soc., 118, 10684–10692. [Google Scholar]
- 20.Chepanoske C.L., Porello,S.L., Fujiwara,T., Sugiyama,H. and David,S.S. (1999) Substrate recognition by Escherichia coli MutY using substrate analogs. Nucleic Acids Res., 27, 3197–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng L., Schärer,O.D. and Verdine,G.L. (1997) Unusually strong binding of a designed transition-state analog to a base-excision DNA repair protein. J. Am. Chem. Soc., 119, 7865–7866. [Google Scholar]
- 22.Bulychev N.V., Varaprasad,C.V., Dorman,G., Miller,J.H., Eisenberg,M., Grollman,A.P. and Johnson,F. (1996) Substrate specificity of Escherichia coli MutY protein. Biochemistry, 35, 13147–13156. [DOI] [PubMed] [Google Scholar]
- 23.Chepanoske C.L., Langelier,C.R., Chmiel,N.H. and David,S.S. (2000) Recognition of the nonpolar base 4-methylindole in DNA by the DNA repair adenine glycosylase MutY. Org. Lett., 2, 1341–1344. [DOI] [PubMed] [Google Scholar]
- 24.Porello S.L., Leyes,A.E. and David,S.S. (1998) Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry, 37, 14756–14764. [DOI] [PubMed] [Google Scholar]
- 25.Michaels M.L., Pham,L., Nghiem,Y., Cruz,C. and Miller,J.H. (1990) MutY, an adenine glycosylase active on G-A mispairs, has homology to endonuclease III. Nucleic Acids Res., 18, 3841–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katcher H.L. and Wallace,S.S. (1983) Characterization of the Escherichia coli X-ray endonuclease, endonuclease III. Biochemistry, 22, 4072–4081. [DOI] [PubMed] [Google Scholar]
- 27.Hatahet Z., Kow,Y.W., Purmal,A., Cunningham,R.P. and Wallace,S.S. (1994) New substrates for old enzymes. J. Biol. Chem., 269, 18814–18820. [PubMed] [Google Scholar]
- 28.Breimer L.H. and Lindahl,T. (1984) DNA glycosylase activities for thymine residues damaged by ring saturation, fragmentation or ring contraction are functions of endonuclease III in Escherichia coli. J. Biol. Chem., 259, 5543–5548. [PubMed] [Google Scholar]
- 29.Manuel R.C., Czerwinski,E.W. and Lloyd,R.S. (1996) Identification of the structural and functional domains of MutY, an Escherichia coli DNA mismatch repair enzyme. J. Biol. Chem., 271, 16218–16226. [DOI] [PubMed] [Google Scholar]
- 30.Gogos A., Cillo,J., Clarke,N.D. and Lu,A.-L. (1996) Specific recognition of A/G and A/7,8-dihydro-8-oxoguanine (8-oxoG) mismatches by Escherichia coli MutY: removal of the C-terminal domain preferentially affects A/8-oxo-G recognition. Biochemistry, 35, 16665–16671. [DOI] [PubMed] [Google Scholar]
- 31.Guan Y., Manuel,R.C., Arvai,A.S., Parikh,S.S., Mol,C.D., Miller,J.H., Lloyd,R.S. and Tainer,J.A. (1998) MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nature Struct. Biol., 5, 1058–1064. [DOI] [PubMed] [Google Scholar]
- 32.Manuel R.C. and Lloyd,R.S. (1997) Cloning, overexpression and biochemical characterization of the catalytic domain of MutY. Biochemistry, 36, 11140–11152. [DOI] [PubMed] [Google Scholar]
- 33.Noll D.M., Gogos,A., Granek,J.A. and Clarke,N.D. (1999) The C-terminal domain of the adenine-DNA glycosylase MutY confers specificity of 8-oxoguanine-adenine mispairs and may have evolved from MutT, an 8-oxo-dGTPase. Biochemistry, 38, 6374–6379. [DOI] [PubMed] [Google Scholar]
- 34.Li X., Wright,P.M. and Lu,A.-L. (2000) The C-terminal domain of MutY glycosylase determines the 7,8-dihydro-8-oxo-guanine specificity and is crucial for mutation avoidance. J. Biol. Chem., 275, 8448–8455. [DOI] [PubMed] [Google Scholar]
- 35.Volk D.E., House,P.G., Thiviyanathan,V., Luxon,B.A., Zhang,S., Lloyd,R.S. and Gorenstein,D.G. (2000) Structural similarities between MutT and the C-Terminal domain of MutY. Biochemistry, 39, 7331–7336. [DOI] [PubMed] [Google Scholar]
- 36.Chepanoske C.L., Golinelli,M.-P., Williams,S.D. and David,S.S. (2000) Positively charged residues within the iron-sulfur cluster loop of E. coli MutY participate in damage recogniton and removal. Arch. Biochem. Biophys., 380, 11–19. [DOI] [PubMed] [Google Scholar]
- 37.Williams S.D. and David,S.S. (2000) A single engineered point mutation in the adenine glycosylase MutY confers bifunctional glycosylase/AP lyase activity. Biochemistry, 39, 10098–10109. [DOI] [PubMed] [Google Scholar]
- 38.Miller J.H. (1992) A Short Course in Bacterial Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 39.Cupples C.G. and Miller,J.H. (1989) A set of LacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc. Natl Acad. Sci. USA, 86, 5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Golinelli M.-P., Chmiel,N.H. and David,S.S. (1999) Site-directed mutagenesis of the cysteine ligands to the [4Fe-4S] cluster of Escherichia coli MutY. Biochemistry, 38, 6997–7007. [DOI] [PubMed] [Google Scholar]
- 41.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 42.Carey J. (1991) Gel retardation. Methods Enzymol., 208, 103–117. [DOI] [PubMed] [Google Scholar]
- 43.Schärer O.D., Nash,H.M., Jiricny,J., Laval,J. and Verdine,G.L. (1998) Specific binding of a designed pyrrolidine abasic site analog to mulitple DNA glycosylases. J. Biol. Chem., 273, 8592–8597. [DOI] [PubMed] [Google Scholar]
- 44.Zharkov D.O. and Grollman,A.P. (1998) MutY DNA glycosylase: base release and intermediate complex formation. Biochemistry, 37, 12384–12394. [DOI] [PubMed] [Google Scholar]
- 45.Gerstle J.T. and Fried,M.G. (1993) Measurements of binding kinetics using the gel electrophoresis mobility shift assay. Electrophoresis, 14, 725–731. [DOI] [PubMed] [Google Scholar]
- 46.Stivers J.T., Pankiewicz,K.W. and Watanabe,K.A. (1999) Kinetic mechanism of damage site recognition and uracil flipping by Escherichia coli uracil DNA glycosylase. Biochemistry, 38, 952–963. [DOI] [PubMed] [Google Scholar]
- 47.Savva R., McAuley-Hecht,K.E., Brown,T. and Pearl,L. (1995) The structural basis of specific base-excision repair by uracil-DNA glycosylase. Nature, 373, 487–493. [DOI] [PubMed] [Google Scholar]
- 48.Mol C.D., Arvai,A.S., Slupphaug,G., Kavli,B., Alseth,I., Krokan,H.E. and Tainer,J.A. (1995) Crystal structure and mutational analysis of human uracil-DNA glycosylase: structural basis for specificity and catalysis. Cell, 80, 1–20. [DOI] [PubMed] [Google Scholar]
- 49.Thayer M.M., Ahern,H., Xing,D., Cunningham,R.P. and Tainer,J.A. (1995) Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J., 14, 4108–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kung H.C. and. Bolton.,P.H. (1997) Structure of a duplex DNA containing a thymine glycol residue in solution. J. Biol. Chem., 272, 9227–9236. [DOI] [PubMed] [Google Scholar]
- 51.McAuley-Hecht K.E., Leonard,G.A., Gibson,N.J., Thomson,J.B., Watson,W.P., Hunter,W.N. and Brown,T. (1994) Crystal structure of a DNA duplex containing 8-hydroxydeoxyguanine-adenine base pairs. Biochemistry, 33, 10266–10270. [DOI] [PubMed] [Google Scholar]
- 52.Bjellqvist B., Basse,B., Olsen,E. and Celis,J.E. (1994) Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis, 15, 529–539. [DOI] [PubMed] [Google Scholar]
- 53.Wilkins M.R., Gasteiger,E., Bairoch,A., Sanchez,J.-C., Williams,K.L., Appel,R.D. and Hochstrasser,D.F. (1998) Protein identification and analysis tools in the ExPASy Server. In Link,A.J. (ed.), 2-D Proteome Analysis Protocols. Humana Press, NJ. [DOI] [PubMed]
- 54.Hickerson R.P., Chepanoske,C.L., Williams,S.D., David,S.S. and Burrows,C.J. (1999) Mechanism-based DNA-protein cross-linking of MutY via oxidation of 8-oxoguanosine. J. Am. Chem. Soc., 121, 9901–9902. [Google Scholar]
- 55.Kouchakdijian M., Bodepudi,V., Shibutani,S., Eisenberg,M., Johnson,F., Grollman,A.P. and Patel,D.J. (1991) NMR structural studies of the ionizing radiation adduct 7-hydro-8-oxodeoxyguanosine (8-oxo-7H-dG) opposite deoxyadenosine in a DNA duplex. 8-Oxo-7H-dG(syn)-dA(anti) alignment at lesion site. Biochemistry, 30, 1403–1412. [DOI] [PubMed] [Google Scholar]